

Abstract
With a prevalence of 1 in 2500 people, inherited peripheral nerve diseases, collectively called Charcot-Marie-Tooth disease (CMT), are among the most common inherited neurologic disorders. Patients with CMT typically present with chronic muscle weakness and atrophy in limbs, sensory loss in the feet and hands, and foot deformities. Clinical similarities between patients often require genetic testing to achieve a precise diagnosis. In this article, the author reviews the clinical and pathologic features of CMT, and demonstrates how electrodiagnostic and genetic tools are used to assist in the diagnosis and symptomatic management of the diseases. Several cases are presented to illustrate the diagnostic processes.
Keywords: Charcot-Marie-Tooth disease, neuropathy, dysmyelination, demyelination, axonal degeneration, nerve conduction study, sural nerve biopsy, PMP22, MPZ, mitofusin-2, genetics, DNA testing
Cell bodies of sensory and motor neurons in the spinal cord , and dorsal root ganglia extend their long axons to communicate with the skin and muscle. These axons are insulated by Schwann cell membranes that can either wrap the axons in multiple layers to form myelinated nerve fibers or wrap axons in only one layer to form the nonmyelinated nerve fibers. Mutations in many human genes cause pathologic lesions in the peripheral nervous system resulting in a group of neuropathies collectively called Charcot-Marie-Tooth disease (CMT), in honor of the investigators who initially described the disorder in the late 1800s. CMT has a prevalence of 1 in 2500 and ranks among the most common inherited neurologic disorders. The majority of CMTs are autosomal dominant, although other subtypes are X-linked or autosomal recessive.
CMT typically presents with muscle weakness and sensory loss in the feet and hands, foot deformities (high arched feet and/or hammer toes), and absent deep tendon reflexes. Although clinical similarities between patients often make the differential diagnosis seem difficult, a combination of family history, neurologic examination, and electrophysiologic findings often focuses the differential drastically. In this article, we will review the clinical, electrodiagnostic, genetic, and pathologic features of CMT, then follow with a discussion regarding the management of these patients.
Classification
Autosomal dominant CMT is classified into two categories based on the electrophysiologic and pathologic features. CMT type I, also known as hereditary motor and sensory neuropathy type I (HMSN-I), refers to de-/dysmyelinating neuropathies, whereas CMT type II (HMSN-II) refers to axonal neuropathies.1CMTX refers to X-linked neuropathies, whereas CMT4 is linked to recessively inherited neuropathies. Each of the four groups is further subdivided based on the historical order in when the mutations or linkage was initially discovered (A, B, C, etc.). For instance, the first identified mutation causing CMT1 was a duplication of chromosome 17p11.2 (CMT1A) and the second involved mutations in the myelin protein zero (MPZ) gene (CMT1B). A simplified version of the current classification system is shown in Table 1.
Table 1.
Classification of Charcot-Marie-Tooth Disease (CMT)
| Type* | Locus/gene | Prominent phenotype |
|---|---|---|
| CMT1: | ||
| CMT1A | 17p11.2/PMP22 duplication | Prototype of CMT1 |
| CMT1B | 1q21–23/MPZ | May present as an axonal neuropathy |
| CMT1C | 16p12–13/SIMPLE/LITAF | Almost indistinguishable from CMT1A |
| CMT1D | 10q21–22/EGR2 | Severe dysmyelination with cranial nerve involvement |
| CMT1F | 8p21/NEFL | Lacks de/dysmyelination, but CV is in the range of CMT1 |
| CMT with nerve susceptibility to mechanical stress: | ||
| HNPP | 17p11.2/PMP22 deletion | Focal sensory loss and weakness; tomacula |
| Others? | ||
| CMTX: | ||
| CMTX1 | X-chromosome/GJB1 | Intermediate range of CV |
| CMT2: | ||
| CMT2A | 1p36/MFN2 | Early & late onset; optic atrophy/hearing loss |
| CMT2B | 3q21/RAB7 | Prominent sensory loss and foot ulcers |
| CMT2C | 12q23/TRPV4 | Vocal cord paralysis; skeletal deformities |
| CMT2D | 7p15/GARS | Motor deficits in upper limbs |
| CMT2E | 8p21/NEFL | Lacks de/dysmyelination, but CV is in the range of CMT1 |
| CMT2F | 7q/HSP27 | Classical CMT2 or distal SMA |
| CMT2G | 12q12–13/unknown | |
| CMT2L | 12q24/HSP27 | Classical CMT2 or distal SMA |
| CMT4: | ||
| CMT4A | 8q13/GDAP1 | Demyelination or axonal; vocal cord paralysis |
| CMT4B1 | 11q22/MTMR2 | Myelin folding |
| CMT4B2 | 11p15/MTMR13 | Myelin folding |
| CMT4C | 5q32/SH3TC2(KIAA1985) | |
| CMT4D | 8q24/NDRG1 | Severe; hearing loss; CNS involvement |
| CMT4E | 10q21/EGR2 | Severe dysmyelination or amyelination |
| CMT4F | 19q13/PRX | Myelin folding—tomacula |
| CMT4H | 12q11/FGD4 | |
| CMT4J | 6q21/FIG4 | Rapidly progressive asymmetric weakness |
| ARCMT2A | 1q21/LMNA | Proximal muscle weakness; muscular dystrophy; cardiomyopathy |
| DI-CMT: | ||
| DI-CMTA | 10q24 | |
| DI-CMTB | 19q12/DNM2 | |
| DI-CMTC | 1p34/YARS | |
| Inherited brachial plexopathy | ||
| HNPP | 17p11.2/PMP22 deletion | No pain/unilateral arm weakness |
| HNA | 17q25/SEPT9 | Severe pain/arm weakness & atrophy |
CNS, Central nervous system; SMA, spinal muscular dystrophy; CV
Note:
Dejerine-Sottas Disease (DSD) has been defined clinically with onset by 2 years of age, delayed motor milestones, and severe motor/sensory and skeletal deficits. DSD is caused by autosomal dominant mutations in PMP22, MPZ, and EGR2, thus should not be viewed as a subtype of CMT due to the heterogeneity of genetic causes and inconsistent pathology ranging from severe dysmyelination to predominant axonal loss. Due to its unique phenotypic features, we recommend listing hereditary neuropathy with liability to pressure palsies (HNPP) as a separate group.
CMT1 (De-/Dysmyelinating Neuropathies with Autosomal Dominant Inheritance)
Clinical Features
CMT1A is the prototypic form of CMT1 and will be discussed in detail. It is the most common form of CMT, accounting for ~50% of all CMT patients.2,3 Most CMT1A patients (85%) become symptomatic in the first two decades of life. They are slow runners in childhood and are often called “clumsy kids.” Foot deformities, such as high arches or hammertoes, often become conspicuous during teenage years and may necessitate surgical correction. Ankle weakness is common and can result in tripping or ankle sprain. Many patients require ankle/foot orthotics. Hand weakness typically develops about 10 years after foot weakness, resulting in difficulties with buttoning shirts or using utensils.
