

Abstract
Increasing studies have demonstrated a small proportion of cancer stem cells (CSCs) exist in the cancer cell population. CSCs have powerful self-renewal capacity and tumor-initiating ability and are resistant to chemotherapy and radiation. Conventional anticancer therapies kill the rapidly proliferating bulk cancer cells but spare the relatively quiescent CSCs, which cause cancer recurrence. So it is necessary to develop therapeutic strategies acting specifically on CSCs. In recent years, studies have shown that therapeutic agents such as metformin, salinomycin, DECA-14, rapamycin, oncostatin M (OSM), some natural compounds, oncolytic viruses, microRNAs, cell signaling pathway inhibitors, TNF-related apoptosis inducing ligand (TRAIL), interferon (IFN), telomerase inhibitors, all-trans retinoic acid (ATRA) and monoclonal antibodies can suppress the self-renewal of CSCs in vitro and in vivo. A combination of these agents and conventional chemotherapy drugs can significantly inhibit tumor growth, metastasis and recurrence. These strategies targeting CSCs may bring new hopes to cancer therapy.
Keywords: therapy, target, cancer stem cell
Introduction
In the past decades, even though some progress has been made in anticancer treatment, patients dying from tumor recurrence are still common. The cancer stem cell hypothesis proposes that only a small proportion of cancer cells have self-renewal capacity and tumor-initiating ability, known as CSCs or tumor-initiating cells(TICs).1,2 CSCs share similar cell surface markers and self-renewal pathways with normal stem cells, have potent differentiation capacity and are resistant to chemotherapy and radiation.1,2 Up to the present, the differences between normal stem cells and CSCs remain poorly defined except that the self-renewal of normal stem cells is in control while that of CSCs is out of control.
CSCs were first identified in acute myeloid leukemia (AML).3,4 Till now, CSCs have been isolated in breast cancer,5,6 lung cancer,7 brain tumor,8-10 colon cancer,11-14 prostate cancer,15 pancreatic cancer,16 ovarian cancer,17 liver cancer,18 melanoma,19 and so on. Conventional anticancer therapies kill the rapidly proliferating non-CSCs while have less effect on CSCs1,2. CSCs resist chemotherapy and radiation by their powerful self-renewal capacity, drug effluxion, antiapoptotic ability and other unknown mechanisms.1,2,20
However, there are some doubts about the CSC hypothesis. Some researchers believe that cancer initiation and progression are better explained by the stochastic (or clonal evolution) model,21 in which most or all cancer cells have inherent tumorigenic potential. Some propose a more dynamic model, the Yin-Yang mode, in which cancer cells are heterogeneous and consist of varying growing or replicating populations (Yang) and non-dividing or slow growing populations (Yin) and the two populations may inter-convert.22 Some believe that different cancers may follow different models; for example, some cancers may follow the CSC model while others may follow the stochastic model.21 Despite these different insights, there is no doubt that some cancer cells are resistant to chemotherapy and radiation and cause anti-cancer treatment failure and tumor recurrence. The CSC hypothesis prompts the exploration of strategies targeting CSCs, which are resistant to conventional anti-cancer therapies. These strategies will make it possible to eradicate cancer cells (Fig. 1).
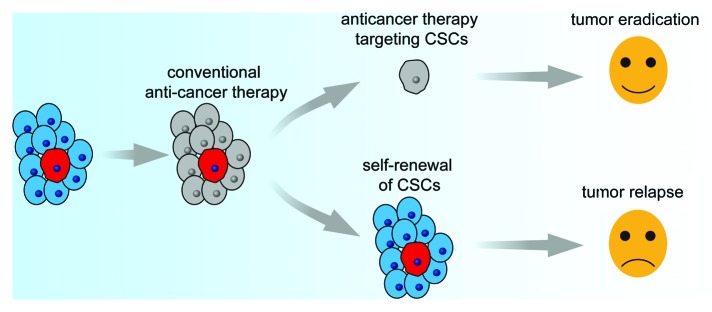
Figure 1. Anti-cancer therapy may consist of two steps, the first step for killing rapidly proliferating bulk cancer cells, which are sensitive to chemotherapy and radiotherapy, the second step for targeting CSCs in a resting state, which are resistant to conventional anticancer therapies. After these two steps, all cancer cells will be eradicated.
Recently, metformin, salinomycin, DECA-14, rapamycin, Oncostatin M (OSM), some natural compounds, oncolytic viruses, microRNAs, signaling pathway inhibitors, TNF-related apoptosis inducing ligand (TRAIL), interferon (IFN), telomerase inhibitors, All-trans retinoic acid (ATRA) and monoclonal antibodies have been shown to inhibit the self-renewal of CSCs in vitro and in vivo. A combination of these agents and conventional chemotherapy drugs can significantly inhibit tumor growth, metastasis and recurrence. In this paper, therapeutic strategies targeting CSCs in recent years are reviewed. Cancer cells with cancer stem-like cell characteristics such as CD133-positive cells, side population (SP) cell,aldehyde dehydrogenase (ALDH)-positive cells, radioresistant cells and chemoresistant cells are all considered as cancer stem-like cells in this paper.
Metformin
Metformin is a standard drug for type 2 diabetes treatment and has been used for years. Epidemiological research shows that the cancer risk in patients with type 2 diabetes taking metformin was lower than that in other patient groups.23 It has been showed that in vivo and in vitro metformin can inhibit cancer cell growth of breast cancer,24-26 ovarian cancer,27 endometrial cancer,28 prostate cancer,29,30 and pancreatic cancer.31 Chemotherapy-resistant ovarian cancer cells can also be killed by metformin.27 In a recent study by Hirsch et al., contrary to the chemotherapeutic drug doxorubicin, metformin selectively killed CD44high/CD24low breast CSCs while having less effect on CD44low/CD24high breast non-CSCs cells,32 and had synergistic effect with doxorubicin on killing breast CSCs in vitro and in vivo. When breast cancer xenografts were treated with doxorubicin and metformin, the CSCs proportion rapidly decreased to undetectable levels. After treatment was stopped, the animals remained in tumor remission for at least 60 d, meaning that the tumors were almost completely eliminated. However, treatment with doxorubicin alone increased the percentage of CSCs, and the mouse xenografts re-grew rapidly 20 d after the treatment was stopped.32 The molecular mechanism by which metformin inhibits the self-renewal of breast CSCs is unclear and requires further study. It has been shown that in cancer cells, metformin can activate the AMP-activated protein kinase (AMPK) enzyme,24-31 arrest the cell cycle, decrease cyclin D1 expression, increase p21 protein expression, attenuate mTOR-S6RP phosphorylation, inhibit protein-translational and lipid biosynthetic pathways27 and disrupt crosstalk between the insulin/IGF-1 receptor and GPCR signaling.31 Metformin has been used in antidiabetic treatment for more than 50 y and is now believed to be the most widely prescribed antidiabetic drug in the world. It causes few adverse effects and prevents the cardiovascular complications of diabetes. The proper dose for metformin in contributing to killing CSCs in humans is unknown, and lactic acidosis may be caused by an overdose of metformin. So, although metformin is a promising agent in targeting CSCs, more research is needed.
