

Drosophila Arfaptin is a novel dynactin complex–associated protein identified in a screen for proteins that specify dynactin complex function in motor neurons. Arfaptin mediates the binding of the dynactin complex to Golgi membranes and specifies normal dynactin complex function during synapse growth independent of axonal transport.
Abstract
Mutations in DCTN1, a component of the dynactin complex, are linked to neurodegenerative diseases characterized by a broad collection of neuropathologies. Because of the pleiotropic nature of dynactin complex function within the neuron, defining the causes of neuropathology in DCTN1 mutants has been difficult. We combined a genetic screen with cellular assays of dynactin complex function to identify genes that are critical for dynactin complex function in the nervous system. This approach identified the Drosophila homologue of Arfaptin, a multifunctional protein that has been implicated in membrane trafficking. We find that Arfaptin and the Drosophila DCTN1 homologue, Glued, function in the same pathway during synapse growth but not during axonal transport or synapse stabilization. Arfaptin physically associates with Glued and other dynactin complex components in the nervous system of both flies and mice and colocalizes with Glued at the Golgi in motor neurons. Mechanistically, membrane binding by Arfaptin mediates membrane association of the dynactin complex in motor neurons and is required for normal synapse growth. Arfaptin represents a novel dynactin complex–binding protein that specifies dynactin complex function during synapse growth.
INTRODUCTION
Dynamic cellular processes, such as membrane trafficking, are predicted to use the precise spatial and temporal regulation of a vast array of proteins, including motor proteins such as kinesin and dynein. The mechanisms underlying the precise regulation of motor protein function in neurons are incompletely described. One strategy used to specify motor function is illustrated by the kinesins, which constitute a large and diverse family of proteins that have unique motor properties (Hirokawa et al., 2009). Thus the expression of different sets of kinesin genes can endow the cell with a unique collection of kinesin motor functions. For dynein, the diversity of dynein family members is less, and the specification of function within cells, including neurons, appears to use the binding of a diverse group of proteins to the dynein motor complex that serve to specify dynein function. A critical protein complex known to be involved in the specification of dynein function within cells and neurons is the dynactin complex. An emerging model is that the regulation of dynein function within the cell is achieved by the binding of a diverse set of adaptor proteins to the dynactin complex and that these individual adaptor proteins provide the specificity of dynein function (Kardon and Vale, 2009). This predicts that there exist adaptor proteins that specify dynactin complex function in neurons during established dynactin-dependent processes such as synapse growth and axonal transport.
In humans, mutations in the DCTN1 gene, a component of the dynactin complex, preferentially affect the nervous system resulting in a spectrum of adult-onset neurodegenerative diseases, including spinal and bulbar muscular atrophy, frontotemporal dementia, and Perry syndrome (Hafezparast et al., 2003; Münch et al., 2004, 2005; Puls et al., 2005; Farrer et al., 2009). Studies of the nervous system in dynactin complex mutants in mice and flies revealed a number of common neuronal pathologies, including reduced synapse growth, decreased synaptic stability, and impaired axonal transport (Eaton et al., 2002; LaMonte et al., 2002; Chevalier-Larsen et al., 2008; Laird et al., 2008). One interpretation of these results is that these neural pathologies are a secondary consequence of a primary etiology such as impaired axonal transport (Chevalier-Larsen and Holzbaur, 2006; Duncan and Goldstein, 2006). A number of dynactin-associated proteins have been implicated to have important roles in axonal transport (Engelender et al., 1997; Bowman et al., 1999; Liu et al., 2003; Magnani et al., 2007; Zhang et al., 2009). An alternative interpretation is that the neuropathologies observed in dynactin complex mutants reflect the diverse set of cellular processes within the neuron supported by dynein function and that neurodegeneration is the result of a combined effect of these distinct pathologies. In support of this model, data from a DCTN1-mutant mouse model suggest that the effects of dynactin complex mutations on synaptic defects are independent of the effects on axonal transport (Chevalier-Larsen et al., 2008). Only a few candidate molecules have been identified that specify dynactin complex function in neurons independent of axonal transport.
Here we present the results of a forward genetic enhancer/suppressor screen designed to identify genes that function to specify dynactin complex function within the motor neuron during development. From a collection of >3600 individual P-element insertions we isolated 15 insertions that had significant effects on specific cellular phenotypes observed in dynactin complex mutants. Thus our screen data support a model in which neurodegeneration in dynactin complex mutants are the result of a combination of genetically distinct neuronal processes. To extend these genetic studies, we characterized the Drosophila homologue of Arfaptin2 (arfaptin), which was identified in our screen as an enhancer of specific synaptic growth phenotypes. Arfaptin2 is a multifunctional protein that can bind small GTPases and contains a BAR domain that is capable of inducing membrane curvature, leading to the suggestion that this protein functions during membrane trafficking (Tarricone et al., 2001; Peter et al., 2004; Habermann et al., 2004). A role in membrane trafficking is supported by the findings that Arfaptin2 can bind to both Arf and Arf-like small GTPases known to be involved in vesicle formation (Man et al., 2011; Van Valkenburgh et al., 2001). In addition, recent data demonstrated that Arfaptin2 is localized to the trans-Golgi network (TGN) in HeLa cells, supporting a role during Golgi vesicle biogenesis (Man et al., 2011). Finally, Arfaptin2 also binds the Rho-family small GTPase Rac1, suggesting that Arfaptin2 could function to coordinate membrane trafficking and cytoskeletal reorganization during vesicle formation (Shin and Exton, 2001). The association of Arfaptin2 with any motor protein has not been previously demonstrated.
We find that Drosophila arfaptin (arfip) is required within motor neurons for dynactin-dependent synapse growth but not synapse stability or axonal transport. In addition to the genetic interactions observed between arfip and glued during synapse growth, we find that Arfaptin (Arfip) also biochemically interacts with dynactin complexes in flies and mice demonstrating a conserved biochemical interaction. Mechanistically, Arfip is a membrane-binding protein localized at the Golgi that is required for binding of the dynactin complex to membranes in the motor neuron. This membrane-binding property of Arfip is required for normal synapse growth, supporting a role for Arfip and the dynactin complex at the Golgi for normal motor neuron development and function.
RESULTS
Enhancer/suppressor screen for modifiers of dynactin complex function in motor neurons
A genetic screen of 3663 P-element insertion lines was performed to identify P-element insertions that specify dynactin complex function in motor neurons. To enrich for the recovery of insertions required for normal motor neuron function, we performed initial viability screens in backgrounds in which the function of the dynactin complex is impaired only in motor neurons. Specifically, F1 flies were generated harboring a single-copy P-element insertion in-trans to a recombinant third chromosome harboring both the UAS-DNGluedIIIA transgene and the D42 Gal4 driver (D42, DNGlued; Figure 1A; Parkes et al., 1998; see Materials and Methods). Glued is the Drosophila homologue of the P150/DCTN1 component of the dynactin complex (Waterman-Storer and Holzbaur, 1996). Expression of the UAS-DNGlued in fly motor neurons using the D42-Gal4 driver results in animals with delayed larval development, reduced eclosion rates, and decreased survival, providing useful metrics for screening. Because a portion of the P-elements screened have the ability to drive the expression of nearby genes in response to the presence of the D42 Gal4, the recovery of both loss-of-function and gain-of-function alleles was possible (Rørth et al., 1998). F1 animals were scored for enhanced lethality within the following lethal phases: 1) embryonic/larval, 2) pupal, and 3) eclosion/early adult. Note that although lethality is not an overly specific phenotype, in the context of our sensitized motor neuron backgrounds, the effects of these insertions on motor neuron health is strong enough to result in reductions in survival and therefore should identify candidate molecules that are critical for normal dynactin complex function in neurons.
FIGURE 1:

CG17184 encodes Arfaptin, a modifier of dynactin complex function during synapse growth. (A) An enhancer-suppressor screen of 3663 individual P-element insertion lines identified 40 candidate insertions that enhance or suppress the toxicity of dominant dynactin complex mutations in the motor neuron (D42, DNGlued). Candidates were subsequently tested for specific effects on dynactin-dependent processes in the motor neuron. (B) Immunofluorescence images of the NMJs on muscle 4 stained with an antibody recognizing the presynaptic DVGluT synaptic vesicle protein in EY11874/+ (top), D42, DNGlued/+ (middle), and EY11874/D42, DNGlued (bottom) third-instar larvae. Scale bar, 10 μm. (C) Bar graph of the average number of boutons at the NMJ on muscle 4 in third-instar larvae of the indicated P-elements in-trans to the D42, DNGlued allele. The two P-elements residing in CG17184 (EY11874 and d04252) are indicated. The P-elements resulting in significant reductions (enhancers; p < 0.05; green bars) or increases (suppressors; p < 0.05; red bars) of bouton numbers are indicated. The P-element insertions that also result in a significant enhancement (E, p < 0.05) or suppression (S, p < 0.05) of synaptic stability (footprints) are indicated above the bars. Bouton numbers for the D42, DNGlued/+ heterozygote control background are indicated by the gray bar. Significance was determined using Student's t test for each line vs. control. Dashed line in C represents the wild-type synapse growth levels.
Insertion lines were categorized as either enhancers or suppressors based on the effects on the lethality compared with D42, DNGlued heterozygote controls. We performed two rounds of primary screening under low- and high-stringency conditions, resulting in the identification of 65 insertion lines that enhanced or suppressed viability in D42, DNGlued mutants. Control screens found that 25 lines interacted only with the Gal4 driver (D42) line, and these were removed from further consideration. The resulting 40 insertions represent mutations in candidate genes for the regulation of dynactin complex function within the motor neuron (Figure 1A; see later discussion).
Genetic dissection of dynactin complex functions in motor neurons
Our hypothesis is that the neuropathologies observed in glued-mutant motor neurons represent a combination of genetically distinct dynactin-dependent neuronal processes. To test this possibility, we analyzed all 40 candidates as single-copy insertions in-trans to the heterozygote D42, DNGlued allele for effects on known dynactin-dependent processes within the motor neuron: synaptic growth (determined by counting the number of 1b boutons at the neuromuscular junction [NMJ]), synaptic stability (determined by quantifying the number of presynaptic retraction events), and axonal transport (determined by quantifying the extent axonal blockage). These analyses found that 14 insertions significantly enhanced the synapse growth phenotype, resulting in significantly fewer boutons compared with the heterozygote D42,DNGlued control (Figure 1, B and C, and Table 1). Five of these also enhanced the number of synaptic retractions (footprints), a measure of synapse stability and an established synaptic phenotype of dynactin complex mutations in flies and mice (Figure 1C, indicated by E above bar; Table 1; Eaton et al., 2002; Pielage et al., 2005; Chevalier-Larsen et al., 2008). We predict that the mutations in these genes have a generalized effect on dynactin complex function in the motor neuron. Six of the lines that enhanced synaptic growth dysfunction had no effect on stability, and three of these enhancer lines slightly suppressed the defect in synaptic stability (Figure 1C, suppression indicated by S above bar). Note that we believe that a certain amount of synapse growth is required for accurate analysis of retractions, so it is possible that severe impairment of synapse growth can indirectly suppresses retractions. Nonetheless, synaptic growth and synaptic stability are uncoupled in these six insertion lines. These alleles represent the best candidate loci for genes involved in specifying unique functions of the dynactin complex in the motor neuron.
