

Abstract
NMDA receptors are glutamate-activated, Ca2+-permeable ion channels with critical roles in synaptic transmission and plasticity. The shape and size of their current is modulated by several kinase/phosphatase systems, and numerous residues located on the receptors’ intracellular C-termini are phosphorylated in vivo. To investigate the mechanisms by which phosphorylation may control channel gating, we examined the single-channel behaviors of receptors carrying the S900A or S929A substitution in their GluN2A subunits and thus were rendered resistant to phosphorylation at those sites. We found that the mutations reduced channel open probability primarily by increasing the frequency of desensitized events. The kinetic models we developed revealed complex but similar changes in mechanism for the two mutants, leading to the view that dephosphorylation at either site may cause receptors to activate slower, deactivate faster and desensitize more frequently. This modulatory mechanism is consistent with the proposed roles for these residues in Ca2+-dependent desensitization and calcineurin-mediated reduction of current during brain development.
Keywords: ionotropic glutamate receptors, NMDA receptors, reaction mechanism, single-channel kinetics, protein phosphorylation
Introduction
NMDA receptors (NRs) are prominent members of the ionotropic glutamate receptor family whose activation initiates synaptic plasticity at a majority of synapses in the central nervous system and also mediates neurotoxic actions of glutamate. They have high Ca2+ permeability and characteristically slow gating kinetics. Consistent with their central roles in health and disease, NR activity is controlled by numerous cellular mechanisms including reversible covalent modification by kinase/phosphatase pathways.
Functional receptors are tetramers of two GluN1 (N1) and two GluN2 (N2) subunits.1 Each subunit spans the synaptic membrane and projects significantly into both the extra- and the intra-cellular fields. The large N-termini are organized into eight distinct globular domains; whereas C-terminal domains (CTDs), which represent about one third of the receptor’s mass, are believed to be intrinsically disordered.1,2 Substantial functional diversity arises from differential splicing of the N1 transcript combined with regulated expression of four separate N2 subunits, denoted A through D.3 In addition, both the level and the kinetics of the NR-generated current are controlled by mechanisms with remarkable diversity. Numerous diffusible ligands bind at specific extracellular sites and control receptor kinetics with allosteric mechanisms, while several inorganic divalent cations and organic small molecules bind within the membrane-spanning pore to affect the level and composition of ionic flux.1 Further, covalent modifications, mainly by phosphorylation of intracellular residues, have been proposed to control receptor permeation and gating.4-6
We have recently discovered that, relative to wild-type receptors, NRs engineered to lack the CTDs of N1 subunits (N1Δ838/N2A) produce currents with larger unitary conductance but similar kinetics, whereas receptors engineered to lack the CTD of N2A subunits (N1/N2AΔ844) produce currents of similar unitary conductance but dwell significantly longer in open and desensitized states. In addition, we found that the gating effect of N2A CTD truncation is mediated solely by the membrane-proximal region, residues 844 to 944, of the C-terminal tail.7 Based on these results, we hypothesized that the C-termini of N2A subunits may exert their effects on receptor gating through specific sites that may be subject to phosphorylation by cellular kinases. Based on available literature, two residues in this region, Ser-900 and Ser-929, stand out in functional significance. These were identified by alanine scanning mutagenesis to be responsible for the Ca2+-dependent calcineurin-mediated increase in use-dependent desensitization of the NR currents.8 Importantly, one of these residues, Ser-900, is a direct target for dephosphorylation by calcineurin in vivo and represents the mechanism that causes the rapid reduction in NR current decay observed during early development of the visual system.9
We recorded currents from individual channels and used statistical analyses and kinetic modeling to examine how perturbations at residues Ser-900 or Ser-929 control N1/N2A receptor activity. Our results show that modifications at these positions impose substantial change on receptor open probability and delineate how these perturbations at either of these two sites modulate the extent of macroscopic desensitization and the duration of NR current decay, both of which are biologically important attributes of the NR signal.