Salinomycin
Salinomycin, a highly selective potassium ionophore and an antibiotic extensively used for coccidiosis, has been shown to selectively kill breast CSCs.33 In a study by Gupta et al., 16,000 different chemical compounds were screened, and only 32 compounds targeted CSCs.33 Among these compounds, salinomycin killed CSCs of breast cancer at least 100 times more effectively than paclitaxel in mice. In vitro, the percentage of CD44high/CD24low breast CSCs increased 18-fold in breast cancer cells treated with paclitaxel while it decreased 20-fold in salinomycin treatment. Furthermore, the tumorigenicity of breast cancer cells pretreated with salinomycin decreased more than 100-fold compared with cells pretreated with paclitaxel. In animal models, salinomycin intraperitoneal injection inhibited mouse xenografts' growth, induced apoptosis, necrosis, epithelial differentiation and suppressed stemness gene expression of cancer cells. In addition, salinomycin can overcome ATP-binding cassette (ABC) transporter-mediated multidrug resistance and apoptosis resistance of KG-1a cells (human leukemia stem-like cells), restoring the sensitivity of these cells to chemotherapeutic drugs.34 The mechanism(s) by which salinomycin induces breast CSCs specific toxicity remains unclear. In the same study, nigericin, another potassium ionophore bearing structural similarity to salinomycin, also exhibited similar selective toxicity on breast CSCs, suggesting that salinomycin killing CSCs may due to its action as a potassium ionophore.33 However, salinomycin has high toxicity which may limit its clinical use.
DECA-14 and Rapamycin
In a study by Smith et al., 51 compounds that selectively targeted patient-derived neuroblastoma TICs (NB TICs) were identified by a high-throughput cell-based screening assay. Among the compounds, dequalinium analog, C-14 linker (DECA-14), an antimicrobial agent used in mouthwashes and throat lozenges and rapamycin, a specific inhibitor of the mammalian target of rapamycin(mTOR), were characterized in detail.35 In vitro DECA-14 treatment induced apoptosis of NB TICs isolated from multiple patients, while had less effect on established NB cell lines and little effect on normal pediatric stem cells (skin-derived precursors, SKPs), indicating that DECA-14 selectively targeted NB TICs. Gene expression analysis showed that most significantly altered transcripts in DECA-14 treatment were the mitochondrial NADH dehydrogenase subunits that compose complex I of the mitochondrial electron transport chain, indicating that DECA-14 induced apoptosis of NB TICs by affecting mitochondria electron transport. Intraperitoneal injections of DECA-14 significantly inhibited subcutaneous xenograft tumor growth and tumor-initiating capacity of NB TICs in vivo. Except mild body weight reduce, no other significant toxicities were noted in DECA-14 treated animals. In NB TICs, p70S6K and S6 ribosomal protein (S6RP), two proteins in the mTOR signaling pathway, were hyperphosphorylated, indicating that mTOR signaling pathway is constitutively activated. Rapamycin treatment inhibited cell proliferation and induced apoptosis of NB TICs and had little effect on SKPs and established NB cell lines, which are similar with DECA-14 treatment. The phosphorylation of p70S6K and S6RP in NB TICs was rapidly inhibited by rapamycin treatment, indicating the activity of mTOR signaling pathway was reduced by rapamycin. Rapamycin treatment reduced NB TICs xenograft tumor weight by 74.0% to 82.6%, while vinblastine, a chemotherapy used to treat NB, only reduced tumor weight 43.4% relative to vehicle-treated controls, indicating that rapamycin was much more effective than vinblastine. For the advantage of being selectively toxic to NB TICs while sparing normal pediatric stem cells, DECA-14 and rapamycin will be promising agents targeting CSCs in vivo, especially in pediatric cancers.
Oncostatin M (OSM)
Oncostatin M (OSM), a multifunctional interleukin 6-related cytokine, has been shown to inhibit the proliferation of various solid tumor.36 In a study by Yamashita et al., OSM receptor (OSMR) was detected in hepatocellular carcinoma (HCC) cells and had a high frequency in epithelial cell adhesion molecule (EpCAM)–positive HCC cells, which had CSCs characteristics.37 In vitro OSM treatment on HuH1 and HuH7 HCC cells led to a decrease in stemness gene expression, EpCAM expression, α-fetoprotein and cytokeratin 19 protein expressions and an increase in albumin protein expression, indicating differentiation of the HCC cells. OSM is known to enhance hepatocytic differentiation of hepatoblasts by inducing the activation of the signal transducer and activator of transcription 3 (STAT3) pathway. Incubation of HCC cells with OSM resulted in the induction and nuclear accumulation of phosphorylated STAT3, indicating that differentiation of EpCAM+ HCC cells induced by OSM is also in a STAT3-dependent manner. More over, although OSM treatment alone showed weak tumor-suppressive effects in primary EpCAM+ AFP+ HCC xenograft tumors in NOD/SCID mice, the combination of OSM with 5-FU showed a marked inhibition of tumor growth compared with 5-FU alone, indicating that OSM treatment increases the chemosensitivity of EpCAM+ HCC cells. 5-FU treatment alone can induce Annexin V+ and activated caspase 3+ cells and diminish EpCAM− non-CSCs, while enrich the EpCAM+ CSCs in HCC. Although OSM treatment alone had a slight effect on apoptosis induction of HCC cells, it can decrease EpCAM+ cell population and significantly enhance the activation of caspase 3 in 5-FU treatment. The apoptosis induction and tumor suppression of OSM combined with 5-FU is much more effective compared with OSM or 5-FU alone indicates that the combination of some agents targeting CSCs and conventional chemotherapies may produce one plus one is greater than two effects.
Natural Compounds
Some natural compounds isolated from vegetables, fruits and other plants have many biological activities including killing cancer cells. Recently, it was shown that some natural compounds can also target CSCs or sensitize CSCs to anticancer drugs. Sulforaphane, an isothiocyanate enriched in broccoli, can inhibit cell proliferation, induce apoptosis, suppress mammosphere formation and inhibit the ALDH-positive cells of breast cancer cell lines in vitro, reduce ALDH-positive cells by > 50% in NOD/SCID mice xenograft tumors and abrogat tumor growth after the reimplantation of primary tumor cells into the secondary recipient mice.38 Downregulation of the Wnt/β-catenin pathway may be one of the possible mechanisms for sulforaphane inhibiting self-renewal of breast CSCs.38 In human prostate cancer cell lines, epigallocatechin gallate (EGCG), the most abundant catechin in green tea, can inhibit cell growth, self-renewal, migration, invasion and induce apoptosis of CSCs by engaging cell-intrinsic pathway of apoptosis, suppress epithelial-mesenchymal transition (EMT) of CSCs by inhibiting the Wnt/β-catenin pathway and the expression of the transcription factors slug, snail, which are required for induction of EMT39. Quercetin, a plant-derived flavonoid found in fruits, vegetables, leaves and grains, can inhibit the self-renewal and EMT of pancreatic CSCs and revert apoptotic resistance of the pancreatic CSCs in vitro.40 It can also suppress pancreatic CSC-enriched xenograft growth through reduced cell proliferation, apoptosis induction and angiogenesis inhibition; moreover, has synergistic effects with sulforaphane and have no pronounced toxicity to normal cells or animals.40 In addition to having synergistic effects with sulforaphane, quercetin, combined with chemotherapeutic drugs, is effective in blocking antiapoptosis pathway of lung CSCs.41 Curcumin, a popular food spice, can inhibit the SP phenotype of the rat C6 glioma cell line,42 and berberine, a quaternary ammonium salt from the protoberberine, can suppress the self-renewal of SP cells and ABCG2 transporter expression in human MCF-7 breast cancer cells.43 Gamma-tocotrienols (gamma-T3), one of the vitamin-E constituents, can inhibit the tumorigenicity of prostate cancer cells and kill docetaxel-resistant CD133-enriched PC-3 prostate cancer cells.44 Parthenolide, a sesquiterpene lactone well known in natural medicine, can reverse the vinorelbine resistance of SP cells in MCF-7 breast cancer cells.45 A combination of stealthy liposomal vinorelbine and stealthy liposomal parthenolide can fully inhibit the MCF-7 cell xenograft growth.45 Moreover, some phytoestrogens/flavonoids, such as genistein, naringenin, acacetin and kaempferol, which have less effect on drug-sensitive leukemia K562 cells, can potentiate the cytotoxicity of SN-38 and mitoxantrone in breast cancer resistance protein (BCRP)-transduced K562 (K562/BCRP) cells, indicating these phytoestrogens/flavonoids may reverse multidrug-resistance protein (MRP)-mediated chemotherapy resistance.46 All these results show that some natural compounds are promising candidates in targeting CSCs for their several anticancer mechanisms.