TABLE 1:
Genetic modifiers of synapse growth in dynactin complex mutants.
| Insertion genotypea | Boutons (average)b | nc | Footprints (%)d | ne | Axonsf | ng | Viabilityh |
|---|---|---|---|---|---|---|---|
| Control backgrounds | |||||||
| wild type (w1118) | 17.7 | 24 | 3.1 | 102 | — | 12 | — |
| D42,DNGlued/+ | 10.63 | 16 | 44 | 148 | ** | 8 | — |
| Enhancersh | |||||||
| P{EP}CG17184EY11874 | 6.66 | 16 | 26 | 119 | * | 8 | Enhancer |
| PBac{PB}fanc04756 | 6.67 | 12 | 78 | 61 | *** | 6 | Enhancer |
| P{EP}EY20330 | 6.88 | 8 | 37 | 47 | * | 5 | Enhancer |
| P{EP}CoopEY13293 | 7.06 | 16 | 26 | 77 | * | 8 | Suppressor |
| PBac{PB}Sirt2c03323 | 7.5 | 16 | 66 | 74 | ** | 8 | Enhancer |
| P{EP}CG4562EY09703 | 7.71 | 14 | 61 | 70 | * | 7 | Suppressor |
| P{EP}B4EY14645 | 7.71 | 21 | 55 | 82 | ** | 11 | Suppressor |
| P{EP}nesdEY11086 | 7.75 | 16 | 47 | 78 | * | 8 | Suppressor |
| P{EP}bbgEY05191 | 7.76 | 17 | 15 | 77 | ** | 8 | Enhancer |
| P{XP}CG17184d04252 | 7.78 | 12 | 38 | 64 | ** | 7 | Enhancer |
| P{EP}EY12448 | 7.78 | 14 | 68 | 68 | ** | 7 | Enhancer |
| P{EP}klarEY01576 | 7.81 | 16 | 41 | 72 | *** | 9 | Enhancer |
| P{EP}CG42575EY04050 | 7.81 | 16 | 48 | 73 | ** | 8 | Suppressor |
| P{EP}TM9SF4EY00960 | 7.83 | 12 | 85 | 64 | * | 6 | Suppressor |
| Suppresssorsh | |||||||
| P{EP}EY06888 | 14 | 9 | 15 | 54 | *** | Enhancer | |
| P{EP}GRHREY11371 | 16.37 | 8 | 9 | 45 | * | Enhancer | |
All data from transheterozygotes consisting of single-copy insertion in-trans to the D42,DNGlued recombinant chromosome. See Supplemental Table S1 for more molecular and genetic information on inserts listed.
aGenotype for each P-element insertion obtained from FlyBase (http://flybase.org/). Enhancer and suppressor refer to the effects of the insert on bouton numbers.
bValues represent the average number of 1b boutons per synapse at the larval NMJ on muscle 4, segment 3, in transheterozygotes. All values listed are significantly different from the D42,DNGlued/+ control background. p < 0.05, Student's T-test.
cTotal number of synapses counted for bouton number averages.
dThe percentage of all NMJs on muscles 6 and 7 from each animal that has a synaptic footprint as visualized by costaining NMJs with anti-VGluT and anti-Dlg. These events reflect instability of the presynaptic nerve terminal (Eaton et al., 2002; Pielage et al., 2005). Bold values are significantly different from the D42,DNGlued/+ heterozygote control background using a test of proportions (z).
eTotal number of muscle 6/7 synapses scored for synaptic footprints.
fRefers to the severity of axonal blockages observed in transheterozygotes compared with D42,DNGlued/+ controls. ***enhancement, **no change, *suppression of the blockage phenotype.
gTotal number of larvae inspected for axon blockages and bouton counts.
hRefers to the effects of inserts on viability when in-trans to D42,DNGlued/+ at 25°C. Enhancers (E) enhance the toxicity of the D42,DNGlued mutation, leading to reduced viability, and suppressors (S) reduce the effects of D42,DNGlued on viability.
The arfaptin gene encodes a modifier of dynactin-dependent synapse growth
Our screen recovered two P-element insertions, P{EP}EY11874 (P1) and P{XP}d04252 (P2), that map independently to the gene CG17184 (Figures 1C and 2A and Table 1). BLAST analysis of the peptide sequence obtained from the translation of the putative gene product of CG17184 found that this gene encodes a homologue of the mammalian Arfaptin2 protein (see later discussion of Figure 3A; Kanoh et al., 1997)). Thus we refer to this gene as arfaptin (arfip) and the gene product as Arfaptin (Arfip). To confirm the results of our screen identifying arfip as an important modifier of dynactin complex function during synapse growth, we first repeated the genetic conditions of the screen using outcrossed arfipP1 and arfipP2 insertions and found that both insertions still significantly enhanced the defect in synapse growth observed in D42, DNGlued heterozygotes (Figure 2D). Analysis of arfip expression using in situ hybridization demonstrates that this gene is expressed within the developing nervous system, consistent with a role for Arfip in the nervous system during development (Figure 2B).
FIGURE 2:
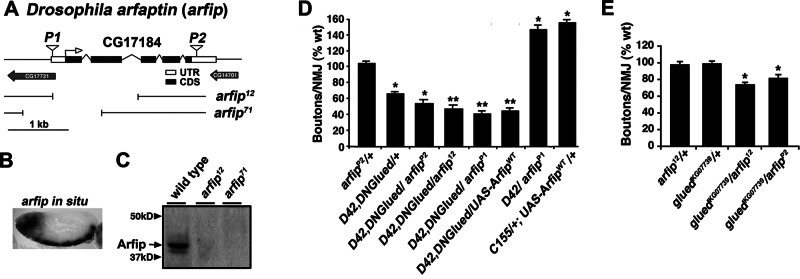
CG17184 encodes Drosophila arfaptin (arfip), a novel gene that functions with glued during synapse growth but not synapse stability. (A) A schematic of the CG17184 (arfaptin) locus including coding sequences (CDS), untranslated regions (UTR), and the locations of the two P-element insertions isolated in the screen. Breakpoints for arfaptin (arfip) deletion mutations arfip12 and arfip71 are indicated. (B) In situ hybridization of stage 15 Drosophila embryo showing mRNA expression of arfip within the developing nervous system. (C) Immunoblot analysis of wild-type (w1118) and homozygous arfip12 and arfip71 mutants probed with anti-Arfip antibody. (D) Graph represents the average number of boutons per NMJ (n = 16–20) at muscle 4 for the indicated genotypes. *p < 0.01 vs. ArfipP2/+ using one way analysis of variance (ANOVA); **p < 0.05 vs. D42, DNGlued/ArfipP2 using Student's t test. Error bars, SEM; n = 16–20 NMJs. (E) Graph of the average number of boutons per NMJ (n = 16–20) at muscle 4 for the indicated genotypes. *p < 0.05. Significance determined vs. wild type using one-way ANOVA.
FIGURE 3:

Arfaptin associates biochemically with dynactin complexes. (A) Schematic of Arfip protein structure reveals two functional domains: Bin-amphiphysin-Rvs (BAR) and GTPase-binding domain (GBD). Percentages shown below protein schematic represent amino acid identity between Arfip and human Arfaptin2. (B) Immunoblots of coimmunoprecipitation analysis from S2 cells coexpressing FLAG-Glued and Arfip-HA fusion proteins incubated with anti-FLAG–antibody coated beads (lane 3). Inclusion of a competing FLAG peptide during incubation with beads completely abolished binding of Arfip-HA (lane 4). S2 cells only expressing Arfip-HA when incubated with FLAG beads showed no binding of Arfip-HA to FLAG beads (lane 5). Cell lysate of S2 cells coexpressing FLAG-Glued and Arfip-HA (lane 1) or Arfip-HA only (lane 2) shows equivalent amount of protein materials added in each coimmunoprecipitation experiment. (C) Immunoblots of coimmunoprecipitation analysis from mouse brain homogenate show that Arfaptin2, but not Arfaptin1, binds to mouse P150 (compare lanes 2–5). Mouse brain homogenate containing endogenous P150, Arfaptin2, and Arfaptin1 proteins (lanes 1 and 4) was incubated with anti-P150 antibody–coated beads (lanes 2 and 5) or control beads coated with mouse IgG (lanes 3 and 6). (D) Immunoblot analysis of gradient fractionation of Drosophila S2 cells. Sedimentation of the dynactin subunits was monitored by immunoblot of dynein heavy chain (DHC), dynein IC (DIC), Glued, and Arp1. Fraction numbers are indicated above blots, and sedimentation is monitored with protein standards for 10S and 17S sizes. Immunoblot of Arfip demonstrates the presence of Arfip in the 17S fractions known to contain dynactin complexes (fractions 9–11). Line graph represents the total protein load in each lane as determined by Coomassie protein staining of protein gels. Sedimentation values were determined by running parallel protein standards. (E) Immunoblot analysis of gradient fractionation of Drosophila heads. Sedimentation of the dynactin subunits was monitored by immunoblot of Glued and Arp1. Fraction numbers are indicated above blots, and sedimentation is monitored with protein standards for 10S and 17S sizes. Immunoblot of Arfip demonstrates the presence of Arfip in the 17S fractions known to contain dynactin complexes (fractions 9–11). Line graph represents the total protein load in each lane. (F) Heads of flies expressing Arfip-HA in the nervous system were subjected to sucrose gradient sedimentation. Fractions 9–11 from gradients of heads from flies expressing Arfip-HA in all neurons (+Tg; lane 1) or fractions from control flies (-Tg; lane 2) were incubated with anti-HA beads. Immunoblot analysis of endogenous Glued and Arp-1 was used to evaluate specific coIP with Arfip-HA (lane 3 vs. lane 4).
The arfipP1 P element resides in the 5′ untranslated region (UTR) of arfip gene and could potentially drive expression of arfip when supplied Gal4 (Figure 2A). To investigate this possibility, we performed immunoblot analysis using an anti-Arfip antibody, which indicated that arfipP1 could drive the expression of arfip in the presence of Gal4 (Supplemental Figure S1). This suggests that arfipP1 could function as a gain of function in our screen. In support, we found that crossing the arfipP1 insert to the D42-Gal4 driver resulted in a similar effect on synapse growth as overexpression of transgenic Arfip (UAS-ArfipWT; Figure 2D). We also observed that the UAS-ArfipWT transgene behaved as a strong enhancer in-trans to the D42, DNGlued allele, similar to what was observed with arfipP1 (Figure 2D). Thus the arfipP1 insertion phenocopies the effects of the UAS-ArfipWT transgene on synapse growth and supports the finding that arfipP1 was isolated as a gain of function in our screen.
The arfipP2 insertion maps to the 3′ UTR of the gene and is predicted to be a loss of function. To improve the analysis of arfip loss-of-function phenotypes, we generated null mutations of arfip using P-element mobilization of P{EP}EY11874 (arfip71; Figure 2A). A second loss-of-function excision allele was obtained from the Rothenfluh lab (arfip12; Figure 2A). Both of these mutations are semilethal, with a portion of mutants dying during pupal development. Nonetheless, viable homozygotes can be isolated that are fertile and can reproduce. Newly eclosed homozygote arfip mutant animals have no obvious neurological phenotypes and are able to fly and wall climb. Western blot analysis of Arfip levels in flies homozygous for these mutations using a polyclonal antibody generated against full-length Arfip fail to detect Arfip protein, supporting the notion that these mutations are indeed null alleles (Figure 2C). To confirm the genetic interaction between arfip and glued, we analyzed synapse growth in a line harboring one copy of the null arfip12 allele in-trans to one copy of the strong loss-of-function gluedKG07739 allele. Although these alleles have no effect on synapse growth as heterozygotes, the transheterozygote combination of these alleles results in a significant reduction in synapse growth (Figure 2E). We also observe that arfip12 behaves as an enhancer in-trans to the D42, DNGlued allele, similar to what we observe for arfipP2 (Figure 2B). Taken together, these data support the notion that glued and arfip work in the same genetic pathway within the motor neuron during synapse growth.
