

Abstract
Glucocorticoids are essential for maintaining homeostasis and regulate a wide variety of physiological processes. Therapeutically, synthetic glucocorticoids are widely prescribed for the treatment of inflammation, autoimmune disorders, and malignancies of lymphoid origin. In this review we examine emerging evidence highlighting both proinflammatory and anti-inflammatory actions of glucocorticoids on both the innate and adaptive immune systems. We incorporate these findings into the more traditional anti-inflammatory role attributed to glucocorticoids, and propose how the two seemingly disparate processes seamlessly work together to resolve cellular responses to inflammatory stimuli. These ideas provide a framework by which glucocorticoids ready and reinforce the innate immune system, and repress the adaptive immune system, to help to resolve inflammation and restore homeostasis.
Inflammation and the glucocorticoids
Inflammation is a physiological response to the detection of a foreign antigen or pathogen. Increases in the release or expression of cytokines, chemokines, adhesion molecules, receptors, and enzymes are critical steps for both vascular changes and leukocyte infiltration that occur in response to inflammatory stimuli [1]. Although initially beneficial, an unrestrained or chronic inflammatory condition can also be detrimental, often requiring pharmacological intervention [2]. It was recognized in the 1940s that glucocorticoids have potent anti-inflammatory properties and, as such, both natural and synthetic glucocorticoids have become one of the most prescribed classes of anti-inflammatory medications worldwide [3].
Under normal physiological conditions, glucocorticoids act on nearly every tissue in the body and are important regulators of carbohydrate, fat, and protein metabolism. In addition, glucocorticoids impact upon the cardiovascular, immune, reproductive, and central nervous systems [4], and are critical for lung development [5]. Glucocorticoids exert their anti-inflammatory actions via a complex interplay between glucocorticoid receptor (GR)-mediated transcriptional regulation and signal transduction within target tissues [6]. A major focus of recent studies on the anti-inflammatory actions of GR and glucocorticoids has centered on their ability to tether to and inhibit the activity of transcription factors, but without physically binding to DNA, a process known as ‘transrepression’ (see Glossary). Here we review what is known about the anti-inflammatory actions of glucocorticoids and highlight how recent discoveries have provided evidence for additional unrecognized proinflammatory actions of these steroids. Readers are referred to recent publications for a more comprehensive review of the anti- [6-8] and proinflammatory effects of glucocorticoids in the central nervous system [9,10].
Glucocorticoids ready the innate immune system and repress adaptive immunity
In mammals, the immune system is divided into two parts: the innate and the adaptive immune systems. The innate immune system is critical for the initial immune response upon infection or tissue damage. Through invariant pattern-recognition receptors (PRRs), the innate immune system is activated immediately upon detection of evolutionarily conserved structures known as pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) [11]. By contrast, the adaptive immune system serves as a second line of defense, relying on the expansion of antigen-specific T and B cells that effectively neutralize and remove specific pathogens and help to form immunological memory [12]. There is considerable crosstalk between the innate and adaptive immune system that helps to shape the nature and duration of the inflammatory response [13].
Owing to their lipophilic nature, glucocorticoids diffuse freely across the plasma membrane and exert their effects through activation of GR, a member of the nuclear receptor (NR) superfamily of ligand-dependent transcription factors [14]. Glucocorticoids, by virtue of almost ubiquitous GR expression, can affect nearly every cell of the immune system, depending on differentiation or state of activation [8,15]. Glucocorticoids target specific cell populations to combat hyperactivation of the immune system or systemic infections, at both the transcriptional and cellular level (Box 1). For example, by blocking the expression of cyclooxygenase-2 (Cox-2), glucocorticoids target T cells to control hyperactivation in response to excessive T cell receptor (TCR) binding or superantigen [16]. Glucocorticoid-induced apoptosis of T cells reduces inflammation associated with experimental autoimmune encephalomyelitis [16,17]. By contrast, glucocorticoids target macrophages to ensure survival in response to lipopolysaccharide (LPS)-induced sepsis, or to suppress inflammation associated with contact allergy [18,19]. Collectively, these actions are in line with the classical notion that glucocorticoids are able to repress and resolve inflammatory conditions, ultimately restoring homeostasis.
Box 1.
Glucocorticoid biology, the basics
Glucocorticoid receptor (GR) structure: structurally, GR consists of three modular domains: an N-terminal transactivation domain (NTD), a central DNA-binding domain (DBD), and a C-terminal ligand-binding domain (LBD) [71]. The NTD interacts with cofactors and components of the basal transcriptional machinery via a transcriptional activation function (AF1) exposed upon ligand binding [71]. The DBD, which shares the highest degree of sequence identity with other members of the nuclear receptor (NR) superfamily, contains two zinc-finger motifs responsible for binding DNA at glucocorticoid response elements (GREs) [71]. In addition, the second zinc finger of the DBD contains residues that constitute the dimerization or D-loop and that aids in receptor dimerization following activation [71]. Following a short hinge region, the 12 α-helices and four β-sheets of the LBD form a hydrophobic pocket that allows high-affinity binding of glucocorticoids [72]. This region also contains residues that contribute to receptor dimerization [72] and a second activation function domain (AF2) that interacts with various coregulators in a ligand-dependent manner.
Glucocorticoid signaling: in the absence of ligand, GR is primarily located in the cytoplasm as part of a large multiprotein complex [73]. Upon activation, GR is actively transported into the nucleus in an importin-dependent manner [73,74]. It was generally thought that a conformational change triggered in GR upon glucocorticoid binding resulted in dissociation of GR from the complex, exposure of nuclear localization sequences, and import into the nucleus [75]. However, emerging evidence suggests that components of the chaperone complex are also needed for efficient nuclear translocation [74]. Once in the nucleus, GR homodimers bind to GREs on target genes and stimulate transcription. However, not all GR–DNA interactions induce gene expression. In fact, whole-genome microarray analysis in vivo and in vitro has shown that ~50% of the glucocorticoid-regulated genes are repressed [66,67], partly due to GR interacting with negative GREs to suppress gene activation [76]. Further fine-tuning of the GR response is accomplished through structural changes induced by the ligand and the GRE sequence itself [77,78], which help to coordinate the recruitment of coregulators and chromatin-remodeling complexes that influence Pol II-dependent transcription [4].
Interestingly, not all glucocorticoid-regulated genes contain GREs [79] because GR is also able to positively or negatively influence gene expression by physically interacting with other transcription factors, in other words through protein–protein interactions, a mechanism known as tethering [4]. For other transcription factors there is a composite regulation where GR needs to bind both to a GRE and to the transcription factor bound at an adjacent promoter site to effect gene transcription [4].
GR signaling can be directly attributed to the genomic effects described above. However, nongenomic (i.e., occurring independently of gene transcription) effects also exist. These effects are thought to occur via either nonspecific interaction of glucocorticoids with membrane components or perhaps through membrane-bound GR [80,81]. Additionally, GR is able to interact with and alter the activity of kinases such as JNK [82], Src [83], ERK [84,85], and PI3K [86,87], affecting signaling pathways independently of gene transcription [40]. Although the mechanisms and physiological outcomes of nongenomic glucocorticoid signaling are not well-defined, these responses could have important roles in the overall actions of glucocorticoids.