Sensory symptoms are more insidious and typically negative (loss of feeling) rather than positive (paresthesias, pain). However, hip and knee pain is common, likely due to joint injury secondary to chronic weakness and altered biomechanics. Ankle weakness and decreased proprioception may result in poor balance. Nevertheless, most patients remain ambulatory throughout their life. A small group of patients with CMT1A present with gait ataxia, postural tremor, and kyphoscoliosis, a constellation often referred to as Roussy-Levy syndrome.4
CMT1B (due to mutations in the MPZ gene) shares a similar phenotype with CMT1A. However, CMT1B usually clusters into two groups, early or late-onset. Patients in the early-onset group have severe sensory and motor deficits in infancy, whereas the clinical presentation of the late-onset group is closer to that of CMT1A.5–7 CMT1C (due to point mutation in SIMPLE/LITAF gene) is indistinguishable from CMT1A.8 Finally, phenotypes of CMT1E patients possessing point mutations of the PMP22 gene vary from a severe childhood onset Dejerine-Sottas disease to a mild form of hereditary neuropathy with liability to pressure palsies, which is described below. Other subtypes of CMT1 are rare and will not be discussed.
Cranial nerve examination is usually normal in CMT1 patients. In addition to sensory loss and foot deformities, most patients have absent deep tendon reflexes. Sensory examination shows reduced pinprick, touch, and/or vibration sensation in feet and hands. Muscle weakness and atrophy involve distal muscles. Proximal muscle weakness can be observed in patients with the severe early-onset phenotype.
Genetic
CMT1A is caused by a duplication of a DNA segment on chromosome 17p11.2, which contains the gene encoding peripheral myelin protein 22kD (PMP22).9,10 About 80% of CMT1 patients carry this mutation. Interestingly, a heterozygous deletion of exactly the same chromosomal 17p11.2 region causes an entirely different disorder, hereditary neuropathy with liability to pressure palsies (HNPP).11 PMP22 is primarily expressed in the compact myelin of the peripheral nervous system. However, during the embryonic stage, PMP22 is detectable in the central nervous system as well as in nonneural tissues.12–14 Immunologic electron microscopic studies in patients with CMT1A demonstrate an increased level of PMP22 protein in the myelin sheath.15–18 In contrast, patients with HNPP have a decreased PMP22 level in nerve myelin.18–20 Taken together, these studies suggest that the level of PMP22 plays a critical role in peripheral nerve myelination and axonal survival, and should be tightly regulated.
Between −1600 and −2100bp in the PMP22 gene promoter, there are two sites that can bind with a cAMP response element binding (CREB) protein, which silences the PMP22 promoter. This inhibition can be removed by cAMP, an effect that may be counteracted by vitamin C to reduce PMP22 expression.21,22 These findings provided the theoretical basis for treatment trial in CMT1A. Administration of vitamin C to decrease PMP22 expression resulted in clinical and pathologic improvement in CMT1A animal models,22,23 a finding that led to ongoing human clinical trials.24 Unfortunately, data from recently completed trials shows no clinical benefit.25 Although the negative trial does not necessarily reject the vitamin C effect in the regulation of PMP22, it does question additional confounding factors that may diminish the effectiveness of vitamin C in CMT1A patients.
Missense or nonsense mutations in PMP22, although rare, may cause variable phenotypes ranging from severe early-onset dysmyelinating neuropathy such as Dejerine-Sottas disease (CMT1E) to HNPP that may be caused by a nonsense mutation in the PMP22 gene.19,20
CMT1B is caused by missense mutations in the MPZ gene on chromosome 1.26 MPZ accounts for ~50% of all myelin proteins. It is a member of the immunoglobulin superfamily, has a single transmembrane domain, and is necessary for the adhesion between myelin layers.27 However, a toxic gain-of-function by the mutant MPZ protein stresses the endoplasmic reticulum, which appears more relevant to the pathogenesis of CMT1B.28
CMT1C is caused by missense mutations in a gene encoding SIMPLE/LITAF. Although the molecular functions of this protein are largely unknown, similarities in the clinical presentation are striking between CMT1A and CMT1C. This raises the question of whether PMP22 and LITAF are functionally related at the molecular level.
Missense mutations in the early growth response 2 gene (EGR2, also called krox20), located on chromosome 10, cause CMT1D.29 EGR2 is a transcription factor that is directly involved in the transcriptional regulation of myelin protein encoding genes Schwann cells. Thus, this gene is instrumental in understanding the molecular mechanism regulating the myelination.
Nerve Conduction Testing
Nerve conduction studies (NCSs) are helpful for differentiating CMT1 from CMT2 as the former has slowed conduction velocities, whereas the latter causes reduced amplitudes of sensory and motor responses with preserved conduction velocity. A striking electrophysiologic feature of CMT1 is uniformly slowed conduction velocities. This contrasts with acquired demyelinating neuropathies that cause nonuniform or asymmetric slowing with motor conduction block and abnormal temporal dispersion of motor responses.30 However, caution should be taken because nonuniform slowing can be seen in some subtypes of CMT, such as HNPP or CMTX1 (31).
The distinction between demyelinating and axonal forms using NCS is challenging in a subset of patients with conduction velocities in the intermediate range (30–40 m/s), called “intermediate CMT1.” CMTX1 that is caused by mutations in the connexin-32 gene falls into this category. In addition, temporal dispersion and conduction block have also been observed in patients with CMTX1,31 which may make it even more difficult to differentiate from acquired demyelinating neuropathy on electrodiagnostic grounds alone.
Pathology
Onion bulbs in the nerves of patients with CMT1 were named due to their resemblance to the layered structure of an onion sliced in half. Multiple Schwann cells concentrically extend cytoplasmic membrane to encircle the axon, but fail to form compact myelin. In the most-inner layers of the processes, one of the Schwann cells establishes a one-to-one relationship with the axon and forms compact myelin with reduced thickness (Fig. 1). Onion bulbs occur in many forms of CMT1.32
Figure 1.
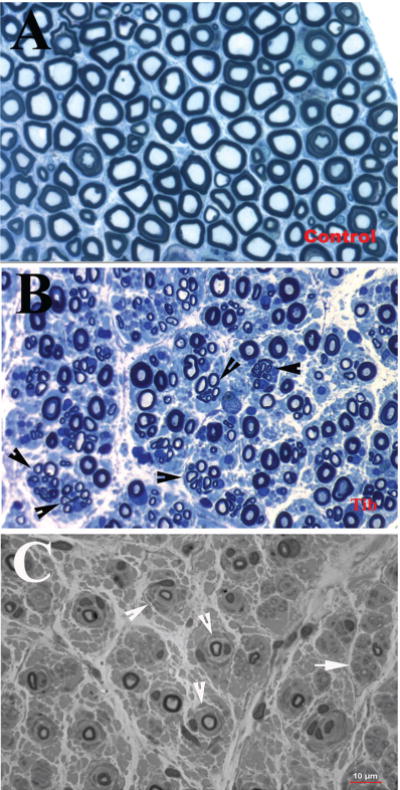
(A) Sciatic nerve section from normal control autopsy, stained with Toluidine blue, contains numerous myelinated nerve fibers. (B) In contrast, a semithin section from the tibial nerve of a patient with axonal form of MPZ mutation, also stained with Toluidine blue, showed a reduced density of myelinated nerve fibers with many regenerating clusters (arrowheads). These features are consistent with axonal type of neuropathy. (C) A semithin section from a sural nerve biopsy of a patient with CMT1B showed numerous onion bulbs (arrowheads) with severely reduced densityof myelinated nervefibers. These features are typical for CMT1. (Reprinted with permission from Li J et al6 and Bai et al7)
Axonal loss is almost an inevitable feature of patients with CMT1. The degree of axonal loss, and not conduction velocity slowing, correlates with weakness and disability.2,33 Focal sausage-like thickenings of the myelin sheath (tomacula) may be present in various types of CMT1. Tomacula are very common in, and are the pathologic hallmark of HNPP, which used to be referred to as “tomaculous neuropathy.”
