

Abstract
Multiple studies have confirmed the contribution of rare de novo copy number variations (CNVs) to the risk for Autism Spectrum Disorders (ASD).1-3 While de novo single nucleotide variants (SNVs) have been identified in affected individuals,4 their contribution to risk has yet to be clarified. Specifically, the frequency and distribution of these mutations has not been well characterized in matched unaffected controls, data that are vital to the interpretation of de novo coding mutations observed in probands. Here we show, via whole-exome sequencing of 928 individuals, including 200 phenotypically discordant sibling pairs, that highly disruptive (nonsense and splice-site) de novo mutations in brain-expressed genes are associated with ASD and carry large effects (OR=5.65; CI: 1.44-22.2; p=0.01 asymptotic test). Based on mutation rates in unaffected individuals, we demonstrate that multiple independent de novo SNVs in the same gene among unrelated probands reliably identifies risk alleles, providing a clear path forward for gene discovery. Among a total of 279 identified de novo coding mutations, there is a single instance in probands, and none in siblings, in which two independent nonsense variants disrupt the same gene, SCN2A (Sodium Channel, Voltage-Gated, Type II, Alpha Subunit), a result that is highly unlikely by chance (p=0.005).
We completed whole-exome sequencing in 238 families from the Simons Simplex Collection (SSC), a comprehensively-phenotyped ASD cohort consisting of pedigrees with two unaffected parents, an affected proband, and, in 200 families, an unaffected sibling.5 Exome sequences were captured with NimbleGen oligonucleotide libraries, subjected to DNA sequencing on the Illumina platform, and genotype calls were made at targeted bases (Supplementary Information).6,7 On average, 95% of the targeted bases in each individual were assessed by ≥8 independent sequence reads; only those bases showing ≥20 independent reads in all family members were considered for de novo mutation detection. This allowed for analysis of de novo events in 83% of all targeted bases and 73% of all exons and splice sites in RefSeq hg18 (Table S1; Supplementary_Data_S1). Given uncertainties regarding the sensitivity of detection of insertion-deletions, case-control comparisons reported here consider only single base substitutions (Supplementary Information). Validation was attempted for all predicted de novo SNVs via Sanger sequencing of all family members, with sequence readers blinded to affection status; 96% were successfully validated. We determined there was no evidence of systematic bias in variant detection between affected and unaffected siblings through comparisons of silent de novo, non-coding de novo, and novel silent transmitted variants (Fig. 1a; Fig. S1-5; Supplementary Information).
Figure 1. Enrichment of non-synonymous de novo variants in probands compared with sibling controls.
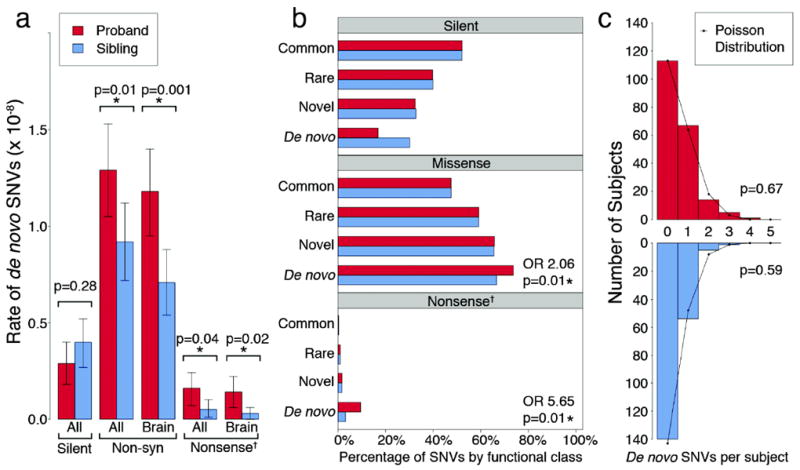
a) The rate of de novo variants is shown for 200 probands (red) and matched unaffected siblings (blue). ‘All’ refers to all RefSeq genes in hg18, ‘Brain’ refers to the subset of genes that are brain-expressed23 and ‘Non-syn’ to non-synonymous SNVs (including missense, nonsense and splice site SNVs). Error bars represent the 95% CI and p-values are calculated with a two-tailed binomial exact test. b) The proportion of transmitted variants in brain-expressed genes is equal between 200 probands (red) and matched unaffected siblings (blue) for all mutation types and allele frequencies, including common (≥1%); rare (<1%), and novel (single allele in one of the 400 parents); in contrast both non-synonymous and nonsense de novo variants show significant enrichment in probands compared to unaffected siblings (73.7% vs. 66.7%, p=0.01, asymptotic test and 9.5% vs. 3.1%, p=0.01 respectively). c) The frequency distribution of brain-expressed non-synonymous de novo SNVs is shown per sample for probands (red) and siblings (blue). Neither distribution differs from the Poisson distribution (black line) suggesting that multiple de novo SNVs within a single individual do not confirm ASD risk.