Oncolytic Viruses
Oncolytic viruses can infect tumor cells and replicate in the cells, eventually lysis tumor cells and release more viruses to infect other tumor cells. These viruses cannot replicate in normal cells, so they do not kill normal cells47; for this advantage some oncolytic viruses have been used in clinical.47 If specific promoters are used, oncolytic viruses will be more specific, effective and safe in anticancer therapy. In a study by Bauerschmitz et al., oncolytic viruses featuring cyclo-oxygenase 2 (Cox-2), telomerase (hTERT) and multidrug-resistance (MDR) promoters(Ad5/3cox2LΔ24,Ad5/3-hTERT-Δgp and Ad5/3-mdr-Δ24), destroyed or even completely eliminated CD44+ CD24-/low breast CSCs in vitro and had a significant antitumor effect on CD44+ CD24-/low breast CSCs- derived tumors.48 Fifty percent was necrotic in the tumors treated with Ad5/3-mdr-Δ24 while only 20% was necrotic in the mock-treated tumors, indicating that Ad5/3-mdr-Δ24 had strong cell killing effects on these breast CSCs.48 In another study, a telomerase-specific oncolytic adenoviral vector carrying TRAIL and E1A genes (Ad/TRAIL-E1) preferentially targeted and killed radioresistant human esophageal carcinoma cells; these cells had CSCs’ characteristics and higher telomerase activity than their parent cells. When Ad/TRAIL-E1 was injected intratumorally, the radioresistant tumor cells in xenografts underwent apoptosis and eventually 40% of the mice survived free of tumors for more than 180 d. Moreover, Ad/TRAIL-E1 did not show significant toxicity on normal cells in vitro and in vivo.49 Oncolytic viruses can be genetically modified for targeting specific cells as required and do no harm to normal cells, making them promising therapies for targeting CSCs.
MicroRNAs
MicroRNAs (miRNAs) are a class of endogenous, non-coding 20−22 nt small RNAs that can base pair their target mRNAs to repress their translation or induce their degradation.50 They are crucial post-transcriptional regulators of gene expression in cellular proliferation, differentiation and apoptosis, and they act as oncogenes or tumor suppressor genes contributing to the development of human malignant tumors. Altered expression of miRNAs have been observed in human cancers, such as miR-143 and miR-145 downregulation in colorectal cancer51 and lung cancer,52 and miR-21 upregulation in glioblastoma tumors53 and breast cancer.54,55 Recently, it was indicated that some miRNAs are involved in the self-renewal and survival of CSCs.
The miR-34 family was found to be directly regulated by p53 and target Notch and Bcl-2.56,57 In four human gastric cancer cell lines, Kato III, AGS, N87 and MKN45, p53-mutant Kato III cells have the lowest levels of both pri-miR34a and mature miR-34a and the highest expression levels of miR34 target genes Bcl-2, Notch1 and Notch4.58 MiR-34 restoration in Kato III cells results in inhibition of cell growth and tumorspheres formation, accumulation in G1 phase and caspase-3 activation, downregulates miR-34 target gene Bcl-2 and Bax, Notch1 and HMGA2 and renders the cells 2–3-fold more sensitive to chemotherapeutic agents(doxorubicin, cisplatin, gemcitabine and docetaxel, all of which are used in gastrointestinal cancer chemotherapy).58 p53-mutant human MiaPaCa2 pancreatic cancer cells also have very low miR-34a,b,c expression but high levels of the miR-34 target genes Bcl2 and Notch1.59 In MiaPaCa2 cells, miR-34 restoration inhibits cell clonogenic growth, sensitizes MiaPaCa2 cells to chemotherapy(gemcitabine) and radiation by inducing caspase-3 activation and apoptosis, results a significant reduction of CD44+/CD133+ CSCs proportion and a decreased tumor formation rate in nude mice (miR-34a restoration group 2/10 vs. controal group 10/10). MiR-34 restoration also significantly inhibites the self-renewal of CD44+/CD133+ CSCs and led to an almost 23-fold reduction of Bcl-2 mRNA in CD44+/CD133+ CSCs, while only showing a 43% reduction of MiaPaCa2 total population cells, indicating that miR-34 restoration plays a more critical role in targeting these pancreatic CSCs than in the total population cells.59 These findings suggest that miR-34 mimics may hold significant promise as a novel molecular therapy agent targeting CSCs in p53-deficient human cancers.
In a study of Yang et al., miR145, a tumor-suppressive miRNA, is inversely correlated with the levels of stem gene Oct4 and Sox2 in glioblastomas (GBMs) CD133 positive cells and malignant glioma specimens.60 Expression of miR145 by gene delivery using polyurethane-short branch polyethylenimine (PU-PEI) significantly inhibited tumorigenic potential and CSC-like abilities of GBM-CD133+ cells and induced GBM-CD133+ cells differentiate into CD133--non-CSCs. PU-PEI-miR145 treatment also has synergistic effect with radiation and temozolomide on orthotopic BM-CD133+-transplanted immunocompromised mice by reverseing drug-resistance and apoptosis resistance, indicating that combination with miR145 and radiation or chemotherapy may provide a promising new treatment for targeting CSCs in Glioblastomas. Besides miR34 and miR145, other miRNAs involved in cancer development may also serve as anticancer agents targeted CSCs.
Signaling Pathway Inhibitors
Cell signaling pathways such as Notch, Hedgehog and Wnt/β-catenin play important roles in cancer development and are involved in the self-renewal of CSCs. Li et al. demonstrated that the expression of Sonic hedgehog (Shh) in CD44+ CD24+ ESA+ pancreatic CSCs was 10-fold higher than in bulk pancreatic cancer cells.16 Hedgehog inhibitor cyclopamine, a combination with cyclopamine and gemcitabine or a triplet combination with cyclopamine/CUR199691, rapamycin and chemotherapy can all inhibit Hedgehog signaling pathway, decrease the proportion of CSCs and induce tumor regression in pancreatic cancer xenograft models.61-63 In brain tumor cells, cyclopamine treatment decreased CSCs proportion or even eliminated CSCs and deprived the tumorigenicity of the tumor cells.64 In breast CSCs, activation of the Notch signaling pathway is significantly higher than in bulk cancer cells.65 Knockdown of Notch4 with small interfering RNA (siRNA) significantly inhibited the self-renewal of ESA+/CD44+/CD24low breast CSCs and completely deprived the tumorigenicity of these breast CSCs.65 In human colorectal cancer xenografts, anti-human Notch ligand Delta-like4 antibody (anti-hDLL4) significantly decreased the proportion of ESA+/CD44+/CD166+ CSCs in tumors; a combination of anti-hDLL4 and irinotecan showed synergistic inhibition on tumor growth and recurrence.66 The Wnt/β-Catenin signal pathway is also activated in various tumors and involved in the regulation of the self-renewal of CSCs. In breast cancer, the natural compound sulforaphane inhibiting the self-renewal of ALDH positive cells,38 and oxymatrine suppressing the the self-renewal of SP cells,67 are all associated with downregulation of the Wnt/β-Catenin signaling pathway.