Results
To investigate our hypothesis that lack of endogenous phosphorylation at Ser-900 or Ser-929 of N2A may be responsible for the large gating effects observed in receptors that lack the intracellular C-termini of N2A subunits,7 we produced receptors that cannot be phosphorylated at these sites by substituting alanine residues at these positions: N1/N2A900A and N1/N2A929A. We used the cell-attached patch-clamp technique, which preserves the integrity of the intracellular milieu, and omitted adding Ca2+ to the extracellular recording solution, such that the inward current passed by the channel consisted of only Na+. We reasoned that in these conditions, and for the entire duration of the observation window, the trapped receptor will be in the high-activity form that exists basally before Ca2+ entry initiates desensitization, inactivation or run-down events.8,10-15
We obtained several current traces, each lasting between 10 and 90 min, from patches containing one N1/N2A900A (n = 6) or one N1/N2A929A channel (n = 8). We compared these to previously reported data sets for wild-type receptors (N1/N2A, n = 14) and receptors with truncated N2A CTDs (N1/N2AΔ844, n = 7), which were obtained and analyzed in similar conditions (Fig. 1A).7 All channels examined opened to a single amplitude level that was not different in value across the four data sets (p > 0.05, one-way ANOVA) (Table 1). Relative to N1/N2A receptors, the open probability (Po) of N1/N2A900A and N1/N2A929A, as calculated from entire records, decreased 2- and 3-fold, respectively (p < 0.01). For both mutants, this drop in activity originated mainly from a ~4-fold increase in mean closed time (MCT), with only N1/N2A929A also having shorter (~1.5-fold) mean open time (MOT). Neither mutation reproduced the full phenotype of low activity we observed previously for N1/N2AΔ844, which originated from a combination of much longer MCT (~50-fold) and longer MOT (~3-fold). These results support the interpretation that perturbations to either of the examined serine residues, including by dephosphorylation, will contribute substantially to the low activity observed for the full CTD deletion; however, because the CTD truncations have a much more severe loss of function phenotype, we surmise that yet undetermined sites within the N2A C-termini modulate channel activity by lengthening desensitized and/or open events.

Figure 1. Gating and desensitization mechanism of N1/N2A900A and N1/N2A929A. (A) Representative traces (50 sec) of continuously recorded current from a cell-attached patch containing a single receptor exposed to high concentrations of Glu (1 mM) and Gly (0.1 mM). (B) Individual closed event histograms for entire files. Overlaid are fits to individual closed components (thin lines) and the probability density function (thick lines) calculated by fitting entire files to a kinetic model containing five closed and one open state. For each histogram, time constants (τ) and areas (a) for the five exponential closed components are indicated.
Table 1. Single-channel properties of N1/N2A900A and N1/N2A929A.
| Receptor | Amplitude (pA) | Po | MCT (ms) |
MOT (ms) |
N | Events |
|---|---|---|---|---|---|---|
| N1/N2A |
8.7 ± 0.3 |
0.54 ± 0.04 |
4.7 ± 0.5 |
5.4 ± 0.5 |
14 |
4.9 × 106 |
| N1/N2A900A |
8.3 ± 1.6 |
0.28 ± 0.10* |
18 ± 6*† |
3.9 ± 0.7† |
6 |
1.7 × 106 |
| N1/N2A929A |
8.9 ± 1.6 |
0.18 ± 0.05* |
21 ± 6*† |
3.1 ± 0.4*† |
8 |
1.6 × 106 |
| N1/N2AΔ844 | 9.7 ± 0.3 | 0.22 ± 0.04* | 50 ± 10* | 13 ± 2* | 7 | 5.0 × 105 |
Symbols mark values that are statistically different compared with N1/N2A (*) or N1/N2AΔ844 (†), (p < 0.05, Student’s t-test).
