

Abstract
Usher syndrome (USH) is a leading cause of deaf-blindness in autosomal recessive trait. Phenotypic and genetic heterogeneities in USH make molecular diagnosis much difficult. This is a pilot study aiming to develop an approach based on next-generation sequencing to determine the genetic defects in patients with USH or allied diseases precisely and effectively. Eight affected patients and twelve unaffected relatives from five unrelated Chinese USH families, including 2 pseudo-dominant ones, were recruited. A total of 144 known genes of inherited retinal diseases were selected for deep exome resequencing. Through systematic data analysis using established bioinformatics pipeline and segregation analysis, a number of genetic variants were released. Eleven mutations, eight of them were novel, in the USH2A gene were identified. Biparental mutations in USH2A were revealed in 2 families with pseudo-dominant inheritance. A proband was found to have triple mutations, two of them were supposed to locate in the same chromosome. In conclusion, this study revealed the genetic defects in the USH2A gene and demonstrated the robustness of targeted exome sequencing to precisely and rapidly determine genetic defects. The methodology provides a reliable strategy for routine gene diagnosis of USH.
Introduction
Usher syndrome (USH) is an autosomal recessive disorder characterized by visual loss due to retinitis pigmentosa (RP), sensorineural hearing impairment and variable vestibular dysfunction, with remarkable clinical and genetic heterogeneity [1]. According to the disease severity and progression, three subtypes of USH have been descrobed. Type I (USH1) is the most severe form characterized by prepubertal onset of RP, profound hearing loss, and vestibular dysfunction. Type II (USH2) is characterized by postpuberal onset RP and moderate deafness without vestibular dysfunction. Type III (USH3) is featured as postlingual deafness, teenage-onset RP and varying degree of vestibular dysfunction. To date, at least 12 loci have been mapped for the three types of USH in human chromosomes and 10 genes have been identified [2]. These genes totally comprise 321 coding exons spanning a length of 59,430-nt (Table S1). Reported mutations of USH widely spread over the coding regions of these causative genes. Genetic screening through traditional approaches, such as direct sequencing is therefore difficult. A high-throughout and cost-effective method to detect the genetic defects is needed. Targeted or whole exome sequencing has been proved to be a powerful tool to discover novel disease-related genes or genetic mutations in large genomic regions [3]. With the progresses on next-generation sequencing (NGS) and bioinformatics, it has been demonstrated to have higher efficiency but lower cost comparing with previous methods [4]. In this study, we utilized targeted exome sequencing (TES) to study genetic defects in five USH families and attempted to establish a strategy useful for genetic diagnosis of USH patients.
Materials and Methods
Patient Recruitment
This study adhered to the tenets of the Declaration of Helsinki. The protocol was approved by the ethics committee of The Eye Hospital of Wenzhou Medical College and written informed consent has been obtained from all study subjects. Comprehensive ophthalmic examination, including routine eye tests, perimetry, electroretinography (ERG), and optical coherence tomography (OCT), were carried out in each patient. Detailed family history was obtained through personal interviews with patients and their relatives (Figure 1). Peripheral blood samples were collected from both the affected patients and unaffected kinships.
Figure 1. Families with Usher syndrome.

Pedigrees of the Usher syndrome in this study. Closed symbol represents affected patient and open symbol indicates unaffected subject. The bar over the symbol indicates examined subjects in this study. Arrow indicates proband. Slash represents deceased person. The box labeled with different color indicates different mutation in each pedigree.
Illumina Library Preparation
Genomic DNA was extracted from whole blood using a DNA Extraction kit (TIANGEN, Beijing) using the manufacturer's instructions. The DNA was quantified with Nanodrop 2000 (Thermal Fisher Scientific, DE). A minimum of 3 μg DNA was used for the indexed Illumina libraries according to manufacturer's protocol. The final library size 350 bp–450 bp including adapter sequences was selected.