† ‘Nonsense’ represents the combination of nonsense and splice site SNVs.
Among 200 quartets (Table 1), 125 non-synonymous de novo single nucleotide variants (SNVs) were present in probands and 87 in siblings: 15 of these were nonsense (10 in probands; 5 in siblings) and 5 altered a canonical splice site (5 in probands; 0 in siblings). There were 2 instances in which de novo SNVs were present in the same gene in two unrelated probands; one of these involved two independent nonsense variants (Table 2). Overall, the total number of non-synonymous de novo SNVs was significantly greater in probands compared to their unaffected siblings (p=0.01, two-tailed binomial exact test; Fig. 1a; Table 1) as was the odds ratio of non-synonymous to silent mutations in probands versus siblings (OR=1.93; 95% CI: 1.11-3.36; p=0.02, asymptotic test; Table 1). Restricting the analysis to nonsense and splice site mutations in brain-expressed genes resulted in substantially increased estimates of effect size and demonstrated a significant difference in cases versus controls based either on an analysis of mutation burden (N=13 vs. 3; p=0.02, two-tailed binomial exact test; Fig. 1a; Table 1) or an evaluation of the odds ratio of nonsense and splice site to silent SNVs (OR = 5.65; 95%CI: 1.44-22.2; p=0.01, asymptotic test; Fig. 1b; Table 1).
Table 1.
Distribution of single nucleotide variants (SNVs) between probands and siblings.
| Category | Total number of SNVs a | SNVs per subject | Per base SNV rate (×10-8) | p b | Odds ratio (95% CI) c | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Pro | Sib | Pro | Sib | Pro | Sib | |||||
| N=200 | N=200 | N=200 | N=200 | N=200 | N=200 | |||||
| De novo | All genes | All | 154 | 125 d | 0.77 | 0.63 | 1.58 | 1.31 | 0.09 | NA |
| Silent | 29 | 39 | 0.15 | 0.20 | 0.29 | 0.40 | 0.28 | NA | ||
| All non-synonymous | 125 | 87 | 0.63 | 0.44 | 1.29 | 0.92 | 0.01* | 1.93 (1.11-3.36) | ||
| Missense | 110 | 82 | 0.55 | 0.41 | 1.13 | 0.86 | 0.05 | 1.80 (1.03-3.16) | ||
| Nonsense/splice site | 15 | 5 | 0.08 | 0.03 | 0.16 | 0.05 | 0.04* | 4.03 (1.32-12.4) | ||
| Brain-expressed genes | All | 137 | 96 | 0.69 | 0.48 | 1.41 | 1.01 | 0.01* | NA | |
| Silent | 23 | 30 | 0.12 | 0.15 | 0.24 | 0.31 | 0.41 | NA | ||
| All non-synonymous | 114 | 67 | 0.57 | 0.34 | 1.18 | 0.71 | 0.001* | 2.22 (1.19-4.13) | ||
| Missense | 101 | 64 | 0.51 | 0.32 | 1.04 | 0.68 | 0.005* | 2.06 (1.10-3.85) | ||
| Nonsense/ splice site | 13 | 3 | 0.07 | 0.02 | 0.14 | 0.03 | 0.02* | 5.65 (1.44-22.2) | ||
| Novel transmitted | All genes | All | 26,565 | 26,542 | 133 | 133 | 277 | 277 | 0.92 | NA |
| Silent | 8,567 | 8,642 | 43 | 43 | 90 | 91 | 0.57 | NA | ||
