

Abstract
Polyunsaturated fatty acids such as arachidonic acid (AA) exhibit inhibitory modulation of Kv4 potassium channels. Molecular docking approaches using a Kv4.2 homology model predicted a membrane-embedded binding pocket for AA comprised of the S4-S5 linker on one subunit and several hydrophobic residues within S3, S5 and S6 from an adjacent subunit. The pocket is conserved among Kv4 channels. We tested the hypothesis that modulatory effects of AA on Kv4.2/KChIP channels require access to this site. Targeted mutation of a polar residue (K318) and a nonpolar residue (G314) within the S4-S5 linker as well as a nonpolar residue in S3 (V261) significantly impaired the effects of AA on K+ currents in Xenopus oocytes. These residues may be important in stabilizing (K318) or regulating access to (V261, G314) the negatively charged carboxylate moiety on the fatty acid. Structural specificity was supported by the lack of disruption of AA effects observed with mutations at residues located near, but not within the predicted binding pocket. Furthermore, we found that the crystal structure of the related Kv1.2/2.1 chimera lacks the structural features present in the proposed AA docking site of Kv4.2 and the Kv1.2/2.1 K+ currents were unaffected by AA. We simulated the mutagenic substitutions in our Kv4.2 model to demonstrate how specific mutations may disrupt the putative AA binding pocket. We conclude that AA inhibits Kv4 channel currents and facilitates current decay by binding within a hydrophobic pocket in the channel in which K318 within the S4-S5 linker is a critical residue for AA interaction.
Keywords: voltage-gated K+ channel, arachidonic acid, polyunsaturated fatty acid, homology model, mutagenesis
Introduction
Polyunsaturated fatty acids (PUFAs) contain two or more double bonds in cis positions and are major constituents of plasma membrane phospholipids in nervous tissue1 and cardiac muscle.2
Free PUFAs such as arachidonic acid (AA; 20:4) and docosahexaenoic acid (DHA; C22:6) are enzymatically released during certain cell signaling events. Among other possible targets, PUFAs are known to modulate the fast-inactivating Kv4/KChIP channels3,4 in post-synaptic somatodendritic membranes.5 These channels regulate neuronal firing frequency,6 modify the threshold for dendritic spike initiation,7 and regulate the induction of hippocampal long-term potentiation.8,9 Activation of post-synaptic glutamate receptors evokes AA release10 and inhibition of AA release prevents long-term potentiation,11 while the direct application of PUFAs to hippocampal slices enhances synaptic plasticity.12,13 In addition to their role in synaptic plasticity, Kv4/KChIP channels are important in regulating pain sensitivity14 and cardiac rhythmicity.15 Thus, several important physiological processes are likely to be influenced by PUFA modulation of Kv4/KChIP channel function.
Previously, we characterized the PUFA-mediated inhibition of the peak outward Kv4.2/KChIP1b K+ current and the facilitation of macroscopic inactivation kinetics.16 Because the effects of externally applied AA are prevented by injection of albumin into whole oocytes, we suggested that AA interacts with an internally accessible site.16 Others have found that Kv4 channel activity is inhibited by AA applied to the internal side of the membrane in excised patches.17 The more rapid effects of lower doses of AA when applied internally support the possibility of an internally accessible mechanism of action. We also previously found that AA facilitates inactivation from both open and closed gating states,16 but a structural basis for the PUFA effects remains elusive. A better understanding of the structural determinants of AA inhibition of Kv4 channels may aid in the identification of new targets for the regulation of synaptic strength and will enhance our general understanding of how ion channel function is modified by membrane lipid-derived signaling molecules.
Lipid signaling molecules have been hypothesized to alter the function of ion channels in several ways. The lipid may interact directly with the channel protein and alter its function, perhaps through conformational changes in the protein or through changes in how the channel interacts with other structural components in or near the membrane. The best evidence for this comes from the variety of ion channels that are regulated by phosphatidyl inositol-4,5 bisphosphate.18 Another hypothesis states that the lipid alters properties of the cellular membrane, such as membrane fluidity, curvature, or tension19,20 and thus changes how the channel works through mechanical mechanisms. The best evidence for this comes from the field of mechanosensitive channels,21 but there are other examples of this mechanism, which include voltage-gated ion channels.22 Although these hypotheses are not mutually exclusive, our goal was to test the first hypothesis, that Kv4.2 channel proteins have a specific binding site, which regulates the inhibitory modulation by AA. To test this, we created a Kv4.2 homology model and used molecular docking to identify possible AA binding sites. We located a hydrophobic pocket for AA on the cytosolic side of Kv4.2. The putative binding site is conserved in Kv4 channels and mimics some of the features of AA binding sites in serum albumin, but the structural features are not conserved in the Kv1.2/2.1 chimera, which is also unaffected by AA. Targeted site-directed mutagenesis and electrophysiology further implicate this hydrophobic pocket within the Kv4.2 channel structure in the inhibitory modulation by AA.
Results
Homology model and docking predictions
Our Kv4.2 model consists of four identical subunits that assemble together to form a homotetramer with the K+ permeation pathway in the center (Fig. 1A). Autodock Vina was used to test for the possibility of a docking site for AA within the Kv4.2 channel structure. Regardless of the location of the search grid, the molecular docking results clustered most of the ligand (AA) conformations within a hydrophobic pocket located on the cytosolic half of the transmembrane protein and visible from a lateral view of the channel surface (Fig. 1B). Within each hydrophobic pocket, AutoDock Vina found several docking solutions that differed in the orientation of AA’s 20-carbon chain (Fig. 1C), suggesting that there is not one distinct binding orientation for AA. The pocket is represented four times within the channel structure and is comprised of residues with the S4-S5 linker of one subunit and parts of the S3, S5 and S6 regions of an adjacent subunit (Fig. 1D).

Figure 1. Molecular docking of AA to Kv4.2 homology model. The model was built based on Kv4.2 homology with Kv1.2 and KcSA (see Methods). In this and other figures, all images of the protein surface are color-coded by amino acid side chain property; gray = polar; green = hydrophobic, blue = basic, red = acidic. (A) An intracellular view of the homotetramer. (B) A lateral view of the channel with seven overlapping but unique docking outputs for AA located near the internal surface of the channel. (C) A zoomed-in view of the predicted hydrophobic AA binding pocket. The same seven AA conformations as in (B) are shown for closer inspection. Note that for each docking result, the nonpolar region of AA is embedded in the hydrophobic cavity (green) and the polar part of AA is oriented toward two positively charged residues (blue) lining the pocket. (D) Identification of the residues predicted to comprise the AA binding pocket. Note that four pockets are predicted, each made of residues from two adjacent subunits.
Within the proposed binding pocket, the nature of the cavity is hydrophobic which provides a favorable, nonpolar environment for the aliphatic chain of AA. In addition, all of the AA conformations were oriented with the carboxylate moiety facing two positively charged residues lining the cavity, R311 and K318 (Fig. 1C). A docking experiment with AA and a model of the docking sites within Kv4.1 and Kv4.3 channels showed that this candidate site is completely conserved among Kv4 channels (Fig. S1). A positively charged residue (K320 in Kv4.1 and K315 in Kv4.3) orients toward the negatively charged head group of AA, just as K318 does in Kv4.2. Previously, we showed that all of the Kv4 channel subtypes are inhibited in a similar fashion by AA.23 Thus, our analysis is consistent with the hypothesis that an AA binding pocket exists in all Kv4 channels. The hydrophobicity of the proposed AA binding pocket within the Kv4 channels and the orientation of the ligand for favorable electrostatic interactions are also similar to the features of AA binding sites identified in the crystal structures of human serum albumin24-26 (see Fig. S1) and other fatty acid binding proteins27,28 (see Discussion).
