

Abstract
Cancer treatments using ionizing radiation (IR) therapy are thought to act primarily through the induction of tumor cell damage at a molecular level. However, a new concept has recently emerged, suggesting that the immune system is required for effective IR therapy. Our work here has identified interferon gamma (IFN-γ) as an essential cytokine for the efficacy of IR therapy. Local IR (15 Gy) to mice bearing Colon38, a colon adenocarcinoma, decreases tumor burden in wild-type animals. Interestingly, IR therapy had no effect on tumor burden in IFNγKO mice. We further determined that intratumoral levels of IFN-γ increased 2 days following IR, which directly correlated with a decrease in tumor burden that was not a result of direct cytotoxic effects of IFN-γ on tumor cells. T cells from IR-treated tumors exhibited a far greater capacity to lyse tumor cells in a 51Cr release assay, a process that was dependent on IFN-γ. CD8+ T cells were the predominant producers of IFN-γ, as demonstrated by IFN-γ intracellular staining and studies in IFN-γ reporter mice. Elimination of CD8+ T cells by antibody treatment reduced the intratumoral levels of IFN-γ by over 90%. More importantly, elimination of CD8+ T cells completely abrogated the effects of radiation therapy. Our data suggest that IFN-γ plays a pivotal role in mediating the antitumor effects of IR therapy.
Historically, ionizing radiation (IR) therapy was thought to control cancer predominantly by inducing tumor cell death through DNA damage. However, a new paradigm is emerging, strongly suggesting that the immune system mediates many of the antitumor effects of radiotherapy. Our laboratory has previously demonstrated that local radiation therapy in a melanoma mouse model results in tumor cell death, facilitating the release of tumor antigen.1 Freed antigen can be processed by antigen-presenting cells and used to stimulate effector cells within the draining lymph node. Consequently, these effector cells were able to traffic to the tumor where they could recognize and lyse malignant cells, thereby reducing tumor growth. Additional reports have supported the concept that the immune system is pivotal in determining the effectiveness of radiation therapy.2–5 Lee et al3 demonstrated that CD8+ T cells partially mediated the therapeutic effects of radiation, whereas Apetoh et al2 identified Toll-like receptor 4–positive (TLR4+) dendritic cells as potent immunostimulatory cells capable of processing and presenting antigen from dying irradiated tumor cells. As a whole, there is increasing evidence of immune involvement in the anticancer effects of radiotherapy; however, the exact mechanisms governing this response are largely unknown and may differ from one malignancy to another.
Irradiation of normal tissue, and now more recently described, tumor tissue, induces an inflammatory state often resulting in the secretion of cytokines into the microenvironment.6–9 This is likely the result of danger signals released by damaged or dying cells in response to DNA strand breaks.8 Whereas once largely ignored, it is now appreciated that these cytokines may impact the effectiveness of radiotherapy, either positively or negatively, by contributing to radioresistance or radiosensitivity as well as inducing or suppressing a radiation-mediated immune response. An example of a cytokine with opposing effects on tumors is interferon gamma (IFN-γ). IFN-γ, an inflammatory cytokine that we determined was up-regulated following radiation in a melanoma model,10 has conventionally been viewed as antagonistic to tumor growth.11 The biological functions of this cytokine include direct cytotoxic/antiproliferative effects on tumor cells as well as stimulation of the adaptive arm of the immune system against tumor antigens.10 However, recent reports have questioned the efficacy of IFN-γ as an antitumor agent, instead citing situations where IFN-γ appears to promote tumor progression.11 Depending on the dose, IFN-γ was shown to promote metastases of B16 to the lung as well as stimulate the proliferation of NIH-3T3 cells.12,13 Additionally, under certain circumstances, IFN-γ inhibited the antitumor function of NK cells and stimulated immunosuppression through the development of T regulatory (Treg) and/or myeloid-derived suppressor cells along with the production of inhibitory molecules like indoleamine 2,3-dioxygenase (IDO).14–17 Therefore, in a unique situation such as tumor radiotherapy in which the efficacy depends on a potent immune response, we addressed whether IFN-γ is detrimental or beneficial with regard to modulating the immune system following radiotherapy.
In this report, we determined that IFN-γ was not only beneficial, but was essential in mediating the antitumor effects of radiation in a mouse colon adenocarcinoma tumor.18 IFN-γ was up-regulated in the tumor microenvironment 2 days following radiation, and CD8+ T cells were the chief producers of this cytokine. Elimination of CD8+ T cells, not only greatly reduced the intratumoral (i.t.) levels of IFN-γ, but also abrogated any antitumor effect of radiation. Furthermore, we demonstrated that although IFN-γ had no direct effect on tumor cells in promoting tumor control, it enhanced the cytolytic capacity of T cells, possibly in an autocrine manner, resulting in a decrease of tumor burden.