Disease Genes Enrichment and Sequencing
A total of 144 disease genes (Table S2) were selected by a gene capture strategy, using GenCap custom enrichment kit (MyGenostics, Beijing) based on previously described technologies [5], [6]. The biotinylated single-strand capture probes were designed to tile along the exonic non-repeated regions of the genes (Table S3). The capture experiment was conducted according to manufacturer's protocol. In brief, 1 μg DNA library was mixed with Buffer BL and GenCap probe (MyGenostics, Beijing), heated at 95°C for 7 min and 65°C for 2 min on a PCR machine; 23 μl of the 65°C prewarmed Buffer HY (MyGenostics, Beijing) was then added to the mix, and the mixture was hold at 65°C with PCR lid heat on for 22 hours for hybridization. 50 μl MyOne beads (Life Technology) was washed in 500 μL 1Xbinding buffer for 3 times and resuspended in 80 μl 1Xbinding buffer. Sixty-four μl 2Xbinding buffer was added to the hybrid mix, and transferred to the tube with 80 μl MyOne beads. The mix was rotated for 1 hour on a rotator. The beads were then washed with WB1 buffer at room temperature for 15 minutes once and WB3 buffer at 65°C for 15 minutes three times. The bound DNA was then eluted with Buffer Elute. The eluted DNA was finally amplified for 15 cycles using the following program: 98°C for 30 s (1 cycle); 98°C for 25 s, 65°C for 30 s, 72°C for 30 s (15 cycles) and 72°C for 5 min (1 cycle). The PCR product was purified using SPRI beads (Beckman Coulter) according to manufacturer's protocol. The enrichment libraries were sequenced on Illumina Solexa HiSeq 2000 sequencer for paired read 100bp.
Bioinformatics Analysis
After Solexa HiSeq 2000 sequencing, high-quality reads were retrieved from raw reads by filtering out the low quality reads and adaptor sequences using the Solexa QA package [7] and the cutadapt program (http://code.google.com/p/cutadapt/), respectively. SOAPaligner program [8] was then used to align the clean reads to the reference human genome (hg19). After the PCR duplicates were removed by the Picard software, [9] the SNPs was firstly identified using the SOAPsnp program, [8] Subsequently, we realigned the reads to the reference genome using BWA [10] and identified the insertions or deletions (InDels) using the GATK program [11], The identified SNPs and InDels were annotated using the Exome-assistant program (http://122.228.158.106/exomeassistant). MagicViewer [12] was used to view the short read alignment and validate the candidate SNPs and InDels. Nonsynonymous variants were evaluated by four algorithms, PolyPhen, SIFT, PANTHER and Pmut as described previously, to determine pathogenicity [13]. Sequencing data were deposited in NIH Short Read Archive (PRJNA189497).
Expanded Validation
DNA samples of all the family members (Figure 1) were taken for the same targeted exome sequencing and filtering strategy. Furthermore, coding regions of the mutations identified as described above were amplified by polymerase chain reaction (PCR) for conventional direct sequencing. Purified PSR products were cycle-sequenced on an ABI 3500 Genetic Analyzer (Applied Biosystems, CA). Sanger sequencing results were analyzed by Mutation Surveyor (Softgenetics, PA) and reconfirmed by the same procedure.
Results
Phenotypic Determination
Typical RP signs in the fundi, including bone-spicule hyperpigmentation and attenuated arteries, were observed in all patients. ERG showed extinguished response of the rods and OCT clearly displayed thinner retinal thickness and disorganized inner or outer segment structure (Figure 2). Three proband patients complained progressive night blindness and mild deafness, indicating a diagnosis of type II Usher syndrome. However, another two probands clearly showed a vertigo symptom, supporting a type III diagnosis (Table 1). Among these patients, more severe visual dysfunction was present in the elders. These clinical evidences suggested diagnosis of Usher syndrome type II or III.
Figure 2. Clinical examination.