| All non-synonymous | 17,998 | 17,900 | 90 | 90 | 188 | 187 | 0.61 | 1.01 (0.98-1.05) | ||
| Missense | 17,348 | 17,250 | 87 | 86 | 181 | 180 | 0.60 | 1.01 (0.98-1.05) | ||
| Nonsense/splice site | 650 | 650 | 3.3 | 3.3 | 7 | 7 | 1.00 | 1.01 (0.90-1.13) | ||
| Brain-expressed genes | All | 20,942 | 20,982 | 105 | 105 | 219 | 220 | 0.85 | NA | |
| Silent | 6,884 | 6,981 | 34 | 35 | 72 | 74 | 0.42 | NA | ||
| All non-synonymous | 14,058 | 14,001 | 70 | 70 | 147 | 146 | 0.74 | 1.02 (0.98-1.06) | ||
| Missense | 13,588 | 13,525 | 68 | 68 | 142 | 141 | 0.71 | 1.02 (0.98-1.06) | ||
| Nonsense/splice site | 470 | 476 | 2.3 | 2.4 | 5 | 5 | 0.87 | 1.00 (0.88-1.14) | ||
An additional 15 de novo variants were seen in the probands of 25 trio families; all were missense and 14 were brain-expressed.
The p-values compare the number of variants between probands and siblings using a two-tailed binomial exact test (Supplementary Information).
The odds ratio calculates the proportion of variants in a specific category to silent variants and then compares these ratios in probands versus siblings.
The sum of silent and non-synonymous variants is 126, however one nonsense and two silent de novo variants were indentified in KANK1 in a single sibling, suggesting a single gene conversion event. This event contributed a maximum count of one to any analysis.
Table 2.
Loss of function mutations in probands.
| Gene Symbol | Gene Description | Mutation Type |
|---|---|---|
| ADAM33 | ADAM metallopeptidase domain 33 | Nonsense |
| CSDE1 | Cold shock domain containing E1, RNA-binding | Nonsense |
| EPHB2 | EPH (Ephrin) receptor B2 | Nonsense |
| FAM8A1 | Family with sequence similarity 8, member A1 | Nonsense |
| FREM3 | FRAS1 related extracellular matrix 3 | Nonsense |
| MPHOSPH8 | M-phase phosphoprotein 8 | Nonsense |
| PPM1D | Protein phosphatase 1D magnesium-dependent, delta isoform | Nonsense |
| RAB2A | RAB2A, member RAS oncogene family | Nonsense |
| SCN2A | Sodium channel, voltage-gated, type II, alpha subunit | Nonsense |
| SCN2A | Sodium channel, voltage-gated, type II, alpha subunit | Nonsense |
| BTN1A1 | Butyrophilin, subfamily 1, member A1 | Splice Site |
| FCRL6 | Fc receptor-like 6 | Splice Site |
| KATNAL2 | Katanin p60 subunit A-like 2 | Splice Site |
| NAPRT1 | Nicotinate phosphoribosyltransferase domain containing 1 | Splice Site |
| RNF38 | Ring finger protein 38 | Splice Site |
| SCP2 | Sterol carrier protein 2 | Frameshift a |
| SHANK2 | SH3 and multiple ankyrin repeat domains 2 | Frameshift a |
Frameshift de novo variants are not included in any of the reported case-control comparisons (Supplementary Information).
