

The AMPKβ2 subunit can be modified by sumoylation carried out by the E3-SUMO ligase PIASy, which attaches SUMO2 but not SUMO1 moieties. This posttranslational modification is specific to AMPKβ2 and enhances the activity of the AMPK complex. Sumoylation of AMPKβ2 is antagonistic and competes with ubiquitination of the same subunit.
Abstract
AMP-activated protein kinase (AMPK) is a sensor of cellular energy status. It is a heterotrimer composed of a catalytic α and two regulatory subunits (β and γ). AMPK activity is regulated allosterically by AMP and by the phosphorylation of residue Thr-172 within the catalytic domain of the AMPKα subunit by upstream kinases. We present evidence that the AMPKβ2 subunit may be posttranslationally modified by sumoylation. This process is carried out by the E3-small ubiquitin-like modifier (SUMO) ligase protein inhibitor of activated STAT PIASy, which modifies the AMPKβ2 subunit by the attachment of SUMO2 but not SUMO1 moieties. Of interest, AMPKβ1 is not a substrate for this modification. We also demonstrate that sumoylation of AMPKβ2 enhances the activity of the trimeric α2β2γ1 AMPK complex. In addition, our results indicate that sumoylation is antagonist and competes with the ubiquitination of the AMPKβ2 subunit. This adds a new layer of complexity to the regulation of the activity of the AMPK complex, since conditions that promote ubiquitination result in inactivation, whereas those that promote sumoylation result in the activation of the AMPK complex.
INTRODUCTION
AMP-activated protein kinase (AMPK) is a serine/threonine protein kinase that acts as a sensor of cellular energy status. It is a heterotrimer composed of three different subunits: α, β, and γ. AMPKα is the catalytic subunit of the AMPK complex; it contains a highly conserved kinase domain located at the N-terminus of the protein, followed by an autoinhibitory domain (Pang et al., 2007) and a C-terminal domain required for interaction with the AMPKβ subunit (Xiao et al., 2007). Two isoforms of the catalytic subunit have been described, α1 and α2. The AMPKγ subunit contains four repeats in tandem of a structural module called a CBS motif, described initially in the enzyme cystathionine β-synthase (Bateman, 1997), involved in AMP binding (Scott et al., 2004; Xiao et al., 2007). Three isoforms have been described for this subunit: γ1, γ2, and γ3. Finally, the AMPKβ subunit functions as a scaffold to assemble α and γ subunits and also may affect subcellular localization and substrate specificity of the complex. Two isoforms of the β-subunit (β1 and β2) have been described; they differ in their N-terminal regions, but they interact with the same efficiency with AMPKα and AMPKγ subunits (Gimeno-Alcaniz and Sanz, 2003). AMPK activity is regulated by allosteric activation by AMP and the phosphorylation of Thr-172 residue within the catalytic domain of α subunit by upstream kinases (for review see Sanz, 2008; Carling et al., 2012). In addition to allosteric regulation, AMP prevents the action of phosphatases that promote dephosphorylation of the Thr-172 residue by making AMPK a poorer substrate for these phosphatases (Davies et al., 1995; Sanders et al., 2007).
Although phosphorylation of AMPK is key to regulating AMPK activity, recently other posttranslational modifications have been described that affect the function of the AMPK complex. The AMPKα catalytic subunit may be acetylated by p300 acetyltransferase. Acetylation of AMPKα regulates the interaction between AMPK and the upstream kinase LKB1, providing a direct mechanism for regulating phosphorylation of AMPK by LKB1 (Lin et al., 2012).
In addition, recent studies have identified different mechanisms by which the ubiquitin proteasome system regulates AMPK activity. Ubiquitination of AMPKα1 has been reported. Of interest, the polyubiquitin chains present were made by atypical (K29 and K33) ubiquitin linkages, but the possible effect of this modification on AMPK activity was not studied (Al-Hakim et al., 2008). Other work indicated that AMPKβ subunits were ubiquitinated by a cell death–inducing, DFFA-like effector A (CIDEA)–dependent E3-ubiquitin ligase, which targeted the AMPKβ subunits for proteasomal degradation (Qi et al., 2008). Our group also demonstrated the ubiquitination of the AMPKβ subunits carried out by a complex formed by the glucan phosphatase laforin and the E3-ubiquitin ligase malin. However, in this case the modification did not target the AMPKβ subunits for proteasomal degradation, since the polyubiquitin chains were mainly made by K63-ubiquitin linkages, with this modification related to nondegradative functions (Moreno et al., 2010).
In this article, we present evidence that sumoylation is a novel posttranslational modification of AMPK complex. Sumoylation consists in a reversible covalent attachment of a member of the small ubiquitin-like modifier (SUMO) family of ubiquitin-related peptides to lysine residues in target proteins in a process similar to ubiquitination (Gareau and Lima, 2010; Wilkinson and Henley, 2010). Whereas yeast possesses a single SUMO protein, Smt3p, there are three human SUMO paralogues: SUMO1, SUMO2, and SUMO3. SUMO2 and SUMO3 are 96% identical to each other and are functionally redundant. However SUMO1 is only 45% identical to SUMO2/3 and has different properties when attached to a particular substrate (Gareau and Lima, 2010; Wilkinson and Henley, 2010). Of interest, SUMO2 and SUMO3 can form SUMO chains, whereas SUMO1 is only able to form monosumoylated derivatives, or it can be attached to the end of a SUMO2/3 chain (Tatham et al., 2005; Hay, 2007; Gareau and Lima, 2010). The sumoylation reaction requires the SUMO-activating E1 enzyme Aos1/Uba2 and the SUMO-conjugating E2 enzyme Ubc9 (Gareau and Lima, 2010; Wilkinson and Henley, 2010). Ubc9 recognizes a minimal sumoylation motif in many known targets (ΨKXD/E, with Ψ any hydrophobic residue and X any residue). Although target proteins interact at least transiently with Ubc9, their efficient conjugation often requires E3-SUMO ligases (Muller et al., 2004). There are three types of E3-SUMO ligase (Muller et al., 2004): RanBP2/Nup358 (Pichler et al., 2004), the Polycomb member Pc2 (Kagey et al., 2003), and the protein inhibitor of activated STAT (PIAS) family, which were originally identified as transcriptional coregulators of the JAK-STAT pathway (Rytinki et al., 2009). Mammals have at least five members in the PIAS family: PIAS1, PIAS3, PIASxα, PIASxβ, and PIASy (Rytinki et al., 2009).
In contrast to ubiquitination, poly-SUMO modification does not usually trigger protein degradation. Instead, sumoylation may affect subcellular localization, protein–protein interaction, transcriptional activation, and protein stability of the corresponding target (Gareau and Lima, 2010; Wilkinson and Henley, 2010). Owing to the great similarities between ubiquitination and sumoylation systems, a growing number of proteins are being described as substrates for both ubiquitination and sumoylation, sometimes in the same lysine residues. These two types of posttranslational modification are sometimes viewed as antagonists and control the properties of the targeted protein in a competitive manner (Ulrich, 2005; Wilson and Heaton, 2008; Bergink and Jentsch, 2009). For example, sumoylation on the Lys-21 residue of IκBα protein blocks its ubiquitination and degradation by direct competition for the modification site (Desterro et al., 1998), and sumoylation of aryl hydrocarbon receptor modulates its activity and stability by inhibiting its ubiquitination (Xing et al., 2012).
In this work we show that AMPKβ2 but not AMPKβ1 subunit can be modified by sumoylation, with this process being carried out by the E3-SUMO ligase PIASy. This posttranslational modification enhances AMPK total activity and protects the AMPKβ2 subunit from being modified by ubiquitination.
RESULTS
AMPK interacts physically with PIASy
In a previous yeast two-hybrid screening using AMPKβ2 as bait, we identified PIASy as a putative interaction partner. To study the possible functional relationship between AMPK and PIASy, we quantified by yeast two-hybrid assay the interaction between different AMPK subunits and PIASy. In the assays, we used the catalytic AMPKα2 subunit, the scaffolding AMPKβ1 and β2 subunits, and the regulatory AMPKγ1. As shown in Figure 1A, we detected a robust two-hybrid interaction between PIASy and α2 catalytic and β1 and β2 regulatory subunits of the AMPK complex but not with the regulatory AMPKγ1 subunit. To confirm the physical interaction between AMPK subunits and PIASy, we coexpressed in mammalian HEK293 cells FLAG-PIASy and a combination of AMPKα2, AMPKβ2, and AMPKγ1 in order to produce a heterotrimeric AMPK complex. As shown in Figure 1B, when FLAG-PIASy was immunoprecipitated with anti-FLAG antibodies, AMPKα2, AMPKβ2, and AMPKγ1 were also recovered in the immunoprecipitates, indicating that the interaction of PIASy with AMPK subunits was able to immunoprecipitate the whole AMPK complex. In the same way, if myc-AMPKβ2 was immunoprecipitated using antibodies against the protein tag (myc), in addition to the other two AMPK subunits (AMPKα2 and AMPKγ1, which confirmed the formation of heterotrimeric complexes upon overexpression of the three subunits), we also recovered FLAG-PIASy in the immunoprecipitates (Supplemental Figure S1). To confirm the interaction at endogenous levels, we used human U2OS cells, since they express the highest levels of endogenous AMPKβ2 subunit among all cell types tested (HEK293, CHO, COS7, SH-SY5Y, HeLa; data not shown). These cells were treated with MG132 (a proteasomal inhibitor) to prevent the degradation of endogenous PIASy, and then cell extracts were immunoprecipitated using anti-PIASy antibodies. As shown in Figure 1C, endogenous AMPK subunits (α, β1, β2, and γ1) were recovered in the immunoprecipitate, but this reaction did not occur when preimmune serum was used in the assay. These results confirmed the physical interaction between the AMPK complex and PIASy.