Although glucocorticoids are clearly anti-inflammatory in situations of ongoing inflammation, their role in the normal physiology of the immune system is less understood. The circadian and ultradian changes in the circulating levels of glucocorticoids lead to a dynamic pattern of chromatin occupancy by GR and transcriptional ‘bursts’ that are lost with chronic hormone treatment or upon administration of synthetic hormone [20]. How this changes the transcriptional output of GR is only now beginning to be understood. However, it stands to reason that, under basal conditions, glucocorticoids may well be protective by ensuring the immune system is ready to respond to pathogens. This appears to be the case, at least with respect to the innate immune system where glucocorticoids are not strictly immunosuppressive [21].
Toll-like receptors (TLR1–10) are well-known PRRs that play a critical role in the detection and subsequent reaction to PAMPs. Activation of TLRs induces an intracellular signaling cascade that culminates in the activation of the AP-1, NF-κB, and IRF family of transcription factors. In general, glucocorticoids act to suppress TLR-mediated signaling through the induction of endogenous inhibitors (e.g., MKP-1 and GILZ) or through inhibition of AP-1, NF-κB, and IRF [22]. Interestingly, glucocorticoids induce the expression of TLR2, which is enhanced by the presence of proinflammatory cytokines (e.g., TNF-α or IL-1β) or Haemophilus influenzae [22]. Based on tissue-specific increases in TLR2, glucocorticoids can be viewed as being both pro- and anti-inflammatory and, as such, are crucial for the initiation and resolution of the inflammatory response. The increase in TLR2 on epithelial cells enhances the secretion of IL-6 and IL-8 [22] (Figure 1a), cytokines critical for induction of the acute-phase response and chemotaxis of a variety of cells, respectively. By contrast, the induction of TLR2 (and TLR4) within the adrenal gland is directly involved in the release of cortisol and corticosterone [22] (Box 2), providing a positive feedback loop for the resolution of the inflammatory process. Therefore, the balance between pro- and anti-inflammatory effects of the glucocorticoids in response to bacterial infection would be predicted to depend on the phase of the response in which the glucocorticoids are introduced.
Figure 1.
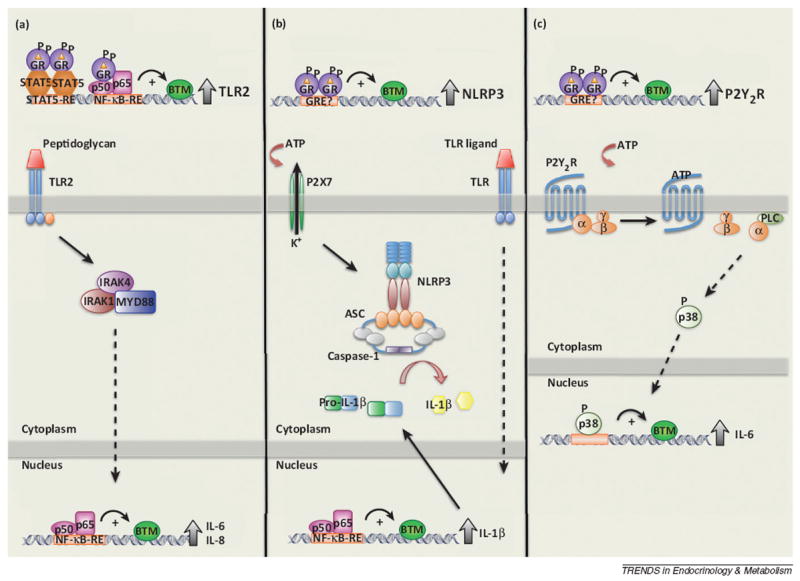
Glucocorticoids ready the innate immune system. Glucocorticoids induce the expression of proteins involved in responding to the detection of microbial products and cellular damage. (a) Proposed mechanism of Toll-like receptor (TLR)-2 induction. Glucocorticoids are able to induce the expression of TLR2 alone, or by acting synergistically with STAT5 and NF-κB activated by interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α). This synergistic induction of TLR2 enhances interleukin-6 (IL-6) and IL-8 production induced by peptidoglycan, a TLR2 agonist. (b,c) Glucocorticoids enhance the ability of cells to respond to cellular damage through induction of NLRP3 and P2Y2R, respectively, via an unknown mechanism. (b) The induction of the intracellular pattern-recognition receptor, NLRP3, sensitizes macrophages to extracellular ATP, a known danger signal, enhancing the secretion of IL-1β in the presence of TLR activation. (c) Glucocorticoids induce the expression of the purinergic receptor, P2Y2R. P2Y2R, a G-protein-coupled receptor, is activated by extracellular ATP, and glucocorticoids enhance the ATP-dependent activation p38, enhancing the expression and secretion of IL-6.
Box 2.
The hypothalamic–pituitary–adrenal axis and glucocorticoids
To maintain homeostasis, organisms have developed an exquisite means to cope with various stressors. Key to this is the activation of the hypothalamic–pituitary–adrenal (HPA) axis, culminating in the synthesis and secretion of cortisol (in humans) or corticosterone (in rodents) [88]. Once secreted, the majority of circulating cortisol (~90%) is bound to corticosteroid-binding globulin (CBG). Free or loosely bound cortisol (~10%) diffuses across cellular membranes and exerts the biological effects of glucocorticoids. However, there is evidence that CBG-bound cortisol provides a means for delivery to particular microenvironments [89]. In healthy individuals, the synthesis and secretion of cortisol displays a circadian rhythm following the pulsatile changes in ACTH, totaling ~10 mg/day, and can increase ~10-fold in response to stress [90]. Importantly, several proinflammatory cytokines are known to activate the HPA axis, resulting in the secretion of glucocorticoids [91].
The bioavailability of cortisol is regulated by the 11β-hydroxysteroid dehydrogenase (11β-HSD) enzymes in a process known as ‘prereceptor ligand metabolism’. The conversion of cortisol to cortisone, the inactive metabolite, is catalyzed by 11β-HSD type 2 (11β-HSD2). By contrast, 11b-HSD type 1 (11β-HSD1) catalyzes the reverse reaction, converting cortisone to cortisol [92]. These enzymes are differentially expressed in vivo, with 11β-HSD2 expression being highest in the kidney to prevent cortisol from binding to the mineralocorticoid receptor, which has a higher affinity for cortisol than does the glucocorticoid receptor (GR) [90,93]. By contrast, 11β-HSD1 expression is highest in the glucocorticoid-responsive tissues (i.e., liver, adipose, and muscle) to ensure intracellular cortisol bioavailability [94].