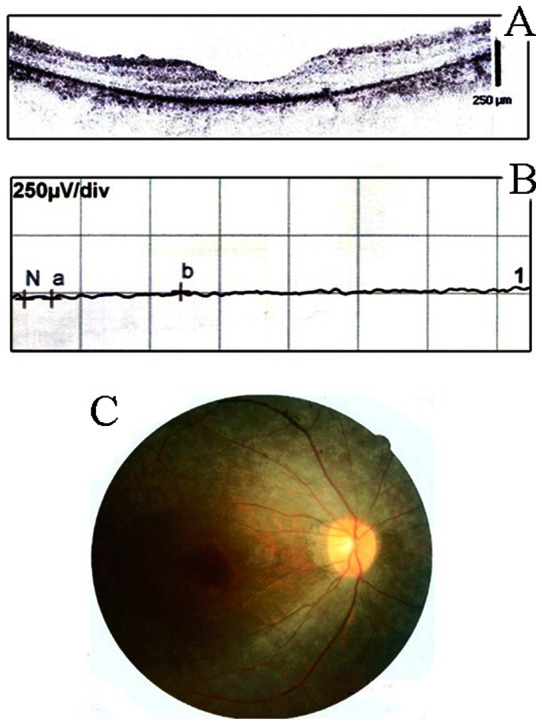
A. OCT examination demonstrated thinned retina in the proband patient with Usher syndrome (F3-III-15); B. ERG testing showed extinguished rod response; C. representative fundography of the patient.
Table 1. Clinical manifestations of the proband patients.
| Proband | M/F | Age | Onset | NB | Deaf | Vertigo | BCVA (R/L) |
| F1-III-2 | M | 29 | 5 | + | + | − | 0.7/0.8 |
| F2-III-2 | F | 33 | <5 | + | + | − | 0.3/0.3 |
| F3-III-15 | F | 31 | 10 | + | + | + | 0.15/0.15 |
| F4-III-4 | F | 40 | <5 | + | + | − | 0.2/0.3 |
| F5-II-4 | F | 65 | <5 | + | + | + | FC/0.1* |
M, male; F, female; NB, night blindness; BCVA, best corrected visual acuity; R, right eye, L, left eye. Asterisk indicates BCVA after cataract surgery in the left eye.
Targeted Exome Sequencing Identified Candidate Mutations
We performed targeted exome resequencing of 144 genes implicated in inherited retinal degeneration. For the samples subjected to TES, the average sequencing depths on the targeted regions were yielded from 104.38 to 140.13 (Table S4). Each sample had more than 98.0% targeted regions covered. Meanwhile, coverage of targeted exons for >10 reads were ranged from 90.7% to 92.3% and >20 reads from 81.6% to 85.0%. By using the SOAPsnp program [8], an average of 474 to 650 variants was identified for each sample. Among them, non-synonymous variants were ranged from146 to 178 including missense, nonsense and splicing variants. The amounts were further narrowed down to 15–27 respectively through excluding the variants reported in the HapMap 28 and the SNP release of the 1000 Genome Project with a MAF>0.05. In case of the missense variants, computational prediction by four algorithms (PolyPhen, SIFT, PANTHER and Pmut) and consistency of genetic transmission mode further confined the number of candidate mutations to less than 8. For the 6–27 coding InDels initially identified in the samples using the GATK program [11], we totally identified 2 variants based on the sample filtering strategy. By the stepwise approach as described, candidate mutations in all patients were successfully identified.