Arfip interacts biochemically with the dynactin complex in cells and neurons
Our genetic data suggested that Arfip might physically interact with the dynactin complex in the motor neuron. To investigate this possibility, we took a biochemical approach. We first performed coimmunoprecipitation experiments using Drosophila S2 cells coexpressing FLAG-tagged Glued and hemagglutinin (HA)-tagged Arfip. Cells were lysed, and FLAG-Glued was immunoprecipitated using anti-FLAG–coated beads (Figure 3B, lane 1, top). We found that Arfip was coimmunoprecipitated with FLAG-Glued from these cells (Figure 3B, lane 1, bottom). The coimmunoprecipitation of HA-Arfip depended on the coexpression of FLAG-Glued (Figure 3B, lane 3) and was completely inhibited by inclusion of a FLAG peptide during the immunoprecipitation (Figure 3B, lane 2). A similar interaction was observed between Arfip and the truncated DNGlued protein (Supplemental Figure S1). To investigate the specificity of this interaction, we extended the coimmunoprecipitation analyses to mouse brain, which expresses both Arfaptin1 and Arfaptin2 (Figure 3C). We observed that endogenous Arfaptin2 (Figure 3C, lane 2), but not Arfaptin1 (Figure 3C, lane 5), could be coimmunoprecipitated with mouse P150/DCTN1, demonstrating the specificity and conservation of the robust interaction between P150/Glued and Arfaptin2. These data support the notion that Arfaptin and Glued can interact biochemically, supporting the observed genetic interaction.
To investigate whether Arfip associates with the dynactin complex, we first investigated the presence of Arfip in isolated dynactin complexes. The large size of the dynactin complex allows for the purification of intact complexes from fly tissues using sedimentation gradients (Haghnia et al., 2007). Gradient fractions are then analyzed using immunoblotting for the presence of dynactin complex components in the fractions corresponding to the predicted sedimentation of intact complexes (∼17S, fractions 9–11; Haghnia et al., 2007). Gradient fractionation of Drosophila S2 cells, which we find express Arfip, demonstrates that a portion of endogenous Arfip cosediments with dynactin complex components in the 17S fractions (Figure 3D). Note that the observed distribution of dynactin complex components in our gradients is similar to what was demonstrated for the sedimentation of dynactin complex components from Drosophila (Haghnia et al., 2007). Arfip is also found in 17S fractions from fly heads, supporting the notion that Arfip is associated with dynactin complexes in both cells and neurons (Figure 3E). To confirm this analysis, we investigated whether we could coimmunoprecipitate components of the dynactin complex with Arfip from the 17S gradient fractions from homogenized fly heads. We subjected gradient fractions 9–11 from homogenized heads from flies expressing Arfip-HA in the nervous system (Figure 3F, lane 1) or from control flies lacking transgenic Arfip-HA expression (Figure 3F, lane 2) to immunoprecipitation using anti-HA coated beads. Immunoblot analysis demonstrated the coimmunoprecipitation of both the Glued and Arp1 components of the dynactin complex specifically from the flies expressing the Arfip-HA transgene in the nervous system (Figure 3F, lane 3) compared with control flies (Figure 3F, lane 4). Taken together, these results support the notion that a portion of Arfip associates with the dynactin complex in the neurons of flies and mice.
arfaptin is required within the motor neuron for normal synapse growth
Our genetic data and biochemical data suggest a model in which Arfip works with the dynactin complex specifically during synapse growth but not during synapse stabilization or axonal transport. This model would predict that Arfip is required within the motor neuron for normal synapse growth but be dispensable for axonal transport and synapse stabilization. To test this model, we first examined whether Arfip is required within the motor neuron for normal synapse growth. The chromosome deficiency, Df ED5518, lacks the region containing the arfip gene and was used as a null allele in these studies. We found that heterozygote mutations in arfip (Df ED5518/+, arfip71/+, and arfip12/+) have wild-type numbers of boutons at their respective NMJs (Figures 2E and 4F). We next observed that arfip71/Df ED5518, arfip12/71, and arfip12 mutant NMJs have significant reductions in bouton numbers compared with controls (Figure 4, B, D, and F). In these analyses, no difference in muscle size was observed in arfip mutants compared with controls (Figure 4H). We also observed reductions in synapse growth in second-instar larvae, supporting the notion that Arfip function is required throughout larval development for normal synapse growth (Figure 4I). In addition, the equivalent effect of these allelic combinations on synapse growth support the idea that arfip71 and arfip12 both function as null mutations, consistent with analysis of protein levels (Figure 2C). Although synapse growth is reduced, we observed little change in amount or morphology of a number of presynaptic proteins at mutant synapses, including VGluT, CSP, and FasII (Figure 4, A and B, insets; data not shown).
FIGURE 4:

Arfaptin functions presynaptically during synapse growth. (A, B) Immunofluorescence image of NMJ at muscle 6/7 in wild-type and arfip12/71 mutant third-instar larvae costained with anti-VGluT (green channel) and anti–Discs-large (red channel) antibodies. High magnification of the marked regions stained for either (i) VGluT or (ii) Discs-large are shown (right). (C–E) immunofluorescence images of NMJ at muscle 4 in driver control line (C; C155; arfip12), transgene control line (D; UAS-ArfipWT/+; arfip12/71), and arfip rescue line (E; C155; UAS-ArfipWT/+; arfip12/71) stained for DVGuT. (F, G) Graphs of the average number of boutons per NMJ at muscle 4 for the indicated genotypes. *p < 0.01 vs. wild type determined using one-way ANOVA. **p < 0.001 rescue vs. transgene control as indicated determined by Student's t test. (H) Graph of the quantification of the area of muscle 4 in third-instar larvae between wild type and arfip12/71 mutant. n = 8–10 muscles. (I) Graph of the average number of boutons/μm2 of muscle in early second-instar larvae between wild type and arfip12/71 mutant. Significance determined by Student's t test (p < 0.05).
To confirm that the reduction in synapse growth observed in the arfip mutants is due to the loss of Arfip function in the motor neuron, we performed rescue experiments by expressing a UAS-ArfipWT transgene in motor neurons of arfip12/71 mutants using both a panneuronal Elav driver (C155; Robinow and White, 1988) and a motor neuron–specific driver (OK6; Sanyal, 2009). We found that neuronal expression of wild-type Arfip (ArfipWT) with either Gal4 driver line in arfip12/71 mutants robustly rescues the defect in synapse growth found in arfip mutants (Figure 4G). The ability of Arfip expression to rescue past wild-type levels is consistent with the ability of Arfip overexpression to enhance synapse growth in wild-type neurons (Figure 2D) and is due to overexpression of Arfip in the rescue background. These data demonstrate that Arfip functions within the neuron during synapse growth.
arfaptin is not required for dynactin-dependent axonal transport or synaptic stability
We next investigated whether arfip was required for normal axonal transport. We first assayed the presence of axonal blockages in arfip mutant axons (Figure 5A). These blockages were used previously to detect defects in axonal transport in motor protein mutants, including dynein and kinesin mutants, and can easily be combined with genetic pathway analysis (Bowman et al., 1999; Martin et al., 1999; Gunawardena et al., 2003; Lee et al., 2004; Lorenzo et al., 2010). Axonal blockages were analyzed in mutant third-instar larvae using immunofluorescence microscopy with antibodies against the synaptic vesicle protein VGluT (our marker for axonal blockages) and the axonal membrane marker horseradish peroxidase (HRP; Figure 5A). Digital images were then analyzed to quantify the size of VGluT immunoreactive blockages (Figure 5, B and C). Using this approach, we found no difference in the size of VGluT puncta between controls and the homozygous arfip 12/71 mutants or the gluedKG07739/arfip12 transheterozygote mutant (Figure 5C). Of importance, we previously demonstrated that both the arfip 12/71 and gluedKG07739/arfip12 genetic backgrounds had significant reductions in synapse growth (Figures 2E and 4F). We also determined the number of VGluT puncta per unit length of axon and did not see any significant difference between arfip mutants and controls (data not shown).
FIGURE 5:

Arfip is not required for normal axonal transport. (A) Inverted images of axonal segments at the midline in A4 of third-instar larvae of indicated genotypes stained with the anti-VGluT antibody. Axonal blockages are characterized by large aggregates of VGluT puncta as seen in D42, DNGlued/+ larvae. Scale bar, 10 um. (B, C) Frequency distribution of VGluT puncta surface area by indicated genotypes. Only D42, DNGlued/+ axons have significant increases in puncta size compared with all other genotypes. *p < 0.01, Mann–Whitney test between wild type and D42,DNGlued/+. (D) Kymograph of inverted still images from live imaging of syb-GFP in axons of motor neurons projecting from the VNC, demonstrating the retrograde (diamonds) and anterograde (triangles) movement of syb-GFP vesicles in wild-type larvae (note: other particles are also observed moving in these frames). (E) Normalized frequency distribution of instantaneous retrograde velocities of syb-GFP axonal transport in wild-type (black bars) and arfip12/71 (gray bars) larvae determined from analysis of live imaging. Inset graphs represent the average retrograde velocity of the distribution. For these analyses, 53 particles from eight animals for wild type and 68 particles from 11 animals for arfip mutant were analyzed. There is no significant difference in the average (as determined by Student's t test) or the distribution of velocities (as determined by a two-sample Kolmogorov–Smirnov test). Error bars, SEM. (F) Normalized frequency distribution histogram of retrograde run length of syb-GFP –containing vesicles in wild type (black bars) and arfip12/71 (gray bars) axons. Inset graphs represent the average retrograde run length for the distribution. For these analyses, 49 particles from eight animals for wild type and 64 particles from 11 animals for arfip mutant were analyzed. Error bars, SEM. There is no significant difference in the average (as determined by Student's T-test) or the distribution of velocities (as determined by a two-sample Kolmogorov–Smirnov test). (G) Graphs represent the average maximum instantaneous retrograde velocity of syb-GFP in wild-type (black bars) and arfip12/71 axons. (H) Graphs represent the average maximum instantaneous anterograde velocity (top) or average anterograde velocity (bottoms) of syb-GFP in wild-type (black bars) and arfip12/71 axons. For these analyses, 22 particles from eight animals for wild type and 22 particles from 11 animals for arfip mutant were analyzed. Error bars, SEM. There is no significant difference for either average anterograde velocity as determined by Student's t test.
Because blockage analysis does not directly measure motor protein function, we directly analyzed the axonal transport of vesicles in arfip mutants using live video microscopy. Axonal tracts from control and arfip-mutant third-instar larvae expressing synaptobrevin-green fluorescent protein (syb-GFP) in the motor neuron were imaged live using video microscopy to generate videos of the axonal transport of vesicles along the axon in both the anterograde and retrograde directions (Supplemental Movies S1 and S2 and Figure 5D). In general, the episodic nature of microtubule-based axonal transport was observed, and run lengths and velocities were easily determined using particle-tracking software (Susalka et al., 2000). We did not observe any differences in the density of syb-GFP punta or in the numbers of puncta moving in either the anterograde or retrograde direction in arfip12/71-mutant axons compared with controls (Supplemental Table S2). Because of the known role of the dynactin complex during retrograde axonal transport, we mostly focused on retrograde transport. This analysis found no significant difference in the average or instantaneous maximum retrograde velocities of syb-GFP transport in arfip-mutant axons compared with wild-type axons (Figure 5, E and G). In addition, no difference was observed between wild type and arfip12/71 mutants for average retrograde run lengths (Figure 5F). We also observe a normal distribution of both retrograde velocities and run lengths in arfip mutants (Figure 5, E and F). Finally, no difference was observed for anterograde transport between arfip mutants and wild type (Figure 5H). These analyses find no requirement for arfip during retrograde or anterograde axonal transport and support the idea that the effects of arfip mutants on synapse growth are not due to impaired axonal transport. This is supported by the lack of a tail-lift phenotype in arfip-mutant larvae, which is characteristic of mutations in motor proteins in larvae (Martin et al., 1999).