The NOD-like receptors (NLR), an intracellular family of PRRs, respond to both PAMPs and DAMPs. Of the 22 members, only three (NLRP1, NLRP3, NLRC4) are able to form the central component of the inflammasome, which is responsible for the maturation of IL-1β from its pro- to mature form [23]. It was recently demonstrated that glucocorticoids positively regulate the expression of NLRP3 in both cultured and primary macrophages [24]. The NLRP3 inflammasome is activated by a wide variety of molecules, including PAMPs, DAMPS, and particulate matter (e.g., aluminum). The glucocorticoid-dependent increase in NLRP3 expression sensitized macrophages to extracellular ATP, a DAMP commonly released following cellular damage or necrosis (Figure 1b). In addition to sensitizing cells to respond to lower concentrations of ATP, glucocorticoids significantly enhance the ATP-dependent secretion of the proinflammatory cytokines, IL-1β, TNF-α, and IL-6 [24]. Interestingly, glucocorticoids also induce the expression of the purinergic receptor P2Y2R, enhancing downstream signaling and subsequent IL-6 secretion from endothelial cells following stimulation with ATP [25] (Figure 1c). Thus, the induction of TLR2, NLRP3, and P2Y2R provides a mechanism by which glucocorticoids ready the innate immune system for rapid activation and enhancement of the acute-phase response (Figure 1).
Coactivation of GR and proinflammatory transcription factors reinforces the inflammatory response
Recent evidence suggests that the interaction between GR and proinflammatory transcription factors is much more complex. Chromatin immunoprecipitation assay coupled to deep-sequencing (ChIP-Seq) analysis has shown that, in the absence of inflammatory stimuli, AP-1 can direct the binding of GR following hormone activation by regulating chromatin accessibility [26]. In fact, 51% of the GR-bound sites were co-occupied by AP-1 and included both composite [glucocorticoid response elements (GRE) and AP-1 sites] and non-composite interactions (AP-1 sites only). It remains to be determined whether or not these interactions are transcriptionally functional. Surprisingly, loss of AP-1 activity significantly reduced GR occupancy at the co-bound sites, and attenuated ~50% of the glucocorticoid-regulated genes, suggesting a functional role for AP-1 in transcriptional pathways activated by GR [26]. Though not directly tested, this data has broader implications for GR-mediated signaling following activation of AP-1 by proinflammatory molecules because the GR-mediated inhibition of AP-1 is selective for some, but not all, of the AP-1 regulated genes, and vice versa [6]. How the timing of AP-1 or GR activation affects this cooperativity at composite or non-composite elements warrants further investigation.
Does the cooperation between GR and proinflammatory transcription factors allow the regulation of glucocorticoid-responsive genes not previously accessible? Some insight into this question comes from the recent ChIP-Seq analysis of GR and NF-κB binding sites in HeLa cells following treatment with the synthetic glucocorticoid triamcinolone acid (TA), TNF-α, or both [27]. Compared to either treatment alone, the majority of identified binding sites were conserved upon cotreatment with TA and TNF-α. Interestingly, a significant proportion of GR and NF-κB binding sites were identified only when TA and TNF-α were administered concomitantly. Occupancy at these ‘gained’ GR and NF-κB sites was highly dependent on the presence of NF-κB and GR, respectively. In fact, NF-κB and AP-1 response elements were enriched at the sites that were co-occupied by GR and NF-κB. In line with the anti-inflammatory actions of GR, a subset of TNF-α-regulated genes were downregulated by cotreatment with TA. However, a subset of genes, including proinflammatory genes, were also synergistically regulated by the combined treatment. One caveat to this study is that GR was activated before NF-κB, and this could alter the transcriptional output compared to simultaneous stimulation or the initiation of TNF-α signaling before GR activation. For example, glucocorticoids could have a prophylactic effect when administered before NF-κB activation, skewing the response towards being anti-inflammatory. By contrast, activating NF-κB (and/or AP-1) before or at the same time as GR may shift the transcriptional profile of repressed/induced genes and expand the synergistically regulated subset. Further characterization of these genes and the functional significance of the observed synergism are warranted.
Interestingly, whole-genome microarray analysis identified ~900 genes that are regulated by concomitant administration of glucocorticoids and TNF-α [28]. Of these genes, more than two-thirds were coregulated by glucocorticoids and TNF-α. Of particular interest was serpinA3, a secreted acute-phase protein involved in several inflammatory diseases [29]. The expression of serpinA3 was induced by both glucocorticoids and TNF-α alone. However, coadministration of glucocorticoid and TNF-α led to a synergistic increase in the level of mRNA and protein in vitro and in vivo [28]. Despite having predicted GREs, the synergistic regulation of serpinA3 appears to occur through enhanced recruitment of GR and RNA polymerase II (Pol II) to the transcription start site. The question of whether or not NF-κB has a similar role to AP-1 in regulating GR-mediated signaling in the basal state remains open.
As indicated in Box 2, the 11β-hydroxysteroid dehydrogenase (HSD) enzymes are critical in controlling the bioavailability of intracellular glucocorticoids. Interestingly, obesity and proinflammatory cytokines induce a significant increase in the expression and activity of 11β-HSD1 in macrophages and in pre- and mature adipocytes [30,31]. Surprisingly, the expression and activity of 11β-HSD1 is necessary for cytokine and LPS-induced secretion of several proinflammatory cytokines. In adipocytes specifically, glucocorticoid reamplification enhanced NF-κB and MAPK activity and may contribute to the persistent inflammation seen in obesity [31].
The observation that GR co-occupies a significant proportion of binding sites with proinflammatory transcription factors is not restricted to AP-1 and NF-κB. To determine the crosstalk between STAT3 and GR, whole-genome tiling array analysis was performed in cells stimulated with leukemia inhibitory factor (LIF), a member of the IL-6 family, and/or dexamethasone (Dex). This analysis identified hormone-activated GR at a significant number of LIF-activated STAT3 binding sites [32]. In fact, glucocorticoids significantly potentiated the number of genes induced by LIF, which alone regulated very few genes. In particular, one cluster of late-onset genes stood out. These genes, which comprise the cell defense response, were only regulated when both LIF and glucocorticoids were administered. Moreover, in vivo, the extent to which this cluster of genes was coregulated by LIF and Dex was similar to their induction by LPS. Furthermore, glucocorticoids also enhanced IL-6-activated STAT3 activity [32,33], potentially by both enhancing STAT3 binding [32] and repressing suppressor of cytokine signaling 3 (SOCS3) expression [33]. Classically, it is recognized that GR tends to repress the activity of these transcription factors (see below). However, the ability of these transcription factors to shape the glucocorticoid response (Figure 2) provides evidence that the initial burst of glucocorticoids elicited by an inflammatory response could actually be to reinforce the proinflammatory environment, ensuring that proper clearance and removal of pathogen is achieved.
Figure 2.