Physiological tests of the docking predictions
Based on the docking results, we prepared point mutations to test the importance of specific residues within the proposed AA binding pocket of Kv4.2. We selected residues that might interact electrostatically with the carboxylate head of AA or that might impair access of AA to the positively charged residues or to hydrophobic interactions. Wild-type (WT) or mutant Kv4.2 RNA was always co-injected into Xenopus oocytes with an equimolar concentration of KChIP1b and K+ currents were studied by two-electrode voltage clamp. Channel expression and gating in the absence of AA were comparable for WT and most mutant channels (but see K318E and K318D below), suggesting that these mutations did not grossly alter structure or unregulated function of the channel. Figure 2A compares the magnitude of the inhibition of the peak K+ current following a 10-minute exposure to 10 µM AA. WT channels were inhibited by 30% whereas the mutant channels V261F, G314F and K318Q were inhibited by only 18%, 22% and 12%, respectively. A V261F/K318Q double mutant did not show an additive effect of two mutations. The charge-conserving mutation, K318R, preserved the strong inhibition by AA (Fig. 2A). Surprisingly, changing the charge on K318 from positive to negative (K318E) did not impair the inhibition of current by AA (Fig. 2A; see Discussion) but these current amplitudes were 50% of those measured for WT channels. K318D mutant channels did not express measurable currents in oocytes (see Methods). And, despite the appearance of a close proximity of R311 to the carboxylate head group of AA in the docking site (Fig. 1C), R311Q, unlike K318Q, did not significantly disrupt the inhibitory effects of AA (Fig. 2A). To further examine the specificity of the K318 residue, we also tested two positively charged residues located near but outside the docking site. Neither R256Q nor R259Q disrupted the fatty acid inhibition of Kv4.2 current (Fig. 2). Thus, among the positively charged residues near the identified hydrophobic pocket, K318 has a particularly important role. Overall, the results suggest that V261, G314 and K318 are key residues regulating the modulatory effects of AA on Kv4.2 channels. Residues G314 and K318 are within the S4-S5 linker, while V261 is within the innermost portion of transmembrane domain S3 (Fig. 2B).

Figure 2. Residues V261, G314 and K318 are important to the inhibitory effects of AA on Kv4.2. (A) Peak current inhibition by 10 μM AA of wild-type and mutant Kv4.2 channels. K+ currents were recorded with voltage steps to +40 mV from a holding potential of −90 mV. The bar graphs show the mean current intensity after 10 min of exposure to 10 μM AA, relative to the intensity before application of AA. Numbers represent the number of cells tested for each mutant; * indicates p < 0.01. Dotted line designates the mean wild-type effect. N.E. = no expression. (B) Mutated residues located on or in close proximity of the S4-S5 linker and their relative importance in AA binding based on results in (A). The radii of the filled circles are proportional to the relative K+ current amplitude after treatment with AA. The inset shows the six transmembrane α-helices of a single Kv4.2 subunit. Residues 256 and 259 are predicted to be outside the proposed AA binding pocket.
The activation properties of three of the mutants that impacted the AA modulation of Kv4 channels (V261F, K318Q, V261F/K318Q) were compared with WT channels (Fig. 3). Small changes in the parameters of the fitted Boltzmann functions were not significantly different between WT and mutant channels. In the absence of AA, we measured a −6 to +4 mV shift in the fitted midpoint (V1/2) of the conductance-voltage relationship for three mutant channels, when compared with WT channels. In the presence of AA, all of the channels showed a rightward shift in the fitted Boltzmann function (+7 mV for WT (Fig. 3A) and an average shift of +5.7 mV for the three mutant constructs (Figs. 3B‒D). A fourth construct, K318E, retained AA inhibition and also showed the largest change in V1/2 compared with WT with a −10 mV shift. K318E retained the +5 mV shift the in V1/2 in the presence of AA, which inhibited the current (Fig. 3C).

Figure 3. Conductance-voltage plots for wild-type and three mutant channels. Peak conductances were calculated from G = I/(Vm-Vh) with Vh = -100 mV and were normalized to the maximal conductance (Gmax) for each cell and plotted vs. the step membrane potential (Vm). The conductance-voltage relationships are plotted in the absence (open symbols) and presence (closed symbols) of 10 μM AA for (A) wild-type (WT) Kv4.2 (n = 14), (B) V261F (n = 8), (C) K318Q (n = 8), K318E (n = 3–6), and (D) V261F/K318Q (n = 6). Solid lines are the best fits of the data to a first order Boltzmann function. In each panel, the black curve is without AA and the gray curve is with AA. The fitted parameters for the slope factor (k) and V1/2 values for the curve without AA and the curve with AA were, respectively: (A) 9.5, −20.3 mV; 12.1, −13.4 mV, (B) 9.2, −20.8 mV; 10.9, −17.1 mV, (C) K318Q: 10.3, −15.6 mV; 11.3, −8.6 mV, K318E: 7.9, −30.7, 7.1, −25.2, (D) 7.3, −26.4 mV; 8.4, −19.1 mV. All fitted curves had a coefficient of determination (R2) ≥ 0.998.
K+ current inhibition by AA of WT Kv4.2 and three mutations that disrupted the inhibition by AA were further examined by measuring the saturating effects for each concentration of AA. Visual inspection of the voltage-activated currents elicited in the presence of increasing concentrations of AA (Fig. 4A) demonstrates this lowered sensitivity to AA of the mutant channels when compared with WT channels. The data were fit to a Hill equation with the Hill slope (nH) = 1.0 (Fig. 4B). The concentration-response curve for each mutant was right-shifted when compared with WT. The fitted Kd value for AA inhibition of WT channels was 21.1 µM, whereas the Kd values for each of the mutants ranged from 37.7 to 57.4 µM (Fig. 4B).
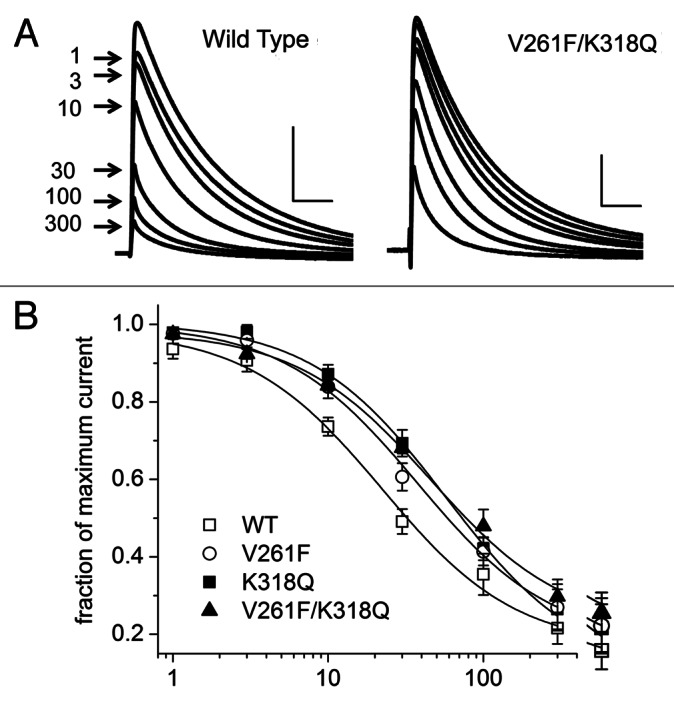
Figure 4. Concentration-dependence of the current inhibition by AA. (A) Representative current traces elicited by voltage steps from −90 mV to +40 mV before and following the application of 1 to 300 μM AA until saturation for wild-type (WT) channels and one mutant construct. Arrows designate peak current amplitude for each test concentration. Horizontal and vertical scale bars represent 25 ms and 1 μA, respectively. (B) Concentration-response curves for AA inhibition of peak Kv4 currents for WT and mutant channels (8–19 cells/point). Data were fitted to a Hill equation with the Hill slope (nH) = 1. All fitted curves had a coefficient of determination (R2) ≥ 0.991. Fitted Kd values were: WT, 21.1 μM; V261F, 37.7 μM; K318Q, 57.4 μM; V261F/K318Q, 45.7 μM.