FIGURE 1:
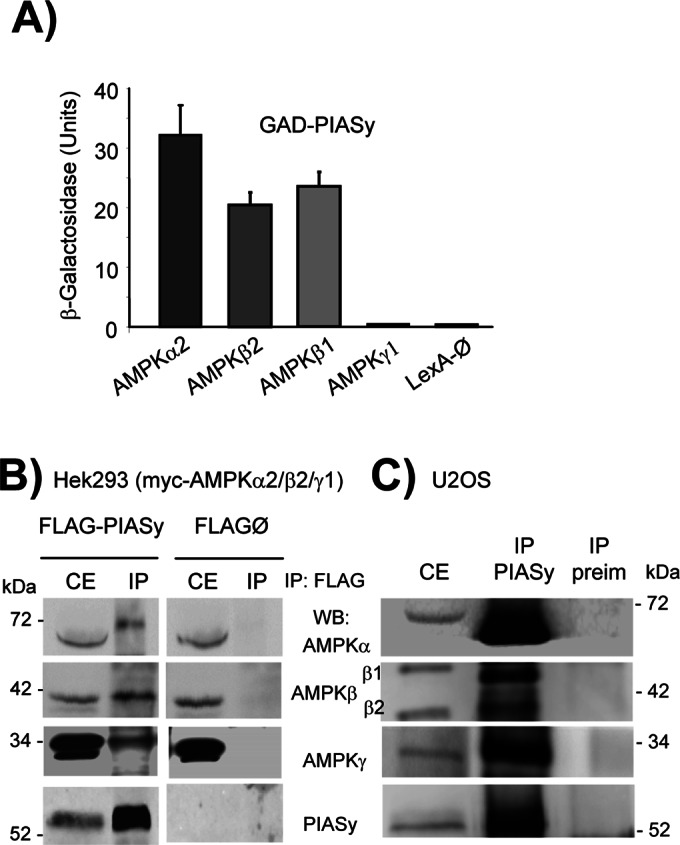
AMPK interacts physically with PIASy. (A) Yeast two-hybrid analysis. Yeast CTY10.5d cells transformed with plasmid pBTM116 (empty, ∅), pBTM-AMPKα2, pBTM-AMPKβ2, or pBTM-AMPKγ1 were cotransformed with plasmid pACT2-PIASy. Protein interaction was estimated by using the yeast two-hybrid system by measuring the β-galactosidase activity. Values correspond to means from four to six different transformants (bars, ±SD). (B) The three subunits of the AMPK complex coimmunoprecipitate with PIASy. Protein extracts (1.2 mg) were prepared from HEK293 cells cotransfected with plasmids pCMV-myc-AMPKα2/β2/γ1 and pFLAG-PIASy or pFLAG (empty, ∅). One microliter of anti-FLAG was used to immunoprecipitate the extracts (IP). Pelleted proteins and proteins in the input crude extracts (CE; 40 μg) were analyzed by SDS–PAGE and immunodetected (WB) with anti-AMPKα, anti-AMPKβ, anti-AMPKγ, and anti-PIASy antibodies. Molecular-size markers are indicated. (C) Human U2OS cells were treated with 6 μM MG132 for 8 h to prevent the degradation of endogenous PIASy. Then protein extracts (1.2 mg) were immunoprecipitated with 1 μl of anti-PIASy (IP PIASy) or preimmune serum (IP preim). Pelleted proteins and proteins in the input crude extracts (CE; 40 μg) were analyzed by SDS–PAGE and immunodetected as in B.
PIASy, an E3-SUMO ligase, promotes SUMO2-specific sumoylation of AMPKβ2 subunit
Because PIASy is a member of the PIAS family of E3-SUMO ligases (Rytinki et al., 2009), we decided to examine whether PIASy was able to promote the sumoylation of the AMPK subunits. The sumoylation event can be carried out by the addition of different SUMO isoforms, with SUMO1, SUMO2, and SUMO3 the most abundant moieties. Because SUMO2 and SUMO3 are highly similar (Rytinki et al., 2009), we set up an assay for the analysis of sumoylated proteins based on the expression of a hexahistidine (6xHis)-tagged version of SUMO1 or SUMO2. Cell extracts were prepared in the presence of the chaotropic agent guanidinium hydrochloride to inhibit desumoylating enzymes, and sumoylated proteins were purified by metal-affinity chromatography. This purified fraction was analyzed by SDS–PAGE and Western blotting using specific antibodies against the protein of interest. We coexpressed in HEK293 cells the three AMPK complex subunits (α2, β2, and γ1) and 6xHis-SUMO1 or 6xHis-SUMO2 in the presence or absence of FLAG-PIASy. As shown in Figure 2A, PIASy was not able to promote the sumoylation of AMPKα2 in any condition (the observed band corresponds to nonspecific binding of AMPKα2 to the column). However, we observed an efficient PIASy-dependent sumoylation of AMPKβ2 subunit, but only when 6xHis-SUMO2 was present (lack of sumoylation using SUMO1 was not due to a deficient SUMO1 moiety, since the same construct allowed an efficient sumoylation of p53; data not shown). Because we observed a two-hybrid interaction of PIASy with both AMPKβ1 and β2 subunits (Figure 1A), we extended our analysis of PIASy-dependent sumoylation to the AMPKβ1 subunit. Surprisingly, we found that PIASy was not able to promote the sumoylation of this isoform with either SUMO1 or SUMO2 (Figure 2B). To discard the possibility that sumoylation of the AMPKβ2 subunit could be nonspecific and independent of PIASy activity, we repeated the experiment by expressing a form of PIASy with decreased activity (PIASy-CI, containing a double C342A/C347A mutation in the catalytic site (Mabb et al., 2006), which diminishes its activity). As shown in Figure 2C, PIASy-CI was not able to carry out an efficient sumoylation of AMPKβ2 subunit compared with wild-type PIASy. Finally, we analyzed the PIASy-dependent sumoylation of the endogenous AMPK complex. Human U2OS cells transfected with plasmids expressing 6xHis-SUMO2 in the presence or absence of FLAG-PIASy were treated with 6 μM MG132 for 8 h (a treatment that improves the sumoylation reaction since, as indicated earlier, by inhibiting proteasome activity, it prevents PIASy degradation). Then sumoylated proteins were purified as described. As shown in Figure 2D, PIASy was also able to promote the sumoylation of endogenous AMPKβ.
FIGURE 2:

PIASy promotes the sumoylation of AMPKβ2 subunit. (A) PIASy promotes the SUMO2-dependent sumoylation of AMPKβ2 but does not affect AMPKα2. HEK293 cells were cotransfected with plasmids pCMV-6xHis-SUMO1 or pCMV-6xHis-SUMO2, pCMV-myc-AMPKα2/β2/γ1, and pFLAG-PIASy or pFLAG (empty). Cell extracts were obtained and sumoylated proteins purified by metal-affinity chromatography as described in Materials and Methods. Clarified extract (CE; 40 μg) and the material bound to the metal-affinity chromatography column (Bound) was analyzed by SDS–PAGE and Western blotting using anti-AMPKα (left) and anti-AMPKβ (right) antibodies. (B) PIASy does not promote the sumoylation of the AMPKβ1 subunit. HEK293 cells were transfected with plasmids pCMV-6xHis-SUMO1 or pCMV-6xHis-SUMO2, pCMV-myc-AMPKα2/γ1, pCMV-myc-AMPKβ1 instead of AMPKβ2, and pFLAG-PIASy or pFLAG (empty). Cell extracts were analyzed as described using anti-AMPKβ antibody. (C) Sumoylation of AMPKβ2 is dependent on PIASy activity. HEK293 cells were cotransfected with plasmids pCMV-6xHis-SUMO2, pCMV-myc-AMPKα2/β2/γ1, and pFLAG-PIASy or pFLAG-PIASy-CI (expressing a form of PIASy with reduced catalytic activity) or pFLAG (empty). Cell extracts were analyzed as described using anti-AMPKβ antibody. (D) Human U2OS cells were transfected with plasmids pCMV-6xHis-SUMO2 and pFLAG-PIASy or pFLAG (empty) and treated with 6 μM MG132 for 8 h. Cell extracts were analyzed as described using anti-AMPKβ antibody. (E) Sumoylation of free AMPKβ2 subunit is independent of PIASy expression. HEK293 cells were cotransfected with plasmids pCMV-6xHis-SUMO2, pCMV-myc-AMPKβ2, and pFLAG-PIASy or pFLAG (empty). Cell extracts were analyzed as described using anti-AMPKβ antibody. Molecular-size markers are indicated on the side; the presence of poly/multisumoylated proteins is indicated.
Free AMPK subunits are less stable than when they form part of a heterotrimeric complex (Crute et al., 1998). Because we reported that ubiquitination, a posttranslational modification similar to sumoylation, affects AMPK subunits in a different way depending on whether they form part of a heterotrimeric complex or are expressed as free subunits (Moreno et al., 2010), we analyzed the sumoylation of AMPKβ2 subunit in cells overexpressing only this subunit. As shown in Figure 2E, sumoylation of free AMPKβ2 subunit occurred, but it was independent of PIASy. This result confirms previous observations that individual subunits of the AMPK complex may follow alternative routes of posttranslational modifications that render them more unstable.
Taken together, these results indicate that PIASy can promote the selective sumoylation of AMPKβ2 subunit in the AMPK complex and that this modification is specific for SUMO2 isoform.