Iinhibition of other cell signal pathwayws can also target CSCs. Platelet-derived growth factor (PDGF) plays a central role in the development of glioma, inhibition of PDGF-B by siRNA leads to glioma-derived cancer-initiating cells (GICs) stop proliferating, lose their self-renewal ability and tumor-initiating capacity and differentiate into normal tissue cells to a certain extent.68 C-Kit (stem cell factor receptor) signaling also play an important role in development of glioma. Inhibition of PDGF receptor (PDGFR) and c-Kit by imatinib mesylate, the tyrosine kinase inhibitor or by siRNA, induces differentiation of glioma CSCs and inhibits the tumorigenicity of the glioma CSCs.69
Recently, a phase I clinical trial of a combination therapy that targets both bulk tumor cells and CSCs in relapsed head and neck cancer patients has been started. National Institutes of Health (NIH) founded this national program which focuses on anti-CSC therapies in the human body for the first time.70 The clinical trial will test the combination of the conventional chemotherapy drug cetuximab with the anti-CSC drug IPI-926, a novel, potent, oral molecule that inhibits Smoothened, which is a key component of the Hedgehog pathway. In the clinical trial program, cetuximab targets the bulk head and neck tumor cells and IPI-926 disrupts the Hedgehog signaling pathway in CSCs.70 The trial will move this therapy from the lab to the clinic.
Tumor Necrosis Factor-Related Apoptosis Inducing Ligand (TRAIL)
TRAIL is a member of the tumor necrosis factor (TNF) super family, which is also called Apo2 ligand (Apo2L). Either membrane-bound or soluble TRAIL can rapidly induce apoptosis of cells expressing TRAIL-specific receptors. An advantage of TRAIL is that this agent can specifically kill cancer cells while sparing normal cells due to the protection of decoy receptors.71 CD133-positive or CD133(high) cancer stem-like cells in Jurkat T-lymphoma cells,72 glioma cells,73 and primary colon cancer cells,13 are more resistant to TRAIL than their CD133-negative or CD133(low) counterparts. It is shown that the high expression of FLIP (an inhibitor of death receptor-mediated apoptosis),72 low levels of caspase-8 expression,73 and production of interleukin-413 cause these CSCs' TRAIL resistance. While there have been different results concerning CSCs' sensitivity to TRAIL, the SP cells in human colon cancer cell lines were shown to be more sensitive to TRAIL than the non-SP cells, so is in several breast cancer cell lines and ovarian carcinoma cell lines.74 After TRAIL treatment, the percentage of SP cells decreased by at least 50% in the above cancer cell lines and even by more than 90% in the SW480 human colon cancer cell line. The pro-apoptotic TRAIL receptor DR4 expression in SW480 SP cells is 10-fold higher than non-SP cells, and SP cells are more sensitive to TRAIL-induced apoptosis than non-SP cells. Moreover, these SW480 SP cells express a higher level of c-Myc than non-SP cells and c-Myc activates DR4 transcription through E-box DNA-response elements located in the DR4 promoter, which increase the expression of cell-surface pro-apoptotic death receptors in these SP-cells.74 Mesenchymal stem cells (MSCs) have been applied as a targeted-delivery vehicle in cancer gene therapy for they having a tendency to distribute at the site of tumors.75 It has been shown that TRAIL-expressing MSCs can significantly inhibit tumor growth and induce significant survival benefits in animal models76,77; moreover, MSCs expressing TRAIL can cause apoptosis or death and reduce colony formation of SP cells in quamous (H357) and lung (A549) cancer cell lines and act in synergy with conventional chemotherapy.78 The sensitivity of cancer stem-like cells to TRAIL is various may be due to their different genetic background. In the above study, all those cells sensitive to TRAIL were SP cancer cells, while those resistant to TRAIL were CD133-positive or CD133 high cancer cells. Even though all these cells have CSCs characteristics, they are not exactly the same. If more studies confirm that CSCs are sensitive to TRAIL, then TRAIL-expressing MSCs will be a promising anticancer agent targeting CSCs for their’s no harm to normal cells and spontaneous migration to the site of tumors in vivo.
Interferon (IFN)
IFN is a cytokine used in treatment for virus diseases and malignant tumors. Its anti-tumor mechanisms include induction of apoptosis and differentiation of cancer cells, suppression of cell proliferation and angiogenesis and immune regulation.79,80 In a study by Moserle et al.,81 IFN-α dramatically reduced the proliferation of ovarian primary cancer cells with high SP proportions in vitro while had less effect or no effect on ovarian primary cancer cells with low SP cell proportions, indicating that SP cells of ovarian primary cancer cells are more sensitive to IFN-α than non-SP cells. In vivo IFN-α gene therapy showed a similar result, mice injected with ovarian primary cancer cells with a high SP proportion (53.1%) showed a good response to IFN-α treatment, while low SP proportions (1.7%) ovarian cell-injected animals did not. After IFN-α treatment, some gene transcripts were strongly upregulated in SP cells but not in non-SP cells, such as IFI16, USP18, PLSCR-1, SAMD9, GBP1 and IFIH1, which are involved in the defense/immune response, cell proliferation, apoptosis and angiogenesis. These may be the molecular mechanisms for SP cells in ovarian primary cancer cells being sensitive to IFN-α. IFN-α can also suppress the self-renewal of SP cells in HT29 colorectal cancer cells and Daoy medulloblastoma cells.81 In another study, it was showed that SP cells are responsible for the paclitaxel (PTX)-resistance of three ovarian cancer cell lines and the PTX-resistance can be overcome by INF-α.82 Therefore, INF-α may improve the anticancer effect of PTX in ovarian cancer and may avoid inducing PTX-resistant cells. In glioma, IFN-β can suppress the cell proliferation, self-renewal and tumorigenesis of CSCs; induce the terminal differentiation of CSCs to mature oligodendroglia-like cells and exhibit synergistic cytotoxicity with temozolomide on glioma CSCs.83 All the above results indicate that IFN may be a potential therapeutic agent targeting CSCs.