X-Linked Charcot-Marie-Tooth Disease (CMTX)
CMTX1 is caused by missense mutations in the connexin 32 (Cx32) gene located on the X-chromosome.34 It is the second most common form of CMT35 representing 16% of all patients. Cx32 is localized in the uncompacted myelin of the paranodal loops and Schmidt-Lanterman incisures and is thought to function as a gap junction protein permitting the passage of small molecules and ions between adjacent loops of the paranode or incisures.36 Over 200 different mutations of Cx32 have been identified. Because men have only a single X-chromosome, they tend to develop more severe symptoms than women. Women, probably due to X-inactivation of the abnormal chromosome, usually have a milder disease. CMTX patients usually develop symptoms in their late teenage years or young adulthood. Abnormalities are slowly progressive, limited to the distal legs and hands, and do not shorten the patient’s lifespan.
As aforementioned, nerve conductions in CMTX1 show features31 that closely resemble those of acquired demyelinating diseases such as CIDP, although the clinical features are usually distinct. Occasionally, female patients present in adulthood with a CIDP-like neuropathy.37
Sural nerve biopsies from patients with Cx32 mutations show axonal loss and demyelination with frequent widening at the nodal gap. Onion bulbs are infrequent (unlike CMT1A).38
Inherited Neuropathies with Nerve Susceptibility to Mechanical Stresses
Hereditary neuropathy with liability to pressure palsies (HNPP) is caused by the heterozygous deletion of the chromosomal region 17p11.2.19,20 Homozygous deletion of the DNA segment is extremely rare presumably due to embryonic lethality and has only recently been reported in a single child.39 Patients with HNPP present with reversible focal sensory loss and weakness that often develop when peripheral nerves are challenged by mechanical stress including stretching, compression, or repetitive movement of the affected limbs.40 Strenuous physical activity can result in limb paralysis with significant axonal damage.41 Young patients often have a minimally abnormal examination. Significant sensory loss, weakness, and muscle atrophy in the hands and/ or feet are often observed in the elderly. Occasionally, a brachial plexopathy may be the presenting symptom. However, HNPP is a distinct disorder from hereditary neuralgic plexopathy (described in a later section) and shows minimal to no pain during an episode of plexopathy.
Tomacula are pathologic hallmarks of HNPP.32 The tomacula are formed by excessive myelin folding, predominantly in the paranodal regions.42 Axons encased by the tomacula are often deformed.43 Although segmental demyelination and remyelination have been described in nerve biopsies from patients with HNPP, the pathology is much less prominent when compared with CMT1A, 1C, or 1E. This is consistent with mildly reduced conduction velocities found in patients with HNPP.44 Immunologic electron microscopic studies of sural nerve and skin biopsies have demonstrated the predicted underexpression of PMP22.16,18
Electrophysiologic findings in patients with HNPP are unique. Nerve conduction studies show accentuated slowing of motor distal latencies at sites susceptible for mechanical pressure, such as the median nerve at the wrist and the peroneal nerve at the ankle. In addition, focal slowing of conduction velocities is almost always seen at sites subject to compression, such as the ulnar nerve around the elbow and the peroneal nerve around the fibular head. In contrast, conduction velocities in other nerve segments are usually normal or only minimally slowed.44 In our experience, 80% of patients fulfilling these electrophysiologic features have positive findings in the evaluation of their DNA for HNPP deletion. Many of the remaining 20% of cases may be caused by another, yet to be discovered mutation.
Axonal CMT with Autosomal Dominant Inheritance
CMT2 represents one third of the known cases of autosomal dominant CMT. Similar to patients with CMT1, those with CMT2 present with distal weakness, atrophy, sensory loss, and foot deformities, but are more likely to maintain their deep tendon reflexes. It is impossible to distinguish CMT1 from CMT2 clinically. It is even more difficult to differentiate subtypes of CMT2 based solely on clinical presentation. Table 1 lists clinical features that are relatively prominent in each subtype of CMT2, but are not completely unique to any particular subtype.
Nerve conduction studies show reduced CMAP and SNAP amplitudes with normal or mildly slowed conduction velocities.45 Sensory or motor nerve responses can be nonresponsive due to the severe loss of axons in some patients. One way to circumvent this problem is to record nerve responses from proximal muscles in the forearm flexors.44 Sural nerve biopsy reveals axonal loss and regenerating clusters without evidence of demyelination (Fig. 1). However, these findings are insufficient to differentiate subtypes of CMT2.
CMT2 is heterogeneous. CMT2A is caused by mutations in the mitofusin-2 gene (MFN2) on chromosome 1p36.46 It is the most common form of CMT2 and accounts for 20% of patients.46 The encoded protein is a regulator for controlled mitochondrial fusion.47 A subset of patients with CMT2A develops optic nerve atrophy or subtle brain abnormalities on magnetic resonance images (MRIs). Although a severe early-onset phenotype is most frequent, a subgroup presents with a milder late-onset phenotype. CMT2B is a predominantly sensory axonal polyneuropathy due to mutations in the RAB7 gene on chromosome 3q13.48 This protein is known to regulate the maturation of intracellular endosomes. CMT2C is a rare disorder in which patients have paresis of vocal cords, prominent weakness, and skeletal deformities in addition to other characteristics of CMT2. The causal mutations have been recently identified in the transient receptor potential vanilloid-4 cation channel (TRPV4) gene on chromosome 12q24.48,49CMT2D is caused by mutations in the glycyl-tRNA synthetase (GARS) gene on chromosome 7p15.50 A majority of these patients present with a motor axonal polyneuropathy that is often regarded as a distal spinal muscular dystrophy (dSMA).51 Similar clinical phenotypes that predominantly involve motor nerves may be seen in patients with CMT2F (caused by mutations in heat shock protein-27)52 and CMT2L (caused by mutations in heat shock protein-22).53CMT2E is linked to mutations in the neurofilament light (NEFL) gene on chromosome 8p21.54 Although NEFL is expressed in axons, patients with CMT2E show significantly slowed conduction velocities that have been attributed to the reduced diameters of myelinated nerve axons. Patients with this mutation tend to develop severe and early-onset phenotypes.