The fitted Kd values for each of the three mutant channels were statistically different from WT, but not from each other. Therefore, to examine the free energy change induced by the mutations, we calculated the fitted Kd value for cells expressing one of these three mutant channels and then grouped them together for comparison to the fitted Kd values from the WT channels. The mean Kd value for the group of mutant channels (46.0 µM) was statistically different from the mean Kd value for the WT channels (21.1 µM). The corresponding free energy of AA binding at room temperature was calculated by ΔG = nHRTlnKd where nH = 1.0, Kd is the fitted binding constant, R is the gas constant, and T is the absolute temperature. The differences in free energy of binding for mutant and wild type channels were then determined by ΔΔG = ΔGMUTANT − ΔGWT. Relative to wild type Kv4.2 channels, the ΔΔG for the AA inhibition of mutant channel currents was 2.75 kcal/mol. These results suggest that the mutations that inhibited the AA effect made the AA-channel interactions thermodynamically less stable than the interactions of AA with WT channels.
Mutant channels impair AA modulation of macroscopic current decay
Figure 5 shows a family of currents elicited in the absence and presence of AA for WT channels and the three mutants that decreased the inhibition by AA of peak outward K+ current. The last column in each row shows overlapped current traces at +40 mV before and after exposure to AA and demonstrates the increased rate of current decay for WT channels, but a smaller effect of AA on the rate of current decay observed in the mutant channels. Insets show the same traces, each normalized to its peak current amplitude, in order to better visualize the impact of AA on the rate of current decay. K318E is included for comparison because it allows current inhibition by AA but its impact on kinetic modulation is less striking. In WT channels, AA inhibition includes modulation of decay kinetics (Fig. 5, top row). The speeding up of the time course of decay was reduced for K318Q, V261F, and the double mutant V261F/K318Q (Fig. 5, rows 2–4, respectively). We quantified the impact of AA on the rate of current decay by measuring the time at which the current was 50% of the maximum and calculated the t1/2 for each concentration of AA as a fraction of the control value in the absence of AA (Fig. 6). Concentration-response curves (Fig. 6B) for each mutant were right-shifted when compared with WT; the fitted Kd value for AA modulation of current decay kinetics for WT channels was 23.7 µM, whereas the Kd value for V261F was 64.0 µM and for K318Q was 60.7 µM. The concentration-response data for the double-mutant V261F/K318Q was fitted with a Kd of 43.5 µM. The fitted Kd values for these three mutant channels were each statistically different from WT, but not from each other.
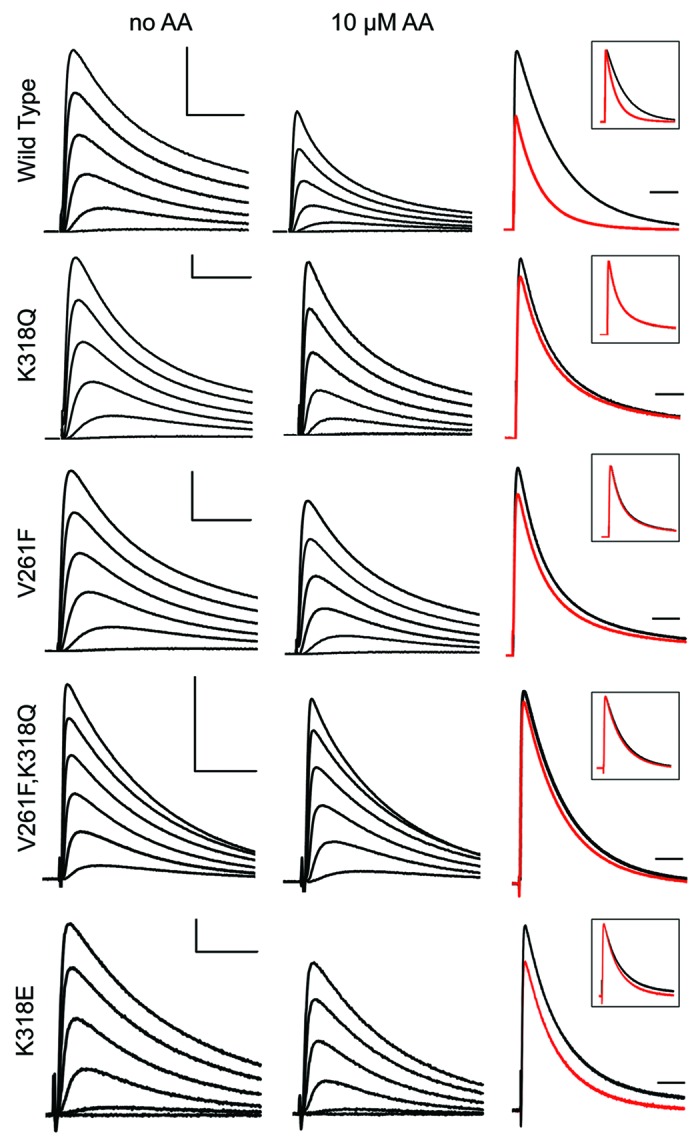
Figure 5. Effects of AA on WT and mutant Kv4 current inactivation. The first two columns show representative Kv4 currents evoked by voltage steps from −90 mV to −40, −20, 0, +20, +40 and +60 mV for wild-type (WT) and selected mutant channels in the absence (first column) and after inhibition by 10 μM AA (second column). The third column displays the current with 10 μM AA (red) superimposed on the current recorded in the absence of AA (black) at +40 mV; inset boxes show the peak of the AA-inhibited current scaled to its control to clarify the effects on the time course of current decay. Horizontal and vertical scale bars represent 25 ms and 0.5 μA, respectively. The scale bars do not apply to the insets.