Sumoylation frequently occurs on a lysine residue within a ΨKXD/E consensus sequence, with Ψ any hydrophobic residue and X any residue (Gareau and Lima, 2010; Wilkinson and Henley, 2010). To identify the lysine residues sumoylated in AMPKβ2 by PIASy, we initially followed a bioinformatics approach using the SUMOsp-SUMOylation Sites Prediction program (Ren et al., 2009). This program identified two putative type II nonconsensus sites for sumoylation: DSVK71PTQ and DALK167LDS. We mutated lysine residues 71 and 167 to arginine (AMPKβ2 K71R, AMPKβ2 K167R, and double mutant AMPKβ2 K71R/K167R) and coexpressed these mutants with the rest of AMPK subunits (AMPKα2, AMPKγ1) to make a heterotrimeric complex and with 6xHis-SUMO2 and FLAG-PIASy. As shown in Figure 3, when the AMPKβ2 mutated forms were used in the assay, a clear PIASy-dependent sumoylation of these modified subunits was detected. These results suggest that these residues are not major sites of sumoylation. The results presented in Figure 3 also suggest that sumoylation of the mutated forms is more efficient than for wild type. Perhaps the introduction of these mutations produces a conformational change in the protein that makes other lysine residues more accessible for sumoylation.
FIGURE 3:
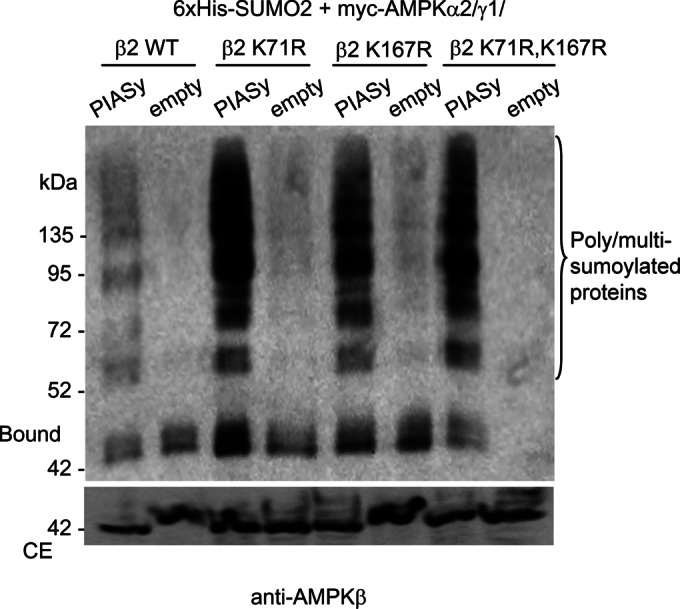
Mutation in consensus sumoylation sites of AMPKβ2 does not decrease its modification. HEK293 cells were cotransfected with plasmids pCMV-6xHis-SUMO2, pFLAG-PIASy or pFLAG (empty), pCMV-myc-AMPKα2/γ1 and pCMV-myc-AMPKβ2 or pCMV-myc-AMPKβ2 K71R, or pCMV-myc-AMPK K167R or the double mutant pCMV-myc-AMPKβ2 K71R, K167R. Cell extracts were analyzed as described in Figure 2 by using anti-AMPKβ antibody. Molecular-size markers are indicated on the left. Presence of poly/multisumoylated proteins is indicated.
To identify the lysine residues involved in the sumoylation process, and due to the fact that AMPKβ2 contains 20 lysine residues in its protein sequence, we decided to map the regions involved in the PIASy-dependent sumoylation by constructing two truncated forms of AMPKβ2, one containing an N-terminal fragment (residues 1–185; molecular weight, 20 kDa) comprising the glycogen-binding domain (GBD) and the other including the C-terminal region (residues 186–272; 10 kDa) containing the so-called “association with Snf1 complex domain” (ASC) involved in binding to both AMPKα and AMPKγ subunits. As shown in Figure 4A, when we coexpressed these forms with the rest of the AMPK subunits (AMPKα and AMPKγ), we detected sumoylated products in the ASC fragment in the presence of FLAG-PIASy. The molecular weight of these modified products corresponded to the incorporation of one to five SUMO2 moieties (11 kDa each). However, in the case of the GBD fragment, although the sumoylation reaction also occurred, it was independent of PIASy coexpression, probably because, since this fragment has lost its capacity to interact with AMPKα2 and AMPKγ1 to make a trimeric complex (as reported in Iseli et al., 2005), its sumoylation becomes PIASy independent, as indicated earlier for the sumoylation of free AMPKβ2 subunit (Figure 2E). To find which lysine residue of the ASC domain was sumoylated, and because we indicated earlier that only AMPKβ2 but not AMPKβ1 follows a PIASy-dependent sumoylation, we compared the protein sequence of AMPKβ1 and AMPKβ2 in order to find lysine residues present in AMPKβ2 but not in AMPKβ1 (Figure 4B). The ASC domain contained six lysine residues present in both AMPKβ1 and AMPKβ2, but K203 was present only in AMPKβ2 (instead, an arginine residue is present in AMPKβ1; Figure 4B). We mutated this residue to arginine (K203R), but the truncated form containing this mutation became very unstable, precluding further analysis. Thus we analyzed the sumoylation of a full-length AMPKβ2 K203R mutant and observed that it was still able to support a PIASy-dependent sumoylation (Figure 5). This result indicated that AMPKβ2, in addition to a putative sumoylation site at the C-terminus, should contain multiple sumoylation sites at the N-terminus. In fact, when we compared the protein sequences of AMPKβ1 and AMPKβ2, we observed four lysine residues in the GBD domain of AMPKβ2 that were absent in the corresponding domain of AMPKβ1 (K31-D, K57-E, K107-R, and K167-M; Figure 4B).
FIGURE 4:
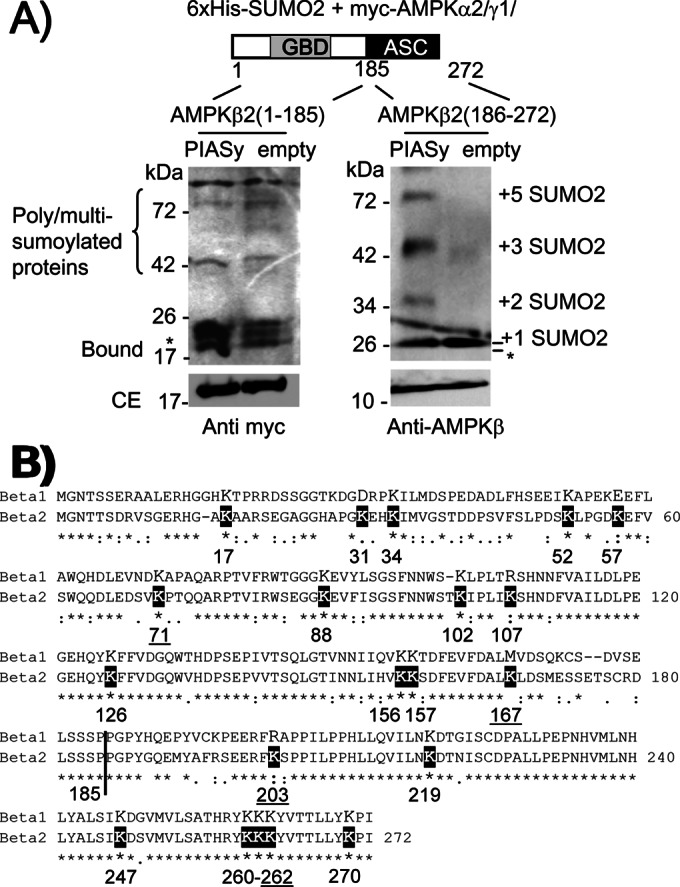
PIASy is able to promote the sumoylation of a C-terminal fragment of AMPKβ2. (A) HEK293 cells were cotransfected with pCMV-6xHis-SUMO2, pCMV-myc-AMPKα2/γ1, and pCMV-myc-AMPKβ2 (1–185), expressing the N-terminal domain, or pCMV-myc-AMPKβ2 (186–272), expressing the C-terminal domain, and pFLAG-PIASy or pFLAG (empty). Cell extracts were analyzed as described in Figure 2 by using anti-AMPKβ or anti-myc antibodies. The calculated size of SUMO-modified forms is indicated. Asterisk indicates possible degradation products. ASC, association with Snf1 complex domain; GBD, glycogen-binding domain. The presence of poly/multisumoylated proteins is indicated. (B) Alignment of human AMPKβ1 vs. AMPKβ2 protein sequences. The lysine residues in AMPKβ2 are indicated in a black box. Lysine residues mutated to arginine analyzed in this work are underlined.
FIGURE 5:
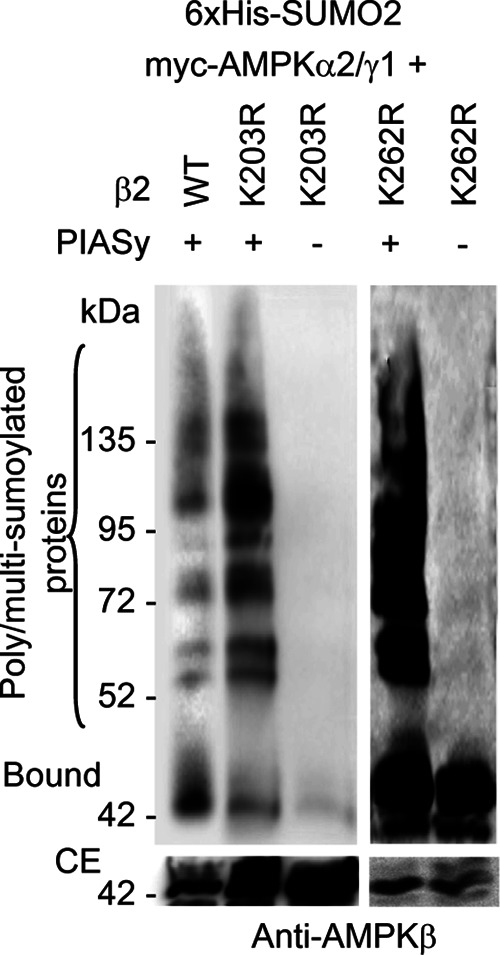
PIASy-dependent sumoylation of AMPKβ2 K203R and K262R mutants. HEK293 cells were transfected with plasmids pCMV-6xHis-SUMO2, pFLAG-PIASy (+) or pFLAG (–), pCMV-myc-AMPKα2/γ1, and pCMV-myc-AMPKβ2 or pCMV-myc-AMPKβ2 K203R or pCMV-myc-AMPKβ2 K262R mutants. Cell extracts were analyzed as described in Figure 2 by using anti-AMPKβ antibodies. The presence of poly/multisumoylated proteins is indicated.