Telomerase Inhibitors
Shortening of telomeres with each cell division eventually leads to cell cycle arrest and apoptosis. Telomere length can be maintained by telomerase, a reverse transcriptase which consists of an RNA primer sequence and a telomere reverse transcriptase (TERT). Telomerase activity has been detected in almost 90% of human cancers while most normal somatic cells are telomerase-negative.84 Telomerase plays an important role in cancer development and has been a target for cancer therapy. Imetelstat, a synthetic lipid-conjugated 13-base oligonucleotide N3′P5′-thio-phosphoramidate, is complementary to the template region of telomerase RNA and acts as a competitive enzyme inhibitor by binding and blocking the active site of the enzyme. This specific telomerase inhibitor has been used in clinical for treatment of multiple myeloma (MM), chronic lymphocytic leukemia, non-small cell lung cancer and breast cancer85. In a study by Brennan et al., long treatment of imetelsta (for two weeks) resulted in telomerase inhibition, decreased cell colony formation and shorten telomere length in CD138-negitive CSCs of MM cell lines and primary MM cells. Short-term imetelstat treatment for 72 h, even with no telomere shortening, also resulted in colony formation inhibition of CSCs in MM cell lines and primary cells.86 By short-term imetelstat treatment, the proportion of CD138-negitive CSCs decreased by approximately 40% and the ALDH+ population decreased from 1.3% to less than 0.4% in unsorted NCI-H929 MM cells and the expression of stem genes OCT3/4, NANOG, SOX2 and BMI1, and Notch target gene HES1 were inhibited in CD138-negitive NCI-H929 CSCs, indicating that short-term imetelstat can suppress the self-renewal and induce differentiation of MM CSCs. These results show that short-term imetelstat treatment inhibiting the self-renewal of MM CSCs is independent of telomere length shortening. In vivo, imetelstat treatment significantly prolonged the survival of NOD/SCID mice with MM engraftment injected by NCI-H929 cells.86 In another study, imetelstat treatment resulted in telomerase inhibition and telomere shortening in MCF7 and MDA-MB231 breast cancer cells and PANC1 pancreatic cancer cells; in vitro long imetelstat treatment (several weeks) resulted in depletion of CSCs and cell growth inhibition in these breast and pancreatic cancer cells and pretreatment with imetelstat decrease the tumorigenicity of PANC1 and MDA-MB231 cells.87 In primary glioblastoma TICs, imetelstat treatment can also produce a dose-dependent inhibition of telomerase.88 In a study of Marian et al., in vitro long-term imetelstat treatment on GBM TICs led to telomere shortening, growth arrest and eventual cell death, and had synergic effect with radiation and temozolomide; the average volume of subcutaneous tumors derived from glioblastoma TICs in imetelstat treated animals was more than 10-fold lower than that of the control animals; moreover, by intraperitoneal injection, imetelstat penetrated the blood-brain barrier and inhibited telomerase activity in animals with orthotopic xenograft tumors of glioblastoma TICs.88 Taken together, these studies indicate that imetelstat can target CSCs and being a prospective candidate agent for eradication of cancer.
All-Trans Retinoic Acid
All-trans retinoic acid (ATRA), a naturally occurring compound derived from vitamin A, plays a role in cell growth, differentiation and apoptosis and has been applied in therapy of hematological malignancies and some solid tumors.89 Being a potent differentiating agent, ATRA is a promising drug in eradicating CSCs. It has been shown that low concentrations of ATRA (10 μM) can induce glioblastoma multiforme CSCs differentiate into glial and neuronal lineages and high doses of ATRA (40 μM) can resulte in apoptosis of glioblastoma multiforme CSCs in an MAPK-dependent manner.90 In another study, agonists for the retinoid X receptor, retinoic acid receptor and peroxisome proliferator-activated receptor (PPAR)-γ, reduced the survival of mammospheres generated from breast cancer tissues and breast cancer MCF7 cell line by suppressing the activity of pro-inflammatory Nuclear Factor-κB (NFκB)/Interleukin-6 (IL6) axis which is hyperactive in breast cancer-derived mammospheres, while had no effect on survival of mammospheres from normal mammary gland or non-tumorigenic MCF10 breast cell lines.91 In head and neck squamous carcinoma CSCs(HNSC CSCs), ATRA can suppress the expression of the stem cell markers Oct4, Sox2, Nestin and CD44 and inhibit the proliferation of HNSC CSCs in vitro and in vivo. Furthermore, ATRA treatment can promote the sensitization of HNSC CSCs to cisplatin. Downregulation of Wnt/β-catenin signaling may be one of the molecular mechanisms of ATRA targeting HNSC CSCs.92 These results indicate that ATRA combined with conventional anticancer therapy may be a novel approach to eradicate CSCs.
Monoclonal Antibodies
CSCs express some specific cell surface markers such as CD133, CD24, CD44 and EpCAM etc. An anti-CD133 monoclonal antibody (mAb) showed a dose-dependent cytotoxic effect on FEMX-I melanoma cells which express CD133 while having no effect on human MA-11 breast carcinoma cells which do not express CD133.93 In vitro pretreated with single-walled carbon nanotubes (SWNTs) conjugated with CD133 monoclonal antibody (anti-CD133) and then irradiated with near-infrared laser light, CD133 positive cells in glioblastoma (GBM-CD133+), which display cancer stem cell-like characteristics, were selectively targeted and eradicated,whereas CD133 negative cells in glioblastoma (GBM-CD133-) remained viable.94 More over, the self-renewal and tumorinitating capability of GBM-CD133+ treated with localized hyperthermia was significantly blocked.94 In another study, a bispecific EpCAMxCD3 antibody linking tumor cells and T lymphocytes significantly retarded the tumor growth of BxPC-3 pancreatic carcinoma xenografts.95 Since CD133 and EpCAM are common surface markers of CSCs, these monoclonal antibodies may also have cytotoxic effects on CSCs. It is worth noting that normal stem cells and CSCs share some of the same surface markers; in order to avoid killing normal stem cells, it is necessary to find more specific surface markers of CSCs and perform a topical application for these antibodies.
Self-renewal pathway inhibition by monoclonal antibody also can target CSCs. Notch1 inhibition by a Notch1 monoclonal antibodies (mAbs) specifically binding to the negative regulatory region of human Notch1 leads to decreased self-renewal ability of CSCs and tumor growth inhibition in xenograft models derived from triple negative breast cancer (TNBC) Sum149 cell line and TNBC patient primary cancer cells and has synergistic effect with docetaxel.96 Moreover, Notch1 mAbs also resultes in decreased tumor incidence upon re-implantation and a delay in tumor recurrence of the TNBC cells. TNBC is very aggressive and currently there is no specific therapeutic stratge,inhibition of self-renewal pathway by Notch1 mAbs may provide a novel anticancer therapy on TNBC.