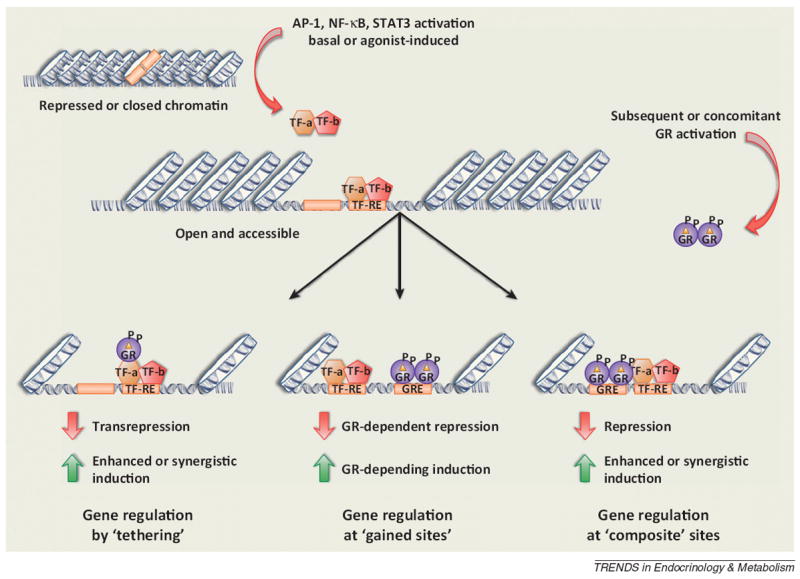
Concomitant activation of proinflammatory transcription factors and the glucocorticoid receptor reinforces inflammation. The ability of the glucocorticoid receptor (GR) to activate gene transcription relies on its ability to interact with glucocorticoid response elements (GREs). (Top) Chromatin, in its inactive state, prevents GR from accessing a GRE and alters the transcriptional response. (Bottom) Proinflammatory transcription factors (TF) promote an open chromatin state in the absence (AP-1) or presence (NK-κB or STAT3) of an inflammatory stimulus, allowing GR to now access chromatin and elicit a response. Some of the glucocorticoid-responsive changes occur as a result of tethering (left) or as a result of a composite regulation (right) where GR is able both to bind to a GRE and tether to the transcription factor. The profile of these complex interactions will shape the immune response and can be either stimulatory or repressive.
Glucocorticoid-mediated repression of transcription factors and signaling pathways involved in inflammation
Proinflammatory molecules released during inflammation initiate signaling cascades that ultimately activate the transcription factors AP-1 and NF-κB. In turn, these transcription factors regulate the synthesis of proinflammatory molecules, providing a positive feedback loop that propagates the inflammatory response [34,35]. The basic tenet and much of the research on the anti-inflammatory effects of the glucocorticoids has focused on GR-mediated repression of the transcriptional activity of AP-1 and NF-κB (Figure 3). However, GR is able to interact with and alter the transcriptional activity of several other factors potentially involved in the inflammatory response, such as the interferon (IFN) regulatory factor 3 (IRF3) [36,37], T-box expressed T cells (T-bet) [38], and GATA3 [39], among others [40]. Additional genome-wide studies are necessary to identify these alternative mechanisms [22,40].
Figure 3.

Glucocorticoid receptor (GR)-mediated repression of the proinflammatory transcription factors AP-1 and NF-κB. (a) Proinflammatory stimuli trigger a signaling cascade that results in the activation of the transcription factor, AP-1, a heterodimer composed of c-Jun and c-Fos. This drives the transcription of several proinflammatory molecules. Activation of the GR results in its translocation to the nucleus where it can now repress AP-1 activity via one of three mechanisms: (i) at some promoters, GR physically interacts with c-Jun in a process known as tethering, which represses the activity of AP-1 and represses the transcription of proinflammatory genes; (ii) at some promoters, GR is able to simultaneously bind to a GRE and tether to c-Jun to repress the transcriptional activity of AP-1; and (iii) GR induces the expression of MKP-1, a phosphatase, that is able to dephosphorylate and inactivate the kinase JNK. (b) Proinflammatory stimuli trigger a signaling cascade that results in the activation of the transcription factor, NF-κB, a heterodimer composed of the p50 and p65 subunits. This drives the transcription of several proinflammatory genes. Although the exact mechanism is not known, there are several theories as to how GR can inhibit NF-κB activity: (i) similarly to AP-1, GR can physically interact with and repress the activity of NF-κB; (ii) GR is able to block the formation of an NF-κB/IRF3 heterodimer, possibly through the recruitment of GRIP; (iii) GR is able to block the recruitment of the C-terminal tail kinase, pTEFb, thus preventing RNA polymerase II (Pol II) phosphorylation and activation; (iv) GR is able to repress NF-κB activity by recruiting a histone deacetylase (HDAC); (v) GR is able to block the ability of NF-κB to interact with p300 and CPB; and (vi) p53 is able to interact with GR, altering its transcriptional activity, and thus preventing NF-κB activity.
AP-1, one of the key mediators of the inflammatory response, functions as a homo- or heterodimer composed of the basic leucine-zipper transcription factors Fos (cFos, Fos B, Fra-1, and Fra-2), Jun (c-Jun, v-Jun, Jun B, and Jun D), activating transcription factor (ATF2, ATF3, B-ATF, JDP-1, and JDP-2), or MAF (MAFA, MAFB, c-MAF, NRL, MAFF, MAFG, and MAFK) [35,41]. These proteins are differentially expressed, altering transcriptional output depending on the subunit composition of the AP-1 dimer [35]. However, the most common form of AP-1 formed downstream of inflammatory cytokine signaling is the c-Fos/c-Jun heterodimer. This heterodimer is formed following activation of the c-Jun N-terminal kinase (JNK), a member of the MAPK family of proteins [42]. Once activated, the AP-1 heterodimer regulates numerous proinflammatory genes by binding to AP-1 response elements. In the classic model of GR-mediated inhibition, the transcriptional activity of AP-1 is suppressed as a result of a direct interaction with the c-Jun subunit of AP-1, which results in the reciprocal antagonism of GR-mediated signaling [43] (Figure 3a). Although the DNA-binding domain (DBD) (Box 1) of GR is necessary for this interaction, it does not seem to require GR binding to GREs within the promoter of the target gene, does not compete for coactivators [44,45], nor does it inhibit AP-1 from binding to its response element within endogenous promoters [6].
Similarly to AP-1, NF-κB plays a crucial role in initiating and amplifying proinflammatory signals. The NF-κB family consists of five members: p65 (RelA), RelB, c-Rel, NF-κB 1 (p50/p105), and NF-κB 2 (P50/p100) [34]. In the canonical pathway (i.e., TNF-α-activated), the transcriptionally active NF-κB dimer (p65–p50) is held in the cytoplasm by a member of the inhibitor of NF-κB (IκB) family [34]. Signaling through proinflammatory stimuli results in the activation of IκB kinase (IKK), phosphorylation and degradation of IκB, and thus the release and nuclear shuttling of NF-κB. Once in the nucleus, NF-κB binds to its response elements to regulate numerous proinflammatory molecules. The exact mechanism by which glucocorticoids are able to inhibit the activity of NF-κB is unclear, although several hypotheses have been proposed (Figure 3b). Similarly to AP-1, GR is proposed to physically interact with RelA and inhibit its transcriptional activity [46]. GR is able to block the formation of the p65/IRF3 complex [36], perhaps by recruiting the GR-interacting protein (GRIP) [37,47]or inhibiting phosphorylation of IRF3 [48]. GR has been reported to block the phosphorylation of the C-terminal domain (CTD) of Pol II by competing with the CTD kinase pTEFb [49,50]. Similarly, GR can recruit histone deacetylases to NF-κB -dependent promoters [51,52], or compete with NF-κB for binding to CREB-binding protein and p300 [53]. Finally, p53 was also shown to be involved in GR-mediated repression of NF-κB activity through the regulation of the transcriptional activity of GR [54].