Materials and Methods
Mice and Cell Lines
C57BL/6J and B6.129S7-Ifngtm1Ts (IFNγ−/−) mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Bicistronic IFN-γ reporter [Yeti (yellow-enhanced transcript for IFN-γ)] mice19 were obtained from Dr. Troy Randall (University of Alabama at Birmingham). All mice were treated in accordance to guidelines approved by the University Committee on Animal Resources. Colon38,18 a murine colon adenocarcinoma, was obtained from Dr. Edward Brown and maintained in MAT/P medium (US patent 4.816.401) supplemented with 100 U/mL penicillin, 100 mg/mL streptomycin, and 2% fetal calf serum. Colon38/dominant negative receptor (DNR) clones (0.1 and 0.2) were generated by stably transfecting parental Colon38 with a plasmid encoding an IFN-γ DNR (pcDNA.mugR) as previously described.10
Tumor Growth and Treatment
Mice were injected with 1 × 105 Colon38 cells i.m. into the left thigh. Tumor size was measured using calipers and mean thigh diameter was calculated (square root of the quantity W × H, where W is the width and H is the height). Mice were restrained and received 15-Gy local irradiation directly to the tumor-bearing leg, 7 days following tumor challenge as previously described.1 Mice were sacrificed for analysis on indicated days. To deplete CD8+ T cells, mice were treated with 200 μg of anti-CD8 (53-6.7) diluted in balanced salt solution (Sigma-Aldrich, St. Louis, MO) i.p. 1 day before tumor challenge and then every 4 days until sacrifice. Rat IgG diluted in balanced salt solution was used as a vehicle control.
Tumor Homogenization and IFN-γ ELISA
To measure i.t. levels of IFN-γ, approximately 200 mg of tumor tissue was removed and placed in 500 μL of Lysis 11 buffer20 containing protease inhibitors (BioVision, Milpitas, CA) and homogenized on ice for 30 to 60 seconds using a Bio-Gen PRO200 tissue homogenizer (PRO Scientific, Oxford, CT). Samples were centrifuged (2300 × g) at 4°C for 8 minutes, and supernatant was collected and tested either for total protein using a BCA kit (Pierce Biotechnology, Rockford, IL) or for IFN-γ by enzyme-linked immunosorbent assay (ELISA) (PeproTech, Rocky Hill, NJ) according to the manufacturer's instructions. IFN-γ concentration was normalized to total protein.
Flow Cytometric Analysis
Tumors were dissociated as previously described.21 Briefly, tumor pieces were weighed, minced, and digested for 45 minutes in collagenase D (Sigma-Aldrich). Single-cell suspensions were counted, blocked using Fc Block, and stained for the various antibodies listed in Table 1 for 30 minutes, and samples were analyzed using a FACSCanto Flow Cytometer (BD Biosciences, San Jose, CA) and FlowJo software version 9.4.11 (Tree Star, Ashland, OR) software. All data in Table 2 are normalized to 100 mg of tumor. For intracellular IFN-γ staining, single-cell suspensions were prepared as described above and directly (no restimulation) fixed/permeabilized using a CytoFix/CytoPerm kit according to the manufacturer's instructions (BD Biosciences). Samples were then stained as described above. Fluorescence minus one (FMO) was used as staining controls. Percentages of immune subsets were determined by first gating on CD45+ cells.
Table 1.
Antibodies Used in Flow Cytometry
| Antibody against | Clone | Manufacturer |
|---|---|---|
| CD4 | L3T4 | BD Biosciences |
| CD8 | 53-6.7 | eBioscience (San Diego, CA) |
| CD11c | HL3 | BD Biosciences |
| CD11b | M1/70 | eBioscience |
| CD19 | ID3 | BD Biosciences |
| CD25 | PC61 | BD Biosciences |
| CD45 | 30-F11 | BD Biosciences |
| CD119 | GR20 | BD Biosciences |
| NK1.1 | PK136 | BD Biosciences |
| F4/80 | BM8 | eBioscience |
| FoxP3 KIT | Cat#.77-5775-40 | eBioscience |
| GR-1 | RB6-8C5 | eBioscience |
| IFNγ | XMB1.2 | BD Biosciences |
| MHC I (H-2kb) | AF6-88.5 | BD Biosciences |
| Fc Block | 2.4G2 | BD Biosciences |
Table 2.