Expanded Familial Validation and Sanger Sequencing Confirmation
Among the 5 USH families, pedigrees F1 and F3 (Figure 1) showed a pseudo-dominant transmission, which is definitely different with previous reports [2]. In family F1, both the proband (F1-III-2) and his father (F1-II-4) were diagnosed as typical USH based on their clinical features. Since the F1-I-1 and F1-I-2 were completely normal, it is thus conceivable that F1-II-4 carried a de novo dominant mutation, which was afterward transmitted to F1-III-2. Family F3 also showed a genetic continuity which was considered as a dominant pedigree. However, the TES results of the two probands were probably indicated to be the USH2A gene defects. To validate the TES results from proband, we extended the same procedure to the parents and a sibling in the F1 pedigree. Interestingly, expanded familial validation successfully revealed two compound heterozygous USH2A mutations (C3416G and R3484X) in the affected father (F1-II-4), a heterozygous USH2A mutation (Q3157X) in the unaffected mother (F1-II-5) and a heterozygous USH2A mutation (R3484X) in the unaffected sibling (F1-III-3), while the proband (F1-III-2) harbored two compound heterozygous USH2A mutations (Q3157X and R3484X) (Figure 1). Sanger sequencing of the coding region further confirmed that the mutation (R3484X) was transmitted from the paternal grandfather (F1-I-1) and the mutation (C3416G) was inherited from F1-I-2. The mutation (Q3157X) in the unaffected F1-II-5 was derived from maternal grandfather (F1-I-3). These results suggested the occurrence of compound heterozygosity in USH2A for USH. Taken together, the two causatives mutations in the proband (F1-III-2) were inherited from the paternal grandfather and maternal grandfather respectively. Furthermore, the affected father (F1-II-4) harbored another mutation in USH2A leading to the disease. Co-segregation analysis was performed to confirm the extracted mutations in the other 4 pedigrees. As a result, all mutations were confirmed to co-segregate well with the disease in these families. In the pseudo-dominant family F3, both the proband (F3-III-15) and the affected sister (F3-III-13) were revealed to have two compound heterozygous mutations, c.538T>C (p.S180P) and IVS48+1G>A. The splice site mutation was transmitted from proband to her unaffected son (F3-IV-2). It was suspected that the deceased affected mother (F3-II-6) carried another unknown mutation in the USH2A gene. In family F2, the proband (F2-III-2) was found to carry three mutations. This rare condition was further confirmed by intra-familial validation. Two heterozygous mutations, c.15427C>T (p.R5143C) and c.5581G>T (p.G1861S), were confirmed in the elder daughter (F2-IV-1) who was unaffected, which indicates the bi-mutation (c.5581G>A and c.15427C>T) in the maternal allele; while another frameshift mutation, c.8602delA, was identified in the unaffected younger daughter (F2-IV-2). In the pedigree F4, two affected siblings (F4-III-2 and F4-III-4) were revealed with the same heterozygous mutations, c.8212G>A (p.D2738N) and c.5528C>T (p.P1843L). In the last pedigree F5, a novel homozygous frameshift mutation (c.4383delT) was identified in the proband (F5-II-4). The unaffected son (F5-III-3) was proved to be a carrier. Taken together, causative mutations (Tables 2, S5 and Figure S1) were successfully finalized in all USH families via expanded TES, Sanger sequencing, and co-segregation analysis.
Table 2. Identified mutations in USH2A gene.
| Family | Proband | Phen otype | Mutation | Type | Amino acid | Reported |
| F1 | III-2 | + | c.10450C>T | hetero | R3484X | Reported |
| c.9469C>T | hetero | Q3157X | Reported | |||
| F2 | III-2 | + | c.5581G>A | hetero | G1861S | Novel |
| c.15427C>T | hetero | R5143C | Novel | |||
| c.8602delA | hetero | frameshift | Novel | |||
| F3 | III-15 | + | c.538T>T | hetero | S180P | Reported |
| IVS48+1G>A | hetero | Splice site | Novel | |||
| F4 | III-4 | + | c.8212G>A | hetero | D2738N | Novel |
| c.5528C>T | hetero | P1843L | Novel | |||
| F5 | II-4 | + | c.4383delT | homo | frameshift | Novel |
In this study a total of 11 identified mutations were presumed to be pathological. Both the nonsense and frameshift mutations were predicted to create a premature stop codon, strongly indicating the protein dysfunction. The splicing site mutation, IVS48+1G>A, is predicted to be damaging in silico. In addition, all missense mutations were located in highly conserved (Figure S2) and functional domains as reported previously [14]. Taken together, these results strongly indicated disease-causing mutations in the USH2A gene.
Mutation Spectrum in the USH2A Gene
The mutation distribution had been summarized in previously reported USH2A mutations [15]–[17] and novel mutations identified in this study (Table 2). It is observed that the mutations spread over the whole region of the gene. Mutations identified in the present study, including the new ones, did not show specific distribution, suggesting that it may be absence of mutation hot spot in the USH2A in the population (Figure 3).
Figure 3. Mutation spectrum in the USH2A gene.