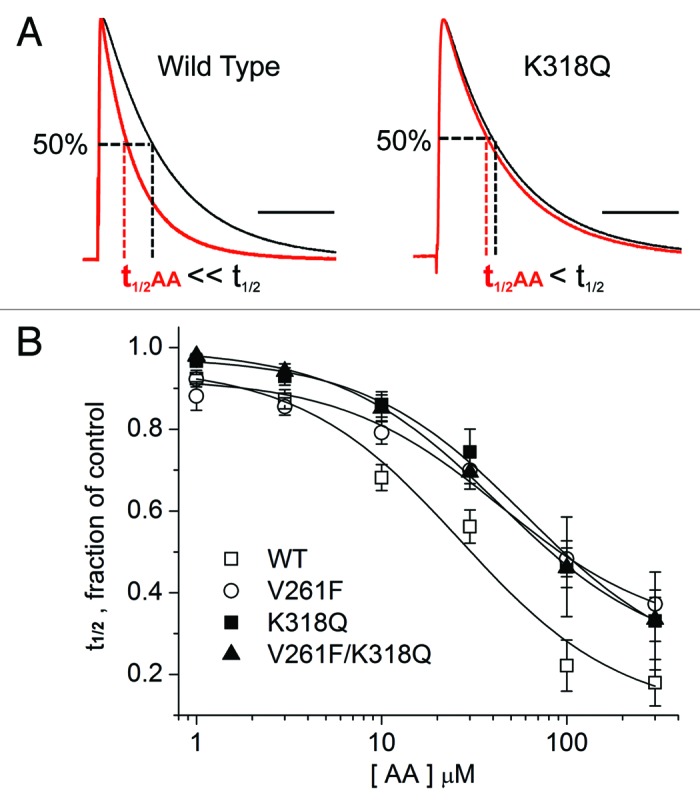
Figure 6. Concentration-dependence of the kinetic modulation by AA. (A) Representative current traces elicited by voltage steps from −90 mV to +40 mV before (black trace) and following the application of 10 μM AA (red trace) for wild-type (WT) channels and one mutant construct. The peak current in the presence of AA was normalized to the pre-AA current in order to measure the effects on current decay. Dashed lines show how t1/2 was measured (time of half-inactivation, measured at +40 mV and normalized to the value measured in the absence of AA). (B) Concentration-response curves for AA-dependent modulation, measured as the fractional reduction in t1/2 Data for 6–12 cells each were fitted to a Hill equation with the Hill slope (nH) = 1; all fitted curves had a coefficient of determination (R2) ≥ 0.986. Fitted Kd values were: WT, 23.7 μM; V261F, 64.0 μM; K318Q, 60.7 μM; V261F/K318Q, 43.5 μM.
Therefore, to examine the free energy change induced by any of these mutations, we calculated the fitted Kd value for the group of cells expressing one of these three mutant channels and then grouped them altogether for comparison to the Kd values from the WT channels. The mean Kd value for the mutant channels (56.1 µM) was statistically different from the mean Kd value for the wild-type channels. Using the same approach as described for the concentration-dependent inhibition of current, we used these Kd values to calculate the corresponding free energy of binding for WT and mutant channels and the binding free energy change (ΔΔG). Like the inhibition of current, the modulation of current decay kinetics is energetically disrupted by mutations at V261F and/or K318Q. Relative to WT channels, the ΔΔG for the kinetic effects of AA on mutant channels was 3.04 kcal/mol.
Interpretation of electrophysiology results using the Kv4.2 homology model
To provide a mechanistic interpretation of our results, we introduced the mutations that affected AA inhibition into our Kv4.2 model and evaluated docking of AA to the mutated channel. Two of the residues lining the hydrophobic pocket in the wild type protein are basic, R311 and K318. In the docking configurations, the carboxylate head of AA is oriented toward these positive charges and might have a stabilizing effect or might even form a salt bridge with AA (Fig. 7A). In our model (Fig. 7B), both R311 and K318 are within hydrogen-bonding distance to the oxygen atom in AA as they are predicted to be less than the 3.4 Å maximum length of a hydrogen bond.29 The hydrophobic tail of the fatty acid is embedded in the cavity and is most likely involved in nonpolar interactions with one or more of the hydrophobic residues lining the pocket.

Figure 7. AA docking specificity. (A) Kv4.2 wild-type (WT) channel model with AA docked; residues of interest are noted. (B) Zoomed-in view of WT channel model predicting the molecular distances of hydrogen bonds with AA. (C–E) Virtual mutants and docked AA. (F) Representative time course of peak outward K+ current amplitudes before and during application of AA (10 μM) for WT Kv4.2 (blue; +40 mV test potential) and Kv1.2/2.1 (red; +110 mV test potential. Inset: Docking result showing the lack of a plausible AA binding site on the Kv1.2/Kv2.1 chimera (PDB: 2R9R63). The same docking and search parameters were used in attempts to dock AA to WT or mutant Kv4.2 channels and the Kv1.2/Kv2.1 chimera.
The mutant V261F impaired the AA effect whereas V261T did not (Fig. 2A). In the proposed binding site, V261 orients close to the carboxylate head group of AA, but is not one of the residues that surround the aliphatic tail of the fatty acid. This suggests that V261 may be important for the access of AA to its proposed interaction site. Steric hindrance by the bulky phenylalanine residue (Fig. 7C), but not the smaller threonine, may disrupt the stability of AA binding. The larger side chain of phenylalanine may partially impede the interaction between the hydrogen bond acceptor (oxygen on AA) and donor (nitrogen on K318). This structural interpretation is consistent with the reduction of the effect of AA on the inhibition of peak V261F K+ current and the reduction in the modulation of current decay kinetics. In a similar fashion, the G314F mutant introduces a bulky side chain that impaired the inhibitory effects of AA (Fig. 2A); docking of AA to the virtual mutant (Fig. 7E) suggests that G314F impairs access of the carboxylate head group of AA to K318.
The K318Q mutant results in a lower AA sensitivity that may be attributed to loss of the positive charge with a concomitant loss of strong electrostatic interaction with the carboxylate of the AA head group and also a shortening of the side chain which may present a less than optimal interaction between the mutant polar side chain and the carboxylate group of AA (Fig. 7D).
While we also expected K318E to be disruptive to the AA modulatory effects due to charge repulsion, it was not. Our model is not sufficient to explain this result, but a hypothesis is offered in the Discussion. In our model of the WT channel, R311 is also close enough to form a hydrogen bond with AA in the binding pocket, but R311Q does not impair the AA inhibition. Therefore, we conclude that R311 does not form the primary interaction with the AA head group. Overall, the results suggest that V261, G314 and K318 are key residues regulating the interaction of AA with the Kv4.2 channel.
Are the structural features of the Kv4.2 hydrophobic pocket specific enough to account for the modulatory effects of AA? We tested this using another channel whose structure has been determined by X-ray crystallography. Standard docking approaches identified a cavity containing AA within the structure of the Kv1.2/Kv2.1 chimeric channel (Fig. 7F), but we found that the Kv1.2/2.1 K+ currents are not modulated by AA. The chimeric K+ channels showed a 1.1 ± 2.1% increase in current in the presence of 10 µM AA (n = 8) compared with a 30.7 ± 1.8% decrease in Kv4.2 currents (n = 3), tested in the same batch of oocytes (Fig. 7F). The cavity identified near S4-S5 within Kv1.2/Kv2.1 (Fig. 7F, inset) may not be viable for AA interaction as it lacks nearby positive charges to stabilize the AA head group, a feature important to the Kv4.2 docking experiments and to the known AA binding sites in albumin24-26 and other fatty acid binding proteins.27,28
Discussion
Using a new homology model for Kv4.2, we identified a hydrophobic pocket where AA may bind to the Kv4 family of voltage-gated potassium channels. The pocket is comprised of Kv4.2 residues 308–318 in the S4-S5 linker on one subunit and, on an adjacent subunit, residues 261262 in S3, 330–335 in S5, and 390–394 in S6. The targeted mutations V261F, G314F and K318Q impaired the effects of AA on Kv4.2. The lack of significant effects of these mutants on voltage-dependence of Kv4.2 activation in the presence or absence of AA suggests that AA affects a voltage-independent or only weakly voltage-dependent gating process. Thus, the physiological and docking results are generally consistent with the role of the S4-S5 linker in coupling voltage sensing to the opening of an internal activation gate.30-33 AA also facilitates inactivation,34 a process that has been shown in other channels to be affected by certain mutations of the S4-S5 linker.34-36 Furthermore, the S4-S5 linker is involved in both anesthetic and alkanol sensitivity of Kv3 channels.37 These drugs share a common binding site comprised of the S4-S5 linker and the cytoplasmic end of S6.38 Together with these results, our study suggests a common mechanism by which amphiphilic ligands modulate the function of certain Kv channels by interacting with a discrete hydrophobic binding pocket near the channel’s internal activation gate.