The prevalence of presynaptic retractions (footprints) is a quantitative measure of changes in synapse stability and is increased in dynactin complex mutants (Eaton et al., 2002; Pielage et al., 2005; Koch et al., 2008). To investigate a role for arfip during synapse stabilization, we determined the number of footprints in arfip12/71 mutants and found no difference in the number of footprints observed in arfip mutants compared with wild type (data not shown). Taken together, these data support that Arfip functions with the dynactin complex specifically during synapse growth.
Arfaptin colocalizes with the dynactin complex at the Golgi
To determine where arfip and the dynactin complex function together in neurons, we next sought to determine the subcellular localization of Arfip within the motor neuron. Immunofluorescence microscopy of third-instar larvae with an anti-Arfip antibody revealed specific staining within all neurons of the nervous system, including all neurons of the brain, sensory neurons, motor neurons, and developing neurons within the optic lobes that was absent in arfip mutants (Figure 6, A and B; and data not shown). Higher magnification of the motor neuron soma showed that Arfip is found near the nuclei in punctate structures that are distributed throughout the soma (Figure 6, C and D). We were unable to detect any endogenous Arfip at the NMJ in wild-type larvae with our antibody (data not shown). We believed that the perinuclear staining represented localization to the Golgi, so we investigated the colocalization of Arfip with the Golgi markers GM-130 and Lava lamp. We found that Arfip is juxtaposed to the GM-130 in motor neuron cell bodies (Figure 6, E and F) and S2 cells (see later discussion of Figure 9B). A three-dimensional reconstruction of Arfip and GM-130 staining in the ventral nerve cord (VNC) from a z-stack clearly demonstrates the close opposition of these two proteins in motor neuron soma (Figure 6F). These data suggest that Arfip is primarily localized to cisternae distal to the cis-Golgi. This model is supported by Arfip staining in S2 cells, which reveals significant colocalization of Arfip with the TGN-resident protein Lava lamp (Figure 7G; Papoulas et al., 2005; Repnikova et al., 2010). Pixel-based colocalization analysis of high-magnification (160×) deconvolved three-dimensional (3D) images of S2 cells allows for the generation of a Manders overlap coefficient (R), a measure of the colocalization of two overlying channels (Manders et al., 1993). The R for the costaining of Arfip and Lava lamp within the Golgi region of S2 cells supports the partial colocalization of Arfip and Lava lamp at the Golgi (R = 0.71 ± 0.019; n = 9). Taken together, these data support the idea that Arfip in S2 cells and neurons is localized primarily to distal cisternae of the Golgi, likely the TGN.
FIGURE 6:
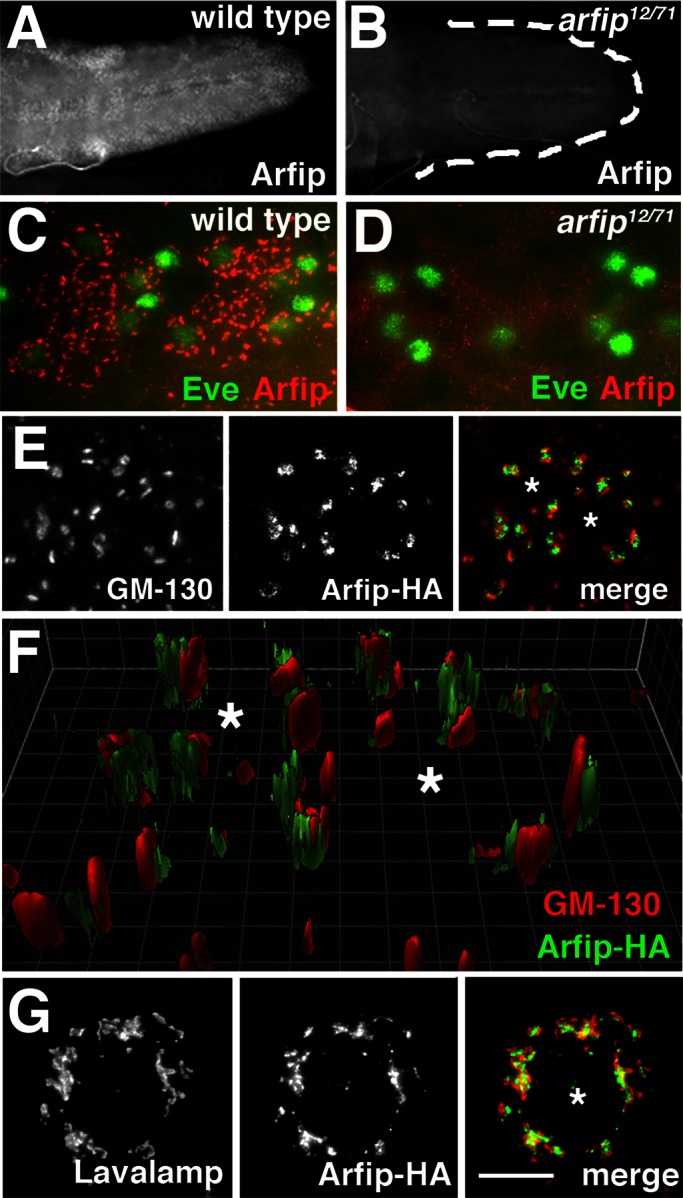
Arfaptin is localized to the distal Golgi in motor neurons. (A, B) Immunofluorescence images of the larval VNC from wild type (A) and arfip12/71 mutants (B) stained with Arfip antibody, demonstrating the expression of Arfip within the VNC. Lack of Arfip staining in arfip12/71 mutants demonstrates the specificity of the Arfip antibody. (C, D) Higher magnification of ventral ganglions from wild-type (C) and Arfip12/71 mutants (D), showing the costaining of Arfip (red) and Eve (green), a protein that stains pools of motor neuron nuclei. (E) Immunofluorescence images of motor neuron soma from larvae expressing Arfip-HA in motor neurons costained for Arfip-HA (middle; green in merge) and GM-130 (left; red in merge). Right, merged image. Asterisks indicate position of Eve-positive nuclei. (F) Three-dimensional reconstruction of z-stack images of the motor neuron cell bodies shown in E, demonstrating the exclusive localization of GM-130 and Arfip. Asterisks indicate position of Eve-positive nuclei. Grid lines are separated by 1 μm. (G) Immunofluorescence images from an S2 cell expressing Arfip-HA costained for Arfip-HA (middle; green in merge) and Lava lamp (left; red in merge). Right, merged image, showing partial colocalization of Arfip and Lava lamp. Asterisk indicates nucleus. Scale bar, 5 μm.
FIGURE 9:

The BAR domain of Arfip is required for Golgi localization and synapse growth. (A) Schematic of Arfip protein, indicating region of the BAR domain containing four key residues (red in Arfip) previously shown in human Arfaptin2 to be required for membrane binding (Peter et al., 2004). We made similar substitutions in the BAR domain of Arfip (R243E, Q244E, R249E, and R256E, highlighted in red). Percentage amino acid identity shown is for Drosophila Arfip vs. human Arfaptin2. (B, C) Immunofluorescence images of arfip RNAi–treated S2 cells expressing HA-tagged wild-type Arfip (Arfipwt; B) or an HA-tagged Arfip harboring the BAR mutations shown in A (ArfipBAR; C) show that ArfipBAR disrupted localization. Golgi staining is normal in these cells, as demonstrated by GM-130 staining (red). Asterisks indicate nuclei. (D, E) Immunofluorescence images of HA-tagged Arfipwt (D) and HA-tagged ArfipBAR (E) expressed in the motor neuron cell bodies of third-instar arfip mutant larvae. (F) Immunoblot analysis of dynactin complex components (Glued, DIC) in membrane flotation assays from arfip RNAi–treated S2 cells transfected with wild-type (WT) Arfip or BAR domain mutant Arfip, demonstrating that membrane binding of Arfip is required to reestablish membrane binding of Glued and DIC in arfip RNAi–treated S2 cells. (G) Graph of the average number of boutons per NMJ at muscle 4 in third-instar larvae for the indicated genotypes. Rescue was performed in the arfip12/71-mutant background using the indicated Gal4 driver and arfip transgene, demonstrating the lack of rescue of synapse growth by the arfipBAR transgene (ArfipBAR). *p < 0.01 vs. C155/+;;Arfip12/+. Significance determined by one-way ANOVA.
FIGURE 7:

Arfip and Glued colocalize at the Golgi and are required for Golgi integrity. (A, B) Deconvolved fluorescence images from S2 cells are shown expressing FLAG-Glued and costained with anti-FLAG (A, left) and anti-Arfip antibody (A, middle) or with anti-FLAG (B, left) and anti–Lava lamp (B, middle). Merged images (A and B, right) representing the staining for FLAG-Glued (green channel) and endogenous Arfip (red channel) reveal partial colocalization at the juxtanuclear region between FLAG-Glued and Arfip (A, right) and between FLAG-Glued and Lava lamp (B, right). Insets show higher magnification of the regions indicated with arrows in A and B. Scale bar, 10 μm. (C, D) Immunofluorescence images of third-instar larval VNC costained with anti–Lava lamp (Golgi marker; green channel) and anti-Eve antibody (motor neuron nuclei; red channel), demonstrating that the Golgi appears smaller and more fragmented in arfip12/71 mutants (D) compared with wild type (C). (ii) Surface views of the deconvolved images from the top panels (i) are presented to highlight changes in Golgi integrity. (E) Example of a 3D surface rendering of Lava lamp staining in wild-type soma used to determine the size of Golgi stacks (voxels). Examples 1–6 are indicated on image. On the basis of our image, we consider structures >1200 voxels as Golgi stacks in this analysis. (F, G) Analysis of Golgi stacks (>1200 voxels) from Eve-positive soma within the VNC demonstrates a significantly reduction in the average number of large Golgi structures in both arfip mutants (G, wild type vs. arfip12/71) and in Glued1 mutants (H, wild type vs. Glued1). *p < 0.05, significance of averages vs. wild type determined by a student's t test. Error bars, SEM. For these analyses, total particle number is 428 for wild type, 700 for arfip12/71 mutants, and 1164 for Glued1 mutants from three ventral ganglia for each genotype.
These localization data suggest the likely possibility that Arfip functions with the dynactin complex at the Golgi. To investigate this model, we used immunofluorescence microscopy in S2 cells to determine whether Arfip and Glued colocalize at the Golgi. This analysis revealed partial colocalization of Arfip and Glued at the Golgi in S2 cells (Figure 7A). This is supported by colocalization analysis in S2 cells costained for endogenous Arfip and transfected Glued, which found significantly higher R near the Golgi region (R = 0.64 ± 0.018; n = 7) than in the cytosol (p < 0.001; R = 0.296 ± 0.025). We further observed that Golgi morphology within the motor neuron soma is altered in arfip mutants compared with controls (Figure 7, C and D). These images suggested that the Golgi in the motor neurons were becoming fragmented, a phenotype reported within the motor neurons of mice harboring a dominant mutation in the dynactin complex (Laird et al., 2008; Teuling et al., 2008). We used segmentation analysis of deconvolved 3D fluorescence images of Golgi staining to determine the size of Golgi stacks in wild-type and arfip mutants and observed that arfip mutants have a reduction in the number of normal-sized Golgi, consistent with a substantial change in Golgi morphology (Figure 7, E and F). This phenotype is similar to what we observe for the Golgi in motor neurons of Glued1 hypomorphic mutants (Figure 7G). These data demonstrate that Arfip and Glued colocalize at the Golgi and that both proteins are required for maintenance of normal Golgi morphology in motor neurons.