In addition to the aforementioned mutant, by serendipity we also constructed the AMPKβ2 K262R mutant, affecting one lysine residue at the C-terminal part of the protein (Figure 4B). When we analyzed the PIASy-dependent sumoylation of this mutant we observed that the efficiency of the modification was higher than for wild type (Figure 5) and the highest among the mutants tested in this work. For this reason we considered it as a PIASy-dependent “hypersumoylable” mutant. Perhaps the change of this lysine residue by an arginine modified the conformation of the protein, making it more accessible to sumoylation.
Effect of PIASy-dependent sumoylation of AMPKβ2 on the stability and function of the AMPK complex
We next studied the effect of the PIASy-dependent sumoylation of AMPKβ2 on the stability and function of the AMPK complex. We first analyzed whether the sumoylation event affected the half-life of the protein. With this aim, we expressed in HEK293 cells the heterotrimeric AMPK complex (AMPKα2/β2/γ1) and 6xHis-SUMO2 in the presence or absence of FLAG-PIASy. Then we treated cells with cycloheximide to inhibit new protein synthesis and measured degradation rates of the protein by Western blot. As shown in Figure 6, PIASy-dependent sumoylation of AMPKβ2 did not affect the half-life of this protein. Therefore the sumoylation of AMPKβ2 does not affect the stability of this protein.
FIGURE 6:
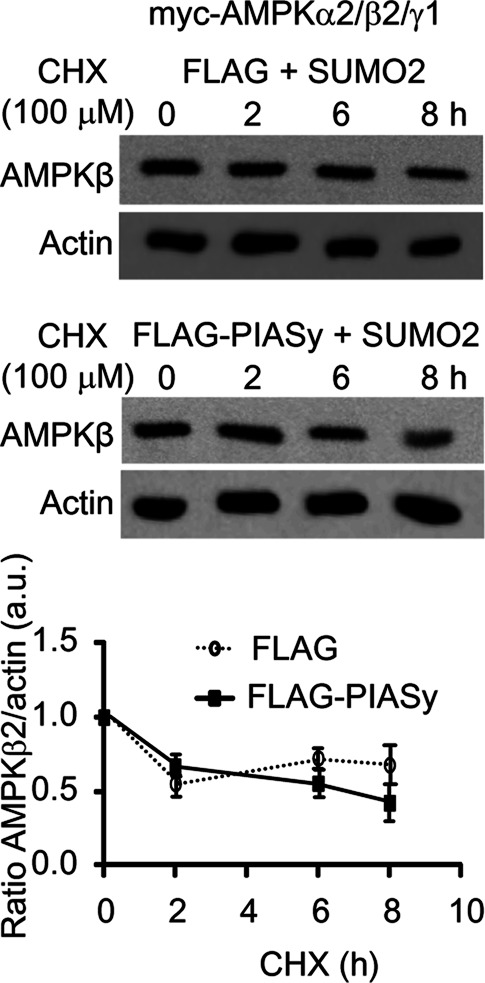
Degradation rates of AMPKβ2 in the presence of PIASy and SUMO2. HEK293 cells were cotransfected with plasmids pCMV-myc-AMPKα2/β2/γ1, pCMV-6xHis-SUMO2, and pFLAG-PIASy or pFLAG (empty). Twenty-four hours after transfection, cycloheximide (CHX; 100 μM) was added to the cultures, and cell extracts were analyzed by Western blotting at the indicated times, using anti-AMPKβ (top) and anti-actin (bottom) antibodies. The relative intensity of the AMPKβ2 bands with respect to the corresponding actin band is plotted as a function of time. Diagram shows means of three independent experiments; bars, SD.
We also studied whether sumoylation affected the subcellular distribution of AMPKβ2. Consistent with previous results (Moreno et al., 2010), the expression in HEK293 cells of the heterotrimeric AMPK complex (α2/β2/γ1) resulted in a spotted distribution of the AMPKβ2 protein throughout the cell (Figure 7A, top). However, the coexpression of PIASy and SUMO2 produced a distinct pattern of cytoplasmic distribution: AMPKβ2 subunit was detected in intracellular deposits dispersed through the cells (Figure 7A, bottom). On the contrary, PIASy showed a prominent nuclear localization (Figure 7A, bottom). We repeated the same experiments with endogenous levels of AMPK complex but were unable to detect any alteration of AMPK localization upon PIASy overexpression, probably due to the low levels of endogenous AMPKβ2 (data not shown). Given that the AMPK complex travels continuously between the nucleus and the cytosol (Kazgan et al., 2010; Supplemental Figure S2A), it is probable that AMPKβ2 is modified by PIASy when it is in the nuclear compartment and then is exported to the cytosol, where accumulates in the form of cytoplasmic deposits. Consistent with this hypothesis, treatment of cells with leptomycin B, an inhibitor of the CRM1-dependent nuclear protein export pathway, resulted in the appearance of AMPKβ2-containing granules inside the nucleus (Supplemental Figure S2B).
FIGURE 7:

PIASy-dependent sumoylation of AMPKβ2 promotes its aggregation. (A) HEK293 cells were cotransfected with plasmids expressing the AMPK subunits (pCMV-myc-AMPKα2/β2/γ1), pCMV-6xHis-SUMO2, and pFLAG-PIASy or pFLAG empty vector. The subcellular localization of AMPKβ2 subunit was carried out as described in Materials and Methods by using anti-AMPKβ as a primary and anti-rabbit Alexa Fluor 488 as a secondary antibody. The same samples were also immunodetected with anti-FLAG as a primary and anti-mouse Texas red as a secondary antibody to determine the localization of PIASy. All the samples were treated with Topro3 to stain the nucleus, and the three images were subjected to a merge analysis. (B) Similar samples were used to immunodetect AMPKβ2 subunit using anti-myc as a primary and anti-mouse Texas red as a secondary antibody and also to immunodetect SUMO-modified proteins using anti-SUMO2 as a primary and Alexa Fluor 488 as a secondary antibody. (C) Quantification of cells expressing AMPKβ2 and showing a granular pattern of intracellular inclusions. One hundred cells expressing AMPKβ2 from each of the indicated conditions were used to estimate the proportion of cells with intracellular inclusions. Values are mean ± SD; statistical significance was considered at *p < 0.05, **p < 0.01, and ***p < 0.001.
We quantified the cells containing AMPKβ2 deposits and observed that when PIASy and SUMO2 were coexpressed with the three AMPK subunits, 60% of the cells contained these inclusions (Figure 7C). The appearance of these deposits was dependent on the presence of PIASy, since only 30% of cells presented these inclusions if only SUMO2 was coexpressed with the trimeric AMPK complex (Figure 7C). Similar basal levels of these deposits (30%; Figure 7C) were observed in cells transfected with PIASy and SUMO1, indicating that the increase in the appearance of the inclusions was due to an efficient SUMO2-dependent sumoylation of AMPKβ2 (as indicated earlier, PIASy does not carry out an efficient sumoylation of AMPKβ2 using SUMO1; Figure 2A). The observed inclusion bodies contained SUMO2, as they were stained with anti-SUMO2 antibodies (Figure 7B). All of these results confirmed that PIASy promotes the SUMO2-dependent sumoylation of AMPKβ2, resulting in the formation of cytoplasmic protein deposits.
Finally, we analyzed the effect of PIASy-dependent sumoylation of AMPKβ2 on the activity of the AMPK complex. AMPK activity was assessed by its ability to phosphorylate acetyl-CoA carboxylase (ACC), one of its specific substrates, and also by the phosphorylation status of AMPKα2 catalytic subunit at Thr-172, a sign of activation. In this assay we used the hypersumoylable AMPKβ2 K262R mutant described earlier (Figure 5) in order to magnify possible differences in activity. First we checked that the expression of AMPKβ2 K262R in combination with AMPKα2 and AMPKγ1 resulted in an AMPK complex that was activated by phenformin similar to wild type (Figure 8A, left and middle). Treatment of the cells with this drug resulted in higher levels of both phospho–Thr-172 AMPKα2 and endogenous phospho–Ser-79 ACC after 30–60 min in both wild-type and AMPKβ2 K262R–containing AMPK complexes. However, if in the latter cells we coexpressed in addition PIASy and SUMO2, we observed a constitutive activation of the AMPK complex: higher levels of both phospho–Thr-172 AMPKα2 and endogenous phospho–Ser-79 ACC were detected in untreated cells, and treatment with phenformin did not increase significantly the levels of these phosphorylated proteins (Figure 8A, right). These results suggested that the PIASy-dependent sumoylation of AMPKβ2 results in constitutive activation of the AMPK complex. To confirm these results, we measured the kinase activity of the corresponding AMPK complexes by in vitro kinase assays. We immunoprecipitated the AMPK complexes in cells expressing the trimeric complex (AMPKα2, AMPKβ2 [wild type or K262R], AMPKγ1) in the presence or absence of PIASy and SUMO2. These immunoprecipitates were used in in vitro kinase assays to measure the intrinsic kinase activity of the corresponding complex. In agreement with the results presented earlier, we detected higher kinase activity (more than twofold) in AMPK complexes containing AMPKβ2 K262R that had been sumoylated (Figure 8B). Taken together, these results indicate that sumoylation of AMPK complex enhances its catalytic activity.