Gene level overview of the summarized USH2A mutations. The gene is comprised of 71 exons which harbored 1000 distinct mutations so far have been reported. The new identified mutations are marked as arrows in the schema. Each color of the box in the figure represents a mutation type: orange, nonsense mutation; black, splice site mutation; green, indel; pink, frameshift mutation; sky blue, mutation in 3′UTR; blue, intronic mutation; purple, missense mutation. Red asterisk indicates novel mutation discovered in this study. Het-one, single heterozygous mutation; homo, homozygous mutation.
Discussion
Usher syndrome is a severe disease with significant vision and hearing impairments. The prevalence worldwide ranged from 1/12,500 to 1/29,000 as previous studies have been shown [1]. Based on the phenotypic characterization, the disease has been classified into three subtypes. Seven loci (USH1B-H) have been mapped and five genes have been identified for type 1, four loci (USH2A-D) have been mapped and three genes have been identified for type 2, and two genes (USH3A, USH3B) have been identified for type 3. However, a number of studies have demonstrated phenotypic variation and crossover of the phenotype-genotype subtyping [18]. Together with the fact that a large number of coding exons exist in these genes, traditional screening of each region is infeasible for clinical application. In this study, we thus attempted to target on a group of genes responsible for inherited retinal degeneration, including RP and USH. As a result, we have proved deep exome sequencing of target 144 known causative genes of inherited retinal degeneration can serve as a fast and efficient way to perform the diagnosis. The cost was much lower compared with direct sequencing in a conservative estimation, while the amount of work is saved even more. Notably, only a single sample from the proband in each family is sufficient to identify causative mutation and intra-familiar mutation validation and co-segregation analysis can enhance the finalization.
In our USH families, mutations in the USH2A gene predispose the disease, suggesting the high prevalence of the USH2A mutation in Chinese USH population. Meantime, unaffected individuals, such as F1-II-5 from a completely normal family, were eventually found to carry a heterozygous mutation. It is worthwhile to be underlined that eight mutations have not yet been reported so far among the 11 mutations identified in this study. As there are few reports of molecular diagnosis on Chinese USH families [19], our results indicate a distinctive mutation spectrum in the population, which may require further investigation in more cohorts.
It is notable that in our strategy revealed genetic mutations in several extraordinary cases in this study. USH is well known as an autosomal recessive disease, however, among our families, pedigree F1 showed a successive transmission in a pseudo-dominant trait. This unusual case raised a question whether the patient F1-II-4 carried a de novo dominant mutation. Our results well addressed an autosomal recessive trait based on the discovery of a heterozygous mutation which is easily ignored in the proband's mother (F1-II-5). In family F2, we identified three distinct USH2A mutations in the proband (F2-III-1). In these cases, targeted exome sequencing allows excavating complete information of genetic defects which may be undetectable by traditional methods.
In the present study, we have successfully performed genetic diagnosis of Usher syndrome by utilization of NGS and have proved that it can serve as a rapid, high-throughput and efficient screening strategy. In brief, targeted exome sequencing of the 144 known genes is sufficient and clinically utilizable to comprehensively reveal genetic defects in patients with genetic retinal disease.
Supporting Information
Identified mutations confirmed by Sanger sequencing. Corresponding chromatograms showing mutant and wild-type alleles are as indicated.
(TIF)
Conserved amino acid sequence. Conservation of amino acid residue across species is highlighted.
(TIF)
Known causative genes responsible for Usher syndrome.
(DOC)
List of the genes captured in the present study.
(DOC)
List of all probes used to enrich for the target genes.
(XLSX)
Data summary of the targeted exome resequencing. Asterisk indicates proband patient.
(DOC)
Expanded familial validation.
(DOC)
Acknowledgments
We thank all patients and family members for their participation in this study.
Funding Statement
This study was supported by the National Natural Science Foundation of China (H1208/81170879), the National Key Basic Research Program (2013CB967502), and the MOST Major Projects (2012YQ12008004). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Wlelber RG, Gregory-Evans K (2006) Retinitis Pigmentosa and Allied Disorders. In: Ryan SJ, ed. Retina, Fourth ed: Elsevier-Mosby, p395–498.