In saturating concentrations of agonists and the absence of divalent cations, as employed in this study (1 mM Glu, 0.1 mM Gly and 1 mM EDTA), traces recorded from wild-type NRs have event durations with complex multi-exponential distributions.16 The closed event distributions of the mutants we examined had five exponential components, E1–E5, as described previously for wild-type NRs (Fig. 1B). The three shorter components (E1–E3) occur largely within bursts and reflect receptor states visited along the activation pathway, whereas the two longest components, E4 and E5, represent largely closures that terminate bursts and inform about desensitized receptor states.17 The closed event durations measured from N1/N2A900A and N1/N2A929A revealed that the longer MCT values could be attributed to slightly (< 2-fold) longer intra-burst, E1–E3 events, 10-fold longer E4-type closures and a 6-fold larger relative area for the E5 component (Table 2). Notably, the time constant of E5 was not significantly different from wild-type for either mutant (p > 0.05), and was ~3 sec for each. This was a conspicuous difference from N1/N2AΔ844, whose longest closures (E5) were significantly longer (p < 0.05) with a time constant of ~8 sec. These results support the hypothesis that dephosphorylation of either serine would lengthen specific time components in a similar manner.
Table 2. Closed components of N1/N2A900A and N1/N2A929A.
| Receptor | τE1 (ms) |
aE1 (%) |
τE2 (ms) |
aE2 (%) |
τE3 (ms) |
aE3 (%) |
τE4 (ms) |
aE4 (%) |
τE5 (ms) |
aE5 (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| N1/N2A |
0.14 ± 0.01 |
45 ± 3 |
1.4 ± 0.1 |
24 ± 2 |
4.4 ± 0.2 |
30 ± 3 |
22 ± 4 |
1.5 ± 0.3 |
3,000 ± 400 |
0.08 ± 0.01 |
| N1/N2A900A |
0.17 ± 0.01* |
31 ± 4*† |
2.1 ± 0.2* |
38 ± 4*† |
5.6 ± 0.6 |
29 ± 6 |
100 ± 50* |
1.2 ± 0.7 |
3300 ± 500† |
0.5 ± 0.1* |
| N1/N2A929A |
0.18 ± 0.01* |
32 ± 6* |
2.1 ± 0.2* |
30 ± 3*† |
6.3 ± 0.7* |
36 ± 6† |
230 ± 80* |
1.3 ± 0.3† |
3400 ± 700† |
0.5 ± 0.1* |
| N1/N2AΔ844 | 0.15 ± 0.02 | 46 ± 4 | 2.0 ± 0.3 | 46 ± 4* | 7 ± 1 | 11 ± 2* | 900 ± 400* | 0.5 ± 0.1* | 8,000 ± 1,000* | 0.7 ± 0.1* |
Symbols mark values that are statistically different compared with N1/N2A (*) or N1/N2AΔ844 (†), (p < 0.05, Student’s t-test).
Although the lower Po exhibited by the S900A and S929A mutations was only partially attributed to a reduction in mean open time, we went on to examine the open duration distributions for the two mutants to learn about possible changes in modal gating. Aside from gating and desensitization transitions, NRs also experience modal transitions, which change the receptor’s activity pattern for seconds or minutes at a time.16 For N1/N2A receptors, three modes can be queried experimentally by determining how many types of openings occur in a record generated by an individual channel. In any mode, defined as a period of uniform kinetics, N1/N2A receptors generate two types of openings, one short and one long. Shifts in modes are characterized by changes in duration of the long openings, while length of the short openings remains constant across all modes. Thus, the open duration distribution will reveal two, three or four open components for records that capture one, two or three modes, respectively. The molecular determinants of NR modal gating are not yet known; however phosphorylation of intracellular residues has been mentioned as a possible mechanism for modal shifts.18 We found that the open interval distributions of both N1/N2A900A and N1/N2A929A receptors had four components and that their time constants and relative areas were not statistically different from those calculated for N1/N2A receptors (data not shown). This result demonstrates that neither Ser-900 nor Ser-929 influences modal gating of N1/N2A receptors.