Cell Number of Intratumoral Immune Subsets per 100 mg of Tumor (×106)
| Tumor Cells | Total CD45+ | CD8+ T cells | Total CD4+ T cells | Tregs | NK cells | Macrophages | Monocytes | Dendritic cells | |
|---|---|---|---|---|---|---|---|---|---|
| Day 7 | 13 ± 2 | 11 ± 1 | 0.3 ± 0.04 | 0.2 ± 0.03 | 0.1 ± 0.01 | 0.6 ± 0.08 | 4.8 ± 0.7 | 2.5 ± 0.6 | 0.4 ± 0.06 |
| Day 9 UT | 14 ± 2 | 11 ± 2 | 0.7 ± 0.2 | 0.4 ± 0.09 | 0.3 ± 0.03 | 0.4 ± 0.08 | 5.7 ± 1 | 2.9 ± 0.8 | 0.4 ± 0.08 |
| Day 9 IR | 8.7 ± 1 | 9 ± 1 | 0.3 ± 0.05 | 0.1 ± 0.02∗ | 0.07 ± 0.004∗ | 0.3 ± 0.06 | 4.4 ± 0.4 | 2.5 ± 0.7 | 0.3 ± 0.05 |
| Day 11 UT | 16 ± 1 | 13 ± 2 | 2.4 ± 0.6 | 0.4 ± 0.06 | 0.3 ± 0.04 | 0.4 ± 0.07 | 6.7 ± 0.9 | 2.2 ± 0.4 | 0.2 ± 0.04 |
| Day 11 IR | 4.7 ± 0.8∗ | 18 ± 4 | 2.7 ± 1 | 0.3 ± 0.06 | 0.1 ± 0.04∗ | 0.6 ± 0.1 | 6.8 ± 1 | 4.3 ± 0.9 | 0.4 ± 0.1 |
Cell populations are defined as follows: tumor cells: CD45−, forward scatterhi; CD8+ T cells: CD45+, CD8+; CD4+ T cells: CD45+, CD4+; Tregs: CD45+, CD4+, FoxP3+; NK cells: CD45+, NK1.1+; macrophages: CD45+, F480hi, forward scatterhi; monocytes: CD45+, F480intermediate, forward scatterlow; dendritic cells: CD45+, F480-, CD11b+, CD11c+. Data were generated from at least six individual tumors per group to determine the means ± SEM for each subset except in Tregs, where three individual tumors were used. Comparisons were made between the UT and IR values at either the day 9 or day 11 time points in each group (but not across groups).
IR, irradiated; UT, unirradiated.
P < 0.05 as determined by one-way analysis of variance and the Bonferroni post hoc test.
Immunoblots
Protein (15 to 20 μg) from tumor homogenate was loaded per lane and subjected to SDS-PAGE and immunoblotted as previously described.20 Antibodies used for immunoblots were as follows: phosphorylated signal transducer and activator of transcription 1 (pSTAT1) (sc-7988; Santa Cruz Biotechnology, Santa Cruz, CA), total STAT1 (sc-345; Santa Cruz Biotechnology), interferon regulatory factor 1 (IRF1) (sc-640; Santa Cruz Biotechnology), and β-actin (Sigma-Aldrich).
FACSort and Real-Time Quantitative RT-PCR
Tumors were dissociated into single-cell suspensions as described above. Preparations were stained as described above with anti–CD8-PE, anti–CD45-PerCP, and anti–CD3-FITC. CD45+, CD3+, CD8+ cells were sorted using a FACSAria fluorescence-activated cell sorter (FACS) (BD Biosciences) and immediately lysed in Buffer RLT lysis buffer (Qiagen, Valencia, CA). RNA was purified using an RNeasy mini kit (Qiagen) according to the manufacturer’s instructions and quantified using a spectrophotometer. mRNA for Tbet, FasL, CTLA4, and Prf1 (perforin) was measured by real-time quantitative RT-PCR as previously described20 using nested primers designed to span an intron (CTLA-4: 5′-ATTCTGACTTCCTCCTTTGG-3′; 5′-CCTGTTGTAAGAGGACTTCTT-3′. FasL: 5′-CACCAACCAAAGCCTTAAAG-3′; 5′-ATATGTGTCTTCCCATTCC-3′. GAPDH: 5′-CATTGCTCTCAATGACAACT-3′; 5′-GGGTTTCTTACTCCTTGGAG-3′. Perforin: 5′-AAGACCTATCAGGACCAGTA-3′; 5′-CTGTGGAGCTGTTAAAGTTG-3′. Tbet: 5′-GATCATCACTAAGCAAGGAC-3′; 5′-ACATCCACAAACATCCTGTA-3′). Data were normalized to GAPDH values and expressed as fold increase over control.