CMT4 with Autosomal Recessive Inheritance
CMT4 represents autosomal recessively inherited neuropathies. CMT4 is severe. Many patients may have nonneural symptoms, such as cataracts and skeletal deformities. The key clinical features of each CMT4 subtype are listed in Table 1. CMT4 can occur in either demyelinating or axonal forms. CMT4B1 and CMT4B2 are caused by recessive mutations in the MTMR2 and MTMR13 genes, respectively. These genes encode phosphatases with specificity toward the 5′ phosphate of the inositol ring of hosphatidylinositol-3,5-diphosphate (PI3,5P2), thereby regulating the concentration of PI3,5P2. Patients present with severe early-onset sensory loss and weakness. Nerve biopsies show focally folded myelin sheaths.55–57
Interestingly, a related 5′ phosphatase, FIG4, is also involved in regulating the level of PI3,5P2. Its mutations are causal for CMT4J. Myelin folding is not prominent in these patients. Instead, loss of FIG4 function results in rapidly progressive asymmetric weakness and denervation. The patients do not complain of sensory symptoms yet sensory abnormalities are conspicuous by nerve conduction studies. Thus, clinically, this presentation resembles motor neuron disease.58,59 However, patients with early onset can present a phenotype like CMT1 with significantly slowed conduction velocities and de-/dysmyelination.60
Another severe form of recessive CMT, CMT4F, is caused by mutations in the periaxin (PRX) gene on chromosome 19. PRX, expressed in Schwann cells, encodes proteins that contain PDZ domains. Conduction velocities of patients with PRX mutations are markedly slowed. Onion bulbs are present in sural nerve biopsies.61
There are a few axonal forms of autosomal recessive CMT’s, including mutations in lamin A/C nuclear-envelope proteins (LMNA) and ganglioside induced differentiation-associated protein-1 (GDAP1) genes. These forms typically cause severe early-onset disability (Table 1). Giant axonal neuropathy (GAN) is a rare autosomal recessive disorder presenting in childhood that progresses to death by the end of the third decade. The genetic cause of the disease has been identified as the mutation of the gigaxonin gene.62 The name of the disorder comes from the characteristically large axons that result from the general disorganization of intermediate filaments. Many patients, though not all, may have kinky hair.
Hereditary Brachial Plexopathy
There are at least two types of genetically defined brachial plexopathies. First, a subset of patients with HNPP mutation may present with acute brachial plexopathy. The sensory and motor deficits are often reversible within a variable length of time. An important clinical feature of this condition is that patients usually do not experience pain during the episode. The other inherited brachial plexopathy is hereditary neuralgic amyotrophy (HNA), which is due to mutations in the SEPT9 gene in the chromosomal 17q25 region. This autosomal dominant disorder presents with episodes of pain, weakness, and sensory loss in the upper extremities. Almost invariably, weakness is preceded by pain in the affected arm. Recovery usually begins several weeks to months after symptom onset, although many patients are left with significant muscle atrophy. Attacks may subsequently occur in the same or opposite arm. Several dysmorphic features, including short stature, hypotelorism, epicanthal folds, and cleft palate that have been associated with HNA are not invariant. Nerve conduction studies have shown either axonal loss, demyelinating changes, or asymmetrically reduced CMAPs and SNAPs.63,64
Hereditary Sensory and Autonomic Neuropathy
Hereditary sensory and autonomic neuropathy (HSAN) is rare and is known to manifest primarily as a sensory neuropathy; autonomic abnormality is often minimal or absent. Motor deficits may also be present. Unlike CMT, patients with HSAN are often afflicted by repetitive limb injuries, presumably due to severe sensory loss. This can lead to ulcerations of the feet, osteomyelitis, and limb amputations.
Known subtypes of HSAN are listed in Table 2. HSAN1 is caused by mutations in the SPTLC1 gene and is one of the most extensively investigated subtypes of HSAN. Patients usually present with neuropathic pain and sensory loss in distal legs with a childhood onset. Interestingly, though autonomic abnormalities are usually absent, distal leg muscle weakness and atrophy can be conspicuous in these patients. Nerve conduction studies and sural nerve pathology in patients with HSAN1 may show mixed axonal and demyelinating features.65 Additional subtypes of HSAN are briefly described in Table 2. Typical autonomic involvement has been mainly found in HSAN-III and -IV, but not in other subtypes.
Table 2.
Classification of Hereditary Sensory and Autonomic Neuropathy (HSAN)
| Type | Gene | Prominent Phenotype |
|---|---|---|
| HSAN with autosomal dominant inheritance | ||
| HSAN-I | HPTLC1 or HPTLC2 or ATL1 | Neuropathic pain; loss of pain/temperature sensation ulcerative mutilations; ± distal muscle atrophy |
| HSAN-I with dementia | DNMT1 | Neuropathic pain; loss of all sensory modalities; ulcerative mutilations; dementia; hearing loss |
| HSAN with autosomal recessive inheritance | ||
| HSAN-II | WNK1 or FAM134B or KIF1A | Loss of sensory functions; mutilating ulcers |
| HSAN-III | IKBKAP | Congenital sensory loss; absence of tongue fungiform Papillae; hyperhidrosis |
| HSAN-IV | NTRK1 | Congenital sensory loss; anhidrosis; fever; skin lesions; joint deformities |
| HSAN-V | NGFB | Congenital sensory loss (pain); bone fracture; joint deformities |
Fabry disease is an X-linked multisystem disease. Its clinical presentation may simulate that of patients with HSAN. Fabry disease is a lysosomal storage disorder caused by mutations in the GLA gene on chromosome Xq22, which result in a deficiency of the enzyme α-galactosidase-A (αGalA). In addition to cerebrovascular accidents, angiokeratomas, and chronic renal and cardiac failure, patients may develop small fiber polyneuropathy with burning pain and dysautonomia (such as anhydrosis). When the neuropathy is a presenting problem, patients are often misdiagnosed. A diagnosis can be inferred from either the presence of typical inclusions in skin and/or nerve biopsies or a reduced activity of αGalA in blood tests. For a definitive diagnosis, a DNA test that reveals mutations in the GLA gene is necessary.66
Diagnosis
Three cases are provided to illustrate the diagnostic process.
Case 1. A 58-year-old man complained of fatigue and limb muscle weakness for decades. At the age of 8 years, he developed difficulty running and sprained his ankles many times. He also noticed his feet became highly arched. These symptoms were stable for decades until about one year earlier, when he started to experience muscle weakness. His job required him to drive and stand on his feet for long hours, which he is no longer able to do. He often tripped over himself and recently developed more numbness and tingling in his feet and legs. Multiple family members had similar symptoms, including his mother, two maternal aunts, one sister, two brothers, and one of his two sons. Muscle strength was reduced in ankle dorsal flexion, eversion, and finger abduction. Muscle atrophy was conspicuous in calf and hand muscles. His feet were highly arched with hammer toes. Sensation in his toes and ankles was decreased to light touch, pinprick, and vibration. Deep tendon reflex was absent in all tendons.
This patient has a typical CMT phenotype. His family history demonstrates male-to-male transmission, which is the most reliable indicator of autosomal dominant inheritance. The next step is performance of nerve conduction studies to characterize the patterns of the neuropathy and to determine if he has CMT1 versus CMT2. Nerve conduction studies revealed uniform slowing of conduction velocities (around 20–30 m/s).
In this situation, the first step is performance of a CMT1A duplication (PMP22 duplication) test. Based on the high prevalence of CMT1A (50% of all patients with CMT), this step provides a specific diagnosis in half of all patients. If this test were negative, a LITAF/SIMPLE gene test would be the next step because CMT1C patients with mutations in SIMPLE have a presentation indistinguishable to CMT1A (see Fig. 2 for a diagnostic algorithm).
Figure 2.
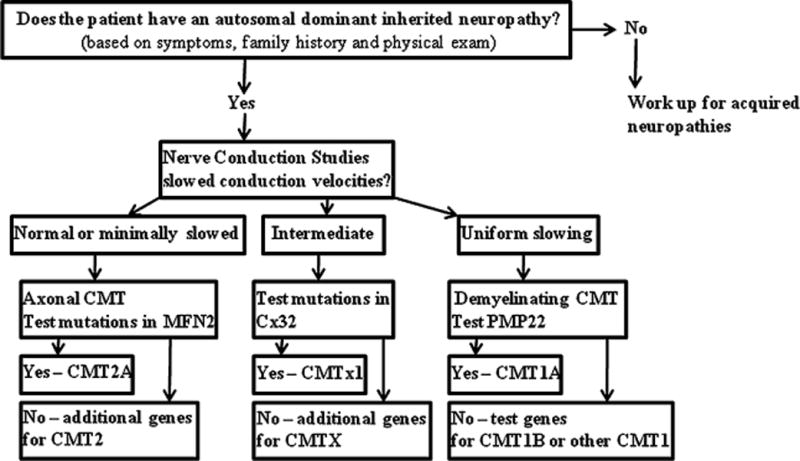
Diagnostic algorithm in patients with Charcot-Marie-Tooth disease.