Next, we summarized the kinetic changes we measured for the two mutants using a commonly used state model for NR gating (Fig. 2).17 In this scheme, all states represent receptors that are fully occupied with glutamate and glycine; C3, C2, and C1 represent a sequence of non-conductive conformations en route to channel open states; C4 and C5 are non-conductive states that represent functionally desensitized receptors; and last, O stands for all active receptor conformations. The rates estimated by fitting this model to entire recordings were largely similar for N1/N2A900A and N1/N2A929A (Fig. 2), as expected from their similar kinetic parameters (Table 1) and duration distributions (Table 2). Relative to wild-type channels, N1/N2A900A and N1/N2A929A had slower activation rates (C3 → C2), slower recovery from the desensitized C4 state (C2 ← C4) and faster access into the most stable desensitized state, C5 (C3 → C5). Relative to the full N2A CTD truncation, the changes in the rate constants for activation (C3 → C2), desensitization (C3 → C5) and resensitization (C4 ← C2) were less pronounced. This may mean that the S900A and S929A substitutions have additive effects, and/or additional site(s) on the N2A proximal CTD can reduce channel activity through similar mechanisms. Interestingly, only the full truncation caused the closing rate, C1 ← O, to become slower. This result strongly suggests that in addition to the modulatory effects described here, N2A C-termini can control channel gating through mechanisms that would increase channel activity above the basal level observed for N1/N2A receptors. Notably, neither the point mutations nor the CTD full deletion affected the second step in the activation pathway C2 ↔ C1. This result may have structural implications, but these are insufficiently determined at this time.19
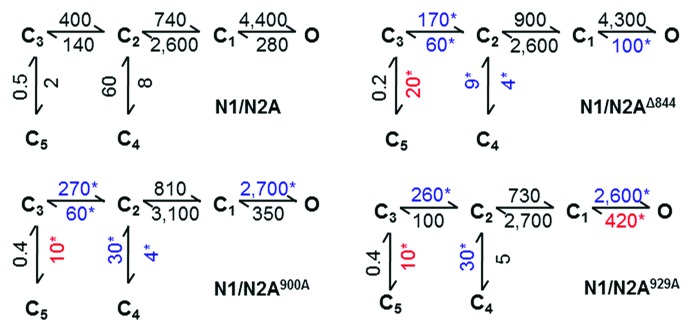
Figure 2. Reaction mechanisms of N1/N2A900A and N1/N2A929A. Rates (s−1) were optimized by fitting the illustrated model directly to idealized single-channel data in each record and are presented as mean values for each data set rounded to the first significant figure. Symbols (*) mark values that are significantly different, larger (red) or smaller (blue) relative to those obtained for N1N/2A receptor (p < 0.05, Student’s t-test).
State models are valuable in that they summarize and organize all observed kinetic changes into a reaction mechanism. More importantly, they have the power to predict channel responses over a broad spectrum of stimulation protocols. This is useful in testing the model experimentally but also in controlling variables that cannot be determined empirically, such as the number of receptors contributing to a macroscopic response. Thus, if the mechanisms we propose here are correct, they should also account for the macroscopic results reported previously for S900A and S929A in recombinant systems,8 as well as the changes in the NR synaptic response upon dephosphorylation at Ser-900 by calcineurin.9 To determine how well our models account for these known effects, we used them to simulate macroscopic currents (100 channels, 10 pA each) in response to desensitizing (5 sec) and synaptic-like (1 ms) pulses of glutamate (1 mM).