To determine whether factors other than diagnosis of ASD could explain our findings, we examined a variety of potential covariates including parental age, IQ, and sex. We found that the rate of de novo SNVs indeed increases with paternal age (p=0.008, two-tailed Poisson regression) and that paternal and maternal ages are highly correlated (p<0.0001, two-tailed linear regression). However, while the mean paternal age of probands in our sample was 1.1 years higher than their unaffected siblings, re-analysis accounting for age did not substantively alter any of the significant results reported herein (Supplementary Information). Similarly, no significant relationship was observed between the rate of de novo SNVs and proband IQ (p≥0.19, two-tailed linear regression, Supplementary Information) or proband sex (p≥0.12, two-tailed Poisson regression; Fig. S6; Supplementary Information).
Overall these data demonstrate that non-synonymous de novo SNVs, and particularly highly disruptive nonsense and splice-site de novo mutations, are associated with ASD. Based on the conservative assumption that de novo single-base coding mutations observed in siblings confer no autism liability, we estimate that at least 14% of affected individuals in the SSC carry de novo SNV risk events (Supplementary Information). Moreover, among probands and considering brain-expressed genes, an estimated 41% of non-synonymous de novo SNVs (95% CI: 21-58%) and 77% of nonsense and splice site de novo SNVs (95% CI: 33-100%) point to bona fide ASD-risk loci (Supplementary Information).
We next set out to evaluate which of the particular de novo SNVs identified in our study confer this risk. Based on our prior work3 we hypothesized that estimating the probability of observing multiple independent de novo SNVs in the same gene in unrelated individuals would provide a more powerful statistical approach to identifying ASD-risk genes versus the alternative of comparing mutation counts in affected vs. unaffected individuals. Consequently, we conducted simulation experiments focusing on de novo SNVs in brain-expressed genes, using the empirical data for per base mutation rates and taking into account the actual distribution of gene sizes and GC content across the genome (Supplementary Information). We calculated probabilities based on a wide range of assumptions regarding the number of genes conferring ASD risk (Fig. 2). Based on 150,000 iterations, we determined that under all models, two or more nonsense and/or splice site de novo mutations were highly unlikely to occur by chance (p=0.005; Supplementary Information; Fig. 2a). Importantly, this threshold was robust both to sample size, anticipating the eventual sequencing of the entire SSC cohort (N=2,648 families), and to variation in our estimates of locus heterogeneity. Similarly, two or more nonsense or splice site de novo mutations remained statistically significant when the simulation was performed using the lower bound of the 95% confidence interval for the estimate of de novo mutation rates (Fig. S7).
Figure 2. Identification of multiple de novo mutations in the same gene reliably distinguishes risk-associated mutations.
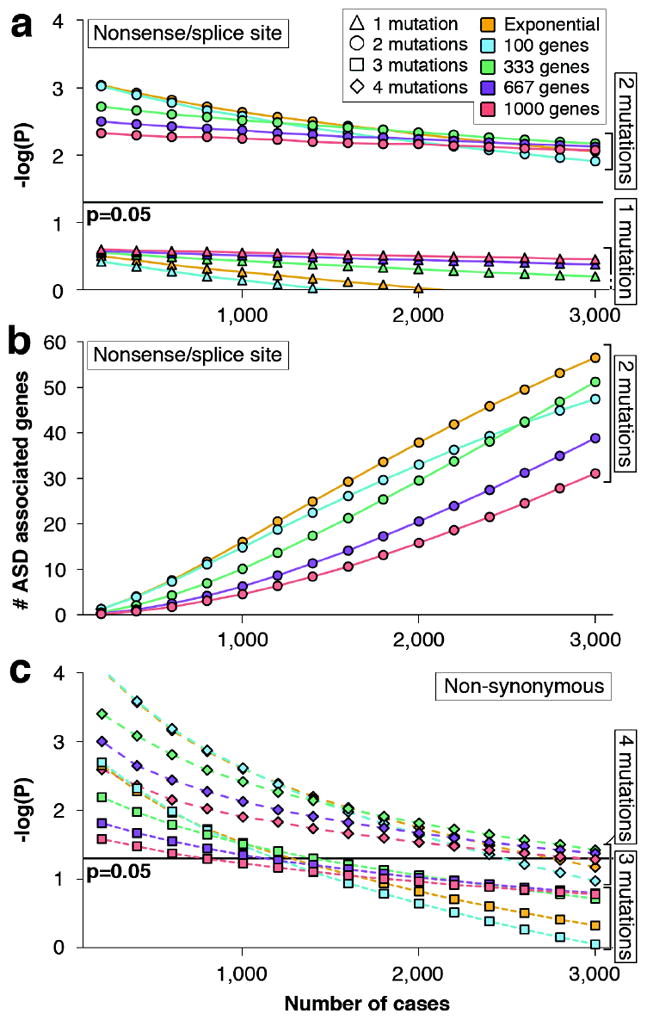