Conclusions
Despite that there is still some debate about the CSC hypothesis, there is no doubt that a small proportion of cancer cells shows stem-like cell characteristics and is resistance to chemotherapy and radiation treatment, resulting in tumor relapse after anticancer therapy. Therapies targeting CSCs combined with chemotherapy and radiation therapy bring new hopes in cancer eradication. This paper reviews recent studies in this area. The strategies in this article include targeting CSCs and those which targeting CD133 positive cells, SP cells, drug-resistant cells, radio-resistant cells or ALDH positive cells; these cells are all regarded as cancer stem-like cells. Although the developed strategies of targeting cancer stem-like cells have great advantages on cancer treatment, several issues need to be extensively studied in the future. Because some signaling pathways and cell surface markers exist in CSCs and normal stem cells are the same, how do we avoid destroying normal stem cells when signaling pathways inhibitors or mAbs specific for cell surface markers are used? What are the mechanisms of each developed strategy targets CSCs? How do we combine these agents targeting CSCs with conventional chemo- or radio-therapies to treat cancers? Differentiation treatment is an ideal strategy in cancer therapy and has been successfully applied in the treatment of leukemia,97 some strategies targeting CSCs can induce the differentiation of CSCs to a certain extent or into non-CSCs, can we induce the differentiation of CSCs to normal tissue cells? Can we convert CSCs to normal stem cells by regulating the self-renewal of CSCs? All these are worthy of exploration in the future. Finally, since most of the recently developed strategies were tested only in vitro and/or in animal models, what are their effective doses and their toxic effects in humans? Clinical trials for these strategies are also required before their clinical application in the future.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/23622
References
- 1.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 2.Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Curr Opin Biotechnol. 2007;18:460–6. doi: 10.1016/j.copbio.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 4.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 5.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ponti D, Costa A, Zaffaroni N, Pratesi G, Petrangolini G, Coradini D, et al. Isolation and in vitro propagation of tumorigenic breast cancer cells with stem/progenitor cell properties. Cancer Res. 2005;65:5506–11. doi: 10.1158/0008-5472.CAN-05-0626. [DOI] [PubMed] [Google Scholar]
- 7.Eramo A, Lotti F, Sette G, Pilozzi E, Biffoni M, Di Virgilio A, et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008;15:504–14. doi: 10.1038/sj.cdd.4402283. [DOI] [PubMed] [Google Scholar]
- 8.Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–83. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–8. [PubMed] [Google Scholar]
- 10.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–21. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 11.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–10. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 12.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, et al. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–5. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 13.Todaro M, Alea MP, Di Stefano AB, Cammareri P, Vermeulen L, Iovino F, et al. Colon cancer stem cells dictate tumor growth and resist cell death by production of interleukin-4. Cell Stem Cell. 2007;1:389–402. doi: 10.1016/j.stem.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 14.Vermeulen L, Todaro M, de Sousa Mello F, Sprick MR, Kemper K, Perez Alea M, et al. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proc Natl Acad Sci U S A. 2008;105:13427–32. doi: 10.1073/pnas.0805706105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005;65:10946–51. doi: 10.1158/0008-5472.CAN-05-2018. [DOI] [PubMed] [Google Scholar]
- 16.Li CW, Heidt DG, Dalerba P, Burant CF, Zhang LJ, Adsay V, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 17.Curley MD, Therrien VA, Cummings CL, Sergent PA, Koulouris CR, Friel AM, et al. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells. 2009;27:2875–83. doi: 10.1002/stem.236. [DOI] [PubMed] [Google Scholar]
- 18.Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153–66. doi: 10.1016/j.ccr.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 19.Monzani E, Facchetti F, Galmozzi E, Corsini E, Benetti A, Cavazzin C, et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur J Cancer. 2007;43:935–46. doi: 10.1016/j.ejca.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 20.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–84. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 21.Rowan K. Are cancer stem cells real? After four decades, debate still simmers. J Natl Cancer Inst. 2009;101:546–7. doi: 10.1093/jnci/djp083. [DOI] [PubMed] [Google Scholar]
- 22.Zhou J, Zhang Y. Cancer stem cells: Models, mechanisms and implications for improved treatment. Cell Cycle. 2008;7:1360–70. doi: 10.4161/cc.7.10.5953. [DOI] [PubMed] [Google Scholar]
- 23.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–5. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zakikhani M, Blouin MJ, Piura E, Pollak MN. Metformin and rapamycin have distinct effects on the AKT pathway and proliferation in breast cancer cells. Breast Cancer Res Treat. 2010;123:271–9. doi: 10.1007/s10549-010-0763-9. [DOI] [PubMed] [Google Scholar]
- 25.Phoenix KN, Vumbaca F, Claffey KP. Therapeutic metformin/AMPK activation promotes the angiogenic phenotype in the ERalpha negative MDA-MB-435 breast cancer model. Breast Cancer Res Treat. 2009;113:101–11. doi: 10.1007/s10549-008-9916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhuang Y, Miskimins WK. Cell cycle arrest in Metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27Kip1 or p21Cip1. J Mol Signal. 2008;3:18. doi: 10.1186/1750-2187-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rattan R, Giri S, Hartmann LC, Shridhar V. Metformin attenuates ovarian cancer cell growth in an AMP-kinase dispensable manner. J Cell Mol Med. 2011;15:166–78. doi: 10.1111/j.1582-4934.2009.00954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cantrell LA, Zhou C, Mendivil A, Malloy KM, Gehrig PA, Bae-Jump VL. Metformin is a potent inhibitor of endometrial cancer cell proliferation--implications for a novel treatment strategy. Gynecol Oncol. 2010;116:92–8. doi: 10.1016/j.ygyno.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ben Sahra I, Tanti JF, Bost F. The combination of metformin and 2 deoxyglucose inhibits autophagy and induces AMPK-dependent apoptosis in prostate cancer cells. Autophagy. 2010;6:670–1. doi: 10.4161/auto.6.5.12434. [DOI] [PubMed] [Google Scholar]
- 30.Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, et al. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–86. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 31.Rozengurt E, Sinnett-Smith J, Kisfalvi K. Crosstalk between insulin/insulin-like growth factor-1 receptors and G protein-coupled receptor signaling systems: a novel target for the antidiabetic drug metformin in pancreatic cancer. Clin Cancer Res. 2010;16:2505–11. doi: 10.1158/1078-0432.CCR-09-2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507–11. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009;138:645–59. doi: 10.1016/j.cell.2009.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fuchs D, Daniel V, Sadeghi M, Opelz G, Naujokat C. Salinomycin overcomes ABC transporter-mediated multidrug and apoptosis resistance in human leukemia stem cell-like KG-1a cells. Biochem Biophys Res Commun. 2010;394:1098–104. doi: 10.1016/j.bbrc.2010.03.138. [DOI] [PubMed] [Google Scholar]