Results of simulations with long stimuli show that both N1/N2A900A and N1/N2A929A receptors produce macroscopic currents that desensitize faster (Fig. 3A). This result is consistent with measurements done by the Westbrook group in that both constructs have pronounced effects on macroscopic desensitization; even though in the previous study the receptors used also lacked the C-termini of N1 subunits (N1Δ838/N2A900A and N1Δ838/N2A929A).8 In addition, our model predicted that both N1/N2A900A and N1/N2A929A will reduce the peak macroscopic response (Fig. 3B), and overall will reduce charge transfer by 40% and 70%, respectively. This information cannot be obtained experimentally because in whole-cell current measurements the total number of channels is unknown; however it is valuable in estimating how the specific serine residues may control the actual charge transfer that generates Ca2+ transient. Our results with brief synaptic-like stimuli predicted that both N1/N2A900A and N1/N2A929A receptors will generate macroscopic currents with lower peaks and shorter decay times (Fig. 3C). Notably, these results match closely previous reports of calcineurin-dependent reduction in synaptic NR current by direct dephosphorylation of Ser-900.9
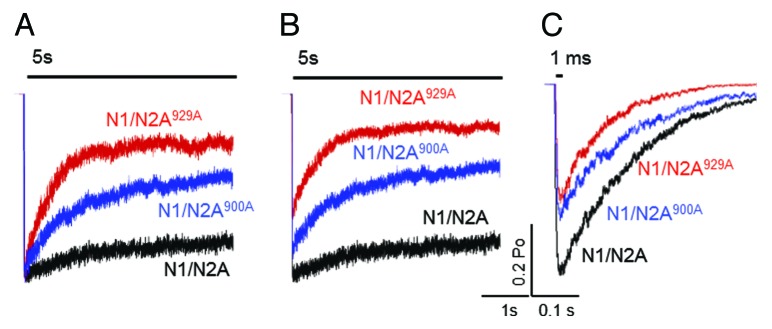
Figure 3. Predicted effects of N2A S900A and S929A substitutions on NR macroscopic responses. Responses to 5 sec (A, B) or 1 ms (C) applications of glutamate (1 mM) were simulated with the models in Figure 2. Current traces are normalized to peak in (A).
Discussion
Based on our previous findings that the N2A CTD has substantial influence over NR gating,7 we investigated how phosphorylation/dephosphorylation cycles may control NR output. Our approach was to examine the gating properties of NRs that were rendered resistant to phosphorylation at two specific sites by serine-to-alanine single-residue substitutions. We recorded currents from individual channels with the cell-attached patch-clamp technique; examined the kinetic distributions of the recorded open and closed intervals; and used kinetic modeling to summarize and interpret the observed changes in terms of a previously proposed reaction scheme. We found that relative to wild-type N1/N2A receptors, N1/N2A900A and N1/N2A929A receptors were substantially less active, and their Po values were low primarily because these channels remained closed for longer periods (longer MCT). Of the five components present in the closed duration distributions, all were longer, except for E5. However, this longest time component, which reflects desensitized intervals, represented a substantially higher fraction of all closures. For N1/N2A929A, but not for N1/N2A900A, we also observed a slight decrease in mean open time; however, the open duration distributions for both mutants were very similar to those of wild-type receptors, an indication that perturbations to Ser-900 or Ser-929 of N2A subunits neither causes nor modulates modal gating. Instead, kinetic modeling of our single channel data indicated changes at several activation and desensitization steps, and predicted that the phosphorylation status at these serine residues may control the kinetics and extent of macroscopic desensitization and also the decay of the synaptic response.
Because in our records we tracked channel activity as pure Na+ currents, and because all our measurements were done with the cell-attached technique, we presume that the receptor trapped within the patch pipette displayed the high level of basal activity that precedes Ca2+-dependent desensitization, inactivation or run down events.8,10-15 This premise was supported by our observation that in the records we obtained, which lasted between 10 and 90 min, channel activity was independent of recording time. For these reasons we assume that the reduction in Po we observed for N1/N2A900A and N1/N2A929A receptors was caused directly by the site-specific perturbations we introduced, and also that the behaviors we characterized for each mutant may inform about the gating mechanism of wild-type N1/N2A receptors that are dephosphorylated at these sites.
Our simulations based on the kinetic models developed from single-channel data of N1/N2A900A and N1/N2A929A agree well with the conclusions by the Westbrook group that these two residues control macroscopic desensitization.8 The shape of our simulated currents, however, was distinct from those reported by the Westbrook group. Namely, they found that N1Δ838/N2A900A had a rapid initial desensitization, reminiscent of wild type receptors that were repeatedly exposed to glutamate in the presence of Ca2+, while N1Δ838/N2A929A were resistant to this time-dependent form of desensitization. Our simulations predicted that N1/N2A900A will have deeper desensitization than wild type receptors, whereas N1/N2A929A will desensitize even more. However, these quantitative discrepancies may originate from the different constructs used (with or without the N1 CTD) and also permeant ions present (with or without Ca2+), and thus may point to the likelihood that in addition to kinase/phosphatase systems acting at the two serine residues examined here, separate biochemical pathways may target sites located elsewhere on NR C-termini.