FIGURE 8:
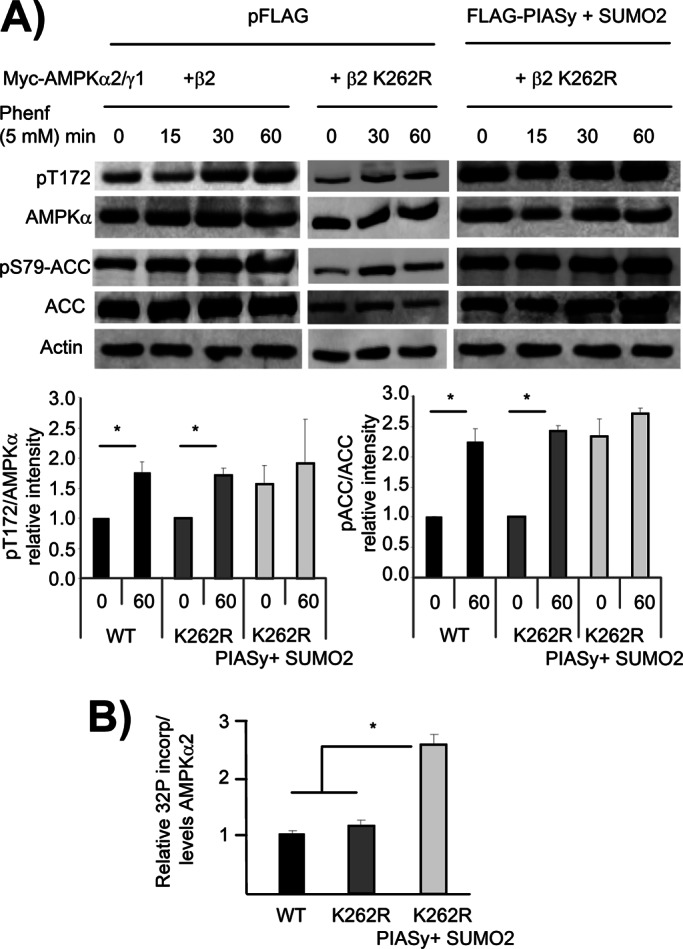
PIASy-dependent sumoylation of AMPKβ2 enhances the activity of the AMPK complex. (A) HEK293 cells were cotransfected with pCMV-myc-AMPKα2/γ1 and pCMV-myc-AMPKβ2 or pCMV-myc-AMPKβ2 K262R and pCMV-6xHis-SUMO2 and pFLAG-PIASy or pFLAG (empty). Twenty-four hours after transfection, cells were treated with 5 mM phenformin for different times. Cells extracts were analyzed by Western blotting using anti–pThr-172AMPKα, anti-AMPKα, anti–pSer-79ACC, and anti-pACC antibodies. The relative intensity of the phosphorylated bands with respect to the amount of the corresponding total protein was plotted as a function of time (time 0 and after 60 min of the treatment). Values are means from three independent assays (bars, ±SD). Statistical significance was considered at *p < 0.05. (B) Cells expressing a combination of plasmids pCMV-HA-AMPKα2, pCMV-myc-AMPKβ2 (WT or K262R), and pCMV-HA-AMPKγ1 were cotransfected or not with plasmids pFLAG-PIASy and pCDNA3–6His-SUMO2. Cell extracts were immunoprecipitated using anti-myc antibody to pull-down the AMPK complex and the in vitro kinase activity measured in the immunoprecipitates as indicated in Materials and Methods. Values indicate the 32P incorporation into GST-ACC relative to the levels of catalytic AMPKα2 in the immunoprecipitates. Statistical significance was considered at *p < 0.05.
Sumoylation and ubiquitination of AMPKβ2 are antagonistic processes
AMPKβ2 can be posttranslationally modified by ubiquitination (Qi et al., 2008; Moreno et al., 2010). Depending on the E3-ubiquitin ligase involved in the process, ubiquitination results in an enhanced degradation of AMPKβ2 (Qi et al., 2008) or in an increase in the steady-state levels of the protein due to the appearance of intracellular aggregates (Moreno et al., 2010). Because both sumoylation and ubiquitination occurs on Lys residues, a possible competition between these two processes could occur, affecting the fate of the protein (Anderson et al., 2012; Xing et al., 2012). To check whether ubiquitination of AMPKβ2 could affect its PIASy-dependent sumoylation, we coexpressed in HEK293 cells the three AMPK subunits (α2/β2/γ1), 6xHis-SUMO2, and FLAG-PIASy in the presence or absence of hemagglutinin (HA)-ubiquitin. As shown in Figure 9A, the presence of HA-ubiquitin greatly reduced the levels of sumoylated AMPKβ2. This could indicate competition of sumoylation and ubiquitination for posttranslational modification of AMPKβ2. To confirm this result, we analyzed the ubiquitination of AMPKβ2 subunit in the presence or absence of PIASy and SUMO2 in cells treated with MG132 (a proteasomal inhibitor) to enhance the accumulation of ubiquitinated proteins. As shown in Figure 9B, the expression of PIASy and SUMO2 decreased the ubiquitination of AMPKβ2 subunit by endogenous E3-ubiquitin ligases. These results indicate clear competition between sumoylation and ubiquitination for posttranslational modification of AMPKβ2 subunit.
FIGURE 9:
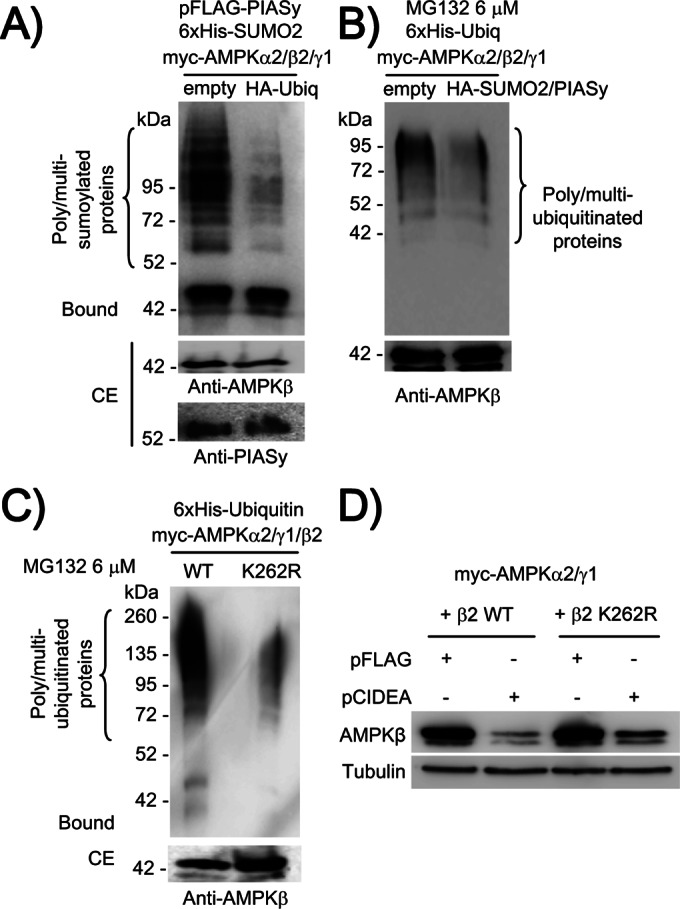
Sumoylation and ubiquitination of AMPKβ2 are antagonistic processes. (A) The coexpression of HA-ubiquitin decreases the levels of PIASy-dependent sumoylated AMPKβ2 protein. HEK293 cells were cotransfected with plasmids pCMV-6xHis-SUMO2, pCMV-myc-AMPKα2/β2/γ1, pFLAG-PIASy, and pCMV-HA-ubiquitin or pCMV-HA (empty) vector. Cell extracts were analyzed as described in Figure 2 by using anti-AMPKβ antibody. (B) The coexpression of HA-SUMO2 decreases the levels of ubiquitinated AMPKβ2 protein. HEK293 cells were cotransfected with plasmids pCMV-6xHis-ubiquitin and pCMV-myc-AMPKα2/β2/γ1, in the presence or absence of pFLAG-PIASy and pCMV-HA-SUMO2. Twenty-four hours after transfection, cells were treated with 6 μM MG132 during 8 h. Then cell extracts were analyzed as described in Figure 2 by using anti-AMPKβ antibodies. (C) Ubiquitination of AMPKβ2 K262R is less efficient than for wild-type protein. HEK293 cells were cotransfected with plasmids pCMV-6xHis-ubiquitin, pCMV-myc-AMPKα2/γ1, and pCMV-myc-AMPKβ2 or pCMV-myc-AMPKβ2K262R mutant. Cells were treated with 6 μM MG132 during 8 h, and cells extracts were analyzed as described in Figure 2 by using anti-AMPKβ antibody. (D) CIDEA-dependent degradation of AMPKβ2 is prevented to some degree in AMPKβ2 K262R mutant. HEK293 cells were cotransfected with pCMV-myc-AMPKα2/γ1 and pCMV-myc-AMPKβ2 or pCMV-myc-AMPKβ2 K262R mutant and FLAG-CIDEA or pFLAG empty vector. Twenty-four hours after transfection, cell extracts (40 μg) were analyzed by SDS–PAGE and immunodetected with anti-AMPKβ and anti-tubulin antibodies.