Arfip mediates membrane binding of the dynactin complex
Drosophila Arfip, like mammalian Arfip2, contains two key functional domains in its C terminus, a GTPase-binding domain and a BAR domain (Figure 3A). Previous work on the BAR domain of mammalian Arfaptin2, including structural analysis, showed that Arfaptin2 forms a dimer that associates lipid membranes via interactions between specific amino acids and lipid head groups (Peter et al., 2004). These previous studies suggested that perhaps Arfip could be mediating the binding of the dynactin complex to internal membranes in neurons and S2 cells. To investigate this possibility, we performed membrane flotation assays to determine the amount of Glued recovered in membrane fractions from S2 cells treated with double-stranded RNA (dsRNA) targeting arfip. S2 cells are ideal for this approach because S2 cells express both Arfip and the dynactin complex, and large amounts of homogeneous RNA interference (RNAi)–treated cells can be easily obtained. Under conditions in which we knocked down Arfip protein levels by ∼90% (Figure 8A), we found that the amount of Glued protein associated with membranes is significantly reduced in S2 cells treated with arfip dsRNA compared with controls (Figure 8, B and C). A similar reduction in membrane binding of the dynein IC was observed in response to arfip dsRNA treatment (DIC; Figure 8, B and C). To determine whether Arfip has a similar function in neurons, we performed membrane flotation analysis on ∼220 dissected larval ventral ganglions from wild-type and arfip mutants. In these assays we also saw significant reduction in the amount of Glued on the membranes in arfip-mutant neurons, similar to what we observed in S2 cells (Figure 8, D and E). These data demonstrate that Arfip binds the dynactin complex to the Golgi membrane in motor neurons.
FIGURE 8:
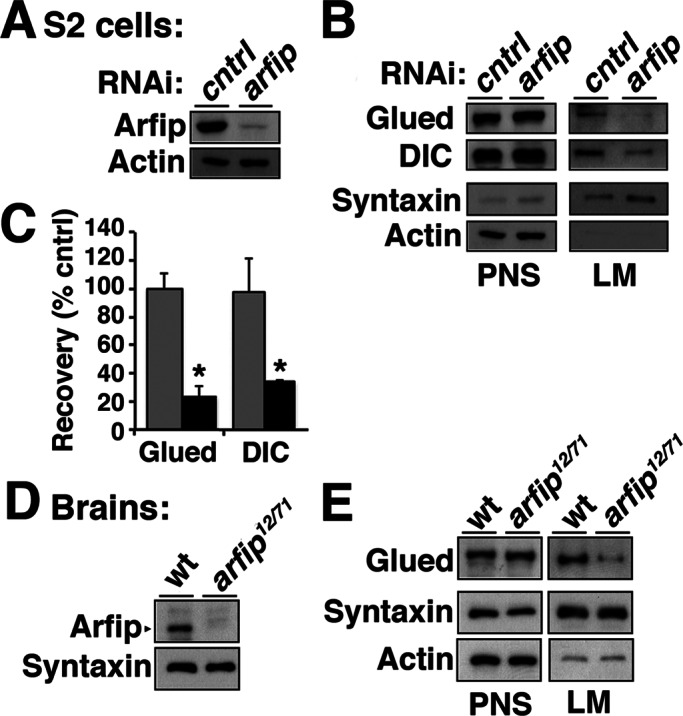
Arfip mediates the membrane association of components of the dynactin complex in S2 cells and neurons. (A) Immunoblot analysis demonstrates reduced Arfip protein level in cell homogenate from arfip RNAi–treated S2 cells compared with control cell homogenate (PNS). (B) Images from immunoblots of membrane flotation analysis of arfip RNAi S2 cell homogenate (PNS), demonstrating the reduced recovery of Glued and dynein IC (DIC) in the light membrane fraction (LM) in arfip mutants. Syntaxin and actin were markers used to monitor fractionation of LMs and normalize protein recoveries during quantification. (C) Graphs of the average mean pixel intensities (m.p.i.) normalized to control of Glued or DIC protein signals recovered in membrane fractions from arfip RNAi–treated S2 cells (black bars) compared with controls (gray bars). Values were normalized for changes in expression (see PNS signal) and for changes in recovery of membrane (syntaxin signal). Values represent the average from three independent membrane flotation assays. *p < 0.01. Significance determined using Student's t test. (D) Immunoblot analysis of homogenate from dissected third-instar ventral ganglion demonstrating loss of Arfip in arfip12/71-mutant ganglions. (E) Images and immunoblots of membrane flotation analysis of ventral ganglions showing reduced Glued level from the LM fraction in arfip12/71 mutants compared to wild type.
Membrane binding of Arfip is required for TGN localization and synapse growth
The foregoing data predict that membrane binding of Arfip will be required for dynactin complex function during synapse growth. Previous work on the BAR domain of Arfaptin2 showed that substituting acidic amino acids for key basic amino acids in the BAR domain completely abolished membrane binding (Peter et al., 2004). We made similar amino acid substitutions in the Arfip BAR domain to investigate the requirement of membrane binding for Arfip function in motor neurons (ArfipBAR; Figure 9A). Because of the possibility of transgenic mutant Arfip dimerizing with the endogenous Arfip, the following studies were performed in S2 cells treated with a dsRNA targeting the arfip 3′ UTR to allow for the transgenic expression of mutant Arfip in the arfip-knockdown cells. We first performed immunofluorescence microscopy on arfip-knockdown S2 cells and found that transgenically expressed wild-type ArfipWT correctly localized to the TGN (Figure 9B) but the transgenically expressed ArfipBAR mutant does not and appears to be diffuse throughout the cytoplasm (Figure 9C). We extended this analysis to the nervous system, where we expressed either the ArfipWT transgene or the ArfipBAR transgene within the motor neurons of an arfip12/71 mutant. Similar to S2 cells, we found that wild-type Arfip localizes properly to the Golgi in arfip-mutant motor neurons (Figure 9D) but the ArfipBAR protein fails to localize to the TGN and is diffuse within the motor neuron soma (Figure 9E). To confirm the effects of the BAR domain mutations on the membrane binding of Arfip, we performed membrane flotation assays on S2 cells and found that wild-type Arfip (ArfipWT) is successfully recovered in the membrane fractions but the BAR domain Arfip mutant (ArfipBAR) is not (Figure 9F). It is unclear why the mobility of the BAR domain mutant is different from the mobility of wild-type Arfip under standard SDS–PAGE gel electrophoresis (Figure 9F). Note that all transgenes used were fully sequenced and verified. We also investigated the effects of BAR mutants on membrane binding of Glued and DIC and observed that rescue with wild-type Arfip restores Glued and DIC binding to normal levels but rescue with the BAR domain mutant is unable to (Figure 9F). Thus membrane binding of Arfip is required for the Golgi localization of Arfip and membrane association of the dynactin complex.
A model in which Arfip-mediated membrane binding by dynactin is required for synapse growth predicts that Arfip harboring the BAR mutations should not be able to support normal synaptic growth. To test this, we expressed ArfipBAR within the motor neuron in an arfip12/71-mutant background and assessed synaptic growth. This analysis revealed that the ArfipBAR mutant protein, in contrast to ArfipWT, does not support synapse growth in these rescue experiments (Figure 9G). It is important to note that this mutation in the BAR domain does not alter the ability of Arfip to bind small GTPases, supporting the notion that the primary effect of this mutation is membrane association (Supplemental Figure S1). These data are consistent with the model that membrane binding of Arfip (and the dynactin complex) at the TGN of the motor neuron is required for normal synapse growth.
DISCUSSION
An unbiased genetic screen in Drosophila designed to recover enhancers and suppressors of mutant Glued (DNGlued) toxicity was performed to identify genes involved in specifying dynactin complex function in motor neurons. This screen recovered two independent heterozygotic insertions in the arfaptin gene (CG17184), the Drosophila homologue of Arfaptin2, which dramatically reduced viability when placed in-trans to the DNGlued mutation, a dominant hypomorphic mutation that impairs dynactin function in the motor neuron (Eaton et al., 2002). Assays of dynactin complex function in motor neurons found that both of these insertions specifically enhanced the synapse growth defects observed in DNGlued mutants but not defects in synapse stability or axonal transport. Analysis of arfaptin mutants confirmed the specific role of Arfaptin during synapse growth and not during axonal transport or synapse stabilization. Similar genetic separation of these dynactin mutant pathologies is observed throughout the results of our screen, which taken together support a model in which multiple molecularly distinct neuropathologies contribute to pathogenesis in dynactin complex mutants. A more complete molecular understanding of the regulation of the dynactin complex within the neuron will be required to define the pathogenic mechanisms associated with these mutant backgrounds.
Our genetic data are consistent with arfip and glued functioning in the same pathway during synapse growth within the motor neuron. In support of a genetic interaction, we find that Arfip is physically associated with Glued and other components of the dynactin complex in the nervous system. Arfip can be coimmunoprecipitated with Glued from both S2 cells and fly brains. In addition, coimmunoprecipitation of Arfaptin2 with P150/Glued, but not Arfaptin1, from mouse brain homogenates demonstrates both the specificity and the conservation of the biochemical interaction between Arfaptins and P150/Glued. Gradient fractionation demonstrates that a portion of Arfip cosediments with dynactin complexes. The combining of sucrose gradient fractionation with coimmunoprecipitation clearly demonstrates that Arfip is associated with dynactin complexes in the fly nervous system. In addition, we find that the membrane association of dynactin complex components is decreased in arfip mutants and restoration of dynactin complex binding requires Arfip membrane binding via the BAR domain, which is also required for Golgi localization of Arfip. Not only do these membrane binding data support the model that Arfip biochemically associates with dynactin complexes, they also support that this interaction results in membrane binding of the dynactin complex. Although the foregoing assays do not specify which population of membranes are being recovered, when combined with our colocalization studies, these data present compelling evidence that Arfip is a novel dynactin-associated protein in neurons that regulates dynactin complex function binding to the Golgi membrane.
Previous data demonstrated that Glued and dynein bind to membranes independently, which is different from what we find here, that membrane binding of both Glued and dynein are reduced in arfip mutants (Holleran et al., 2001; Haghnia et al., 2007). We can imagine a number of explanations for the difference between the previous studies and what we observe in arfip mutants. First, in these previous studies the differences observed in membrane binding were revealed by either biochemical disruption of the dynactin complex (Haghnia et al., 2007) or under conditions of mild protein denaturation (KI treatment; Holleran et al., 2001). This is demonstrated clearly by the change in sedimentation of Glued in Arp-1 mutants (Haghnia et al., 2007). Thus the conditions that previously revealed these differences in binding are very different from what exists in arfip mutants, and the differences in binding of Glued and dynein were aided by disruption of the complex. In addition, there are large reductions in protein levels of Glued in arp-1 mutants but not of dynein (Haghnia et al., 2007). Therefore it is also possible that these differences in expression of Glued and dynein in response to arp-1 mutations contribute to the differences in membrane binding observed in arp-1 mutants compared with arfip mutants, where we do not see any effects on Glued expression levels.