Overall, our simulations with brief pulses of glutamate predicted that substituting an alanine residue at Ser-900 or Ser-929 will cause NR-mediated synaptic currents to become smaller and to decay faster (Fig. 3C). These constructs were developed with the intent of replicating a WT receptor that cannot be phosphorylated at these sites. Our results are consistent with a series of literature reports that show kinetic effects of dephosphorylation at these sites. Tong and Jahr reported that repeated stimulation of synaptic NRs in the presence of physiologic Ca2+ reduces the NR-mediated synaptic current by a mechanism that involves Ca2+ activation of calcineurin.20 At least part of this effect is mediated by dephosphorylation at Ser-900 of N2A subunits.9 Further, inhibiting protein kinase A (PKA) prevents the calcineurin-mediated reduction of NR currents and conversely, stimulating PKA overcomes the ability of calcineurin to reduce NR currents; however, PKA stimulation alone does not elevate NR currents above basal levels.5 These latter observations are salient to our report, in light of biochemical evidence that PKA directly phosphorylates NRs in vivo at multiple sites on both N1 and N2A subunits.21
Together, the available data on NR modulation by kinase/phosphatase systems paint the view that during quiescent periods, synaptic NRs are basally phosphorylated by (at least) PKA action and that the Ca2+ influx that accompanies synaptic stimulation of NRs results in calcineurin-mediated dephosphorylation of specific sites on NR. During development of the visual system, this latter effect causes NR currents to become smaller and to decay faster.9
We should note that in addition to preventing phosphorylation at the examined sites, our serine-to-alanine substitutions may have also perturbed CTD structure in other ways, such as by altering gating directly or by abolishing protein-protein interactions aside from kinases. Despite this, the CTDs of NRs are believed to lack ordered structures,22 and to our knowledge, there is not yet evidence of protein-protein interactions at the examined sites. Given that the CTD of NRs make as much as one third of the receptor’s mass, it is likely that multiple mechanisms exists which can modulate gating through this domain. The role of CTD in NR gating is just beginning to be explored, and additional investigations are necessary to delineate these.
Overall, the state models we developed in this study extend on previous reports regarding changes in macroscopic NR responses caused by dephosphorylation of N2A Ser-900 or Ser-929 by illuminating the kinetic mechanisms responsible for the observed changes.8,9 These models will therefore be instrumental for quantitative investigations into the structural determinates of channel gating, as well as for studies that aim to reveal the mechanisms of NR regulation by kinase/phosphatase systems and their functions in health and disease.
Materials and Methods
DNA constructs and receptor expression
Rat GluN1-1a (U8261) and GluN2A (M91561) were subcloned into pcDNA 3.1(+). Alanine substitutions were introduced into the GluN2A sequence at positions Ser-900 or Ser-929 using standard molecular biology methods. Wild-type or mutated GluN2A subunits were co-expressed with GluN1-1a and GFP after the encoding plasmids were introduced into HEK293 cells with the Ca2+ precipitation method previously described in more detail.17 Transfected cells were cultured in standard medium supplemented with 2 mM Mg2+ and were used for experiments 24 to 48 h post transfection.
Electrophysiology
Steady-state activity was recorded as inward Na+-only currents from cell-attached patches that contained one active channel after applying 100 mV through the recording pipette, which was filled with (extracellular) solution containing (in mM): 150 NaCl, 2.5 KCl, 10 HEPBS, 1 EDTA, 1 glutamate and 0.1 glycine, adjusted to pH 8.0 (NaOH). In all experiments, currents were amplified and low-pass filtered at 10 kHz using Axopatch 200B, were digitally sampled at 40 kHz using National Instruments PCI-6229 A/D board, and were stored into digital files using QuB software (www.qub.buffalo.edu).