Data from clinic interviews and electrodiagnostic testing should be carefully considered to guide genetic testing, which in turn provides a definitive diagnosis. Although there are batteries of DNA tests available commercially, they involve testing a large set of candidate genes and are very expensive. There are a few algorithms that have been published to guide the selection of the genetic tests.67–69 Our simplified approach allows one to reach a diagnosis in > 50% of patients with CMT.
Case 2. A 44-year-old woman developed foot drop and falling in her twenties. The weakness progressed to her hands in the past few years. Her hands were getting thinner. She was able to work full-time on her feet in retail with the use of ankle braces. She complained of foot numbness. Multiple family members had a similar problem including her paternal grandmother, father, a sister, and two of her sons. There was weakness and atrophy of foot and hand intrinsic muscles. Her feet appeared flat. Sensation was diminished to light touch, temperature, and vibration in her toes and ankles. Deep tendon reflexes were absent. Nerve conduction studies showed a nonuniform pattern and an intermediate range of conduction velocity slowing.
Electrophysiologic testing revealed intermediate and nonuniform conduction slowing. Her family history does not show male-to-male transmission and suggests a possible X-linked inheritance. The appropriate next step is DNA sequencing of the Cx32 gene, which showed a V95M point mutation, confirming the diagnosis of CMTX1. She is a symptomatic carrier, presumably due to an inactivation of the normal Cx32 allele. CMTX1 accounts for 10 to 15% CMT patients. Because of the nonuniform slowing, caution should be taken not to misdiagnose CMTX1 with acquired demyelinating neuropathies such as CIDP.
Case 3. This 34-year-old man, recently retired from the army, developed leg weakness over 3 months. He walked more on his heels and even tripped over himself a few times. His feet felt numb, which made it difficult to remain standing.
In retrospect, he was not able to stand on his toes for at least 14 years. He began to lose muscle bulk in his calf 2 to 3 years earlier. He had no upper-extremity symptoms. Multiple family membranes developed a similar condition, suggesting an autosomal dominant inheritance. Neurologic examination showed muscle weakness of ankle dorsal and plantar flexion. Sensation was almost absent to all modalities below the knee. Deep tendon reflexes were absent at the ankles, but normal elsewhere. Nerve conduction studies were consistent with an axonal polyneuropathy.
Given the axonal findings and autosomal dominant inheritance, he has CMT2. Because CMT2A is the most common form of CMT2A, accounting for 20% of all CMT2 patients, the most appropriate initial diagnostic test is a mitofusin-2 gene test, which showed a W740S point mutation, confirming CMT2A.
Together, these simple steps in each of the three cases would diagnose 70% of CMT patients. The remaining patients would have rare forms of CMT. Clinicians can either obtain guidance from published algorithms or seek guidance from physicians who specialize in CMT. New sequencing technology, such as exome and whole genome sequencing, will drastically accelerate the discovery of novel mutations.70
Management
Recent Advances in Treating Inherited Neuropathies
Although there are no disease-altering therapies currently available for CMT, there have been clinical trials. Due to the high prevalence of CMT1A, this disease was targeted first. As described above, ascorbic acid (vitamin C) was shown to suppress the expression of PMP22 both in vitro and in vivo in transgenic mice with overexpression of PMP22.22 These findings led to large clinical trials of ascorbic acid therapies conducted in Europe and the United States. Although the results from the U.S. clinical trial are still pending, the findings of the clinical trial in Europe have raised significant questions regarding the efficacy of ascorbic acid therapy. The clinical trial in Europe enrolled 277 CMT1A patients who were randomized into either an ascorbic acid arm (138 patients) or a placebo arm (133 patients).25 Treatment (1 g or 3 g per day) was well tolerated. The CMT neuropathy score was the primary outcome measure. There were no significant differences found between the two groups at 24 months of treatment. Although these results appear negative, several important lessons can be gleaned from the study. First, the overall disease progression of patients with CMT1A is slow. For example, there was an increase of only 0.5 CMT neuropathy score points over 12 months in the placebo group. This finding indicates that the primary outcome measurement is relatively insensitive for patients with CMT1A and should be improved. Second, a detailed analysis of ascorbic acid pharmacokinetics after large doses, including its absorption rate, is still lacking in human subjects. Finally, it is also unclear how well ascorbic acid is delivered to its molecular targets in myelinating Schwann cells.
The negative trials demand an alternative approach for treating CMT1A. With the advent of large-scale drug-screening facilities, additional therapeutic candidates are expected to emerge for future clinical trials. This approach requires the development of cell-lines expressing a PMP22 reporter, such as a fluorescence protein. Large libraries of candidate compounds (several thousands) are then placed in a robotic system which rapidly screens for drugs that can suppress the level of PMP22 in the cell-lines. Because increases in PMP22 levels are thought to cause CMT1A, newly identified PMP22 suppressing drugs can be selected for future clinical studies.
In the past several years, remarkable progress has been made in understanding the pathogenesis of HSAN1. The SPTLC1 gene encodes a serine palmitoyltransferase that catalyzes the initial step of de novo sphingolipid synthesis. HSAN1 mutations reduce the enzymatic activities and shift the enzymatic substrate preference from canonical L-serine to L-alanine or L-glycine. This leads to the excessive production of toxic intermediates called deoxysphingolipids. Thus, a therapeutic approach has been examined in HSAN1 mice and patients that supplements diets with L-serine. This treatment has significantly improved the phenotype of both mouse model and human subjects.71 This promising advancement is likely to translate into clinical practice after the therapy is successfully verified in a large controlled clinical trial in the near future.
Remarkable therapeutic progress recently has been made in Fabry disease. A treatment that replaces the missing lysosomal enzyme αGalA was approved by the Food and Drug Administration for clinical use in 2003. This treatment is generally well tolerated and safe. Because early initiation of therapy can prevent disease progression, early diagnosis is critical. There are two different preparations of ocGalA available (algasidase-α-Replagal, 0.2 mg/kg, Shire Human Genetic Therapies, Boston, MA; and ß-Fabrazyme, 1.0 mg/kg, Genzyme, Cambridge, MA). Both drugs are administered through infusions (40 min independently of body weight for algasidase-α and 15 mg/h for ß-Fabrazyme). The treatment decreases cardiac mass, improves clearance of lysosomal storage in the skin and kidney, and reduces anhydrosis and painful neuropathy.72
Symptomatic Management
Most CMT patients eventually require some form of physical or occupational therapy. Orthotics or ankle bracing is the cornerstone of foot care and if implemented properly, can help patients ambulate independently throughout their lives. In severely affected patients, surgical intervention may be needed to correct foot deformities. Occupational therapy can help with techniques to aid in buttoning, zippering, and other hand movements requiring dexterity.