Roles for the dynactin complex and Arfip at the Golgi during synapse growth
It has been established in motor neurons that dynactin complex function is required within the axon and nerve terminal for retrograde trophic signaling, which is critical for normal synapse function and neuronal health (LaMonte et al., 2002; Delcroix et al., 2003; Duncan and Goldstein, 2006; Chevalier-Larsen et al., 2008). In Drosophila motor neurons, dynactin-dependent retrograde trafficking of muscle-derived bone morphogenetic protein (BMP) signaling is required for normal synapse growth and stability (McCabe et al., 2003; Eaton and Davis, 2005). Our data extend these observations by providing evidence that the dynactin complex, via its interaction with Arfip at the Golgi, has functions within the soma independent of BMP signaling that are also required for normal synapse growth. In support are observations that mutations in the BMP signaling components mad and wit result in significant increases in synaptic footprints at larval NMJs that are not observed in arfip mutants (Eaton and Davis, 2005). In addition, wit mutants have impaired axonal transport, including increased axonal blockages, which we also do not observe in arfip mutants (Wang et al., 2007). These data demonstrate that the function of Arfaptin and the dynactin complex at the Golgi during synapse growth is independent of role of the dynactin complex within the axon and nerve terminal during BMP signaling.
Recent studies in mammalian cells demonstrated that localization of Arfaptin2 to the membrane of the TGN requires the small GTPase Arl1 and that this interaction is important for the tubularization of Golgi membranes (Man et al., 2011; Nakamura et al., 2012). The disruption of normal Lava lamp staining in the motor neurons of arfip mutants is consistent with a role for Arfip in maintenance of Golgi structure in the nervous system. Of importance, the process of tubularization by BAR domain–containing sorting nexins was proposed to represent a mechanism for the sorting of membrane proteins (Wassmer et al., 2009; Brankatschk et al., 2011; Temkin et al., 2011; van Weering et al., 2012). By extension, this suggests that Arfip in combination with the dynactin complex could play a role in protein sorting at the TGN. Of interest, although arfip-mutant NMJs are smaller, the levels of all of the synaptic proteins we looked at were normal. These observations, in combination with the analysis of synaptobrevin-GFP transport in the axon, support the idea that Arfip functions independently of the mechanisms involved in the delivery of synaptic material to the synapse (Pack-Chung et al., 2007; Goldstein et al., 2008). Thus the effects that arfip mutants have on synapse growth are not due to defects in axonal transport or the delivery of materials to the growing synapse.
Although we do not know the identity of the protein(s) sorted by Arfip at the TGN, we can make some predictions from our present data set. A potentially informative observation is that loss-of-function arfip mutants have reduced synaptic growth and overexpression of Arfaptin results in increased synapse growth. This would be consistent with Arfip functioning at the Golgi to regulate the delivery of specific membrane proteins whose protein levels can directly influence the growth of the synapse. One possible class of proteins fitting this description is growth receptors. Although our data are not consistent with arfip mutants having impaired BMP signaling, there are other signaling pathways implicated in synapse growth that might require Arfip function for normal signaling (Torroja et al., 1999; Packard et al., 2002; Collins et al., 2006).
Another class of membrane proteins that could be affected by arfip mutants is ion channel proteins required for normal neuron function and excitability. Previous data demonstrated that mutations in ion channels can result in both enhanced and reduced synaptic growth independent of axonal transport (Budnik et al., 1990; Lee and Wu, 2010). Although we do not present data supporting changes in ion channel composition in arfip mutants, recent data reported changes in ethanol-induced behavior in arfip-mutant flies consistent with altered neuronal activity (Peru Y Colón de Portugal et al., 2012). In addition, Arl1 mutant yeast have been reported to have defects in potassium homeostasis resulting in a hyperpolarized cell membrane, suggesting that in yeast Arl1 functions to sort membrane proteins important for ion transport (Munson et al., 2004; Marešová and Sychrová, 2010). Nothing is known about the in vivo functions of Arl1 in the nervous system, but data from cell systems and yeast support the notion that Arl1 plays an important role in the sorting of proteins during vesicle formation (Rosenwald et al., 2002; Munro, 2005; Liu et al., 2006). On the basis of our data, we propose that the interaction between the dynactin complex and Arfaptin resulting in membrane binding at the TGN regulates the sorting and delivery of specific proteins to the cell surface, possibly to the soma or dendritic compartments, that is required for normal synapse growth during development.
MATERIALS AND METHODS
Genetics and fly stocks
All fly stocks were maintained at 25°C on normal lab food. C155-Gal4 and OK6-Gal4 lines were used for targeted expression of transgenes in the nervous system. Transgenic UAS-Arfip-HA lines were generated by microinjection into w−1118 (Rainbow Transgenics Flies, Camarillo, CA). arfip-null alleles were generated by mobilizing the P{EPgy2}EY11874 insert in the presence of the Δ2-3 transposase. White-eyed flies indicating P-element excision were screened by genomic PCR for deletions in CG17184 using primers to flanking gene regions, and breakpoints for deletions of both Arfip12 and Arfip71 were determined by sequencing of mutant gene loci.
Molecular biology
A full-length cDNA for arfip (LD44124) was used as a PCR template for directional TOPO cloning into pENTR (Invitrogen, Carlsbad, CA). All subsequent tagging of Arfip was to the N-terminus using the Gateway cloning system (Invitrogen) and appropriate pUAS-based destination vectors for microinjection (Drosophila Genomics Resource Center, Indiana University, Bloomington, IN). For HeLa expression, Arfip-GFP was cloned directly into pcDNA3.1 (Invitrogen). pUAS-Glued-FLAG encodes an N-terminal FLAG epitope attached to full-length Glued protein and was generated by recombinant cloning from the glued cDNA (RE24170) into pUASt.
Antibody production
Full-length recombinant GST-tagged Arfip was purified from bacteria using glutathione beads and injected into rabbits. Arfip-positive serum was affinity purified using a histidine-tagged Arfip bound to nickel column. An N-terminal Glued peptide (amino acids 1–190) tagged with a hexahistidine epitope was purified from bacteria using a nickel column and injected into mice to generate a polyclonal mouse antibody. Immunoglobulin G (IgG) fractions were purified from positive mouse serum. Antibody production was carried out at the Antibody Core at the University of Texas Health Science Center at San Antonio.
Immunocytochemistry
The following antibodies were used for immunofluorescence analyses: 1/5000 rabbit anti-DVGluT (1:5000, peptide antibody made as described; Daniels et al., 2004), mouse anti-DLG (1:200; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), rabbit anti-GM130 (1:400; Abcam, Cambridge, MA), rabbit anti–Lava lamp (1:400; gift of Papoulas lab, University of Texas, Austin), mouse anti-HA (1:500; US Biological, Swampscott, MA), mouse anti-HA (1:1000; Covance, Berkeley, CA), mouse anti-GFP (1:400; Invitrogen), rabbit anti-TGN46 (1:400; Sigma-Aldrich, St. Louis, MO), mouse anti-GM130 (1:200; BD Biosciences, San Diego, CA), mouse anti-FLAG (1:500 and 1:1000; Sigma-Aldrich), and rabbit anti-HA (1:2000; US Biological). Secondary antibodies from goat were coupled to Alexa Fluor 488 or Alexa Fluor 555 and used at 1:400 (Invitrogen). Goat anti-HRP was directly coupled to Alexa Fluor 647 and used at 1:500 (Invitrogen). The following antibodies were used for immunoblot analyses: rabbit anti-Arfip (1:1000), mouse anti-Glued (1:1000), rabbit anti-Arp1 (1:1000; gift of the Goldstein lab, University of California, San Diego), mouse anti-DHC (1:2000; gift of the Hays lab, University of Minnesota), mouse anti-DIC (1:700; Millipore, Billerica, MA), rabbit anti-GFP (1:1000; Invitrogen), rabbit anti-Arfip2 (1:400; Sigma-Aldrich), rabbit anti-Arfip (1:600; Santa Cruz Biotechnology, Santa Cruz, CA), mouse anti-P150 (1:800; BD Biosciences), rabbit anti-FLAG (1:1000; Sigma-Aldrich), and rabbit anti-HA (1:2000; US Biological).
dsRNA interference
Design and treatment of dsRNA constructs followed the protocols described in Rogers and Rogers (2008). Genomic template for arfip dsRNA was isolated by PCR amplification of a 543–base pair region corresponding to 3′ UTR of the predicted Arfip cDNA sequence (National Center for Biotechnology Information accession number NM_141801) using gene-specific primers with the T7 RNA polymerase sequence attached to the 5′ end of each primer to generate a product flanked with T7 binding sites (left primer: 5′-taatacgactcactatagggagaccac-tagacgcagttcgtcccttt-3′; right primer: 5′-taatacgactcactatagggagaccac-agctcgcaaaaacagtttcg-3′). Standard PCR was carried out on wild-type genomic DNA, and 5 μg of reaction products was used directly in an in vitro RNA transcription reaction with T7 polymerase using the T7 RiboMAX Large Scale RNA Production System (Promega, Madison, WI). T7 transcription reaction was incubated in 37°C water bath for 20–24 h. Arfip dsRNA interference in S2 cells was performed on six-well plates. Each well was seeded at 30–40% confluence with 2 ml of complete medium containing 10 μg/ml arfip dsRNA (referred to as dsRNA medium). Fresh dsRNA medium was replaced daily for 5 d. Cells were harvested for analysis on day 6. To determine the effect of BAR mutation, dsRNA-treated cells for 3 d were lifted from six-well plates and reseeded in new wells with 3 ml of fresh dsRNA medium. On day 4, transient transfection was performed as described. On day 5, transfected cells were washed once and allowed to culture in 10-cm plates with dsRNA medium for 48–72 h. Medium was replaced with fresh dsRNA medium daily until harvest on day 7.
Tissue culture and transfection
A polyclonal S2 cell line that constitutively expresses Gal4 was used for all experiments. S2 cells were grown in Schneider's Drosophila medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) and 1/100 dilution of penicillin–streptomycin–amphotericin B (MP Biomedicals, Solon, OH) in a humidified atmosphere at 25°C. Transient transfection of UAS-based fusion protein constructs in S2 cell culture was performed using a calcium phosphate–based method described in the Drosophila Expression System Kits and was used for both immunofluorescence and immunoblot analyses (Invitrogen). Transfected cells were washed once in complete medium and allowed to culture for 48–72 h. HeLa cells were maintained in DMEM (Cellgro, Manassas, VA) supplemented with 10% fetal bovine serum (Invitrogen) and 1/100 dilution of penicillin–streptomycin–amphotericin B (MP Biomedicals) in a humidified 5% CO2 atmosphere at 37°C. For immunofluorescence localization analysis of GFP-Arfip in HeLa cells, cells were transfected using PolyFect Transfection Reagent (Qiagen, Valencia, CA).
Microscopy
All fixed images were captured with a 63× oil Plan-Neofluar (numerical aperture [NA], 1.25) objective using an Orca-2 backcooled charge-coupled device camera (Hamamatsu, Hamamatsu, Japan) attached to a Zeiss Axiovert 200M (Carl Zeiss, Jena, Germany), except for HeLa and S2 cells, which were imaged on the same microscope using a 100× oil Plan-Chromomat (NA 1.4) objective. For analysis of axonal transport of Synaptobrevin-GFP, wild-type and arfip-mutant larvae expressing UAS-synaptobrevin-GFP under control of the OK6 Gal4 driver were dissected and imaged live in Schneider's media supplemented with 7 mM l-glutamic acid. Live imaging was performed with the 100× oil Plan-Chromomat (NA 1.4) objective on an Axiovert 200M microscope equipped with the Orca2 camera set to 2 × 2 binning. Images were captured using a 200-ms exposure at 3.6 Hz. All imaging and postimage analysis was done using Slidebook software (3I, Denver, CO). Syb-GFP particles were tracked manually from coded files to generate “blind” data. For generation of R, z-stack images of S2 cells were generated at 160× and deconvolved using a constrained iterative algorithm that uses a theoretical point-spread function. The 3D deconvolved images were then analyzed pixel by pixel within designated regions of interest to generate R. R = 0 means no overlap of signals, and R = 1 means perfect and compete overlap. All deconvolution and colocalization analyses are built-in features of the Slidebook software that controls our imaging system.