a) This plot shows the results of a simulation experiment modeling the likelihood, measured in −log(P) values, of observing two independent nonsense/splice site de novo mutations in the same brain-expressed gene among unrelated probands. We modeled the observed rate of de novo brain-expressed mutations in probands and siblings and evaluated models of locus heterogeneity, including 100, 333, 667, or 1,000 contributing genes, as well as using the top 1% of genes derived from a model of exponential distribution of risk. A total of 150,000 iterations were run. The identification of two or more independent nonsense/splice site de novo variants in a brain-expressed gene provides significant evidence for ASD association (p<0.05) for all models irrespective of increasing sample size. This observation remained statistically significant when the simulation was repeated using the lower bound of the 95% confidence interval for the estimate of de novo mutation rate (Fig. S7). b) The simulation described in ‘a’ was used to predict the number of genes that will be found to carry two or more nonsense/splice site de novo mutations for a sample of a given size (specified on the x-axis). c) The simulation was repeated for non-synonymous de novo mutations. The identification of three or more independent non-synonymous de novo mutations in a brain-expressed gene provides significant evidence for ASD association (p<0.05) in the sample reported here, however this threshold is sensitive both to sample size and heterogeneity models.
Only a single gene in our cohort, SCN2A (Sodium channel, voltage-gated, type II, alpha) met this threshold (p=0.005; Fig. 2a) with two probands each carrying a nonsense de novo SNV (Table 2). This finding is consistent with a wealth of data showing overlap of genetic risks for ASD and seizure.8 Gain of function mutations in SCN2A are associated with a range of epilepsy phenotypes; a nonsense de novo mutation has been described in a patient with infantile epileptic encephalopathy and intellectual decline,9 de novo missense mutations with variable electrophysiological effects have been found in cases of intractable epilepsy,10 and transmitted rare missense mutations have been described in families with idiopathic ASD.11 Of note, the individuals in the SSC carrying the nonsense de novo SNVs have no history of seizure.
We then considered whether alternative approaches described in the recent literature,4,12 including identifying multiple de novo events in a single individual or predicting the functional consequences of missense mutations, might help identify additional ASD-risk genes. However, we found no differences in the distribution or frequency of multiple de novo events within individuals in the case versus the control groups (Fig. 1c). In addition, when we examined patients carrying large de novo ASD-risk CNVs, we found a trend toward fewer non-synonymous de novo SNVs (Fig. S10; Supplementary Information). Consequently, neither finding supported a “two de novo hit” hypothesis. Similarly, we found no evidence that widely used measures of conservation or predictors of protein disruption such as PolyPhen2,13 SIFT,14 GERP,15 PhyloP,16 or Grantham Score17 differentiated de novo non-synonymous SNVs in probands compared to siblings (Fig. S8; Supplementary Information). Additionally, the de novo SNVs in our study were not significantly over-represented in previously established lists of synaptic genes,18-20 genes on chromosome X, autism-implicated genes,2 intellectual disability genes,2 genes within ASD-risk associated CNVs3 and de novo non-synonymous SNVs identified in schizophrenia probands.12,21 Finally we conducted pathway and protein-protein interaction analyses22 for all non-synonymous de novo SNVs, all brain-expressed non-synonymous de novo SNVs and all nonsense and splice site de novo SNVs (Fig. S8-9; Supplementary Information) and did not find a significant enrichment among cases versus controls that survived correction for multiple comparisons, though these studies were of limited power.