Based on mutagenesis and docking results, we propose that K318 within the S4-S5 linker of Kv4.2 plays a key role in forming a hydrogen bond with the carboxyl group of a bound AA molecule. Likewise, our results suggest that V261 in the S3 region may regulate AA access to K318. The formation of a hydrogen bond between AA and K318 is supported by the differences of free energy of binding between mutant channels plus AA and WT channels plus AA as calculated from concentration response curves for two effects of AA, the inhibition of current and the speeding up of current decay kinetics. The perturbation of the AA effects on mutant Kv4.2 channels was 2.75 to 3.04 kcal/mol less stable than the WT ligand-protein interaction. This energy difference could account for the loss of the hydrogen bond between the amine of K318 and the fatty acid carboxyl head group. The bond energy of a hydrogen bond where nitrogen is the donor and oxygen is the acceptor has been estimated to be 2 to 4 kcal/mol.39-41 Thus, the energetic disruptions of AA inhibition and facilitation of inactivation by K318Q and/or V261F are consistent with the mutagenic disruption of at least one hydrogen bond.
Since neutralization of the positive charge of residue K318 made the channel significantly less sensitive to modulation by AA (Fig. 2A), we expected a K318E mutant to also impair the AA effects. However, this was not the result we observed. To explain this result, we speculate that the presence of AA in the WT channel cavity triggers a conformational change that allows the native R311 to form a salt bridge with the substituted glutamate at residue 318 (K318E). In this scenario, inhibitory modulation of K318E by AA is not significantly different from WT channels, but has the salt bridge between the side chain of R311 and the carboxyl group of the glutamate as its structural basis. In addition, R311 must also provide specificity for AA binding through an interaction with the AA head group, which retains the AA modulation we observed in experiments. One hypothesis is that R311 may neutralize the K318E charge and contribute to coordinating the AA head group. A similar phenomenon where mutation of one of two positively charged residues in a ligand-binding site resulted in altered protein conformation or ligand selection was proposed for the active sites of malate and lactate dehydrogenase.42,43 Future studies will test this hypothesis of dual function at R311 in response to mutations at K318.
The confirmed docking site within Kv4.2 is hydrophobic and likely stabilizes the aliphatic tail of AA through hydrophobic interactions. Although there is no specific fatty acid binding motif within known fatty acid binding proteins, the features of the docking site for AA within Kv4.2 mimic the structural features identified in other proteins. Eight AA binding sites on serum albumin have been determined by co-crystallization of the ligand and protein,24-26 and all of the sites have a hydrophobic lining with at least one basic or polar amino acid in close proximity of the carboxyl group of the bound AA. Basic or polar amino acids located within AA binding sites in serum albumin, including R410, K525 and Y411, form electrostatic interactions with the carboxyl group of bound AA.26 We propose a similar role for the interaction of K318 in the Kv4.2 channel with AA. Residue G314, lining the center of the proposed binding pocket in Kv4.2, is one of the amino acids that may interact with AA’s aliphatic tail via hydrophobic interactions. In support of this hypothesis, replacement of glycine with a bulky residue (G314F) hinders the access of AA to the cavity (Fig. 7E). Molecular docking with the virtual mutant suggests that the substituted phenylalanine hides K318 from AA and may perturb the spatial nature of the hydrophobic residues lining the pocket (Fig. 7E). Other hydrophobic amino acids lining the pocket are L310, L313, G314, L317, L331, M333, A334, I335, L392, G394 and V395. While G314F displayed a decreased inhibitory modulation by AA, mutant L313F retained its sensitivity to AA (Fig. 2A). This observation suggests that either not all hydrophobic amino acids in the pocket participate in the interaction with AA, or the individual van der Waals contacts are weak, but have a summative effect in stabilizing the fatty acid tail. The docking region forms a large hydrophobic patch that may accommodate multiple conformations of the hydrophobic tail, but specific interactions have not yet been mapped. Future mutational analysis of the hydrophobic residues in the docking site or a crystal structure might better define the specificity of the nonpolar interactions with AA.
Our docking results showed multiple possible locations for hydrophobic interactions between AA and binding pocket residues in WT Kv4.2 (Fig. 1C). Although the pocket is only large enough to accommodate one AA molecule at a time (Fig. 7A), our results suggest that the attributes of the non-polar environment are perhaps more important than the specific structure of the hydrophobic side chains of residues lining the pocket. In addition, the flexible position of AA within the channel’s hydrophobic pocket is consistent with the principle that long carbon chains and multiple carbon-carbon double bonds enhance the flexibility of fatty acids.44,45
Ethanol, another amphiphilic molecule, has been co-crystallized with a Drosophila odorant binding protein46 and with the inwardly rectifying Kir3.2 channel.47 In both proteins, the ethanol-binding site is a hydrophobic cavity with polar residues that make one or more hydrogen bonds with the amphiphilic ligand. Mutations within the alcohol-binding pocket of Kir3.2 have confirmed the importance of specific polar residues in the pocket.47 Our results support the conclusion that the general features identified as important to AA binding to serum albumin and ethanol binding to two structurally distinct proteins are also found in the AA binding pocket we have implicated for inhibitory modulation of Kv4.2.
Other studies have investigated the modulatory effects of PUFAs on related voltage-gated K+ channels. AA induces rapid inactivation of delayed rectifier K+ channels such as Kv1.1, Kv3.1 and Kv3.2.48 For these channels, AA was equally effective in facilitating inactivation when applied to the external or internal side of the membrane and the onset of effects was slow and independent of channel opening.48 Furthermore, lack of competition between AA and external or internal TEA supports a mechanism that differs from open channel block.48 A more recent study supports an alternative mechanism of open channel block by AA to explain the inhibition and fast inactivation induced in Kv1.1 channels.49 RNA editing of a critical residue in the Kv1 pore region appears to regulate this mechanism since I400 in Kv1.1 is AA-sensitive whereas V400 is not. Indeed, the residue at position 400 in Kv1.1 is implicated in a direct hydrophobic interaction with the inactivation gate; an Ile at this position strengthens the interaction with the N-terminus of the channel during fast, N-type inactivation, whereas a Val at this position weakens it.49
However, there may be some differences in the structural basis and functional mechanism of AA inhibition and facilitation of inactivation in Kv1 and Kv4 channels. For example, whereas Kv1 channels are not inhibited by anandamide (AEA) applied externally,48 we find that external AEA mimics the effects of AA on current inhibition and kinetic modulation of Kv4.2 (data not shown). Furthermore, in a test of the open channel block mechanism, internal TEA was found to compete with AA for inhibition of Kv1.1 in one study49 but not another.50 Likewise, Kv4 channels are unique among the voltage-gated K+ channels in possessing a closed-state inactivation mechanism,51-53 and we previously found that AA facilitates both open-state inactivation and closed-state inactivation of Kv4 channels.16 The location of the proposed binding site for AA is within the regions previously identified as important for closed state inactivation and channel closing.52,53 Our project focused on the importance of a proposed hydrophobic binding pocket for the inhibitory modulation by AA, but the data do not resolve whether AA enhances inactivation or stabilizes channels in a closed state with subsequent accumulation of inactivation.