In addition to transrepression, glucocorticoids are able to induce the expression of proteins that can antagonize proinflammatory processes at the post-transcriptional level. The induction of MAPK phosphatase-1 prevents the phosphorylation and activation of JNK [55-58]. Although initial evidence suggested that glucocorticoids could induce the expression of IκBα to sequester p65/p50 in the cytoplasm, this response seems to be cellspecific and is not a universal mechanism to regulate NF-κB signaling [59]. Finally, glucocorticoids influence mRNA stability through the induction of tristetraprolin (TTP), which stimulates the degradation of transcripts with AU-rich elements such as TNF-α [60]. In the context of inflammation, glucocorticoids then repress the activity of these and other proinflammatory transcription factors directly, as well as by inducing proteins that antagonize inflammatory signaling pathways. In doing so, glucocorticoids ensure that there is not a prolonged or exaggerated production of inflammatory cytokines, allowing the ultimate resolution of inflammation and restoration of a homeostatic environment.
Glucocorticoids, resolution, and tissue restoration following an inflammatory response
The final phase of inflammation, resolution, is an active process involving multiple biochemical pathways [61]. Some of the key hallmarks of resolution are the active repression of neutrophil recruitment to the inflammatory site, the secretion of bioactive lipids known to have proresolving actions, and the nonphlogistic recruitment of monocytes which play a role in tissue repair [61]. Glucocorticoids affect each of these processes at the cellular and molecular level. For example, glucocorticoids suppress the expression of both endothelial- and neutrophil-expressed adhesion molecules, thus preventing extravasation [8]. Additionally, glucocorticoids induce the expression of Annexin-1, which induces neutrophil apoptosis [62]. Finally, prolonged glucocorticoid exposure promotes an antiinflammatory phenotype with enhanced phagocytic activity in resident macrophages, contributing to the resolution of inflammation by removing apoptotic cells and tissue repair [8] (Figure 4).
Figure 4.

Glucocorticoids promote the resolution of inflammation and restore homeostasis. Glucocorticoids affect nearly every cell type by virtue of nearly ubiquitous expression of the glucocorticoid receptor (GR). During the course of inflammation, glucocorticoids are able to promote resolution by repressing the expression of adhesion molecules, preventing rolling adhesion and extravasation of neutrophils. Glucocorticoids also induce the expression and secretion of Annexin-1, which is able to induce apoptosis of neutrophils at the site of inflammation. Prolonged glucocorticoid exposure induces tissue resident macrophages (MΦ) to undergo a phenotypic change to become M2-like or anti-inflammatory. These macrophages no longer produce proinflammatory cytokines. Instead they produce interleukin-10 (IL-10), have enhanced phagocytic activity to remove apoptotic cells, and promote tissue healing. Glucocorticoids also act on naïve and differentiated T cells that have been recruited to the inflammatory site by blocking T helper 1 (Th1)- and Th2-derived cytokine production as well as inducing apoptosis.
Concluding remarks
As a pharmacological intervention, glucocorticoids are the first line of defense to treat chronic inflammatory diseases. However, how to reconcile the physiological role of glucocorticoids as major factors released in response to stress (inflammation included) with their anti-inflammatory actions is a question that remains to be resolved. The ability of molecules to have both a pro- and anti-inflammatory role in both normal physiological and pathological conditions is not unprecedented. For example, both IL-6 and leptin are known to have pro- and anti-inflammatory properties [63-65]. It is becoming clear that the actions of glucocorticoids are much more pleiotropic than previously thought, and thus cannot be simply categorized as antiinflammatory (Figure 5). The regulation of components of the innate immune system, namely TLR2, NLRP3, and P2Y2R [22,24,25], provides evidence that glucocorticoids ready the immune system to respond quickly to both bacterial infection and tissue damage. In addition, the complete absence of glucocorticoids (i.e., adrenalectomy) renders animals much more susceptible to LPS-induced systemic inflammation [66], suggesting that glucocorticoids are necessary both to respond to infection as well as to prevent a massive release of cytokines and prevent the adaptive immune system from over-reacting.
Figure 5.

The glucocorticoid receptor (GR) acts a cellular rheostat to ensure the proper response is elicited by the immune system. Glucocorticoids, acting through GR, affect all phases of the immune response. By enhancing the expression of TLR2, P2Y2R, and NLRP3, glucocorticoids ready the innate immune system to respond to microbial products and tissue injury. Additionally, glucocorticoids increase the levels of circulating bone marrow-derived neutrophils. Glucocorticoids reinforce the immune system by cooperating with the proinflammatory transcription factors AP-1, NF-κB, and STAT3. This is accomplished by repressing their activity at specific promoters, inhibiting the production of proinflammatory cytokines. However, GR can also act synergistically with these transcription factors, enhancing the expression and activation of some proinflammatory responses. Glucocorticoids are able to promote the resolution of inflammation, and restore homeostasis, by stimulating the secretion of proresolving molecules (Annexin-1), shifting T cell signaling towards a Th2 response, inducing neutrophil and T cell apoptosis, promoting a wound-healing and antiinflammatory phenotype in macrophages (MΦ), and promoting the removal of apoptotic cells. The ability of GR to accomplish these pleiotropic actions will depend on several factors including post-translational modifications, extracellular environment, ligand availability and duration of signaling, cell type-specific cofactors and binding partners, and chromatin accessibility.
We hypothesize that the shift in the ability of glucocorticoids to regulate pro- versus anti-inflammatory gene programs lies in the concomitant signals received by the cell, the duration and magnitude of glucocorticoid signaling, and the duration and magnitude of the proinflammatory stimulus. The inflammatory process is critical for an organism to respond to and remove pathogens. It is also crucial that the process proceeds unfettered through to resolution. Therefore, rather than acting strictly as an anti-inflammatory mediator, GR should be considered as a cellular rheostat. Specifically, in response to inflammatory stimuli, the transcriptional output elicited by glucocorticoids is fine-tuned by the microenvironment [27,32,67], ligand bioavailability [31], receptor expression [4], concomitant hormonal input [66], chromatin state [26,68], receptor dynamics [69], and cellular binding partners [26,27,32,54,70] (Figure 5). Thus, glucocorticoids ready and reinforce the innate immune system to respond quickly (proinflammatory actions), but also act systemically to repress the adaptive immune system and help restore homeostasis (anti-inflammatory actions). Deciphering these mechanisms will provide a great deal of insight into the overall regulation of transcriptional profiles and physiological responses elicited by glucocorticoids.
Acknowledgments
We would like to extend our apologies to those colleagues whose work we were unable to cite owing to space limitations. We would like to thank and acknowledge these individuals for all of their work that significantly contributed to our current understanding of GR action.