Kinetic analysis and modeling
Current amplitude levels and event durations were measured directly from the single-channel record using QUB software with methods described in detail elsewhere.17 Briefly, records were first minimally preprocessed to correct for baseline drifts and noise artifacts and were idealized with the SKM algorithm after imposing a 12 kHz Gaussian digital filter.23 Kinetic modeling was done with the MIL algorithm by fitting the illustrated models directly to the idealized data after imposing a dead time of 0.075 ms.24 This procedure also calculated time constants and areas for the exponential components present in the closed and open event distributions.
Simulations
To simulate glutamate-elicited currents we appended the illustrated kinetic models, which represent saturation gating, with glutamate association steps leading into the closed fully liganded state C3. We used association and dissociation rate constants previously determined for N1/N2A receptors in conditions similar to those described here (1.7 × 10 M−1 s−1 and 60 sec−1, respectively).22 Macroscopic responses were calculated as the time-dependent occupancy of the open state after exposing 100 resting channels (10 pA/each) to pulses (5 sec or 1 ms) of glutamate (1 mM).
Statistical tests
Results are reported as means ± SEM, unless otherwise indicated. Statistical significance of differences was evaluated with ANOVA or Student’s t-test, as indicated, and was considered significant for p values < 0.05).
Acknowledgments
We thank Eileen Kasperek for help with molecular biology and tissue culture. This work was supported by the National Institutes of Health National Institute of Neurological Disorders and Stroke (grant 052669 to G.K.P.).
Glossary
Abbreviations:
- NMDA
- NR
NMDA receptor
- CTD
C-terminal domain
- N1
GluN1
- N2
GluN2
- Po
open probability
- MOT
mean open time
- MCT
mean closed time
- SKM
- MIL
Disclosure of Potential Conflicts of Interest
No conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/23968
References
- 1.Traynelis SF, Wollmuth LP, McBain CJ, Menniti FS, Vance KM, Ogden KK, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–96. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choi UB, Xiao S, Wollmuth LP, Bowen ME. Effect of Src kinase phosphorylation on disordered C-terminal domain of N-methyl-D-aspartic acid (NMDA) receptor subunit GluN2B protein. J Biol Chem. 2011;286:29904–12. doi: 10.1074/jbc.M111.258897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cull-Candy S, Brickley S, Farrant M. NMDA receptor subunits: diversity, development and disease. Curr Opin Neurobiol. 2001;11:327–35. doi: 10.1016/S0959-4388(00)00215-4. [DOI] [PubMed] [Google Scholar]
- 4.Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, et al. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci. 2006;9:501–10. doi: 10.1038/nn1664. [DOI] [PubMed] [Google Scholar]
- 5.Raman IM, Tong G, Jahr CE. Beta-adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron. 1996;16:415–21. doi: 10.1016/S0896-6273(00)80059-8. [DOI] [PubMed] [Google Scholar]
- 6.Urushihara H, Tohda M, Nomura Y. Selective potentiation of N-methyl-D-aspartate-induced current by protein kinase C in Xenopus oocytes injected with rat brain RNA. J Biol Chem. 1992;267:11697–700. [PubMed] [Google Scholar]
- 7.Maki BA, Aman TK, Amico-Ruvio SA, Kussius CL, Popescu GK. C-terminal domains of N-methyl-D-aspartic acid receptor modulate unitary channel conductance and gating. J Biol Chem. 2012;287:36071–80. doi: 10.1074/jbc.M112.390013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krupp JJ, Vissel B, Thomas CG, Heinemann SF, Westbrook GL. Calcineurin acts via the C-terminus of NR2A to modulate desensitization of NMDA receptors. Neuropharmacology. 2002;42:593–602. doi: 10.1016/S0028-3908(02)00031-X. [DOI] [PubMed] [Google Scholar]