Ubiquitination of AMPKβ2 occurs at the C-terminus of the protein (Qi et al., 2008). Because we constructed an AMPKβ2 mutant in this area (K262R; Figure 5), we examined whether this mutant could be properly ubiquitinated by endogenous E3-ubiquitin ligases. With this aim, we coexpressed in HEK293 cells the three AMPK subunits (α2/β2/γ1) containing either wild-type AMPKβ2 or the K262R mutant, together with 6xHis-ubiquitin. Cells were then treated with MG132 to inhibit proteasome activity, and then cell extracts were purified by metal-affinity chromatography. As shown in Figure 9C, we observed a clear ubiquitination of wild-type AMPKβ2. However, in the case of AMPKβ2 K262R, the ubiquitination process was much less efficient. These results suggested that residue K262 affected the ubiquitination of AMPKβ2, either because it was a ubiquitination site (so the K262R mutant would lack this site) or because the K262R mutant promoted a conformational change that affected the ubiquitination of Lys residues in the vicinity. Because ubiquitination of AMPKβ2 mediated by CIDEA results in rapid degradation of the protein, we examined whether the reduced capacity of the AMPKβ2 K262R mutant to be ubiquitinated could prevent CIDEA-mediated degradation. We coexpressed in HEK293 cells the three AMPK subunits (α2/β2/γ1) containing either wild-type AMPKβ2 or the K262R mutant in the presence or absence of FLAG-CIDEA. In agreement with previous results, CIDEA induced the degradation of wild-type AMPKβ2 (Qi et al., 2008). However, CIDEA-induced degradation of AMPKβ2 K262R was prevented to some degree (Figure 9D). Taking all these results together, we suggest that K262 is a residue important for the ubiquitination of AMPKβ2. If ubiquitination at this site is prevented (K262R mutant), then the protein becomes a poorer substrate for ubiquitination, which prevents its proteasomal degradation. At the same time, the protein becomes more competent to be posttranslationally modified by sumoylation (hypersumoylable mutant).
DISCUSSION
We report here that PIASy, an E3-SUMO ligase, interacts physically with and promotes the sumoylation of AMPKβ2 subunit when it forms part of a heterotrimeric AMPK complex. We studied this modification in more detail and observed that PIASy is able to promote the attachment of only SUMO2 conjugates but not SUMO1. This observation is in agreement with other examples of sumoylated proteins that become modified only by one of the SUMO conjugates and not by the other (Saitoh and Hinchey, 2000). However, sumoylation of free AMPKβ2 subunits becomes independent of PIASy, indicating that the E2-SUMO–conjugating enzyme Ubc9 is able to sumoylate the free subunit but requires the help of the E3-SUMO ligase PIASy to gain access to the corresponding Lys residues when AMPKβ2 is in the heterotrimeric AMPK complex.
Although PIASy interacted physically with AMPKβ1 subunit, surprisingly, PIASy was not able to modify this subunit in spite of the high similarity between both AMPKβ subunits. Perhaps AMPKβ1 lacks specific Lys residues present in AMPKβ2. In fact, alignment of the sequence of both subunits highlights the presence of five residues that are present in AMPKβ2 but not in AMPKβ1. We tried to identify the Lys residues involved in the sumoylation reaction, first, by applying a bioinformatics approach to identify putative sumoylation consensus sites, and second, by mapping the region of the AMPKβ2 subunit that is subjected to this modification. However, mutation of the suggested Lys residues (K71 and K167 by the bioinformatics approach, and K203 by the mapping strategy) did not prevent the PIASy-dependent sumoylation of the mutated forms. Thus either other Lys residues are involved in this modification or the absence of one particular Lys residue enables other Lys residues in the vicinity to become competent in the sumoylation reaction.
We also studied the effect of PIASy-dependent sumoylation on AMPK stability and function. Consistent with the idea that poly-SUMO modification does not usually trigger protein degradation (Gareau and Lima, 2010; Wilkinson and Henley, 2010), we show that PIASy-dependent sumoylation of AMPKβ2 does not affect its degradation rate. However, we observed that HEK293 cells coexpressing PIASy, SUMO2, and AMPK subunits produced cytoplasmic deposits that were positive for AMPKβ2 and SUMO2. The fact that the overexpression of PIASy and SUMO1 did not produce these aggregates indicates that sumoylation of AMPKβ2 with SUMO2 triggered the appearance of these deposits. These results suggest that sumoylation produces two pools of AMPKβ2 that might coexist in the cells: one pool forms part of these inclusions bodies, with unknown function, and another pool is soluble and responsible for AMPK activity.
We also found that PIASy-dependent sumoylation of AMPKβ2 enhances the activity of the AMPK complex. Both the phosphorylation status of Thr-172 (a sign of AMPK activation) and the phosphorylation of ACC (a typical AMPK substrate) were enhanced in cells containing sumoylated forms of AMPKβ2. This is a very important result since it indicates that sumoylation of AMPK is another posttranslational modification that renders an active AMPK complex. Therefore conditions that enhance PIASy activity could result in the activation of the AMPK complex. We do not know the molecular mechanism by which sumoylation of AMPKβ2 enhances AMPK activity. Perhaps this posttranslational modification provides a novel change in the conformation of the AMPK complex that either increases the action of upstream kinases or makes it a poorer substrate for specific phosphatases.
Because sumoylation and ubiquitination are very similar processes, both based on the modification of Lys residues, it is likely that in some cases both processes may compete for the same Lys site. In this work we present evidence for a competition between sumoylation and ubiquitination in the posttranslational modification of AMPKβ2. Our results indicate that sumoylation of AMPKβ2 subunit is decreased if ubiquitin is overexpressed and also that ubiquitination of AMPKβ2 is reduced if sumoylation is enhanced. Perhaps sumoylation of AMPKβ2 could provide a conformational change in the protein resulting in a protein structure less competent to be ubiquitinated, or, conversely, sumoylation and ubiquitination may compete for the same target site in AMPKβ2 sequence. Because the final effect of these two posttranslational modifications is different (ubiquitination triggers inactivation of AMPK; sumoylation enhances AMPK activity), we propose that these two processes are competitors and have antagonist effects in AMPK complex activity. This adds an extra layer of complexity to the regulation of the activity of AMPK complex, since conditions that promote one or the other type of these posttranslational modifications may result in different outcomes.
In summary, our results indicate that the AMPKβ2 subunit can be sumoylated in a process dependent on PIASy and SUMO2 and that this modification results in an enhancement of AMPK activity. Of interest, this modification does not occur in the AMPKβ1 subunit, which opens a new way to activate selectively AMPKβ2-containing complexes. In addition, our results indicate that sumoylation and ubiquitination of AMPKβ2 are antagonistic processes.
MATERIALS AND METHODS
Plasmids
Plasmid pACT2-PIASy was a generous gift from Santiago Rodriguez de Cordoba (Centro de Investigaciones Biológicas, Consejo Superior de Investigaciones Científicas, Madrid, Spain). Plasmids pCMV-myc-PIASy and pFLAG-PIASy were obtained by subcloning an EcoRI/SalI fragment from pACT2-PIASy into pCMV-myc and pFLAG vectors. Plasmids pCDNA3–6xHis-SUMO1 and pCDNA3–6xHis-SUMO2 were from R. T. Hay (College of Life Science, University of Dundee, Scotland). Plasmid pCDNA3-myc-PIASy CI (C342/347A; expressing a form with reduced catalytic activity) was from Shigeki Miyamoto (Division of Food Science and Biotechnology, Graduate School of Agriculture, Kyoto University, Kyoto, Japan); this plasmid was digested with EcoRI/XbaI and the resulting fragment subcloned into pFLAG vector to obtain the pFLAG-PIASy CI plasmid. Plasmid pCMV-HA-SUMO2 was constructed by amplifying by PCR a fragment containing the SUMO2 open reading frame, using primers SUMO2-for (5′-CATCAGGCCATGGAGGCCATGTCCGAGGAGAAGCCC-3′), SUMO2-rev (5′-TCTGCAGAATTCCTAACCTCCCGTCTGCTGCTGG-3′), and pCDNA3–6His-SUMO2 as template. The PCR product was digested with EcoRI/SfiI and subcloned into the pCMV-HA vector. pCMV-myc plasmids containing AMPKβ2 mutants were obtained by site-directed mutagenesis using the corresponding pairs of primers: K71Rfor (5′-CAGGATTTGGAGGACTCCGTAAGGCCCACACAGCAGGCCCGGCCC-3′) and K71Rrev (5´-GGGCCGGGCCTGCTGTGTGGGCCTTACGGAGTCCTCCAAATCCTG-3′); K167Rfor (5´-TTTGAGGTGTTCGATGCTTTAAGGTTAGATTCTATGGAAAGTTCT-3′) and K167Rrev (5´-AGAACTTTCCATAGAATCTAACCTTAAAGCATCGAACACCTCAAA-3′); K203Rfor (5´-TTTCGATCTGAGGAAAGATTCAGATCCCCACCCATCCTTCCTCCT-3´) and K203Rrev (5´-AGGAGGAAGGATGGGTGGGGATCTGAATCTTTCCTCAGATCGAAA-3′); and K262Rfor (5´-GCAACCCATCGCTACAAGAAGAGGTATGTTACTACTCTGCTATAC-3´) and K262Rrev (5´-GTATAGCAGAGTAGTAACATACCTCTTCTTGTAGCGATGGGTTGC-3′). Mutations and construct fidelity were confirmed by DNA sequencing. Other plasmids used in this study were pBTM-AMPKα2, pBTM-AMPKβ2, and pBTM-AMPKγ1 (Gimeno-Alcaniz and Sanz, 2003) and pCMV-myc-AMPKα2, pCMV-myc-AMPKβ2, pCMV-myc-AMPKγ1, pCMV-HA-AMPKα2, and pCMV-HA-AMPKγ1 (Solaz-Fuster et al., 2008). Plasmid pCMV-myc-β2-1, containing the N-terminal fragment of AMPKβ2 (residues 1–185), plasmid pCMV-myc-β2-2, containing the C-terminal part of AMPKβ2 (residues 186–271), and plasmids pFLAG-CIDEA, pCMV-HA-ubiquitin, and pCMV-6xHis-ubiquitin were previously described (Moreno et al., 2010).