HNPP exhibits susceptibility to mechanical stress. Nerve dysfunction can be provoked by compression, repetitive use of the affected limbs or stretching. Extraneous physical activities can even induce limb paralysis. These patients should be advised to take precaution in activities that might stress the peripheral nerves.
Pain is rarely an initial symptom; however, most patients suffer from pain during the course of their disease. Pain often arises in the joints or ligaments and neuropathic pain is uncommon (unlike HSAN where it is common). Nonsteroidal antiinflammatory medications can be partially helpful. In addition, the pain may respond to strengthening weakened ankles, correcting foot deformities, and/or using custom-made shoes to restore the balance of gait.
Diseased peripheral nerves in patients with CMT appear particularly sensitive to the side effects of medications. In general, medications that have clear neurotoxic effects, such as vincristine and cisplatin, should be avoided if medically possible because they are likely to exacerbate the existing neuropathy. There have been reports of severe weakness in CMT patients given vincristine.73 For other medications, the situation is less clear. The Charcot-Marie-Tooth Association (CMTA) publishes a list of medications that may exacerbate CMT at the CMTA website. The degree of risk varies with the individual medication. Good judgment by the physician as to the risk/benefit ratio of a given medication can probably serve as a useful guide for the use of these medicines.
Genetic counseling is an important part in the management of patients. Many patients are not well informed about the complicated genetics underlying CMT. There may be concerns about who is at risk in the family and what options are available to parents.
Summary
With advances in genetics and molecular biology, the clinical spectrum of inherited neuropathies has drastically expanded. After the next generation of rapid whole-genome exon sequencing becomes available, the discovery of new mutations causal for CMT is expected to accelerate dramatically. Thus far, there are at least 36 genes known to cause inherited neuropathies and more than 53 distinct loci have been implicated in these diseases. Genetic testing has significantly facilitated the accurate diagnosis of patients with CMT. This large repertoire of human CMT mutations behaves like “a natural library of genetic models.” It will undoubtedly teach us how mutated proteins cause demyelination, axonal degeneration, and alterations of Schwann cell-axonal signaling, which should in turn advance our understanding of the basis of many neurologic diseases. Moreover, increased understanding of the molecular mechanisms underlying these pathways will provide targets for future therapeutic intervention.
Acknowledgments
This material was supported in part by grants from the NIH (R01NS066927-01) and MDA115087.
References
- 1.Dyck PJ, Lambert EH. Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch Neurol. 1968;18(6):603–618. doi: 10.1001/archneur.1968.00470360025002. [DOI] [PubMed] [Google Scholar]
- 2.Thomas PK, Marques W, Jr, Davis MB, et al. The phenotypic manifestations of chromosome 17p11.2 duplication. Brain. 1997;120(Pt 3):465–478. doi: 10.1093/brain/120.3.465. [DOI] [PubMed] [Google Scholar]
- 3.Krajewski KM, Lewis RA, Fuerst DR, et al. Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A. Brain. 2000;123(Pt 7):1516–1527. doi: 10.1093/brain/123.7.1516. [DOI] [PubMed] [Google Scholar]
- 4.Roussy G, Levy G. Sept cas d’une maladie familiale particuliere. Rev Neurol. 1926;45:427–450. [Google Scholar]
- 5.Shy ME, Jáni A, Krajewski K, et al. Phenotypic clustering in MPZ mutations. Brain. 2004;127(Pt 2):371–384. doi: 10.1093/brain/awh048. [DOI] [PubMed] [Google Scholar]
- 6.Li J, Bai YH, Ianakova E, et al. Major myelin protein gene (P0) mutation causes a novel form of axonal degeneration. J Comp Neurol. 2006;498(2):252–265. doi: 10.1002/cne.21051. [DOI] [PubMed] [Google Scholar]
- 7.Bai YH, Ianokova E, Pu Q, et al. R69C Mutation in P0 gene alters myelination and ion channel subtypes. Arch Neurol. 2006;63:1787–1794. doi: 10.1001/archneur.63.12.1787. [DOI] [PubMed] [Google Scholar]
- 8.Street VA, Bennett CL, Goldy JD, et al. Mutation of a putative protein degradation gene LITAF/SIMPLE in Charcot-Marie-Tooth disease 1C. Neurology. 2003;60(1):22–26. doi: 10.1212/wnl.60.1.22. [DOI] [PubMed] [Google Scholar]
- 9.Lupski JR, de Oca-Luna RM, Slaugenhaupt S, et al. DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell. 1991;66(2):219–232. doi: 10.1016/0092-8674(91)90613-4. [DOI] [PubMed] [Google Scholar]
- 10.Raeymaekers P, Timmerman V, Nelis E, et al. HMSN Collaborative Research Group. Estimation of the size of the chromosome 17p11.2 duplication in Charcot-Marie-Tooth neuropathy type 1a (CMT1a) J Med Genet. 1992;29(1):5–11. doi: 10.1136/jmg.29.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chance PF, Alderson MK, Leppig KA, et al. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell. 1993;72(1):143–151. doi: 10.1016/0092-8674(93)90058-x. [DOI] [PubMed] [Google Scholar]
- 12.Parmantier E, Cabon F, Braun C, D’Urso D, Müller HW, Zalc B. Peripheral myelin protein-22 is expressed in rat and mouse brain and spinal cord motoneurons. Eur J Neurosci. 1995;7(5):1080–1088. doi: 10.1111/j.1460-9568.1995.tb01095.x. [DOI] [PubMed] [Google Scholar]
- 13.Parmantier E, Braun C, Thomas JL, Peyron F, Martinez S, Zalc B. PMP-22 expression in the central nervous system of the embryonic mouse defines potential transverse segments and longitudinal columns. J Comp Neurol. 1997;378(2):159–172. [PubMed] [Google Scholar]
- 14.Snipes GJ, Suter U, Welcher AA, Shooter EM. Characterization of a novel peripheral nervous system myelin protein (PMP-22/SR13) J Cell Biol. 1992;117(1):225–238. doi: 10.1083/jcb.117.1.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gouider R, LeGuern E, Gugenheim M, et al. Clinical, electrophysiologic, and molecular correlations in 13 families with hereditary neuropathy with liability to pressure palsies and a chromosome 17p11.2 deletion. Neurology. 1995;45(11):2018–2023. doi: 10.1212/wnl.45.11.2018. [DOI] [PubMed] [Google Scholar]
- 16.Vallat JM, Sindou P, Preux PM, et al. Ultrastructural PMP22 expression in inherited demyelinating neuropathies. Ann Neurol. 1996;39(6):813–817. doi: 10.1002/ana.410390621. [DOI] [PubMed] [Google Scholar]
- 17.Li J, Bai Y, Ghandour K, et al. Skin biopsies in myelin-related neuropathies: bringing molecular pathology to the bedside. Brain. 2005;128(Pt 5):1168–1177. doi: 10.1093/brain/awh483. [DOI] [PubMed] [Google Scholar]