In conclusion, our results support the hypothesis that Kv4.2 channels have a specific binding site that regulates the inhibitory modulation by AA. Two features of the Kv4.2 binding pocket proposed here seem to be essential in the interaction with AA: (1) overall hydrophobicity, which may stabilize the aliphatic lipid tail, and (2) a basic residue in close proximity to the hydrophobic pocket, providing stabilization of the negatively charged carboxylate moiety of the fatty acid. This interpretation is suggested by both steric measurements and changes in binding energy calculated from physiological measurements of current inhibition and the kinetics of inactivation. Our results suggest that K318 within the S4-S5 linker is directly involved in the binding of AA. Access of AA to this critical lysine residue may be regulated by the size and position of V261 and G314 within the non-polar region of the pocket.
Materials and Methods
Model construction and evaluation
A model of Kv4.2 was created based on its homology to Kv1.2 and KcSA using Modeler 9v5.54 Briefly, ClustalW255 was used to align the human Kv4.2 sequence (residues 175–417) with the rat Kv1.2 sequence (residues 153–421), human Kv4.2 (residues 324–409) and S. lividans KcSA (residues 29–114). These alignments encompassed the six predicted transmembrane regions of Kv4.2 and the pore region. Using PDB files for Kv1.2 (2A7956 and 2R9R57) and KcSA, (2QTO58) the alignments were modified to reflect sequences corresponding to the available structural data. Five models were generated by the Modeler software with an average RMSD between chains of 0.451 Å. The models were evaluated by MolProbity version 3.15.59 Model 3, which contained the fewest violations in bond angles, bond lengths, atom overlaps, and phi/psi angles, was used to construct the tetrameric form of Kv4.2, employing the symmetry reported for the Kv1.2 structure. The tetrameric model was subjected to geometry regularization60 and re-evaluated by Molprobity (Ramachandran plot, 98.3% allowed). A PDB file for our Kv4.2 model is provided in the Supplemental Material.
Molecular docking
AutoDock 4 and AutoDock Tools61 were used to protonate the molecular structure of the channel, set the location and the three-dimensional size of the search grid box, and generate docking files for both the ligand and the protein. AutoDock Vina62 was used to predict docking sites and docking conformations of AA on the Kv4.2 protein. The search space characteristics and the other input parameters were varied extensively in order to generate a large pool of possible ligand-protein complexes. The individual docked configurations contained in the output files were separated using VinaSplit. Complexes that involved unfavorable steric or electrostatic interactions were discarded. The docking results also predicted an AA site within the Kv4.2 pore region, but we focused this project on a docking site comprised of hydrophobic residues that could support a favorable energetic environment for the fatty acyl tails of AA. Using PyMOL’s sculpting feature, some ligand-protein interactions were examined for the possibility of hydrogen bonding between AA’s carboxylic head and basic residues in Kv4.2. The residues that were determined by electrophysiology to play a significant role in AA inhibition of Kv4.2 were virtually mutated into the molecular model of the channel. New docking studies were then performed in order to assess the disruption of the binding of AA to the mutant Kv4.2 channel. All model images were generated using the PyMOL Molecular Graphics System of W. DeLano (PyMol, v1.0).
Mutagenesis and expression of channels in oocytes
rKv4.2 channel mutants were constructed using QuikChange (Agilent Technologies, 200523) and confirmed by DNA sequencing. RNA preparation, Xenopus laevis oocyte isolation, and oocyte injection were done using published methods.16 Most studies were done with Kv4.2 WT or mutant channels prepared in the pBluescript vector; 5–10 ng of RNA were injected per oocyte. Mutant K318D did not express to high enough levels to test even when we prepared the mutation in a high expression vector (pGEMHE) and injected up to 40 ng RNA/oocyte.
Animal protocols were approved by the University of Richmond Institutional Animal Care and Use Committee and conform to the requirements in the Guide for the Care and Use of Laboratory Animals from the National Academy of Sciences. Several batches of collagenased oocytes were purchased from EcoCyte Bioscience and yielded the same results. Oocytes were maintained at 17–19°C for 1–5 d post-injection in a solution of (in mM): 96 NaCl, 1 KCl, 1 CaCl2, 2 MgCl2, 10 HEPES, 5 sucrose and 2 Na pyruvate, pH 7.4, with 50 U/ml penicillin G, 50 µg/ml streptomycin and sometimes 50 µg/ml tetracycline.
Electrophysiology
Potassium currents were recorded from oocytes using standard methods of two-electrode voltage clamp16 with a Geneclamp 500B amplifier (Molecular Devices). Experiments were done at room temperature and the chamber was perfused continuously during recordings with a solution containing (in mM): 96 Na methane sulfonate, 2 K methane sulfonate, 2 CaCl2, 1 MgCl2, 10 HEPES, pH 7.4–7.5. Data were recorded and analyzed on Pentium computers equipped with Digidata 1320A A/D hardware and Clampex and Clampfit software. Leak subtraction used P/−4 or P/−6 pulses from the holding potential. Data were also transferred to Microsoft Excel and Microcal Origin for additional analysis and the production of figures. Results are expressed as mean ± SE with n = number of cells tested. In cases with multiple comparisons to a single set of control values, a single factor ANOVA with a post-hoc Dunnett’s test was used with p < 0.01 considered to be significant. Statistical evaluation of the grouped data for multiple mutants (determined by ANOVA to be not different from each other but different from wild-type) vs. wild-type channels used a paired Student’s two-tailed t-test with p < 0.01 considered to be significant.
Reagents
Arachidonic acid from Sigma-Aldrich (A9673) or Nu-Chek Prep (U-72-A), was dissolved in ethanol or DMSO at 1,000–3,000×, stored at −20°C, diluted into recording solution immediately before use and replenished every three hours or less. Vehicle controls were tested for the highest concentrations of diluent and had no measureable effects on the currents. Fatty acid-free bovine serum albumin (Sigma-Aldrich, A7030) was dissolved into the recording solution at 0.5–1.0 mg/ml.
Supplementary Material
Acknowledgments
This work was supported by a grant from the Thomas and Kate Jeffress Foundation (L.M.B.), NIH grant R15-GM096142 (L.M.B.), an undergraduate fellowship from an HHMI grant to the University of Richmond (R.H.), an American Physiological Society summer undergraduate fellowship (R.H.), the Dickinson Award in Biology at the University of Richmond (R.H.), and NIH grant K22-CA122828 (J.K.B.). We thank Rod MacKinnon for the Kv1.2/2.1 plasmid, Ellis Bell for helpful discussions and Diomedes Logothetis and Vasileous Petrou for critical feedback on the manuscript.