Glossary
- Damage-associated molecular patterns (DAMPs)
a diverse set of endogenously derived products that alert the innate immune system to tissue damage
- Extravasation
the movement of cells from the blood vessel into surrounding tissue
- Glucocorticoid response element (GRE)
a short, palindromic DNA sequence that is bound by the liganded receptor, in this case GR. GREs can be found with the promoter sites, introns, or exons of target genes. The DNA sequence is 5′ AGAACAnnnTGTTCT 3′
- Inflammasome
a macromolecular complex responsible for the activation of caspase-1 and -5 and subsequent processing and release of the cytokines interleukin (IL)-1β, IL-18, and IL-33
- NOD-Like receptors
a family of cytoplasmic proteins that can function as pattern-recognition receptors to regulate inflammatory and apoptotic processes
- Nonphlogisitic
the clearance of leukocytes in the absence of or induction of an inflammatory response
- Pathogen-associated molecular patterns (PAMPs)
a diverse set of microbialderived products that share conserved features that alert the innate immune system to intruding pathogens
- Pattern-recognition receptors (PRRs)
germline-encoded receptors that recognize structures conserved among microbes or endogenous molecules released from damaged cells
- Pre-receptor ligand metabolism
a series of enzymatic reactions controlling the intracellular concentration of active hormone, in this case cortisol. Cortisol is converted to its inactive metabolite, cortisone, through the actions of 11β-hydroxysteroid dehydrogenase (HSD) type 2. The reverse reaction, which converts cortisone back to cortisol, is regulated by 11β-HSD type 1
- T cell receptor (TCR) ligation
recognition of an antigen/major histocompatibility complex (MHC) by the TCR. This leads to the activation of signaling pathways that result in clonal expansion, differentiation, and the secretion of cytokine and cytotoxic molecules
- Th1 lymphocytes
a differentiated subset of T cells. T helper (Th)1 cells are primarily made in response to microbes and viruses that activate macrophages and natural killer (NK) cells. They produce cytokines such as interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) to reinforce an inflammatory response by activating effector T cells, natural killer cells, and macrophages
- Th2 lymphocytes
a second differentiated subset of T cells. Th2 cells are primarily made in response to helminthes, allergens, and extracellular microbes and toxins. They produce cytokines such as IL-4, IL-5, and IL-13 and stimulate B cells, eosinophils, and mast cells during allergic responses
- Toll-like receptors
pattern-recognition receptors expressed on cells of the immune system that recognize microbial structures that are evolutionarily conserved and activate the inflammatory response
- Transrepression
repression of gene expression induced by indirect association (tethering) rather than by DNA-specific binding of the nuclear receptor with target genes
References
- 1.Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012;4:a006049. doi: 10.1101/cshperspect.a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140:871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 3.Clark AR, Belvisi MG. Maps and legends: the quest for dissociated ligands of the glucocorticoid receptor. Pharmacol Ther. 2012;134:54–67. doi: 10.1016/j.pharmthera.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 4.Oakley RH, Cidlowski JA. Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem. 2011;286:3177–3184. doi: 10.1074/jbc.R110.179325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cole TJ, et al. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995;9:1608–1621. doi: 10.1101/gad.9.13.1608. [DOI] [PubMed] [Google Scholar]
- 6.Beck IM, et al. Crosstalk in inflammation: the interplay of glucocorticoid receptor-based mechanisms and kinases and phosphatases. Endocr Rev. 2009;30:830–882. doi: 10.1210/er.2009-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barnes PJ. Mechanisms and resistance in glucocorticoid control of inflammation. J Steroid Biochem Mol Biol. 2010;120:76–85. doi: 10.1016/j.jsbmb.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 8.Baschant U, Tuckermann J. The role of the glucocorticoid receptor in inflammation and immunity. J Steroid Biochem Mol Biol. 2010;120:69–75. doi: 10.1016/j.jsbmb.2010.03.058. [DOI] [PubMed] [Google Scholar]
- 9.Sorrells SF, et al. The stressed CNS: when glucocorticoids aggravate inflammation. Neuron. 2009;64:33–39. doi: 10.1016/j.neuron.2009.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sorrells SF, Sapolsky RM. An inflammatory review of glucocorticoid actions in the CNS. Brain Behav Immun. 2007;21:259–272. doi: 10.1016/j.bbi.2006.11.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Akira S, et al. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 12.Pancer Z, Cooper MD. The evolution of adaptive immunity. Annu Rev Immunol. 2006;24:497–518. doi: 10.1146/annurev.immunol.24.021605.090542. [DOI] [PubMed] [Google Scholar]
- 13.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science. 2010;327:291–295. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Evans RM. The steroid and thyroid hormone receptor superfamily. Science. 1988;240:889–895. doi: 10.1126/science.3283939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coutinho AE, Chapman KE. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol. 2010;335:2–13. doi: 10.1016/j.mce.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brewer JA, et al. T-cell glucocorticoid receptor is required to suppress COX-2-mediated lethal immune activation. Nat Med. 2003;9:1318–1322. doi: 10.1038/nm895. [DOI] [PubMed] [Google Scholar]
- 17.Wust S, et al. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J Immunol. 2008;180:8434–8443. doi: 10.4049/jimmunol.180.12.8434. [DOI] [PubMed] [Google Scholar]
- 18.Bhattacharyya S, et al. Macrophage glucocorticoid receptors regulate Toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood. 2007;109:4313–4319. doi: 10.1182/blood-2006-10-048215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tuckermann JP, et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J Clin Invest. 2007;117:1381–1390. doi: 10.1172/JCI28034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stavreva DA, et al. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nat Cell Biol. 2009;11:1093–1102. doi: 10.1038/ncb1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galon J, et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. 2002;16:61–71. doi: 10.1096/fj.01-0245com. [DOI] [PubMed] [Google Scholar]
- 22.Chinenov Y, Rogatsky I. Glucocorticoids and the innate immune system: crosstalk with the toll-like receptor signaling network. Mol Cell Endocrinol. 2007;275:30–42. doi: 10.1016/j.mce.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 23.Strowig T, et al. Inflammasomes in health and disease. Nature. 2012;481:278–286. doi: 10.1038/nature10759. [DOI] [PubMed] [Google Scholar]