These analyses demonstrate that neither the type nor number of de novo mutations in a single individual provides significant evidence for association with ASD. Moreover, we determined that in the SSC cohort at least 3, and most often 4 or more, brain-expressed non-synonymous de novo SNVs in the same gene would be necessary to show a significant association. Unlike the case of highly disruptive nonsense and splice site mutations, this threshold was sensitive to both sample size and heterogeneity models (Fig. 2c; Fig. S7; Supplementary Information).
Finally, at the completion of our study, we had the opportunity to combine all de novo events in our sample with those identified in an independent whole-exome analysis of non-overlapping Simons Simplex families that focused predominantly on trios (O’Roak et al.). From a total of 414 probands, two additional genes were found to carry two highly disruptive mutations each, KATNAL2 (Katanin p60 subunit A-like 2) (our results and O’Roak et al.) and CHD8 (Chromodomain helicase DNA binding protein 8) (O’Roak et al.) thereby showing association with the ASD phenotype.
Overall, our results substantially clarify the genomic architecture of ASD, demonstrate significant association of three genes SCN2A, KATNAL2 and CHD8, and indicate that approximately 25-50 additional ASD-risk genes will be identified as sequencing of the 2,648 SSC families is completed (Fig. 2b). Rare non-synonymous de novo SNVs are associated with risk, with odds ratios for nonsense and splice-site mutations in the range previously described for large multigenic de novo CNVs.3 It is important to note that these estimates reflect a mix of risk and neutral mutations in probands. We anticipate that the true effect size for specific SNVs and mutation classes will be further clarified as more data accumulate. From the distribution of de novo CNVs in probands versus siblings, we previously estimated the number of ASD-risk loci at 234.3 Using the same approach, the current data result in a higher point estimate of 1,034 genes, however the confidence intervals are large and the distribution of this risk among these loci is unknown (Supplementary Information). What is clear is that our results strongly support a high degree of locus heterogeneity in the SSC cohort, involving hundreds of genes or more. Finally, via examination of mutation rates in well-matched controls, we have determined that the observation of highly disruptive de novo SNVs clustering within genes can robustly identify risk-conferring alleles. The focus on recurrent rare de novo mutation described herein provided sufficient statistical power to identify associated genes in a relatively small cohort and despite both a high degree of locus heterogeneity and the contribution of intermediate genetic risks. This approach promises to be valuable for future high-throughput sequencing efforts in ASD and other common neuropsychiatric disorders.
Methods (for on-line version only)
Sample selection
238 families (928 individuals) were selected from the SSC on the basis of: male probands with autism, low NVIQ, and discordant SRS with sibling and parents (n=40); female probands (n=46); multiple unaffected siblings (n=28); probands with known multigenic CNVs (n=15); and random selection (n=109). Thirteen families (6%) did not pass quality control (Supplementary Information) leaving 225 families (200 quartets, 25 trios) for analysis (Supplementary_Data_1). Of the 200 quartets, 194 (97%) probands had a diagnosis of autism and 6 (3%) were diagnosed with ASD; the median non-verbal IQ was 84. Three of these quartets have previously been reported as trios;4 there is no overlap between the current sample and those presented in the companion article.
Exome capture, sequencing and variant prediction
Whole-blood DNA was enriched for exonic sequences (exome capture) through hybridization with a NimbleGen custom array (n=210) or EZExomeV2.0 (n=718). The captured DNA was sequenced using an Illumina GAIIx (n=592) or HiSeq 2000 (n=336). Short read sequences were aligned to hg18 with BWA,6 duplicate reads were removed and variants were predicted using SAMtools.7 The data was normalized across each family by only analyzing bases with at least 20 unique reads in all family members (Supplementary Information). De novo predictions were made blinded to affected status using experimentally verifed thresholds (Supplementary Information). All de novo variants were confirmed using Sanger sequencing blinded to affected status.