Cell culture and immunoblotting analysis
Human embryonic kidney (HEK293) and human osteosarcoma U2OS cells were grown in DMEM (Lonza, Barcelona, Spain) supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM glutamine, and 10% inactivated fetal bovine serum (Invitrogen, Madrid, Spain). We plated 1.5 × 106 or 3 × 106 cells onto 60-mm-diameter (p60) or 100-mm-diameter (p100) culture dishes, respectively, the day before transfection. Cells were transfected with 1 μg (for p60 culture dishes) or 3 μg (for p100 culture dishes) of each plasmid using Lipofectamine 2000 (Invitrogen). Twenty-four hours after transfection, cells were treated when indicated with 5 mM phenformin (Sigma-Aldrich, Madrid, Spain), 6 μM MG132 (Enzo-Biomol, Madrid, Spain), or 100 μM cycloheximide (Sigma-Aldrich). Then cells were scraped on ice in lysis buffer (10 mM Tris-HCl, pH 7.0, 15 mM EDTA, pH 8.0, 50 mM NaF, 0.6 M sucrose, 5 mM Na2P2O7, 150 mM NaCl, 0.5% Nonidet P-40, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 μM microcystine, 1 mM NaVO4, and protease inhibitor mixture [Roche, Barcelona, Spain]). Forty micrograms of total protein from the soluble fraction of cell lysates was analyzed by SDS–PAGE and Western blotting using appropriate antibodies: anti-AMPKα total, anti-AMPKβ total, anti-ACC total, anti–pSer-79 ACC, and anti–pThr-172 AMPKα (Cell Signaling, Barcelona, Spain); anti-PIASy and anti-SUMO2/3 (Abcam, Cambridge, United Kingdom); and anti-myc and anti-actin (Sigma-Aldrich).
Yeast two-hybrid analyses
The yeast strain CTY10.5d was cotransformed with combinations of pACT2-PIASy and different pBTM-AMPKα2, pBTM-AMPKβ2, and pBTM-AMPKγ1 plasmids. Transformants were grown in selective synthetic complete medium, and β-galactosidase activity was assayed in permeabilized cells and expressed in Miller units (Ludin et al., 1998).
Coimmunoprecipitation analyses
HEK293 cells transfected with plasmid pFLAG-PIASy or empty vector and a combination of plasmids pCMV-myc-AMPKα2, pCMV-myc-AMPKβ2, and pCMV-myc-AMPKγ1 were used for coimmunoprecipitation studies. Transfected cells were scraped in ice-cold lysis buffer (10 mM NaCl, 10 mM Tris-HCl, pH 7, 0.5% Nonidet P-40, 5 mM NaF, 5 mM Na2P2O7, 1 mM PMSF, 1 mM NaVO4, and protease inhibitor mixture [Roche]) and then clarified by centrifugation at 5000 × g for 5 min at 4 °C. Then 1.2 mg of total protein from the soluble fraction was used for immunoprecipitation using specific anti-FLAG antibody (Sigma-Aldrich) and protein A/G-plus agarose (Santa Cruz Biotechnology, Barcelona, Spain). Immunoprecipitates were analyzed by Western blotting using polyclonal specific antibodies: anti-AMPKα, anti-AMPKβ, anti-AMPKγ (Cell Signaling Technology), and anti-PIASy (Abcam). HEK293 cells were also transfected with plasmid pFLAG-PIASy and a combination of plasmids pCMV-HA-AMPKα2, pCMV-myc-AMPKβ2, and pCMV-HA-AMPKγ1 and used in immunoprecipitation studies using anti-myc antibody. The resulting immunoprecipitates were analyzed by Western blotting using anti-AMPKα, anti-AMPKβ, anti-AMPKγ (Cell Signaling), and anti-PIASy (Abcam) antibodies.
Human osteosarcoma U2OS cells were treated with 6 μM MG132 (8 h). Then cell extracts were immunoprecipitated using anti-PIASy (Abcam) antibodies and the immunoprecipitates analyzed by Western blot using anti-AMPKα, anti-AMPKβ, and anti-AMPKγ (Cell Signaling) antibodies.
Analysis of in vitro kinase activity
Cells expressing a combination of plasmids pCMV-HA-AMPKα2, pCMV-myc-AMPKβ2 (wild type or K262R), and pCMV-HA-AMPKγ1 were cotransfected or not with plasmids pFLAG-PIASy and pCDNA3–6His-SUMO2. Cell extracts were immunoprecipitated using anti-myc antibody to pull-down the AMPK complex. Immunoprecipitates were divided into two aliquots: one-third was used to determine the amount of the catalytic AMPKα2 subunit present in the pellet by Western blotting using anti-AMPKα antibody, and two-thirds of the immunoprecipitates were used to carry out in vitro kinase activity in the following way: immunoprecipitates were resuspended in 20 μl of kinase buffer (50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM dithiothreitol) containing 0.5 μg glutathione S-transferase (GST)–ACC1 (a generous gift of Mhairi Towler, University of Dundee, Dundee, Scotland). After the addition of 2 μl of [γ-32P]ATP (3000 Ci/mmol) to the samples, reactions were incubated at 30ºC for 30 min and stopped by boiling the mixtures in sample buffer. Samples were analyzed by SDS–PAGE and autoradiography, and gels were subjected to phosphoimaging for the measurement of 32P incorporation. This value was related to the levels of AMPKα2 in the immunoprecipitates and adjusted to the value of wild-type AMPK complex.
Analysis of in vivo sumoylation
To study sumoylation in intact cells, HEK293 cells were transfected with plasmid pCDNA3–6xHis-SUMO1 or pCDNA3–6xHis-SUMO2 (encoding modified forms of SUMO1 and SUMO2, respectively, tagged with six His residues), pCMV-myc plasmids encoding the AMPK subunits (α2, β1/2, and γ1), and pFLAG-PIASy wild type (WT) or pFLAG-PIASy CI (expressing a form with reduced activity) plasmids, by using the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions. After 24 h of transfection, cells were lysed in buffer A (6 M guanidinium-HCl, 0.1 M sodium phosphate, 0.1 M Tris-HCl, pH 8.0). We incubated 1.5 mg of protein of a clarified extract (CE; 12,000 × g for 15 min) in 150 μl of TALON column (Clontech, Madrid, Spain) in the presence of 10 mM imidazole for 2 h at room temperature on a rocking platform to purify His-tagged proteins. The column was then successively washed with 1 ml each of buffer B (buffer A plus 10 mM imidazole) and four more times with buffer C adjusted to pH 8.0 (buffer B but with 8 M urea instead of 6 M guanidinium-HCl). Bound proteins were eluted with 30 μl of 2× Laemmli sample buffer and analyzed by Western blotting using appropriated antibodies.
Analysis of the degradation rates of AMPKβ2 subunit
HEK293 cells were transfected with plasmids pCMV-myc-AMPKβ2, pCMV-myc-AMPKα2, pCMV-myc-AMPKγ1, pCDNA3–6xHis-SUMO2, and pFLAG-PIASy or pFLAG empty vector. Twenty-four hours after transfection, cells were treated with 100 μM cycloheximide and, at the indicated times (from 0 to 8 h), aliquots were taken from the cultures, and cell extracts (40 μg) were analyzed by SDS–PAGE and Western blotting using anti-AMPKβ antibody. The same extracts were analyzed using anti-actin antibody as a loading control. Western blots were analyzed by densitometry using an LAS-3000 electronic reader and Image Gauge, version 4.0, software (Fujifilm, Tokyo, Japan). The levels of the corresponding AMPKβ2 subunit with respect to those of actin at each time point are expressed as a percentage of values at time zero.
Immunofluorescence and confocal microscopy
HEK293 cells transfected with the appropriate plasmids were grown on plates containing coverslips. Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min, and then the fixation was stopped with 10 mM glycine for 5 min. Cells were permeabilized with 0.5% Triton X-100 in PBS for 30 min. Samples were washed three times with PBS and blocked with 10% fetal bovine serum, 5% nonfat dried milk, and 0.1% Triton X-100 in PBS for 1 h at room temperature. Samples were then incubated overnight at 4°C with a 1:200 dilution of anti-AMPKβ total (Cell Signaling Technology) with or without anti-FLAG (1:200) in blocking solution. Samples were washed five times with PBS during 1 h at room temperature and incubated again with 1:500 dilution of appropriated fluorescence-labeled secondary antibodies (anti-rabbit Alexa Fluor 488 and anti-mouse Texas red; Invitrogen). Nuclei were stained with 1:1000 dilution of a Topro3 fluorescent probe in PBS for 30 min. Finally, samples were washed again with PBS and mounted on slices using Fluoromount G. Images were acquired for 20-μm slices with a TCS/SP2 confocal microscope (Leica, Wetzlar, Germany) using a 63× oil immersion objective. Images were treated with ImageJ 1.43c software (National Institutes of Health, Bethesda, MD). More than 100 green cells (therefore containing AMPKβ) were analyzed for each condition and used to determine the presence or absence or large intracellular inclusions. Because in cotransfection experiments some of the green cells may not contain the rest of the plasmids, the quantification of cells containing large inclusions could be underestimated. When indicated, cells were treated with 20 ng/ml leptomycin B (Sigma-Aldrich) for 20 min.