Glossary
Abbreviations:
- PUFA
polyunsaturated fatty acid
- AA
arachidonic acid
- BSA
bovine serum albumin
- G-V
conductance-voltage
- WT
wild type
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/23453
References
- 1.Bazan NG. Synaptic signaling by lipids in the life and death of neurons. Mol Neurobiol. 2005;31:219–30. doi: 10.1385/MN:31:1-3:219. [DOI] [PubMed] [Google Scholar]
- 2.Xiao YF, Sigg DC, Leaf A. The antiarrhythmic effect of n-3 polyunsaturated fatty acids: modulation of cardiac ion channels as a potential mechanism. J Membr Biol. 2005;206:141–54. doi: 10.1007/s00232-005-0786-z. [DOI] [PubMed] [Google Scholar]
- 3.Villarroel A, Schwarz TL. Inhibition of the Kv4 (Shal) family of transient K+ currents by arachidonic acid. J Neurosci. 1996;16:2522–32. doi: 10.1523/JNEUROSCI.16-08-02522.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Holmqvist MH, Cao J, Knoppers MH, Jurman ME, Distefano PS, Rhodes KJ, et al. Kinetic modulation of Kv4-mediated A-current by arachidonic acid is dependent on potassium channel interacting proteins. J Neurosci. 2001;21:4154–61. doi: 10.1523/JNEUROSCI.21-12-04154.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song WJ, Tkatch T, Baranauskas G, Ichinohe N, Kitai ST, Surmeier DJ. Somatodendritic depolarization-activated potassium currents in rat neostriatal cholinergic interneurons are predominantly of the A type and attributable to coexpression of Kv4.2 and Kv4.1 subunits. J Neurosci. 1998;18:3124–37. doi: 10.1523/JNEUROSCI.18-09-03124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liss B, Franz O, Sewing S, Bruns R, Neuhoff H, Roeper J. Tuning pacemaker frequency of individual dopaminergic neurons by Kv4.3L and KChip3.1 transcription. EMBO J. 2001;20:5715–24. doi: 10.1093/emboj/20.20.5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Connor JA, Stevens CF. Voltage clamp studies of a transient outward membrane current in gastropod neural somata. J Physiol. 1971;213:21–30. doi: 10.1113/jphysiol.1971.sp009365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Migliore M, Hoffman DA, Magee JC, Johnston D. Role of an A-type K+ conductance in the back-propagation of action potentials in the dendrites of hippocampal pyramidal neurons. J Comput Neurosci. 1999;7:5–15. doi: 10.1023/A:1008906225285. [DOI] [PubMed] [Google Scholar]
- 9.Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, et al. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 2006;26:12143–51. doi: 10.1523/JNEUROSCI.2667-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taylor AL, Hewett SJ. Potassium-evoked glutamate release liberates arachidonic acid from cortical neurons. J Biol Chem. 2002;277:43881–7. doi: 10.1074/jbc.M205872200. [DOI] [PubMed] [Google Scholar]
- 11.Wolf MJ, Izumi Y, Zorumski CF, Gross RW. Long-term potentiation requires activation of calcium-independent phospholipase A2. FEBS Lett. 1995;377:358–62. doi: 10.1016/0014-5793(95)01371-7. [DOI] [PubMed] [Google Scholar]
- 12.Ramakers GM, Storm JF. A postsynaptic transient K(+) current modulated by arachidonic acid regulates synaptic integration and threshold for LTP induction in hippocampal pyramidal cells. Proc Natl Acad Sci U S A. 2002;99:10144–9. doi: 10.1073/pnas.152620399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujita S, Ikegaya Y, Nishikawa M, Nishiyama N, Matsuki N. Docosahexaenoic acid improves long-term potentiation attenuated by phospholipase A(2) inhibitor in rat hippocampal slices. Br J Pharmacol. 2001;132:1417–22. doi: 10.1038/sj.bjp.0703970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu HJ, Carrasquillo Y, Karim F, Jung WE, Nerbonne JM, Schwarz TL, et al. The kv4.2 potassium channel subunit is required for pain plasticity. Neuron. 2006;50:89–100. doi: 10.1016/j.neuron.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 15.Guo W, Jung WE, Marionneau C, Aimond F, Xu H, Yamada KA, et al. Targeted deletion of Kv4.2 eliminates I(to,f) and results in electrical and molecular remodeling, with no evidence of ventricular hypertrophy or myocardial dysfunction. Circ Res. 2005;97:1342–50. doi: 10.1161/01.RES.0000196559.63223.aa. [DOI] [PubMed] [Google Scholar]
- 16.Boland LM, Drzewiecki MM, Timoney G, Casey E. Inhibitory effects of polyunsaturated fatty acids on Kv4/KChIP potassium channels. Am J Physiol Cell Physiol. 2009;296:C1003–14. doi: 10.1152/ajpcell.00474.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bittner K, Müller W. Oxidative downmodulation of the transient K-current IA by intracellular arachidonic acid in rat hippocampal neurons. J Neurophysiol. 1999;82:508–11. doi: 10.1152/jn.1999.82.1.508. [DOI] [PubMed] [Google Scholar]
- 18.Falkenburger BH, Jensen JB, Dickson EJ, Suh BC, Hille B. Phosphoinositides: lipid regulators of membrane proteins. J Physiol. 2010;588:3179–85. doi: 10.1113/jphysiol.2010.192153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bruno MJ, Koeppe RE, 2nd, Andersen OS. Docosahexaenoic acid alters bilayer elastic properties. Proc Natl Acad Sci U S A. 2007;104:9638–43. doi: 10.1073/pnas.0701015104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lundbaek JA, Andersen OS. Lysophospholipids modulate channel function by altering the mechanical properties of lipid bilayers. J Gen Physiol. 1994;104:645–73. doi: 10.1085/jgp.104.4.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perozo E. Gating prokaryotic mechanosensitive channels. Nat Rev Mol Cell Biol. 2006;7:109–19. doi: 10.1038/nrm1833. [DOI] [PubMed] [Google Scholar]
- 22.Morris CE, Juranka PF. Lipid stress at play: Mechanosensitivity of voltage‐gated channels. In: Hamill OP, ed. Current Topics in Membranes. Academic Press, 2007:297-338. [DOI] [PubMed] [Google Scholar]
- 23.Boland LM, Drzewiecki MM. Polyunsaturated fatty acid modulation of voltage-gated ion channels. Cell Biochem Biophys. 2008;52:59–84. doi: 10.1007/s12013-008-9027-2. [DOI] [PubMed] [Google Scholar]
- 24.Curry S, Mandelkow H, Brick P, Franks N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat Struct Biol. 1998;5:827–35. doi: 10.1038/1869. [DOI] [PubMed] [Google Scholar]
- 25.Petitpas I, Grüne T, Bhattacharya AA, Curry S. Crystal structures of human serum albumin complexed with monounsaturated and polyunsaturated fatty acids. J Mol Biol. 2001;314:955–60. doi: 10.1006/jmbi.2000.5208. [DOI] [PubMed] [Google Scholar]
- 26.Simard JR, Zunszain PA, Ha CE, Yang JS, Bhagavan NV, Petitpas I, et al. Locating high-affinity fatty acid-binding sites on albumin by x-ray crystallography and NMR spectroscopy. Proc Natl Acad Sci U S A. 2005;102:17958–63. doi: 10.1073/pnas.0506440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thumser AEA, Voysey J, Wilton DC. Mutations of recombinant rat liver fatty acid-binding protein at residues 102 and 122 alter its structural integrity and affinity for physiological ligands. Biochem J. 1996;314:943–9. doi: 10.1042/bj3140943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson J, Winter N, Terwey D, Bratt J, Banaszak L. The crystal structure of the liver fatty acid-binding protein. A complex with two bound oleates. J Biol Chem. 1997;272:7140–50. doi: 10.1074/jbc.272.11.7140. [DOI] [PubMed] [Google Scholar]