- 35.Smith KM, Datti A, Fujitani M, Grinshtein N, Zhang L, Morozova O, et al. Selective targeting of neuroblastoma tumour-initiating cells by compounds identified in stem cell-based small molecule screens. EMBO Mol Med. 2010;2:371–84. doi: 10.1002/emmm.201000093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka M, Miyajima A. Oncostatin M, a multifunctional cytokine. Rev Physiol Biochem Pharmacol. 2003;149:39–52. doi: 10.1007/s10254-003-0013-1. [DOI] [PubMed] [Google Scholar]
- 37.Yamashita T, Honda M, Nio K, Nakamoto Y, Yamashita T, Takamura H, et al. Oncostatin m renders epithelial cell adhesion molecule-positive liver cancer stem cells sensitive to 5-Fluorouracil by inducing hepatocytic differentiation. Cancer Res. 2010;70:4687–97. doi: 10.1158/0008-5472.CAN-09-4210. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Zhang T, Korkaya H, Liu S, Lee HF, Newman B, et al. Sulforaphane, a dietary component of broccoli/broccoli sprouts, inhibits breast cancer stem cells. Clin Cancer Res. 2010;16:2580–90. doi: 10.1158/1078-0432.CCR-09-2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang SN, Singh C, Nall D, Meeker D, Shankar S, Srivastava RK. The dietary bioflavonoid quercetin synergizes with epigallocathechin gallate (EGCG) to inhibit prostate cancer stem cell characteristics, invasion, migration and epithelial-mesenchymal transition. J Mol Signal. 2010;18:5–14. doi: 10.1186/1750-2187-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou W, Kallifatidis G, Baumann B, Rausch V, Mattern J, Gladkich J, et al. Dietary polyphenol quercetin targets pancreatic cancer stem cells. Int J Oncol. 2010;37:551–61. doi: 10.3892/ijo_00000704. [DOI] [PubMed] [Google Scholar]
- 41.Hsu HS, Lin JH, Huang WC, Hsu TW, Su K, Chiou SH, et al. Chemoresistance of lung cancer stemlike cells depends on activation of Hsp27. Cancer. 2011;117:1516–28. doi: 10.1002/cncr.25599. [DOI] [PubMed] [Google Scholar]
- 42.Fong D, Yeh A, Naftalovich R, Choi TH, Chan MM. Curcumin inhibits the side population (SP) phenotype of the rat C6 glioma cell line: towards targeting of cancer stem cells with phytochemicals. Cancer Lett. 2010;293:65–72. doi: 10.1016/j.canlet.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim JB, Ko E, Han W, Shin I, Park SY, Noh DY. Berberine diminishes the side population and ABCG2 transporter expression in MCF-7 breast cancer cells. Planta Med. 2008;74:1693–700. doi: 10.1055/s-0028-1088313. [DOI] [PubMed] [Google Scholar]
- 44.Luk SU, Yap WN, Chiu YT, Lee DT, Ma S, Lee TK, et al. Gamma-tocotrienol as an effective agent in targeting prostate cancer stem cell-like population. Int J Cancer. 2011;128:2182–91. doi: 10.1002/ijc.25546. [DOI] [PubMed] [Google Scholar]
- 45.Liu Y, Lu WL, Guo J, Du J, Li T, Wu JW, et al. A potential target associated with both cancer and cancer stem cells: a combination therapy for eradication of breast cancer using vinorelbine stealthy liposomes plus parthenolide stealthy liposomes. J Control Release. 2008;129:18–25. doi: 10.1016/j.jconrel.2008.03.022. [DOI] [PubMed] [Google Scholar]
- 46.Imai Y, Tsukahara S, Asada S, Sugimoto Y. Phytoestrogens/flavonoids reverse breast cancer resistance protein/ABCG2-mediated multidrug resistance. Cancer Res. 2004;64:4346–52. doi: 10.1158/0008-5472.CAN-04-0078. [DOI] [PubMed] [Google Scholar]
- 47.Ries SJ, Brandts CH. Oncolytic viruses for the treatment of cancer: current strategies and clinical trials. Drug Discov Today. 2004;9:759–68. doi: 10.1016/S1359-6446(04)03221-0. [DOI] [PubMed] [Google Scholar]
- 48.Bauerschmitz GJ, Ranki T, Kangasniemi L, Ribacka C, Eriksson M, Porten M, et al. Tissue-specific promoters active in CD44+CD24-/low breast cancer cells. Cancer Res. 2008;68:5533–9. doi: 10.1158/0008-5472.CAN-07-5288. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, Komaki R, Wang L, Fang B, Chang JY. Treatment of radioresistant stem-like esophageal cancer cells by an apoptotic gene-armed, telomerase-specific oncolytic adenovirus. Clin Cancer Res. 2008;14:2813–23. doi: 10.1158/1078-0432.CCR-07-1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–31. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 51.Wang CJ, Zhou ZG, Wang L, Yang L, Zhou B, Gu J, et al. Clinicopathological significance of microRNA-31, -143 and -145 expression in colorectal cancer. Dis Markers. 2009;26:27–34. doi: 10.3233/DMA-2009-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu X, Sempere LF, Galimberti F, Freemantle SJ, Black C, Dragnev KH, et al. Uncovering growth-suppressive MicroRNAs in lung cancer. Clin Cancer Res. 2009;15:1177–83. doi: 10.1158/1078-0432.CCR-08-1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res. 2005;65:6029–33. doi: 10.1158/0008-5472.CAN-05-0137. [DOI] [PubMed] [Google Scholar]
- 54.Yan LX, Huang XF, Shao Q, Huang MY, Deng L, Wu QL, et al. MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis. RNA. 2008;14:2348–60. doi: 10.1261/rna.1034808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–70. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 56.Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol. 2007;17:1298–307. doi: 10.1016/j.cub.2007.06.068. [DOI] [PubMed] [Google Scholar]
- 57.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, et al. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ji Q, Hao X, Meng Y, Zhang M, Desano J, Fan D, et al. Restoration of tumor suppressor miR-34 inhibits human p53-mutant gastric cancer tumorspheres. BMC Cancer. 2008;8:266. doi: 10.1186/1471-2407-8-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ji Q, Hao X, Zhang M, Tang W, Yang M, Li L, et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One. 2009;4:e6816. doi: 10.1371/journal.pone.0006816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang YP, Chien Y, Chiou GY, Cherng JY, Wang ML, Lo WL, et al. Inhibition of cancer stem cell-like properties and reduced chemoradioresistance of glioblastoma using microRNA145 with cationic polyurethane-short branch PEI. Biomaterials. 2012;33:1462–76. doi: 10.1016/j.biomaterials.2011.10.071. [DOI] [PubMed] [Google Scholar]
- 61.Feldmann G, Fendrich V, McGovern K, Bedja D, Bisht S, Alvarez H, et al. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Mol Cancer Ther. 2008;7:2725–35. doi: 10.1158/1535-7163.MCT-08-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jimeno A, Feldmann G, Suárez-Gauthier A, Rasheed Z, Solomon A, Zou GM, et al. A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol Cancer Ther. 2009;8:310–4. doi: 10.1158/1535-7163.MCT-08-0924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mueller MT, Hermann PC, Witthauer J, Rubio-Viqueira B, Leicht SF, Huber S, et al. Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology. 2009;137:1102–13. doi: 10.1053/j.gastro.2009.05.053. [DOI] [PubMed] [Google Scholar]
- 64.Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25:2524–33. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, et al. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70:709–18. doi: 10.1158/0008-5472.CAN-09-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoey T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S, et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell. 2009;5:168–77. doi: 10.1016/j.stem.2009.05.019. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y, Piao B, Zhang Y, Hua B, Hou W, Xu W, et al. Oxymatrine diminishes the side population and inhibits the expression of β-catenin in MCF-7 breast cancer cells. Med Oncol. 2011;28(Suppl 1):S99–107. doi: 10.1007/s12032-010-9721-y. [DOI] [PubMed] [Google Scholar]
- 68.Jiang Y, Boije M, Westermark B, Uhrbom L. PDGF-B Can sustain self-renewal and tumorigenicity of experimental glioma-derived cancer-initiating cells by preventing oligodendrocyte differentiation. Neoplasia. 2011;13:492–503. doi: 10.1593/neo.11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dong Y, Han Q, Zou Y, Deng Z, Lu X, Wang X, et al. Long-term exposure to imatinib reduced cancer stem cell ability through induction of cell differentiation via activation of MAPK signaling in glioblastoma cells. Mol Cell Biochem. 2012;370:89–102. doi: 10.1007/s11010-012-1401-0. [DOI] [PubMed] [Google Scholar]
- 70.Garth S. NIH funds first cancer stem cell clinical trial at University of Colorado Cancer Center. Colorado Cancer Blogs(News and Views from the University of Colorado Cancer). September 19, 2011 .