- 18.Katona I, Wu X, Feely SM, et al. PMP22 expression in dermal nerve myelin from patients with CMT1A. Brain. 2009;132(Pt 7):1734–1740. doi: 10.1093/brain/awp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nicholson GA, Valentijn LJ, Cherryson AK, et al. A frame shift mutation in the PMP22 gene in hereditary neuropathy with liability to pressure palsies. Nat Genet. 1994;6(3):263–266. doi: 10.1038/ng0394-263. [DOI] [PubMed] [Google Scholar]
- 20.Li J, Ghandour K, Radovanovic D, et al. Stoichiometric alteration of PMP22 protein determines the phenotype of HNPP. Arch Neurol. 2007;64:974–978. doi: 10.1001/archneur.64.7.974. [DOI] [PubMed] [Google Scholar]
- 21.Sabéran-Djoneidi D, Sanguedolce V, Assouline Z, Lévy N, Passage E, Fontés M. Molecular dissection of the Schwann cell specific promoter of the PMP22 gene. Gene. 2000;248(1–2):223–231. doi: 10.1016/s0378-1119(00)00116-5. [DOI] [PubMed] [Google Scholar]
- 22.Passage E, Norreel JC, Noack-Fraissignes P, et al. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med. 2004;10(4):396–401. doi: 10.1038/nm1023. [DOI] [PubMed] [Google Scholar]
- 23.Sereda MW, Meyer zu Hörste G, Suter U, Uzma N, Nave KA. Therapeutic administration of progesterone antagonist in a model of Charcot-Marie-Tooth disease (CMT-1A) Nat Med. 2003;9(12):1533–1537. doi: 10.1038/nm957. [DOI] [PubMed] [Google Scholar]
- 24.Pareyson D, Schenone A, Fabrizi GM, et al. CMT-TRIAAL Group A multicenter, randomized, double-blind, placebo-controlled trial of long-term ascorbic acid treatment in Charcot-Marie-Tooth disease type 1A (CMT-TRIAAL): the study protocol [EudraCT no.: 2006-000032-27]. [EudraCT no.: 2006-000032-27] Pharmacol Res. 2006;54(6):436–441. doi: 10.1016/j.phrs.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Pareyson D, Reilly MM, Schenone A, et al. CMT-TRIAAL; CMT-TRAUK groups Ascorbic acid in Charcot-Marie-Tooth disease type 1A (CMT-TRIAAL and CMT-TRAUK): a double-blind randomised trial. Lancet Neurol. 2011;10(4):320–328. doi: 10.1016/S1474-4422(11)70025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hayasaka K, Takada G, Ionasescu VV. Mutation of the myelin P0 gene in Charcot-Marie-Tooth neuropathy type 1B. Hum Mol Genet. 1993;2(9):1369–1372. doi: 10.1093/hmg/2.9.1369. [DOI] [PubMed] [Google Scholar]
- 27.Martini R, Mohajeri MH, Kasper S, Giese KP, Schachner M. Mice doubly deficient in the genes for P0 and myelin basic protein show that both proteins contribute to the formation of the major dense line in peripheral nerve myelin. J Neurosci. 1995;15(6):4488–4495. doi: 10.1523/JNEUROSCI.15-06-04488.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pennuto M, Tinelli E, Malaguti M, et al. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron. 2008;57(3):393–405. doi: 10.1016/j.neuron.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Warner LE, Mancias P, Butler IJ, et al. Mutations in the early growth response 2 (EGR2) gene are associated with hereditary myelinopathies. Nat Genet. 1998;18(4):382–384. doi: 10.1038/ng0498-382. [DOI] [PubMed] [Google Scholar]
- 30.Lewis RA, Sumner AJ. The electrodiagnostic distinctions between chronic familial and acquired demyelinative neuropathies. Neurology. 1982;32(6):592–596. doi: 10.1212/wnl.32.6.592. [DOI] [PubMed] [Google Scholar]
- 31.Gutierrez A, England JD, Sumner AJ, et al. Unusual electrophysiological findings in X-linked dominant Charcot-Marie-Tooth disease. Muscle Nerve. 2000;23(2):182–188. doi: 10.1002/(sici)1097-4598(200002)23:2<182::aid-mus6>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 32.Gabreëls-Festen A, Wetering RV. Human nerve pathology caused by different mutational mechanisms of the PMP22 gene. Ann N Y Acad Sci. 1999;883:336–343. [PubMed] [Google Scholar]
- 33.Krajewski K, Turansky C, Lewis R, et al. Correlation between weakness and axonal loss in patients with CMT1A. Ann N Y Acad Sci. 1999;883:490–492. [PubMed] [Google Scholar]
- 34.Bergoffen J, Scherer SS, Wang S, et al. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262(5142):2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- 35.Reilly MM, Shy ME. Diagnosis and new treatments in genetic neuropathies. J Neurol Neurosurg Psychiatry. 2009;80(12):1304–1314. doi: 10.1136/jnnp.2008.158295. [DOI] [PubMed] [Google Scholar]
- 36.Balice-Gordon RJ, Bone LJ, Scherer SS. Functional gap junctions in the Schwann cell myelin sheath. J Cell Biol. 1998;142(4):1095–1104. doi: 10.1083/jcb.142.4.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tabaraud F, Lagrange E, Sindou P, Vandenberghe A, Levy N, Vallat JM. Demyelinating X-linked Charcot-Marie-Tooth disease: unusual electrophysiological findings. Muscle Nerve. 1999;22(10):1442–1447. doi: 10.1002/(sici)1097-4598(199910)22:10<1442::aid-mus16>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 38.Hahn AF, Ainsworth PJ, Bolton CF, Bilbao JM, Vallat JM. Pathological findings in the X-linked form of Charcot-Marie-Tooth disease: a morphometric and ultrastructural analysis. Acta Neuropathol. 2001;101(2):129–139. doi: 10.1007/s004010000275. [DOI] [PubMed] [Google Scholar]
- 39.Saporta MA, Katona I, Zhang X, et al. Neuropathy in a human without the PMP22 gene. Arch Neurol. 2011;68(6):814–821. doi: 10.1001/archneurol.2011.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Krajewski K, Lewis RA, Shy ME. Loss-of-function phenotype of hereditary neuropathy with liability to pressure palsies. Muscle Nerve. 2004;29(2):205–210. doi: 10.1002/mus.10521. [DOI] [PubMed] [Google Scholar]
- 41.Horowitz SH, Spollen LE, Yu W. Hereditary neuropathy with liability to pressure palsy: fulminant development with axonal loss during military training. J Neurol Neurosurg Psychiatry. 2004;75(11):1629–1631. doi: 10.1136/jnnp.2003.029314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Madrid R, Bradley G. The pathology of neuropathies with focal thickening of the myelin sheath (tomaculous neuropathy): studies on the formation of the abnormal myelin sheath. J Neurol Sci. 1975;25:415–448. [Google Scholar]
- 43.Bai Y, Zhang X, Katona I, et al. Conduction block in PMP22 deficiency. J Neurosci. 2010;30(2):600–608. doi: 10.1523/JNEUROSCI.4264-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li J, Krajewski K, Shy ME, Lewis RA. Hereditary neuropathy with liability to pressure palsy: the electrophysiology fits the name. Neurology. 2002;58(12):1769–1773. doi: 10.1212/wnl.58.12.1769. [DOI] [PubMed] [Google Scholar]