- 9.Townsend M, Liu Y, Constantine-Paton M. Retina-driven dephosphorylation of the NR2A subunit correlates with faster NMDA receptor kinetics at developing retinocollicular synapses. J Neurosci. 2004;24:11098–107. doi: 10.1523/JNEUROSCI.1207-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krupp JJ, Vissel B, Heinemann SF, Westbrook GL. Calcium-dependent inactivation of recombinant N-methyl-D-aspartate receptors is NR2 subunit specific. Mol Pharmacol. 1996;50:1680–8. [PubMed] [Google Scholar]
- 11.Krupp JJ, Vissel B, Thomas CG, Heinemann SF, Westbrook GL. Interactions of calmodulin and α-actinin with the NR1 subunit modulate Ca2+-dependent inactivation of NMDA receptors. J Neurosci. 1999;19:1165–78. doi: 10.1523/JNEUROSCI.19-04-01165.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ehlers MD, Zhang S, Bernhadt JP, Huganir RL. Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunit. Cell. 1996;84:745–55. doi: 10.1016/S0092-8674(00)81052-1. [DOI] [PubMed] [Google Scholar]
- 13.Zhang S, Ehlers MD, Bernhardt JP, Su C-T, Huganir RL. Calmodulin mediates calcium-dependent inactivation of N-methyl-D-aspartate receptors. Neuron. 1998;21:443–53. doi: 10.1016/S0896-6273(00)80553-X. [DOI] [PubMed] [Google Scholar]
- 14.Rosenmund C, Westbrook GL. Calcium-induced actin depolymerization reduces NMDA channel activity. Neuron. 1993;10:805–14. doi: 10.1016/0896-6273(93)90197-Y. [DOI] [PubMed] [Google Scholar]
- 15.Rosenmund C, Westbrook GL. Rundown of N-methyl-D-aspartate channels during whole-cell recording in rat hippocampal neurons: role of Ca2+ and ATP. J Physiol. 1993;470:705–29. doi: 10.1113/jphysiol.1993.sp019884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Popescu G, Auerbach A. Modal gating of NMDA receptors and the shape of their synaptic response. Nat Neurosci. 2003;6:476–83. doi: 10.1038/nn1044. [DOI] [PubMed] [Google Scholar]
- 17.Kussius CL, Kaur N, Popescu GK. Pregnanolone sulfate promotes desensitization of activated NMDA receptors. J Neurosci. 2009;29:6819–27. doi: 10.1523/JNEUROSCI.0281-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Popescu GK. Modes of glutamate receptor gating. J Physiol. 2012;590:73–91. doi: 10.1113/jphysiol.2011.223750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murthy SE, Shogan T, Page JC, Kasperek EM, Popescu GK. Probing the activation sequence of NMDA receptors with lurcher mutations. J Gen Physiol. 2012;140:267–77. doi: 10.1085/jgp.201210786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tong G, Shepherd D, Jahr CE. Synaptic desensitization of NMDA receptors by calcineurin. Science. 1995;267:1510–2. doi: 10.1126/science.7878472. [DOI] [PubMed] [Google Scholar]
- 21.Leonard AS, Hell JW. Cyclic AMP-dependent protein kinase and protein kinase C phosphorylate N-methyl-D-aspartate receptors at different sites. J Biol Chem. 1997;272:12107–15. doi: 10.1074/jbc.272.18.12107. [DOI] [PubMed] [Google Scholar]
- 22.Popescu G, Robert A, Howe JR, Auerbach A. Reaction mechanism determines NMDA receptor response to repetitive stimulation. Nature. 2004;430:790–3. doi: 10.1038/nature02775. [DOI] [PubMed] [Google Scholar]
- 23.Qin F. Restoration of single-channel currents using the segmental k-means method based on hidden Markov modeling. Biophys J. 2004;86:1488–501. doi: 10.1016/S0006-3495(04)74217-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qin F, Auerbach A, Sachs F. Maximum likelihood estimation of aggregated Markov processes. Proc Biol Sci. 1997;264:375–83. doi: 10.1098/rspb.1997.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]