Statistical analyses
Values are given as mean ± SD of at least three independent experiments. Differences between groups were analyzed by two-tailed Student's t tests. The significance is considered at *p < 0.05 and **p < 0.01, as indicated in each case.
Supplementary Material
Acknowledgments
We thank Santiago Rodriguez de Cordoba, R. T. Hay, and Shigeki Miyamoto for plasmids. This work was supported by a grant from the Spanish Ministry of Education and Science (SAF2011-27442) and a grant from Generalitat Valenciana (Prometeo 2009/051). T.R. was supported by a JAE-Predoctoral fellowship from the Consejo Superior de Investigaciones Científicas.
Abbreviations used:
- ACC
acetyl-CoA carboxylase
- AMPK
AMP-activated protein kinase
- CIDEA
cell death–inducing, DFFA-like effector A
- PIAS
protein inhibitor of activated STAT
- SUMO
small ubiquitin-like modifier
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-11-0806) on April 3, 2013.
REFERENCES
- Al-Hakim AK, Zagorska A, Chapman L, Deak M, Peggie M, Alessi DR. Control of AMPK-related kinases by USP9X and atypical Lys(29)/Lys(33)-linked polyubiquitin chains. Biochem J. 2008;411:249–260. doi: 10.1042/BJ20080067. [DOI] [PubMed] [Google Scholar]
- Anderson DD, Eom JY, Stover PJ. Competition between sumoylation and ubiquitination of serine hydroxymethyltransferase 1 determines its nuclear localization and its accumulation in the nucleus. J Biol Chem. 2012;287:4790–4799. doi: 10.1074/jbc.M111.302174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci. 1997;22:12–13. doi: 10.1016/s0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- Bergink S, Jentsch S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature. 2009;458:461–467. doi: 10.1038/nature07963. [DOI] [PubMed] [Google Scholar]
- Carling D, Thornton C, Woods A, Sanders MJ. AMP-activated protein kinase: new regulation, new roles. Biochem J. 2012;445:11–27. doi: 10.1042/BJ20120546. [DOI] [PubMed] [Google Scholar]
- Crute BE, Seefeld K, Gamble J, Kemp BE, Witters LA. Functional domains of the alpha1 catalytic subunit of the AMP-activated protein kinase. J Biol Chem. 1998;273:35347–35354. doi: 10.1074/jbc.273.52.35347. [DOI] [PubMed] [Google Scholar]
- Davies SP, Helps NR, Cohen PT, Hardie DG. 5’-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995;377:421–425. doi: 10.1016/0014-5793(95)01368-7. [DOI] [PubMed] [Google Scholar]
- Desterro JM, Rodriguez MS, Hay RT. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol Cell. 1998;2:233–239. doi: 10.1016/s1097-2765(00)80133-1. [DOI] [PubMed] [Google Scholar]
- Gareau JR, Lima CD. The SUMO pathway: emerging mechanisms that shape specificity, conjugation and recognition. Nat Rev. 2010;11:861–871. doi: 10.1038/nrm3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno-Alcaniz JV, Sanz P. Glucose and type 2A protein phosphatase regulate the interaction between catalytic and regulatory subunits of AMP-activated protein kinase. J Mol Biol. 2003;333:201–209. doi: 10.1016/j.jmb.2003.08.022. [DOI] [PubMed] [Google Scholar]
- Hay RT. SUMO-specific proteases: a twist in the tail. Trends Cell Biol. 2007;17:370–376. doi: 10.1016/j.tcb.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Iseli TJ, Walter M, van Denderen BJ, Katsis F, Witters LA, Kemp BE, Michell BJ, Stapleton D. AMP-activated protein kinase beta subunit tethers alpha and gamma subunits via its C-terminal sequence (186–270) J Biol Chem. 2005;280:13395–13400. doi: 10.1074/jbc.M412993200. [DOI] [PubMed] [Google Scholar]
- Kagey MH, Melhuish TA, Wotton D. The polycomb protein Pc2 is a SUMO E3. Cell. 2003;113:127–137. doi: 10.1016/s0092-8674(03)00159-4. [DOI] [PubMed] [Google Scholar]
- Kazgan N, Williams T, Forsberg LJ, Brenman JE. Identification of a nuclear export signal in the catalytic subunit of AMP-activated protein kinase. Mol Biol Cell. 2010;21:3433–3442. doi: 10.1091/mbc.E10-04-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YY, et al. Functional dissection of lysine deacetylases reveals that HDAC1 and p300 regulate AMPK. Nature. 2012;482:251–255. doi: 10.1038/nature10804. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Ludin K, Jiang R, Carlson M. Glucose-regulated interaction of a regulatory subunit of protein phosphatase 1 with the Snf1 protein kinase in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1998;95:6245–6250. doi: 10.1073/pnas.95.11.6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb AM, Wuerzberger-Davis SM, Miyamoto S. PIASy mediates NEMO sumoylation and NF-kappaB activation in response to genotoxic stress. Nat Cell Biol. 2006;8:986–993. doi: 10.1038/ncb1458. [DOI] [PubMed] [Google Scholar]
- Moreno D, Towler MC, Hardie DG, Knecht E, Sanz P. The laforin-malin complex, involved in Lafora disease, promotes the incorporation of K63-linked ubiquitin chains into AMP-activated protein kinase beta subunits. Mol Biol Cell. 2010;21:2578–2588. doi: 10.1091/mbc.E10-03-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, Ledl A, Schmidt D. SUMO: a regulator of gene expression and genome integrity. Oncogene. 2004;23:1998–2008. doi: 10.1038/sj.onc.1207415. [DOI] [PubMed] [Google Scholar]
- Pang T, Xiong B, Li JY, Qiu BY, Jin GZ, Shen JK, Li J. Conserved alpha-helix acts as autoinhibitory sequence in AMP-activated protein kinase alpha subunits. J Biol Chem. 2007;282:495–506. doi: 10.1074/jbc.M605790200. [DOI] [PubMed] [Google Scholar]
- Pichler A, Knipscheer P, Saitoh H, Sixma TK, Melchior F. The RanBP2 SUMO E3 ligase is neither HECT- nor RING-type. Nat Struct Mol Biol. 2004;11:984–991. doi: 10.1038/nsmb834. [DOI] [PubMed] [Google Scholar]
- Qi J, Gong J, Zhao T, Zhao J, Lam P, Ye J, Li JZ, Wu J, Zhou HM, Li P. Downregulation of AMP-activated protein kinase by CIDEA-mediated ubiquitination and degradation in brown adipose tissue. EMBO J. 2008;27:1537–1548. doi: 10.1038/emboj.2008.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Gao X, Jin C, Zhu M, Wang X, Shaw A, Wen L, Yao X, Xue Y. Systematic study of protein sumoylation: development of a site-specific predictor of SUMOsp 2.0. Proteomics. 2009;9:3409–3412. doi: 10.1002/pmic.200800646. [DOI] [PubMed] [Google Scholar]
- Rytinki MM, Kaikkonen S, Pehkonen P, Jaaskelainen T, Palvimo JJ. PIAS proteins: pleiotropic interactors associated with SUMO. Cell Mol Life Sci. 2009;66:3029–3041. doi: 10.1007/s00018-009-0061-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh H, Hinchey J. Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J Biol Chem. 2000;275:6252–6258. doi: 10.1074/jbc.275.9.6252. [DOI] [PubMed] [Google Scholar]
- Sanders MJ, Grondin PO, Hegarty BD, Snowden MA, Carling D. Investigating the mechanism for AMP activation of the AMP-activated protein kinase cascade. Biochem J. 2007;403:139–148. doi: 10.1042/BJ20061520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz P. AMP-activated protein kinase: structure and regulation. Curr Protein Peptide Sci. 2008;9:478–492. doi: 10.2174/138920308785915254. [DOI] [PubMed] [Google Scholar]
- Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, Norman DG, Hardie DG. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. 2004;113:274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaz-Fuster MC, et al. Regulation of glycogen synthesis by the laforin-malin complex is modulated by the AMP-activated protein kinase pathway. Hum Mol Genet. 2008;17:667–678. doi: 10.1093/hmg/ddm339. [DOI] [PubMed] [Google Scholar]
- Tatham MH, Kim S, Jaffray E, Song J, Chen Y, Hay RT. Unique binding interactions among Ubc9, SUMO and RanBP2 reveal a mechanism for SUMO paralog selection. Nat Struct Mol Biol. 2005;12:67–74. doi: 10.1038/nsmb878. [DOI] [PubMed] [Google Scholar]
- Ulrich HD. Mutual interactions between the SUMO and ubiquitin systems: a plea of no contest. Trends Cell Biol. 2005;15:525–532. doi: 10.1016/j.tcb.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Wilkinson KA, Henley JM. Mechanisms, regulation and consequences of protein SUMOylation. Biochem J. 2010;428:133–145. doi: 10.1042/BJ20100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson VG, Heaton PR. Ubiquitin proteolytic system: focus on SUMO. Expert Rev Proteomics. 2008;5:121–135. doi: 10.1586/14789450.5.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature. 2007;449:496–500. doi: 10.1038/nature06161. [DOI] [PubMed] [Google Scholar]
- Xing X, Bi H, Chang AK, Zang MX, Wang M, Ao X, Li S, Pan H, Guo Q, Wu H. SUMOylation of AhR modulates its activity and stability through inhibiting its ubiquitination. J Cell Physiol. 2012;227:3812–3819. doi: 10.1002/jcp.24092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.