Axonal blockage analysis
Third-instar larvae of indicated genotypes were dissected and fixed as previously described (Eaton et al., 2002). The larvae were stained with rabbit anti-VGluT and goat anti–HRP-Alexa 647. Goat anti-rabbit Alexa 488 was used to visualize VGluT staining. The z-stack images were collected using the 63× objective and deconvolved using a constrained iterative algorithm (Slidebook software). Individual VgluT-positive puncta were masked by intensity values and subjected to quantitative analyses of surface area. From 10 to 14 larvae were dissected and analyzed for each genotype. The quantification analysis consists 950–1200 objects per genotype.
Sucrose density gradient
Sucrose density gradient was performed as previously described (Haghnia et al., 2007) with slight modifications. Flies were killed by freezing in −80°C for a minimum of 5 min. Approximately 250 fly heads were collected on ice and homogenized in 1 ml of PMEG buffer (0.1 M piperazine-N,N′-bis(2-ethanesulfonic acid), pH 6.9, 5 mM ethylene glycol tetraacetic acid [EGTA], 0.9 M glycerol, 5 mM MgSO4, 0.1 mM EDTA, 0.5 mM dithiothreitol, and protease inhibitors; Hays et al., 1994). A high-speed supernatant was prepared by centrifugation of sample at 53,000 rpm for 1 h in a TLA120.2 rotor (Beckman Coulter, Brea, CA). Supernatant was then overlaid on a continuous 5–20% sucrose gradient and centrifuged at 35,000 rpm for 16.5 h in a TH641 rotor (Beckman Coulter). For sucrose density gradient analysis of HeLa cells, three 10-cm dishes (∼80% confluency) of GFP-Arfip–transfected cells were homogenized in 1 ml of buffer A (50 mM Tris, 150 mM NaCl, pH 7.4, and 0.5 mM EDTA) plus protease inhibitors and centrifuged sequentially for 10 min at 1000 × g, 10 min at 10,000 × g, and 50 min at 100,000 × g. The supernatant was overlaid on a continuous 5–20% sucrose gradient prepared in buffer A and centrifuged at 35,000 rpm for 16.5 h in a TH641 rotor. For all gradients, 1-ml fractions were collected, and total proteins were precipitated using trichloroacetic acid (TCA) and analyzed by SDS–PAGE and immunoblot. Protein molecular weight standards for 10S (amylase) and 17S (apoferritin) were run on a parallel gradient to monitor the fractionation profiles of 10S and 17S protein complexes, respectively. TCA precipitation was monitored by adding 5 μg of carbonic anhydrase to each fraction before precipitation and monitored with Ponceau S staining of blots.
Coimmunoprecipitation
For coimmunoprecipitation (coIP) two 80% confluent 10-cm dishes of cells cotransfected with pUAS-Glued-3XFLAG and pUAS-Arfip-3XHA were homogenized in cold IP buffer (IPB; 50 mM Tris, pH 7.4, 50 mM NaCl, 1% TX-100, 1 mM MgCl2, and protease inhibitors). Supernatant from the cell lysate was incubated with preblocked anti-FLAG–antibody coated beads (Sigma-Aldrich) and incubated at 4°C for 2 h on rotation. For FLAG peptide inhibition, excess amounts of FLAG peptides were added to bead incubation. Beads were extensively washed, and bound proteins were analyzed by standard SDS–PAGE and Western blot. For coIP of HA-Arfip and dynactin complexes from fly brains, a sucrose density gradient was first performed as described using ∼300 fly heads from either C155/+; UAS-Arfip-HA/+ or C155/+ control flies. Fractions corresponding to the 17S fractions (fractions 9–11) were collected and combined (∼3 ml). Buffer conditions of the 2.5 ml of combined fractions were supplemented (50 mM Tris, pH 7.4, 100 mM NaCl, 1% TX-100, 1 mM MgCl2, and protease inhibitors) and incubated overnight at 4°C with anti-HA coated beads (US Biologicals). The remaining 0.5 ml was TCA precipitated and reserved for immunoblot analysis. The bound protein material was washed and analyzed by SDS–PAGE and immunoblot. To perform CoIP of P150 and Arfaptin2, half of brain from 2-wk-old mice was homogenized in homogenizing buffer (0.32 M sucrose, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], 2 mM EDTA, pH 7.4). A high-speed supernatant was prepared by centrifugation of sample at 53,000 rpm for 45 min in a TLA120.2 rotor (Beckman Coulter). The resulting supernatant was incubated with either anti-P150 antibody–coated or control IgG-coated Dynabeads Protein G (Invitrogen) according to the manufacturer's protocol. The bound protein material was washed and analyzed by SDS–PAGE and immunoblot.
Membrane flotation
Membrane flotation assay was performed as previously described (Haghnia et al., 2007). Third-instar larvae were individually dissected and ∼200 brains were collected in dissection saline (70 mM NaCl, 20 mM MgCl2, 4.8 mM KCl, 5 mM HEPES, 10 mM NaHCO3, 0.1 M sucrose, 20 mM EGTA, and protease inhibitor). The brains were homogenized in acetate buffer (Haghnia et al., 2007). For flotation analysis of S2 cells, three to four 10-cm dishes of Arfip dsRNA–treated cells and non–dsRNA-treated control cells were homogenized in homogenization buffer (8% sucrose and 3 mM imidazole, pH 7.4). A postnuclear supernatant (PNS) was obtained by centrifuging at 1000 × g for 7 min in standard tabletop microcentrifuge (Labnet, Edison, NJ). The resulting PNS was brought to 40% sucrose, bottom loaded, and overlaid with two cushions of 35 and 8% sucrose. The gradient was centrifuged at 28,000 rpm for 1 h in a TH641 rotor (Beckman Coulter). The fractionation separates proteins into the following classes: light membranous organelles, soluble proteins, and heavy membranes. Equal amounts of protein from each fraction were analyzed by SDS–PAGE and immunoblotting. Protein quantification was performed from scanned autoradiographic films by measuring the pixel intensities of individual bands (corrected for background) with ImageJ (National Institutes of Health, Bethesda, MD). Background was determined by drawing an equal-sized rectangle near the band that was to be quantified. Only subsaturation bands were included in the analysis as determined by sample dilution.
Supplementary Material
Acknowledgments
We thank Rosario Martinez for excellent technical support and fly husbandry and Rebekah Mahoney and Efosa-Dimitar Iyinbor for help with large-scale ganglion isolations. Funding for this project was provided by an RO1 award (NS062811) to B.A.E. from the National Institute of Neurological Disorders and Stroke. L.C. was supported by Grant T32-AG021890 from the National Institute on Aging.
Abbreviations used:
- NMJ
neuromuscular junction
- TGN
trans-Golgi network
- VNC
ventral nerve cord
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-09-0697) on April 17, 2013.
REFERENCES
- Bowman AB, Patel-King RS, Benashski SE, McCaffery JM, Goldstein LS, King SM. Drosophila roadblock and Chlamydomonas LC7: a conserved family of dynein-associated proteins involved in axonal transport, flagellar motility, and mitosis. J Cell Biol. 1999;146:165–180. [PMC free article] [PubMed] [Google Scholar]
- Brankatschk B, Pons V, Parton RG, Gruenberg J. Role of SNX16 in the dynamics of tubulo-cisternal membrane domains of late endosomes. PloS One. 2011;6:e21771. doi: 10.1371/journal.pone.0021771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik V, Zhong Y, Wu CF. Morphological plasticity of motor axons in Drosophila mutants with altered excitability. J Neurosci. 1990;10:3754–3768. doi: 10.1523/JNEUROSCI.10-11-03754.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevalier-Larsen E, Holzbaur ELF. Axonal transport and neurodegenerative disease. Biochim Biophys Acta. 2006;1762:1094–1108. doi: 10.1016/j.bbadis.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen ES, Wallace KE, Pennise CR, Holzbaur ELF. Lysosomal proliferation and distal degeneration in motor neurons expressing the G59S mutation in the p150Glued subunit of dynactin. Hum Mol Genet. 2008;17:1946–1955. doi: 10.1093/hmg/ddn092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins CA, Wairkar YP, Johnson SL, DiAntonio A. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron. 2006;51:57–69. doi: 10.1016/j.neuron.2006.05.026. [DOI] [PubMed] [Google Scholar]
- Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, Krantz DE, DiAntonio A. Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J Neurosci. 2004;24:10466–10474. doi: 10.1523/JNEUROSCI.3001-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcroix JD, Valletta JS, Wu C, Hunt SJ, Kowal AS, Mobley WC. NGF signaling in sensory neurons: evidence that early endosomes carry NGF retrograde signals. Neuron. 2003;39:69–84. doi: 10.1016/s0896-6273(03)00397-0. [DOI] [PubMed] [Google Scholar]
- Duncan JE, Goldstein LSB. The genetics of axonal transport and axonal transport disorders. PLoS Genet. 2006;2:e124. doi: 10.1371/journal.pgen.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton BA, Davis GW. LIM kinase1 controls synaptic stability downstream of the type II BMP receptor. Neuron. 2005;47:695–708. doi: 10.1016/j.neuron.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Eaton BA, Fetter RD, Davis GW. Dynactin is necessary for synapse stabilization. Neuron. 2002;34:729–741. doi: 10.1016/s0896-6273(02)00721-3. [DOI] [PubMed] [Google Scholar]
- Engelender S, Sharp AH, Colomer V, Tokito MK, Lanahan A, Worley P, Holzbaur EL, Ross CA. Huntingtin-associated protein 1 (HAP1) interacts with the p150Glued subunit of dynactin. Hum Mol Genet. 1997;6:2205–2212. doi: 10.1093/hmg/6.13.2205. [DOI] [PubMed] [Google Scholar]
- Farrer MJ, et al. DCTN1 mutations in Perry syndrome. Nat Genet. 2009);41:163–165. doi: 10.1038/ng.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein AY, Wang X, Schwarz TL. Axonal transport and the delivery of pre-synaptic components. Curr Opin Neurobiol. 2008;18:495–503. doi: 10.1016/j.conb.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena S, Her L-S, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LSB. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Habermann B. The BAR-domain family of proteins: a case of bending and binding. EMBO Rep. 2004;5:250–255. doi: 10.1038/sj.embor.7400105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezparast M, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- Haghnia M, Cavalli V, Shah SB, Schimmelpfeng K, Brusch R, Yang G, Herrera C, Pilling A, Goldstein LSB. Dynactin is required for coordinated bidirectional motility, but not for dynein membrane attachment. Mol Biol Cell. 2007;18:2081–2089. doi: 10.1091/mbc.E06-08-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays TS, Porter ME, McGrail M, Grissom P, Gosch P, Fuller MT, McIntosh JR. A cytoplasmic dynein motor in Drosophila: identification and localization during embryogenesis. J Cell Sci. 1994;107:1557–1569. doi: 10.1242/jcs.107.6.1557. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- Holleran EA, Ligon LA, Tokito M, Stankewich MC, Morrow JS, Holzbaur EL. beta III spectrin binds to the Arp1 subunit of dynactin. J Biol Chem. 2001;276:36598–605. doi: 10.1074/jbc.M104838200. [DOI] [PubMed] [Google Scholar]