Whole-Mount Histology
Tumors were examined by whole-mount histology on day 11 as previously described.22,23
51Cr Release Cytotoxicity Assay
Tumors were dissociated as described above, and tumor-infiltrating lymphocytes were magnetically separated using beads conjugated to anti–Thy-1 (clone T24/40.7). Tumor-infiltrating lymphocytes were assayed immediately for the ability to lyse Colon38 target cells in a standard 6-hour 51Cr release assay.
Statistical Analysis
Results are presented as means ± SE from at least three replicates. Data were examined for significance using paired Student's t-test, or a one-way analysis of variance followed by a Bonferroni post hoc test when appropriate.
Results
IFN-γ Mediates the Antitumor Effects of Radiotherapy
Our previous data have demonstrated the necessity of an immune response in mediating the antitumor effects of radiotherapy in a melanoma model.10 To examine the role of IFN-γ in promoting antitumor effects, we injected C57BL/6 mice with 1 × 105 Colon38 cells i.m. into either wild-type (WT) or IFNγKO mice. Tumors were either untreated or treated locally with 15-Gy γ-irradiation 7 days later, and mean thigh diameter was monitored over time. A single high dose of radiation was used because our previous data showed that 15 Gy stimulated a greater antitumor immune response when compared to a 5 × 3-Gy fractionated dose regimen.1 Unirradiated tumors in WT mice grew progressively until animals were sacrificed around day 14 (Figure 1). Radiation therapy did not have an immediate impact on controlling growth in WT mice because tumors grew progressively for 2 days following therapy (day 9). However, after day 9, a significant decrease of tumor burden was seen. Most treated tumors eventually grew, albeit at a slower rate compared to unirradiated control tumors. Interestingly, although unirradiated tumors in both IFNγKO and WT mice grew similarly, radiation treatment was ineffective in controlling tumor growth in IFNγKO mice (Figure 1).
Figure 1.
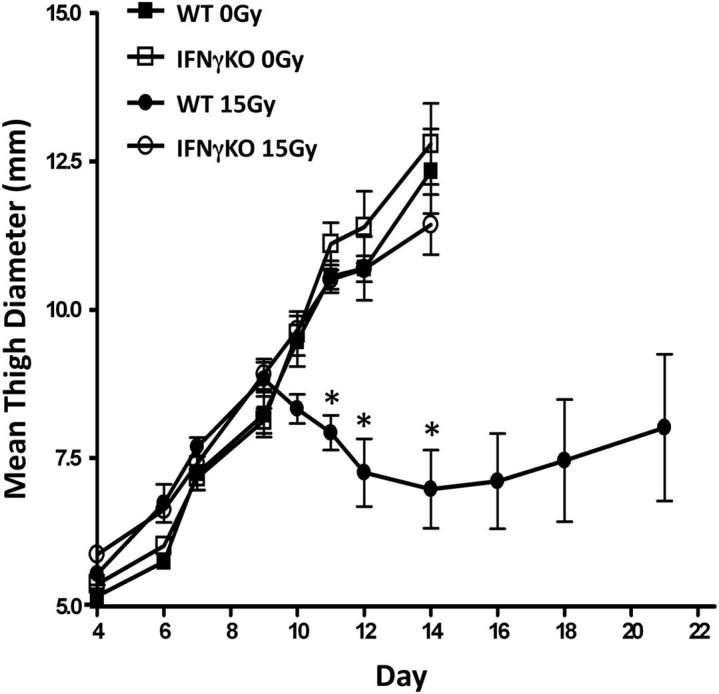
The antitumor effects of radiation therapy are abrogated in IFNγKO mice. A total of 1 × 105 Colon38 tumor cells were injected i.m. into either WT or IFNγKO mice and treated with or without 15-Gy radiation on day 7. Tumor growth was measured and expressed as mean thigh diameter (mm). Mice were sacrificed when tumors reached 11 to 13 mm in size (n = 6). ∗P < 0.001 as determined by analysis of variance and the Bonferroni post hoc test.
To examine whether IFN-γ was present within the tumor microenvironment, tumor homogenates from treated and untreated mice were analyzed for IFN-γ by ELISA. All results were standardized to total protein. IFN-γ was significantly induced following radiotherapy on days 9, 10, and 11 (Figure 2A) when compared to unirradiated control. Figure 2B represents the fold increase of IFN-γ in irradiated over unirradiated tumor samples over time and illustrates that minimal cytokine induction is seen on day 8 (24 hours after IR therapy). However, i.t. levels of IFN-γ are increased on day 9 and remain elevated out to day 11. This rise in the level of IFN-γ correlates with the decrease of tumor burden following radiotherapy at the same time point (Figure 1). These data demonstrate that i.t. levels of IFN-γ are increased following radiotherapy. More importantly, we have identified IFN-γ as an essential cytokine that appears to mediate the antitumor effects of radiation therapy.