- 29.Lipscomb LA, Peek ME, Zhou FX, Bertrand JA, VanDerveer D, Williams LD. Water ring structure at DNA interfaces: hydration and dynamics of DNA-anthracycline complexes. Biochemistry. 1994;33:3649–59. doi: 10.1021/bi00178a023. [DOI] [PubMed] [Google Scholar]
- 30.McCormack K, Joiner WJ, Heinemann SH. A characterization of the activating structural rearrangements in voltage-dependent Shaker K+ channels. Neuron. 1994;12:301–15. doi: 10.1016/0896-6273(94)90273-9. [DOI] [PubMed] [Google Scholar]
- 31.Shieh CC, Klemic KG, Kirsch GE. Role of transmembrane segment S5 on gating of voltage-dependent K+ channels. J Gen Physiol. 1997;109:767–78. doi: 10.1085/jgp.109.6.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Labro AJ, Raes AL, Grottesi A, Van Hoorick D, Sansom MSP, Snyders DJ. Kv channel gating requires a compatible S4-S5 linker and bottom part of S6, constrained by non-interacting residues. J Gen Physiol. 2008;132:667–80. doi: 10.1085/jgp.200810048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanguinetti MC, Xu QP. Mutations of the S4-S5 linker alter activation properties of HERG potassium channels expressed in Xenopus oocytes. J Physiol. 1999;514:667–75. doi: 10.1111/j.1469-7793.1999.667ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barghaan J, Bähring R. Dynamic coupling of voltage sensor and gate involved in closed-state inactivation of kv4.2 channels. J Gen Physiol. 2009;133:205–24. doi: 10.1085/jgp.200810073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Covarrubias M, Vyas TB, Escobar L, Wei A. Alcohols inhibit a cloned potassium channel at a discrete saturable site. Insights into the molecular basis of general anesthesia. J Biol Chem. 1995;270:19408–16. doi: 10.1074/jbc.270.33.19408. [DOI] [PubMed] [Google Scholar]
- 36.Harris T, Shahidullah M, Ellingson JS, Covarrubias M. General anesthetic action at an internal protein site involving the S4-S5 cytoplasmic loop of a neuronal K(+) channel. J Biol Chem. 2000;275:4928–36. doi: 10.1074/jbc.275.7.4928. [DOI] [PubMed] [Google Scholar]
- 37.Shahidullah M, Harris T, Germann MW, Covarrubias M. Molecular features of an alcohol binding site in a neuronal potassium channel. Biochemistry. 2003;42:11243–52. doi: 10.1021/bi034738f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhattacharji A, Klett N, Go RCV, Covarrubias M. Inhalational anaesthetics and n-alcohols share a site of action in the neuronal Shaw2 Kv channel. Br J Pharmacol. 2010;159:1475–85. doi: 10.1111/j.1476-5381.2010.00642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vargas R, Garza J, Dixon D, Hay B. How strong is the C-α-H …O=C hydrogen bond? J Am Chem Soc. 2000;122:4750–5. doi: 10.1021/ja993600a. [DOI] [Google Scholar]
- 40.Vargas R, Garza J, Friesner R, Stern H, Hay B, Dixon D. Strength of the N-H …O=C and CH … O=C bonds in formamide and N-methylacetamide dimers. J Phys Chem A. 2001;105:4963–8. doi: 10.1021/jp003888m. [DOI] [Google Scholar]
- 41.Zhang Y, Wang CS. Estimation on the intramolecular 10-membered ring N-H...O=C hydrogen-bonding energies in glycine and alanine peptides. J Comput Chem. 2009;30:1251–60. doi: 10.1002/jcc.21141. [DOI] [PubMed] [Google Scholar]
- 42.Goward CR, Nicholls DJ. Malate dehydrogenase: a model for structure, evolution, and catalysis. Protein Sci. 1994;3:1883–8. doi: 10.1002/pro.5560031027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McClendon S, Vu DM, Clinch K, Callender R, Dyer RB. Structural transformations in the dynamics of Michaelis complex formation in lactate dehydrogenase. Biophys J. 2005;89:L07–09. doi: 10.1529/biophysj.105.064675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zimmerberg J, Gawrisch K. The physical chemistry of biological membranes. Nat Chem Biol. 2006;2:564–7. doi: 10.1038/nchembio1106-564. [DOI] [PubMed] [Google Scholar]
- 45.Feller SE. Acyl chain conformations in phospholipid bilayers: a comparative study of docosahexaenoic acid and saturated fatty acids. Chem Phys Lipids. 2008;153:76–80. doi: 10.1016/j.chemphyslip.2008.02.013. [DOI] [PubMed] [Google Scholar]
- 46.Kruse SW, Zhao R, Smith DP, Jones DNM. Structure of a specific alcohol-binding site defined by the odorant binding protein LUSH from Drosophila melanogaster. Nat Struct Biol. 2003;10:694–700. doi: 10.1038/nsb960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aryal P, Dvir H, Choe S, Slesinger PA. A discrete alcohol pocket involved in GIRK channel activation. Nat Neurosci. 2009;12:988–95. doi: 10.1038/nn.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oliver D, Lien CC, Soom M, Baukrowitz T, Jonas P, Fakler B. Functional conversion between A-type and delayed rectifier K+ channels by membrane lipids. Science. 2004;304:265–70. doi: 10.1126/science.1094113. [DOI] [PubMed] [Google Scholar]
- 49.Decher N, Streit AK, Rapedius M, Netter MF, Marzian S, Ehling P, et al. RNA editing modulates the binding of drugs and highly unsaturated fatty acids to the open pore of Kv potassium channels. EMBO J. 2010;29:2101–13. doi: 10.1038/emboj.2010.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gonzalez C, Lopez-Rodriguez A, Srikumar D, Rosenthal JJ, Holmgren M. Editing of human K(V)1.1 channel mRNAs disrupts binding of the N-terminus tip at the intracellular cavity. Nat Commun. 2011;2:436. doi: 10.1038/ncomms1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bähring R, Boland LM, Varghese A, Gebauer M, Pongs O. Kinetic analysis of open- and closed-state inactivation transitions in human Kv4.2 A-type potassium channels. J Physiol. 2001;535:65–81. doi: 10.1111/j.1469-7793.2001.00065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bähring R, Covarrubias M. Mechanisms of closed-state inactivation in voltage-gated ion channels. J Physiol. 2011;589:461–79. doi: 10.1113/jphysiol.2010.191965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jerng HH, Shahidullah M, Covarrubias M. Inactivation gating of Kv4 potassium channels: molecular interactions involving the inner vestibule of the pore. J Gen Physiol. 1999;113:641–60. doi: 10.1085/jgp.113.5.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 55.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 56.Long SB, Campbell EB, Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 57.Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–82. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 58.Chen X, Poon BK, Dousis A, Wang Q, Ma J. Normal-mode refinement of anisotropic thermal parameters for potassium channel KcsA at 3.2 A crystallographic resolution. Structure. 2007;15:955–62. doi: 10.1016/j.str.2007.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PIW, Word JM, Prisant MG, et al. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–50. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 60.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morris GM, Huey R, Olson AJ. Using AutoDock for ligand-receptor docking. Curr Protoc Bioinformatics. 2008;Chapter 8(Chapter 8):8–, 14. doi: 10.1002/0471250953.bi0814s24. [DOI] [PubMed] [Google Scholar]
- 62.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–61. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tao X, MacKinnon R. Functional analysis of Kv1.2 and paddle chimera Kv channels in planar lipid bilayers. J Mol Biol. 2008;382:24–33. doi: 10.1016/j.jmb.2008.06.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.