- 24.Busillo JM, et al. Glucocorticoids sensitize the innate immune system through regulation of the NLRP3 inflammasome. J Biol Chem. 2011;286:38703–38713. doi: 10.1074/jbc.M111.275370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding Y, et al. Dexamethasone enhances ATP-induced inflammatory responses in endothelial cells. J Pharmacol Exp Ther. 2010;335:693–702. doi: 10.1124/jpet.110.171975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Biddie SC, et al. Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell. 2011;43:145–155. doi: 10.1016/j.molcel.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rao NA, et al. Coactivation of GR and NFKB alters the repertoire of their binding sites and target genes. Genome Res. 2011;21:1404–1416. doi: 10.1101/gr.118042.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lannan EA, et al. Proinflammatory actions of glucocorticoids: glucocorticoids and TNFalpha coregulate gene expression in vitro and in vivo. Endocrinology. 2012;153:3701–3712. doi: 10.1210/en.2012-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baker C, et al. SERPINA3 (aka alpha-1-antichymotrypsin) Front Biosci. 2007;12:2821–2835. doi: 10.2741/2275. [DOI] [PubMed] [Google Scholar]
- 30.Ishii T, et al. Augmentation of 11beta-hydroxysteroid dehydrogenase type 1 in LPS-activated J774.1 macrophages – role of 11beta-HSD1 in pro-inflammatory properties in macrophages. FEBS Lett. 2007;581:349–354. doi: 10.1016/j.febslet.2006.11.032. [DOI] [PubMed] [Google Scholar]
- 31.Ishii-Yonemoto T, et al. Glucocorticoid reamplification within cells intensifies NF-kappaB and MAPK signaling and reinforces inflammation in activated preadipocytes. Am J Physiol Endocrinol Metab. 2009;298:E930–E940. doi: 10.1152/ajpendo.00320.2009. [DOI] [PubMed] [Google Scholar]
- 32.Langlais D, et al. Regulatory network analyses reveal genome-wide potentiation of LIF signaling by glucocorticoids and define an innate cell defense response. PLoS Genet. 2008;4:e1000224. doi: 10.1371/journal.pgen.1000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dittrich A, et al. Glucocorticoids increase interleukin-6-dependent gene induction by interfering with the expression of the suppressor of cytokine signaling 3 feedback inhibitor. Hepatology. 2012;55:256–266. doi: 10.1002/hep.24655. [DOI] [PubMed] [Google Scholar]
- 34.Kumar A, et al. Nuclear factor-kappaB: its role in health and disease. J Mol Med (Berl) 2004;82:434–448. doi: 10.1007/s00109-004-0555-y. [DOI] [PubMed] [Google Scholar]
- 35.Zenz R, et al. Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res Ther. 2008;10:201. doi: 10.1186/ar2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ogawa S, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reily MM, et al. The GRIP1:IRF3 interaction as a target for glucocorticoid receptor-mediated immunosuppression. EMBO J. 2006;25:108–117. doi: 10.1038/sj.emboj.7600919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liberman AC, et al. The activated glucocorticoid receptor inhibits the transcription factor T-bet by direct protein–protein interaction. FASEB J. 2007;21:1177–1188. doi: 10.1096/fj.06-7452com. [DOI] [PubMed] [Google Scholar]
- 39.Liberman AC, et al. Glucocorticoids inhibit GATA-3 phosphorylation and activity in T cells. FASEB J. 2009;23:1558–1571. doi: 10.1096/fj.08-121236. [DOI] [PubMed] [Google Scholar]
- 40.Kassel O, Herrlich P. Crosstalk between the glucocorticoid receptor and other transcription factors: molecular aspects. Mol Cell Endocrinol. 2007;275:13–29. doi: 10.1016/j.mce.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 41.Eychene A, et al. A new MAFia in cancer. Nat Rev Cancer. 2008;8:683–693. doi: 10.1038/nrc2460. [DOI] [PubMed] [Google Scholar]
- 42.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 43.Schule R, et al. Functional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell. 1990;62:1217–1226. doi: 10.1016/0092-8674(90)90397-w. [DOI] [PubMed] [Google Scholar]
- 44.De Bosscher K, et al. Glucocorticoid repression of AP-1 is not mediated by competition for nuclear coactivators. Mol Endocrinol. 2001;15:219–227. doi: 10.1210/mend.15.2.0591. [DOI] [PubMed] [Google Scholar]
- 45.Heck S, et al. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. 1994;13:4087–4095. doi: 10.1002/j.1460-2075.1994.tb06726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liden J, et al. A new function for the C-terminal zinc finger of the glucocorticoid receptor. Repression of RelA transactivation. J Biol Chem. 1997;272:21467–21472. doi: 10.1074/jbc.272.34.21467. [DOI] [PubMed] [Google Scholar]
- 47.Chinenov Y, et al. Role of transcriptional coregulator GRIP1 in the anti-inflammatory actions of glucocorticoids. Proc Natl Acad Sci U S A. 2012;109:11776–11781. doi: 10.1073/pnas.1206059109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McCoy CE, et al. Glucocorticoids inhibit IRF3 phosphorylation in response to Toll-like receptor-3 and -4 by targeting TBK1 activation. J Biol Chem. 2008;283:14277–14285. doi: 10.1074/jbc.M709731200. [DOI] [PubMed] [Google Scholar]
- 49.Nissen RM, Yamamoto KR. The glucocorticoid receptor inhibits NFkappaB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev. 2000;14:2314–2329. doi: 10.1101/gad.827900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luecke HF, Yamamoto KR. The glucocorticoid receptor blocks P-TEFb recruitment by NFkappaB to effect promoter-specific transcriptional repression. Genes Dev. 2005;19:1116–1127. doi: 10.1101/gad.1297105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Islam KN, Mendelson CR. Glucocorticoid/glucocorticoid receptor inhibition of surfactant protein-A (SP-A) gene expression in lung type II cells is mediated by repressive changes in histone modification at the SP-A promoter. Mol Endocrinol. 2008;22:585–596. doi: 10.1210/me.2007-0412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ito K, et al. Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits interleukin-1beta-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol. 2000;20:6891–6903. doi: 10.1128/mcb.20.18.6891-6903.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKay LI, Cidlowski JA. CBP (CREB binding protein) integrates NF-kappaB (nuclear factor-kappaB) and glucocorticoid receptor physical interactions and antagonism. Mol Endocrinol. 2000;14:1222–1234. doi: 10.1210/mend.14.8.0506. [DOI] [PubMed] [Google Scholar]
- 54.Murphy SH, et al. Tumor suppressor protein (p)53, is a regulator of NF-kappaB repression by the glucocorticoid receptor. Proc Natl Acad Sci U S A. 2011;108:17117–17122. doi: 10.1073/pnas.1114420108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baschant U, et al. Glucocorticoid therapy of antigen-induced arthritis depends on the dimerized glucocorticoid receptor in T cells. Proc Natl Acad Sci U S A. 2011;108:19317–19322. doi: 10.1073/pnas.1105857108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caelles C, et al. Nuclear hormone receptor antagonism with AP-1 by inhibition of the JNK pathway. Genes Dev. 1997;11:3351–3364. doi: 10.1101/gad.11.24.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kleiman A, et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB J. 2011;26:722–729. doi: 10.1096/fj.11-192112. [DOI] [PubMed] [Google Scholar]
- 58.Vandevyver S, et al. Glucocorticoid receptor dimerization induces MKP1 to protect against TNF-induced inflammation. J Clin Invest. 2012;122:2130–2140. doi: 10.1172/JCI60006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clark AR. Anti-inflammatory functions of glucocorticoid-induced genes. Mol Cell Endocrinol. 2007;275:79–97. doi: 10.1016/j.mce.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 60.Smoak K, Cidlowski JA. Glucocorticoids regulate tristetraprolin synthesis and posttranscriptionally regulate tumor necrosis factor alpha inflammatory signaling. Mol Cell Biol. 2006;26:9126–9135. doi: 10.1128/MCB.00679-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Serhan CN, et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007;21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perretti M, D’Acquisto F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol. 2009;9:62–70. doi: 10.1038/nri2470. [DOI] [PubMed] [Google Scholar]