Variant frequency
The frequency of variants in the offspring were determined by comparison with dbSNPv132 and 1,637 whole-exome controls including 400 parents. Variants were classifed as: ‘novel’, if only a single allele was present in a parent and none were seen in dbSNP or the other control exomes; ‘rare’, if they did not meet the criteria for novel and were present in <1% of controls; and ‘common’, if they were present in ≥1% of controls.
Gene annotation
Variants were analyzed against the RefSeq hg18 gene definitions, a list that includes 18,933 genes. Where multiple isoforms gave varying results the most severe outcome was chosen. All nonsense and canonical splice site variants were checked manually and were present in all RefSeq isoforms. A variant was listed as altering the splice site only if it disrupted canonical 2bp acceptor (AG) or donor (GT) sites.
Brain-expressed genes
A list of brain-expressed genes was obtained from expression array analysis across 57 postmortem brains (age 6 weeks post conception to 82 years) and multiple brain regions.23 Using this data 14,363 (80%) of genes were classified as brain-expressed (Supplementary Information).
Rate of de novo SNVs
To allow an accurate comparison between the de novo burden in probands and siblings the number of de novo SNVs found in each sample was divided by the number of bases analyzed (i.e. bases with ≥20 unique reads in all familiy members) to calculate a per base rate of de novo SNVs. Rates are given in Table 1.
Simulation model
The likelihood of observing multiple independent de novo events of a given type for a given sample size in an ASD risk-conferring gene was modeled using gene size and GC content (derived from the full set of brain-expressed RefSeq genes) and the observed rate of brain-expressed de novo variants in probands and siblings. These values were then used to evaluate the number of genes contributing to ASD showing two or more variants of the specified type (Fig. 2); comparing this to the number of genes with similar events not carrying ASD risk gave the liklihood of the specified pattern reflecting association with ASD. The simulation was run through 150,000 iterations across a range of samples sizes and multiple models of locus heterogeneity (Supplementary Information).
Severity scores
Severity scores were calculated for missense variants using web-based interfaces for PolyPhen2,13 SIFT,14 and GERP,15 using the default settings (Supplementary Information). PhyloP16 and Grantham Score17 were determined using an in-house annotated script. For nonsense/splice site variants the maximum score was assigned for Grantham, SIFT, and PolyPhen2; for GERP and PhyloP every possible coding base for the specific protein was scored and the highest value selected.
Pathway analysis
The list of brain-expressed genes with non-synonymous de novo SNVs was submitted to KEGG using the complete set of 14,363 brain-expressed genes as the background to prevent bias. For IPA the analysis was based on human nervous system pathways only, again to prevent bias. Otherwise default settings were used for both tools.
Protein-protein interactions
Genes with brain-expressed non-synonymous de novo variants in probands were submitted to the Disease Association Protein-Protein Link Evaluator (DAPPLE)22 using the default settings.
Comparing de novo SNV counts to gene lists
To assess whether non-synonymous de novo SNVs were enriched in particular gene sets, the chance of seeing a de novo variant in each gene on a given list was estimated based on the size and GC content of the gene. The observed number of de novo events was then assessed using the binomial distribution probabilty based on the total number of non-synonymous de novo variants in probands and the sum of probabilities for de novo events within these genes.