- Kanoh H, Williger BT, Exton JH. Arfaptin 1, a putative cytosolic target protein of ADP-ribosylation factor, is recruited to Golgi membranes. J Biol Chem. 1997;272:5421–5429. doi: 10.1074/jbc.272.9.5421. [DOI] [PubMed] [Google Scholar]
- Kardon JR, Vale RD. Regulators of the cytoplasmic dynein motor. Nat Rev Mol Cell Biol. 2009;10:854–865. doi: 10.1038/nrm2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch I, Schwarz H, Beuchle D, Goellner B, Langegger M, Aberle H. Drosophila ankyrin 2 is required for synaptic stability. Neuron. 2008;58:210–222. doi: 10.1016/j.neuron.2008.03.019. [DOI] [PubMed] [Google Scholar]
- Laird FM, Farah MH, Ackerley S, Hoke A, Maragakis N, Rothstein JD, Griffin J, Price DL, Martin LJ, Wong PC. Motor neuron disease occurring in a mutant dynactin mouse model is characterized by defects in vesicular trafficking. J Neurosci. 2008;28:1997–2005. doi: 10.1523/JNEUROSCI.4231-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M, Van Winkle T, Howland DS, Holzbaur EL. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- Lee J, Wu CF. Orchestration of stepwise synaptic growth by K+ and Ca2+ channels in Drosophila. J Neurosci. 2010;30:15821–15833. doi: 10.1523/JNEUROSCI.3448-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W-CM, Yoshihara M, Littleton JT. Cytoplasmic aggregates trap polyglutamine-containing proteins and block axonal transport in a Drosophila model of Huntington's disease. Proc Natl Acad Sci USA. 2004;101:3224–3229. doi: 10.1073/pnas.0400243101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JJ, Ding J, Kowal AS, Nardine T, Allen E, Delcroix JD, Wu C, Mobley W, Fuchs E, Yang Y. BPAG1n4 is essential for retrograde axonal transport in sensory neurons. J Cell Biol. 2003;163:223–229. doi: 10.1083/jcb.200306075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y-W, Lee S-W, Lee F-JS. Arl1p is involved in transport of the GPI-anchored protein Gas1p from the late Golgi to the plasma membrane. J Cell Sci. 2006;119:3845–3855. doi: 10.1242/jcs.03148. [DOI] [PubMed] [Google Scholar]
- Lorenzo DN, Li M-G, Mische SE, Armbrust KR, Ranum LPW, Hays TS. Spectrin mutations that cause spinocerebellar ataxia type 5 impair axonal transport and induce neurodegeneration in Drosophila. J Cell Biol. 2010;189:143–158. doi: 10.1083/jcb.200905158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnani E, Fan J, Gasparini L, Golding M, Williams M, Schiavo G, Goedert M, Amos LA, Spillantini MG. Interaction of tau protein with the dynactin complex. EMBO J. 2007;26:4546–4554. doi: 10.1038/sj.emboj.7601878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man Z, Kondo Y, Koga H, Umino H, Nakayama K, Shin H-W. Arfaptins are localized to the trans-Golgi by interaction with Arl1, but not Arfs. J Biol Chem. 2011;286:11569–11578. doi: 10.1074/jbc.M110.201442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders EMM, Verbeek FJ, Aten JA. Measurement of colocalization of objects in dual-colour confocal images. J Microsc. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- Marešová L, Sychrová H. Genetic interactions among the Arl1 GTPase and intracellular Na(+)/H(+) antiporters in pH homeostasis and cation detoxification. FEMS Yeast Res. 2010;10:802–811. doi: 10.1111/j.1567-1364.2010.00661.x. [DOI] [PubMed] [Google Scholar]
- Martin M, Iyadurai SJ, Gassman A, Gindhart JG, Jr, Hays TS, Saxton WM. Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport. Mol Biol Cell. 1999;10:3717–3728. doi: 10.1091/mbc.10.11.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe BD, Marques G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O'Connor MB. The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron. 2003;39:241–254. doi: 10.1016/s0896-6273(03)00426-4. [DOI] [PubMed] [Google Scholar]
- Münch C, et al. Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann Neurol. 2005;58:777–780. doi: 10.1002/ana.20631. [DOI] [PubMed] [Google Scholar]
- Münch C, et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology. 2004;63:724–726. doi: 10.1212/01.wnl.0000134608.83927.b1. [DOI] [PubMed] [Google Scholar]
- Munro S. The Arf-like GTPase Arl1 and its role in membrane traffic. Biochem Soc Trans. 2005;33:601–605. doi: 10.1042/BST0330601. [DOI] [PubMed] [Google Scholar]
- Munson AM, Haydon DH, Love SL, Fell GL, Palanivel VR, Rosenwald AG. Yeast ARL1 encodes a regulator of K+ influx. J Cell Sci. 2004;117:2309–2320. doi: 10.1242/jcs.01050. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Man Z, Xie Y, Hanai A, Makyio H, Kawasaki M, Kato R, Shin HW, Nakayama K, Wakatsuki S. Structural basis for membrane binding specificity of the Bin/amphiphysin/Rvs (BAR) domain of Arfaptin-2 determined by Arl1 GTPase. J Biol Chem. 2012;287:25478–25489. doi: 10.1074/jbc.M112.365783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packard M, Koo ES, Gorczyca M, Sharpe J, Cumberledge S, Budnik V. The Drosophila Wnt, wingless, provides an essential signal for pre- and postsynaptic differentiation. Cell. 2002;111:319–330. doi: 10.1016/s0092-8674(02)01047-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pack-Chung E, Kurshan PT, Dickman DK, Schwarz TL. A Drosophila kinesin required for synaptic bouton formation and synaptic vesicle transport. Nat Neurosci. 2007;10:980–989. doi: 10.1038/nn1936. [DOI] [PubMed] [Google Scholar]
- Papoulas O, Hays TS, Sisson JC. The golgin Lava lamp mediates dynein-based Golgi movements during Drosophila cellularization. Nat Cell Biol. 2005;7:612–618. doi: 10.1038/ncb1264. [DOI] [PubMed] [Google Scholar]
- Parkes TL, Elia AJ, Dickinson D, Hilliker AJ, Phillips JP, Boulianne GL. Extension of Drosophila lifespan by overexpression of human SOD1 in motorneurons. Nat Genet. 1998;19:171–174. doi: 10.1038/534. [DOI] [PubMed] [Google Scholar]
- Peru Y Colón de Portugal RL, Acevedo SF, Rodan AR, Chang LY, Eaton BA, Rothenfluh A. Adult neuronal Arf6 controls ethanol-induced behavior with Arfaptin downstream of Rac1 and RhoGAP18B. J Neurosci. 2012;322:17706–17713. doi: 10.1523/JNEUROSCI.1944-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJG, Evans PR, McMahon HT. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- Pielage J, Fetter RD, Davis GW. Presynaptic spectrin is essential for synapse stabilization. Curr Biol. 2005;15:918–928. doi: 10.1016/j.cub.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Puls I, et al. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann Neurol. 2005;57:687–694. doi: 10.1002/ana.20468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repnikova E, Koles K, Nakamura M, Pitts J, Li H, Ambavane A, Zoran MJ, Panin VM. Sialyltransferase regulates nervous system function in Drosophila. J Neurosci. 2010;30:6466–6476. doi: 10.1523/JNEUROSCI.5253-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinow S, White K. The locus elav of Drosophila melanogaster is expressed in neurons at all developmental stages. Dev Biol. 1988;126:294–303. doi: 10.1016/0012-1606(88)90139-x. [DOI] [PubMed] [Google Scholar]
- Rogers SL, Rogers GC. Culture of Drosophila S2 cells and their use for RNAi-mediated loss-of-function studies and immunofluorescence microscopy. Nat Protoc. 2008;3:606–611. doi: 10.1038/nprot.2008.18. [DOI] [PubMed] [Google Scholar]
- Rørth P, et al. Systematic gain-of-function genetics in Drosophila. Development. 1998;125:1049–1057. doi: 10.1242/dev.125.6.1049. [DOI] [PubMed] [Google Scholar]
- Rosenwald AG, Rhodes MA, Van Valkenburgh H, Palanivel V, Chapman G, Boman A, Zhang C-J, Kahn RA. ARL1 and membrane traffic in Saccharomyces cerevisiae. Yeast. 2002;19:1039–1056. doi: 10.1002/yea.897. [DOI] [PubMed] [Google Scholar]
- Sanyal S. Genomic mapping and expression patterns of C380, OK6 and D42 enhancer trap lines in the larval nervous system of Drosophila. Gene Expr Patterns. 2009;9:371–380. doi: 10.1016/j.gep.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Shin OH, Exton JH. Differential binding of Arfaptin2/POR1 to ADP-ribosylation factors and Rac1. Biochem Biophys Res Commun. 2001;285:1267–73. doi: 10.1006/bbrc.2001.5330. [DOI] [PubMed] [Google Scholar]
- Susalka SJ, Hancock WO, Pfister KK. Distinct cytoplasmic dynein complexes are transported by different mechanisms in axons. Biochim Biophys Acta. 2000;1496:76–88. doi: 10.1016/s0167-4889(00)00010-0. [DOI] [PubMed] [Google Scholar]
- Tarricone C, Xiao B, Justin N, Walker PA, Rittinger K, Gamblin SJ, Smerdon SJ. The structural basis of Arfaptin-mediated cross-talk between Rac and Arf signalling pathways. Nature. 2001;411:215–219. doi: 10.1038/35075620. [DOI] [PubMed] [Google Scholar]
- Temkin P, Lauffer B, Jäger S, Cimermancic P, Krogan NJ, von Zastrow M. SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signalling receptors. Nat Cell Biol. 2011;13:715–721. doi: 10.1038/ncb2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teuling E, van Dis V, Wulf PS, Haasdijk ED, Akhmanova A, Hoogenraad CC, Jaarsma D. A novel mouse model with impaired dynein/dynactin function develops amyotrophic lateral sclerosis (ALS)-like features in motor neurons and improves lifespan in SOD1-ALS mice. Hum Mol Genet. 2008;17:2849–2862. doi: 10.1093/hmg/ddn182. [DOI] [PubMed] [Google Scholar]
- Torroja L, Packard M, Gorczyca M, White K, Budnik V. The Drosophila beta-amyloid precursor protein homolog promotes synapse differentiation at the neuromuscular junction. J Neurosci. 1999;19:7793–7803. doi: 10.1523/JNEUROSCI.19-18-07793.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Valkenburgh H, Shern JF, Sharer JD, Zhu X, Kahn RA. ADP-ribosylation factors (ARFs) and ARF-like 1 (ARL1) have both specific and shared effectors. J Biol Chem. 2001;276:22826–22837. doi: 10.1074/jbc.M102359200. [DOI] [PubMed] [Google Scholar]
- van Weering JR, Verkade P, Cullen PJ. SNX-BAR-mediated endosome tubulation is co-ordinated with endosome maturation. Traffic. 2012;13:94–107. doi: 10.1111/j.1600-0854.2011.01297.x. [DOI] [PubMed] [Google Scholar]
- Wang X, Shaw WR, Tsang HTH, Reid E, O'Kane CJ. Drosophila spichthyin inhibits BMP signaling and regulates synaptic growth and axonal microtubules. Nat Neurosci. 2007;10:177–185. doi: 10.1038/nn1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassmer T, et al. The retromer coat complex coordinates endosomal sorting and dynein-mediated transport, with carrier recognition by the trans-Golgi network. Dev Cell. 2009);17:110–122. doi: 10.1016/j.devcel.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, Holzbaur ELF. The product of the Drosophila gene, Glued, is the functional homologue of the P150Glued component of the vertebrate dynactin complex. J Biol Chem. 1996;271:1153–1159. doi: 10.1074/jbc.271.2.1153. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Wang F, Cao J, Shen Y, Huang Q, Bao L, Zhu X. Nudel promotes axonal lysosome clearance and endo-lysosome formation via dynein-mediated transport. Traffic. 2009;10:1337–1349. doi: 10.1111/j.1600-0854.2009.00945.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.