- 45.Lewis RA, Sumner AJ, Shy ME. Electrophysiological features of inherited demyelinating neuropathies: A reappraisal in the era of molecular diagnosis. Muscle Nerve. 2000;23(10):1472–1487. doi: 10.1002/1097-4598(200010)23:10<1472::aid-mus3>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 46.Züchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet. 2004;36(5):449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 47.Chen H, Chan DC. Critical dependence of neurons on mitochondrial dynamics. Curr Opin Cell Biol. 2006;18(4):453–459. doi: 10.1016/j.ceb.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 48.Deng HX, Klein CJ, Yan J, et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat Genet. 2010;42(2):165–169. doi: 10.1038/ng.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Landouré G, Zdebik AA, Martinez TL, et al. Mutations in TRPV4 cause Charcot-Marie-Tooth disease type 2C. Nat Genet. 2010;42(2):170–174. doi: 10.1038/ng.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Antonellis A, Ellsworth RE, Sambuughin N, et al. Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am J Hum Genet. 2003;72(5):1293–1299. doi: 10.1086/375039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sivakumar K, Kyriakides T, Puls I, et al. Phenotypic spectrum of disorders associated with glycyl-tRNA synthetase mutations. Brain. 2005;128(Pt 10):2304–2314. doi: 10.1093/brain/awh590. [DOI] [PubMed] [Google Scholar]
- 52.Evgrafov OV, Mersiyanova I, Irobi J, et al. Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat Genet. 2004;36(6):602–606. doi: 10.1038/ng1354. [DOI] [PubMed] [Google Scholar]
- 53.Irobi J, Van Impe K, Seeman P, et al. Hot-spot residue in small heat-shock protein 22 causes distal motor neuropathy. Nat Genet. 2004;36(6):597–601. doi: 10.1038/ng1328. [DOI] [PubMed] [Google Scholar]
- 54.Mersiyanova IV, Ismailov SM, Polyakov AV, et al. Screening for mutations in the peripheral myelin genes PMP22, MPZ and Cx32 (GJB1) in Russian Charcot-Marie-Tooth neuropathy patients. Hum Mutat. 2000;15(4):340–347. doi: 10.1002/(SICI)1098-1004(200004)15:4<340::AID-HUMU6>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 55.Bolis A, Zordan P, Coviello S, Bolino A. Myotubularin-related (MTMR) phospholipid phosphatase proteins in the peripheral nervous system. Mol Neurobiol. 2007;35(3):308–316. doi: 10.1007/s12035-007-0031-0. [DOI] [PubMed] [Google Scholar]
- 56.Bolino A, Bolis A, Previtali SC, et al. Disruption of Mtmr2 produces CMT4B1-like neuropathy with myelin outfolding and impaired spermatogenesis. J Cell Biol. 2004;167(4):711–721. doi: 10.1083/jcb.200407010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bolis A, Coviello S, Bussini S, et al. Loss of Mtmr2 phosphatase in Schwann cells but not in motor neurons causes Charcot-Marie-Tooth type 4B1 neuropathy with myelin outfoldings. J Neurosci. 2005;25(37):8567–8577. doi: 10.1523/JNEUROSCI.2493-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chow CY, Zhang Y, Dowling JJ, et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448(7149):68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang X, Chow CY, Sahenk Z, Shy ME, Meisler MH, Li J. Mutation of FIG4 causes a rapidly progressive, asymmetric neuronal degeneration. Brain. 2008;131(Pt 8):1990–2001. doi: 10.1093/brain/awn114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nicholson G, Lenk GM, Reddel SW, et al. Distinctive genetic and clinical features of CMT4J: a severe neuropathy caused by mutations in the PI(3,5)P phosphatase FIG4. Brain. 2011;134(Pt 7):1959–1971. doi: 10.1093/brain/awr148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boerkoel CF, Takashima H, Stankiewicz P, et al. Periaxin mutations cause recessive Dejerine-Sottas neuropathy. Am J Hum Genet. 2001;68(2):325–333. doi: 10.1086/318208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bomont P, Cavalier L, Blondeau F, et al. The gene encoding giga-xonin, a new member of the cytoskeletal BTB/kelch repeat family, is mutated in giant axonal neuropathy. Nat Genet. 2000;26(3):370–374. doi: 10.1038/81701. [DOI] [PubMed] [Google Scholar]
- 63.Chance PF, Lensch MW, Lipe H, Brown RH, Sr, Brown RH, Jr, Bird TD. Hereditary neuralgic amyotrophy and hereditary neuropathy with liability to pressure palsies: two distinct genetic disorders. Neurology. 1994;44(12):2253–2257. doi: 10.1212/wnl.44.12.2253. [DOI] [PubMed] [Google Scholar]
- 64.Kuhlenbäumer G, Hannibal MC, Nelis E, et al. Mutations in SEPT9 cause hereditary neuralgic amyotrophy. Nat Genet. 2005;37(10):1044–1046. doi: 10.1038/ng1649. [DOI] [PubMed] [Google Scholar]
- 65.Houlden H, King R, Blake J, et al. Clinical, pathological and genetic characterization of hereditary sensory and autonomic neuropathy type 1 (HSAN I) Brain. 2006;129(Pt 2):411–425. doi: 10.1093/brain/awh712. [DOI] [PubMed] [Google Scholar]
- 66.Toyooka K. Fabry disease. Curr Opin Neurol. 2011;24(5):463–468. doi: 10.1097/WCO.0b013e32834a9433. [DOI] [PubMed] [Google Scholar]
- 67.England JD, Gronseth GS, Franklin G, et al. American Academy of Neurology Practice parameter: evaluation of distal symmetric polyneuropathy: role of laboratory and genetic testing (an evidence-based review). Report of the American Academy of Neurology, American Association of Neuromuscular and Electrodiagnostic Medicine, and American Academy of Physical Medicine and Rehabilitation. Neurology. 2009;72(2):185–192. doi: 10.1212/01.wnl.0000336370.51010.a1. [DOI] [PubMed] [Google Scholar]
- 68.Patzkó A, Shy ME. Update on Charcot-Marie-Tooth disease. Curr Neurol Neurosci Rep. 2011;11(1):78–88. doi: 10.1007/s11910-010-0158-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reilly MM, Shy ME. Diagnosis and new treatments in genetic neuropathies. J Neurol Neurosurg Psychiatry. 2009;80(12):1304–1314. doi: 10.1136/jnnp.2008.158295. [DOI] [PubMed] [Google Scholar]
- 70.Lupski JR, Reid JG, Gonzaga-Jauregui C, et al. Whole-genome sequencing in a patient with Charcot-Marie-Tooth neuropathy. N Engl J Med. 2010;362(13):1181–1191. doi: 10.1056/NEJMoa0908094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garofalo K, Penno A, Schmidt BP, et al. Oral L-serine supplementation reduces production of neurotoxic deoxysphingolipids in mice and humans with hereditary sensory autonomic neuropathy type 1. J Clin Invest. 2011;121(12):4735–4745. doi: 10.1172/JCI57549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schaefer RM, Tylki-Szymańska A, Hilz MJ. Enzyme replacement therapy for Fabry disease: a systematic review of available evidence. Drugs. 2009;69(16):2179–2205. doi: 10.2165/11318300-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 73.Graf WD, Chance PF, Lensch MW, Eng LJ, Lipe HP, Bird TD. Severe vincristine neuropathy in Charcot-Marie-Tooth disease type 1A. Cancer. 1996;77(7):1356–1362. doi: 10.1002/(SICI)1097-0142(19960401)77:7<1356::AID-CNCR20>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 74.Verhoeven K, De Jonghe P, Coen K, et al. Mutations in the small GTP-ase late endosomal protein RAB7 cause Charcot-Marie-Tooth type 2B neuropathy. Am J Hum Genet. 2003;72(3):722–727. doi: 10.1086/367847. [DOI] [PMC free article] [PubMed] [Google Scholar]