- 71.Bonavida B, Ng CP, Jazirehi A, Schiller G, Mizutani Y. Selectivity of TRAIL-mediated apoptosis of cancer cells and synergy with drugs: the trail to non-toxic cancer therapeutics (review) Int J Oncol. 1999;15:793–802. doi: 10.3892/ijo.15.4.793. [DOI] [PubMed] [Google Scholar]
- 72.Zobalova R, McDermott L, Stantic M, Prokopova K, Dong LF, Neuzil J. CD133-positive cells are resistant to TRAIL due to up-regulation of FLIP. Biochem Biophys Res Commun. 2008;373:567–71. doi: 10.1016/j.bbrc.2008.06.073. [DOI] [PubMed] [Google Scholar]
- 73.Capper D, Gaiser T, Hartmann C, Habel A, Mueller W, Herold-Mende C, et al. Stem-cell-like glioma cells are resistant to TRAIL/Apo2L and exhibit down-regulation of caspase-8 by promoter methylation. Acta Neuropathol. 2009;117:445–56. doi: 10.1007/s00401-009-0494-3. [DOI] [PubMed] [Google Scholar]
- 74.Sussman RT, Ricci MS, Hart LS, Sun SY, El-Deiry WS. Chemotherapy-resistant side-population of colon cancer cells has a higher sensitivity to TRAIL than the non-SP, a higher expression of c-Myc and TRAIL-receptor DR4. Cancer Biol Ther. 2007;6:1490–5. doi: 10.4161/cbt.6.9.4905. [DOI] [PubMed] [Google Scholar]
- 75.Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008;15:730–8. doi: 10.1038/gt.2008.39. [DOI] [PubMed] [Google Scholar]
- 76.Choi SA, Hwang SK, Wang KC, Cho BK, Phi JH, Lee JY, et al. Therapeutic efficacy and safety of TRAIL-producing human adipose tissue-derived mesenchymal stem cells against experimental brainstem glioma. Neuro Oncol. 2011;13:61–9. doi: 10.1093/neuonc/noq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mohr A, Lyons M, Deedigan L, Harte T, Shaw G, Howard L, et al. Mesenchymal stem cells expressing TRAIL lead to tumour growth inhibition in an experimental lung cancer model. J Cell Mol Med. 2008;12(6B):2628–43. doi: 10.1111/j.1582-4934.2008.00317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Loebinger MR, Sage EK, Davies D, Janes SM. TRAIL-expressing mesenchymal stem cells kill the putative cancer stem cell population. Br J Cancer. 2010;103:1692–7. doi: 10.1038/sj.bjc.6605952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jonasch E, Haluska FG. Interferon in oncological practice: review of interferon biology, clinical applications, and toxicities. Oncologist. 2001;6:34–55. doi: 10.1634/theoncologist.6-1-34. [DOI] [PubMed] [Google Scholar]
- 80.Naldini A, Carraro F. Role of inflammatory mediators in angiogenesis. Curr Drug Targets Inflamm Allergy. 2005;4:3–8. doi: 10.2174/1568010053622830. [DOI] [PubMed] [Google Scholar]
- 81.Moserle L, Indraccolo S, Ghisi M, Frasson C, Fortunato E, Canevari S, et al. The side population of ovarian cancer cells is a primary target of IFN-alpha antitumor effects. Cancer Res. 2008;68:5658–68. doi: 10.1158/0008-5472.CAN-07-6341. [DOI] [PubMed] [Google Scholar]
- 82.Kobayashi Y, Seino K, Hosonuma S, Ohara T, Itamochi H, Isonishi S, et al. Side population is increased in paclitaxel-resistant ovarian cancer cell lines regardless of resistance to cisplatin. Gynecol Oncol. 2011;121:390–4. doi: 10.1016/j.ygyno.2010.12.366. [DOI] [PubMed] [Google Scholar]
- 83.Yuki K, Natsume A, Yokoyama H, Kondo Y, Ohno M, Kato T, et al. Induction of oligodendrogenesis in glioblastoma-initiating cells by IFN-mediated activation of STAT3 signaling. Cancer Lett. 2009;284:71–9. doi: 10.1016/j.canlet.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 84.Holt SE, Shay JW, Wright WE. Refining the telomere-telomerase hypothesis of aging and cancer. Nat Biotechnol. 1996;14:836–9. doi: 10.1038/nbt0796-836. [DOI] [PubMed] [Google Scholar]
- 85.Röth A, Harley CB, Baerlocher GM. Imetelstat (GRN163L)--telomerase-based cancer therapy. Recent Results Cancer Res. 2010;184:221–34. doi: 10.1007/978-3-642-01222-8_16. [DOI] [PubMed] [Google Scholar]
- 86.Brennan SK, Wang Q, Tressler R, Harley C, Go N, Bassett E, et al. Telomerase inhibition targets clonogenic multiple myeloma cells through telomere length-dependent and independent mechanisms. PLoS One. 2010;5 doi: 10.1371/journal.pone.0012487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Joseph I, Tressler R, Bassett E, Harley C, Buseman CM, Pattamatta P, et al. The telomerase inhibitor imetelstat depletes cancer stem cells in breast and pancreatic cancer cell lines. Cancer Res. 2010;70:9494–504. doi: 10.1158/0008-5472.CAN-10-0233. [DOI] [PubMed] [Google Scholar]
- 88.Marian CO, Cho SK, McEllin BM, Maher EA, Hatanpaa KJ, Madden CJ, et al. The telomerase antagonist, imetelstat, efficiently targets glioblastoma tumor-initiating cells leading to decreased proliferation and tumor growth. Clin Cancer Res. 2010;16:154–63. doi: 10.1158/1078-0432.CCR-09-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Siddikuzzaman GC, Guruvayoorappan C, Berlin Grace VM. All trans retinoic acid and cancer. Immunopharmacol Immunotoxicol. 2011;33:241–9. doi: 10.3109/08923973.2010.521507. [DOI] [PubMed] [Google Scholar]
- 90.Karsy M, Albert L, Tobias ME, Murali R, Jhanwar-Uniyal M. All-trans retinoic acid modulates cancer stem cells of glioblastoma multiforme in an MAPK-dependent manner. Anticancer Res. 2010;30:4915–20. [PubMed] [Google Scholar]
- 91.Papi A, Guarnieri T, Storci G, Santini D, Ceccarelli C, Taffurelli M, et al. Nuclear receptors agonists exert opposing effects on the inflammation dependent survival of breast cancer stem cells. Cell Death Differ. 2012;19:1208–19. doi: 10.1038/cdd.2011.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lim YC, Kang HJ, Kim YS, Choi EC. All-trans-retinoic acid inhibits growth of head and neck cancer stem cells by suppression of Wnt/β-catenin pathway. Eur J Cancer. 2012;48:3310–8. doi: 10.1016/j.ejca.2012.04.013. [DOI] [PubMed] [Google Scholar]
- 93.Rappa G, Fodstad O, Lorico A. The stem cell-associated antigen CD133 (Prominin-1) is a molecular therapeutic target for metastatic melanoma. Stem Cells. 2008;26:3008–17. doi: 10.1634/stemcells.2008-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang CH, Chiou SH, Chou CP, Chen YC, Huang YJ, Peng CA. Photothermolysis of glioblastoma stem-like cells targeted by carbon nanotubes conjugated with CD133 monoclonal antibody. Nanomedicine. 2011;7:69–79. doi: 10.1016/j.nano.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 95.Salnikov AV, Groth A, Apel A, Kallifatidis G, Beckermann BM, Khamidjanov A, et al. Targeting of cancer stem cell marker EpCAM by bispecific antibody EpCAMxCD3 inhibits pancreatic carcinoma. J Cell Mol Med. 2009;13(9B):4023–33. doi: 10.1111/j.1582-4934.2009.00723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Qiu M, Peng Q, Jiang I, Carroll C, Han G, Rymer I, et al. Specific inhibition of Notch1 signaling enhances the antitumor efficacy of chemotherapy in triple negative breast cancer through reduction of cancer stem cells. Cancer Lett. 2013;328:261–70. doi: 10.1016/j.canlet.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 97.Petrie K, Zelent A, Waxman S. Differentiation therapy of acute myeloid leukemia: past, present and future. Curr Opin Hematol. 2009;16:84–91. doi: 10.1097/MOH.0b013e3283257aee. [DOI] [PubMed] [Google Scholar]