Supplementary Material
Acknowledgments
We are indebted to B. Neale and M. Daly for the invaluable discussions regarding de novo variation. We are grateful to all of the families participating in the Simons Foundation Autism Research Initiative (SFARI) Simplex Collection (SSC). This work was supported by a grant from the Simons Foundation. R.P. Lifton is an Investigator of the Howard Hughes Medical Institute. We wish to thank the SSC principal investigators A.L. Beaudet, R. Bernier, J. Constantino, E.H. Cook, Jr., E. Fombonne, D. Geschwind, D.E. Grice, A. Klin, D.H. Ledbetter, C. Lord, C.L. Martin, D.M. Martin, R. Maxim, J. Miles, O. Ousley, B. Peterson, J. Piggot, C. Saulnier, M.W. State, W. Stone, J.S. Sutcliffe, C.A. Walsh, and E. Wijsman; the coordinators and staff at the SSC sites; the SFARI staff, in particular M. Benedetti; Prometheus Research; the Yale Center of Genomic Analysis staff, in particular M. Mahajan, S. Umlauf, I. Tikhonova and A. Lopez; T. Brooks-Boone, N. Wright-Davis and M. Wojciechowski for their help in administering the project at Yale; I. Hart for support; and G.D. Fischbach, A. Packer, J. Spiro, M. Benedetti and M. Carlson for their helpful suggestions throughout. We would also like to acknowledge T. Lehner and the Autism Sequencing Consortium for providing an opportunity for pre-publication data exchange among the participating groups. Approved researchers can obtain the SSC population data set described in this study by applying at https://base.sfari.org.
Footnotes
Author Contributions S.J.S., M.T.M., R.P.L., M.G., D.H.G. and M.W.S. designed the study; M.T.M, A.R.G., J.M., M.R., A.G.E-S., N.M.D., S.M., M.W., G.O., Y.S., P.E., R.M. and J.O. designed and performed high-throughput sequencing experiments and variant confirmations; S.J.S., M.C., K.B., R.B. and N.C. designed the exome-analysis bioinformatics pipeline; S.J.S., A.J.W., N.N.P., J.L.S., N.T., K.A.M., N.S., K.R., D.H.G., B.D. and M.W.S. analyzed the data; S.J.S., A.J.W., K.R., B.D. and M.W.S. wrote the paper; J.M., M.R., A.J.W., A.R.G., A.G.E-S. and N.M.D., contributed equally to the study. All authors discussed the results and contributed to editing the manuscript.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
The authors have no competing financial interests to declare.
References
- 1.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–9. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pinto D, et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–72. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sanders SJ, et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–85. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Roak BJ, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–9. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fischbach GD, Lord C. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron. 2010;68:192–5. doi: 10.1016/j.neuron.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 6.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meisler MH, O’Brien JE, Sharkey LM. Sodium channel gene family: epilepsy mutations, gene interactions and modifier effects. J Physiol. 2010;588:1841–8. doi: 10.1113/jphysiol.2010.188482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamiya K, et al. A nonsense mutation of the sodium channel gene SCN2A in a patient with intractable epilepsy and mental decline. J Neurosci. 2004;24:2690–8. doi: 10.1523/JNEUROSCI.3089-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ogiwara I, et al. De novo mutations of voltage-gated sodium channel alphaII gene SCN2A in intractable epilepsies. Neurology. 2009;73:1046–53. doi: 10.1212/WNL.0b013e3181b9cebc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weiss LA, et al. Sodium channels SCN1A, SCN2A and SCN3A in familial autism. Mol Psychiatry. 2003;8:186–94. doi: 10.1038/sj.mp.4001241. [DOI] [PubMed] [Google Scholar]
- 12.Xu B, et al. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat Genet. 2011;43:864–8. doi: 10.1038/ng.902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 15.Cooper G, et al. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat Methods. 2010;7:250–1. doi: 10.1038/nmeth0410-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper GM, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–13. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–4. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 18.Abul-Husn NS, et al. Systems approach to explore components and interactions in the presynapse. Proteomics. 2009;9:3303–15. doi: 10.1002/pmic.200800767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bayés A, et al. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat Neurosci. 2011;14:19–21. doi: 10.1038/nn.2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collins MO, et al. Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J Neurochem. 2006;97(Suppl 1):16–23. doi: 10.1111/j.1471-4159.2005.03507.x. [DOI] [PubMed] [Google Scholar]
- 21.Girard SL, et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat Genet. 2011 doi: 10.1038/ng.886. [DOI] [PubMed] [Google Scholar]
- 22.Rossin EJ, et al. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet. 2011;7:e1001273. doi: 10.1371/journal.pgen.1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang HJ, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–9. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.