- 63.Gruver AL, Sempowski GD. Cytokines, leptin, and stress-induced thymic atrophy. J Leukoc Biol. 2008;84:915–923. doi: 10.1189/jlb.0108025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hick RW, et al. Leptin selectively augments thymopoiesis in leptin deficiency and lipopolysaccharide-induced thymic atrophy. J Immunol. 2006;177:169–176. doi: 10.4049/jimmunol.177.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scheller J, et al. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813:878–888. doi: 10.1016/j.bbamcr.2011.01.034. [DOI] [PubMed] [Google Scholar]
- 66.Duma D, et al. Sexually dimorphic actions of glucocorticoids provide a link to inflammatory diseases with gender differences in prevalence. Sci Signal. 2010;3:ra74. doi: 10.1126/scisignal.2001077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Galliher-Beckley AJ, et al. Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular stress pathways with nuclear receptor signaling. Mol Cell Biol. 2011;31:4663–4675. doi: 10.1128/MCB.05866-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.John S, et al. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat Genet. 2011;43:264–268. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.John S, et al. Interaction of the glucocorticoid receptor with the chromatin landscape. Mol Cell. 2008;29:611–624. doi: 10.1016/j.molcel.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 70.Johnson TA, et al. Chromatin remodeling complexes interact dynamically with a glucocorticoid receptor-regulated promoter. Mol Biol Cell. 2008;19:3308–3322. doi: 10.1091/mbc.E08-02-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kumar R, Thompson EB. Gene regulation by the glucocorticoid receptor: structure:function relationship. J Steroid Biochem Mol Biol. 2005;94:383–394. doi: 10.1016/j.jsbmb.2004.12.046. [DOI] [PubMed] [Google Scholar]
- 72.Bledsoe RK, et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. 2002;110:93–105. doi: 10.1016/s0092-8674(02)00817-6. [DOI] [PubMed] [Google Scholar]
- 73.Grad I, Picard D. The glucocorticoid responses are shaped by molecular chaperones. Mol Cell Endocrinol. 2007;275:2–12. doi: 10.1016/j.mce.2007.05.018. [DOI] [PubMed] [Google Scholar]
- 74.Vandevyver S, et al. On the trail of the glucocorticoid receptor: into the nucleus and back. Traffic. 2012;13:364–374. doi: 10.1111/j.1600-0854.2011.01288.x. [DOI] [PubMed] [Google Scholar]
- 75.Freedman ND, Yamamoto KR. Importin 7 and importin alpha/importin beta are nuclear import receptors for the glucocorticoid receptor. Mol Biol Cell. 2004;15:2276–2286. doi: 10.1091/mbc.E03-11-0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Surjit M, et al. Widespread negative response elements mediate direct repression by agonist-liganded glucocorticoid receptor. Cell. 2011;145:224–241. doi: 10.1016/j.cell.2011.03.027. [DOI] [PubMed] [Google Scholar]
- 77.Meijsing SH, et al. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–410. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ronacher K, et al. Ligand-selective transactivation and transrepression via the glucocorticoid receptor: role of cofactor interaction. Mol Cell Endocrinol. 2009;299:219–231. doi: 10.1016/j.mce.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 79.So AY, et al. Conservation analysis predicts in vivo occupancy of glucocorticoid receptor-binding sequences at glucocorticoid-induced genes. Proc Natl Acad Sci U S A. 2008;105:5745–5749. doi: 10.1073/pnas.0801551105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lowenberg M, et al. Novel insights into mechanisms of glucocorticoid action and the development of new glucocorticoid receptor ligands. Steroids. 2008;73:1025–1029. doi: 10.1016/j.steroids.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 81.Song IH, Buttgereit F. Non-genomic glucocorticoid effects to provide the basis for new drug developments. Mol Cell Endocrinol. 2006;246:142–146. doi: 10.1016/j.mce.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 82.Bruna A, et al. Glucocorticoid receptor-JNK interaction mediates inhibition of the JNK pathway by glucocorticoids. EMBO J. 2003;22:6035–6044. doi: 10.1093/emboj/cdg590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Croxtall JD, et al. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br J Pharmacol. 2000;130:289–298. doi: 10.1038/sj.bjp.0703272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gutierrez-Mecinas M, et al. Long-lasting behavioral responses to stress involve a direct interaction of glucocorticoid receptors with ERK1/2-MSK1-Elk-1 signaling. Proc Natl Acad Sci U S A. 2011;108:13806–13811. doi: 10.1073/pnas.1104383108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Xiao L, et al. Glucocorticoid rapidly enhances NMDA-evoked neurotoxicity by attenuating the NR2A-containing NMDA receptor-mediated ERK1/2 activation. Mol Endocrinol. 2010;24:497–510. doi: 10.1210/me.2009-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leis H, et al. Glucocorticoid receptor counteracts tumorigenic activity of Akt in skin through interference with the phosphatidylinositol 3-kinase signaling pathway. Mol Endocrinol. 2004;18:303–311. doi: 10.1210/me.2003-0350. [DOI] [PubMed] [Google Scholar]
- 87.Smith E, Frenkel B. Glucocorticoids inhibit the transcriptional activity of LEF/TCF in differentiating osteoblasts in a glycogen synthase kinase-3beta-dependent and -independent manner. J Biol Chem. 2005;280:2388–2394. doi: 10.1074/jbc.M406294200. [DOI] [PubMed] [Google Scholar]
- 88.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids – new mechanisms for old drugs. N Engl J Med. 2005;353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 89.Torpy DJ, Ho JT. Corticosteroid-binding globulin gene polymorphisms: clinical implications and links to idiopathic chronic fatigue disorders. Clin Endocrinol (Oxf) 2007;67:161–167. doi: 10.1111/j.1365-2265.2007.02890.x. [DOI] [PubMed] [Google Scholar]
- 90.Lu NZ, et al. International Union of Pharmacology. LXV. The pharmacology and classification of the nuclear receptor superfamily: glucocorticoid, mineralocorticoid, progesterone, and androgen receptors. Pharmacol Rev. 2006;58:782–797. doi: 10.1124/pr.58.4.9. [DOI] [PubMed] [Google Scholar]
- 91.Rivest S. Interactions between the immune and neuroendocrine systems. Prog Brain Res. 2010;181:43–53. doi: 10.1016/S0079-6123(08)81004-7. [DOI] [PubMed] [Google Scholar]
- 92.Gross KL, Cidlowski JA. Tissue-specific glucocorticoid action: a family affair. Trends Endocrinol Metab. 2008;19:331–339. doi: 10.1016/j.tem.2008.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Naray-Fejes-Toth A, Fejes-Toth G. Novel mouse strain with Cre recombinase in 11beta-hydroxysteroid dehydrogenase-2-expressing cells. Am J Physiol Renal Physiol. 2007;292:F486–F494. doi: 10.1152/ajprenal.00188.2006. [DOI] [PubMed] [Google Scholar]
- 94.Walker BR, Andrew R. Tissue production of cortisol by 11beta-hydroxysteroid dehydrogenase type 1 and metabolic disease. Ann N Y Acad Sci. 2006;1083:165–184. doi: 10.1196/annals.1367.012. [DOI] [PubMed] [Google Scholar]