- 2. Bonnet C, El-Amraoui A (2012) Usher syndrome (sensorineural deafness and retinitis pigmentosa): pathogenesis, molecular diagnosis and therapeutic approaches. Curr Opin Neurol 25: 42–49. [DOI] [PubMed] [Google Scholar]
- 3. Bras J, Guerreiro R, Hardy J (2012) Use of next-generation sequencing and other whole-genome strategies to dissect neurological disease. Nat Rev Neurosci 13: 453–464. [DOI] [PubMed] [Google Scholar]
- 4. Bowne SJ, Sullivan LS, Koboldt DC, Ding L, Fulton R, et al. (2011) Identification of disease-causing mutations in autosomal dominant retinitis pigmentosa (adRP) using next-generation DNA sequencing. Invest Ophthalmol Vis Sci 52: 494–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. He J, Wu J, Jiao Y, Wagner-Johnston N, Ambinder RF, et al. (2011) IgH gene rearrangements as plasma biomarkers in Non- Hodgkin's lymphoma patients. Oncotarget 2: 178–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu J, Matthaei H, Maitra A, Dal Molin M, Wood LD, et al. (2011) Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med 3: 92ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cox MP, Peterson DA, Biggs PJ (2010) SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11: 485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li R, Yu C, Li Y, Lam TW, Yiu SM, et al. (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25: 1966–1967. [DOI] [PubMed] [Google Scholar]
- 9. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, et al. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hou H, Zhao F, Zhou L, Zhu E, Teng H, et al. (2010) MagicViewer: integrated solution for next-generation sequencing data visualization and genetic variation detection and annotation. Nucleic Acids Res 38: W732–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jin ZB, Mandai M, Yokota T, Higuchi K, Ohmori K, et al. (2008) Identifying pathogenic genetic background of simplex or multiplex retinitis pigmentosa patients: a large scale mutation screening study. J Med Genet 45: 465–472. [DOI] [PubMed] [Google Scholar]
- 14. van Wijk E, Pennings RJ, te Brinke H, Claassen A, Yntema HG, et al. (2004) Identification of 51 novel exons of the Usher syndrome type 2A (USH2A) gene that encode multiple conserved functional domains and that are mutated in patients with Usher syndrome type II. Am J Hum Genet 74: 738–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dreyer B, Brox V, Tranebjaerg L, Rosenberg T, Sadeghi AM, et al. (2008) Spectrum of USH2A mutations in Scandinavian patients with Usher syndrome type II. Hum Mutat 29: 451. [DOI] [PubMed] [Google Scholar]
- 16. Nakanishi H, Ohtsubo M, Iwasaki S, Hotta Y, Usami S, et al. (2011) Novel USH2A mutations in Japanese Usher syndrome type 2 patients: marked differences in the mutation spectrum between the Japanese and other populations. J Hum Genet 56: 484–490. [DOI] [PubMed] [Google Scholar]
- 17. Nakanishi H, Ohtsubo M, Iwasaki S, Hotta Y, Mizuta K, et al. (2009) Identification of 11 novel mutations in USH2A among Japanese patients with Usher syndrome type 2. Clin Genet 76: 383–391. [DOI] [PubMed] [Google Scholar]
- 18. Besnard T, Vache C, Baux D, Larrieu L, Abadie C, et al. (2012) Non-USH2A mutations in USH2 patients. Hum Mutat 33: 504–510. [DOI] [PubMed] [Google Scholar]
- 19. Xu W, Dai H, Lu T, Zhang X, Dong B, et al. (2011) Seven novel mutations in the long isoform of the USH2A gene in Chinese families with nonsyndromic retinitis pigmentosa and Usher syndrome Type II. Mol Vis 17: 1537–1552. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Identified mutations confirmed by Sanger sequencing. Corresponding chromatograms showing mutant and wild-type alleles are as indicated.
(TIF)
Conserved amino acid sequence. Conservation of amino acid residue across species is highlighted.
(TIF)
Known causative genes responsible for Usher syndrome.
(DOC)
List of the genes captured in the present study.
(DOC)
List of all probes used to enrich for the target genes.
(XLSX)
Data summary of the targeted exome resequencing. Asterisk indicates proband patient.
(DOC)
Expanded familial validation.
(DOC)