Figure 2.
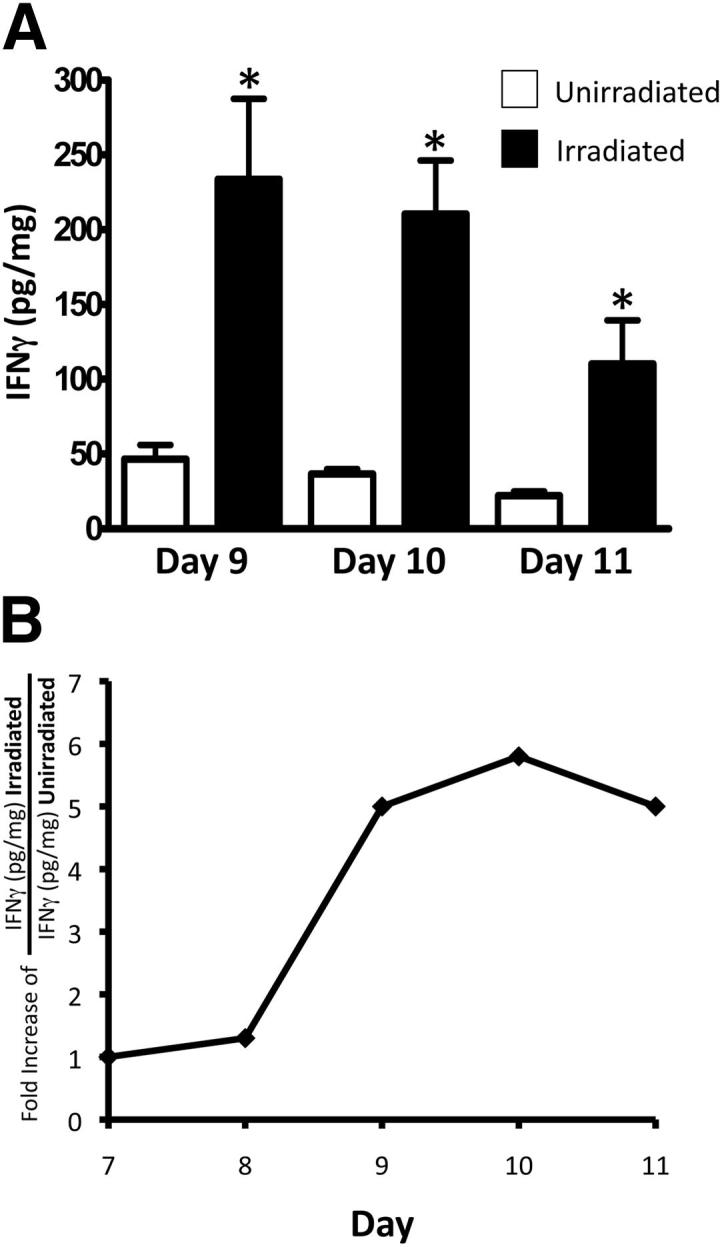
Radiotherapy increases the i.t. levels of IFN-γ. A total of 1 × 105 Colon38 cells were injected i.m. and treated with or without 15-Gy radiation on day 7 of tumor growth. A: Tumor homogenate was examined for IFN-γ protein by ELISA. All data are normalized to total protein. B: Kinetic analysis of IFN-γ protein plotted as fold increase of IFN-γ in irradiated samples over unirradiated samples over time (n = 6). ∗P < 0.05 as determined by Student t-test.
Intratumoral IFN-γ Does Not Act Directly on Tumor Cells to Promote Radiation-Induced Tumor Control
To assess the mechanism by which IFN-γ mediates the antitumor effects of radiotherapy, we examined whether radiation-induced IFN-γ was directly cytotoxic to tumor cells. Parental Colon38 cells were stably transfected with an IFN-γ DNR vector, rendering these cells unresponsive to IFN-γ as demonstrated by their inability to phosphorylate STAT1 or induce IRF1 following 1- and 3-hour treatments of IFN-γ in vitro (Figure 3A). Additionally, Colon38/DNR were unable to induce major histocompatibility complex (MHC) class I following IFN-γ stimulation in vitro (data not shown). DNR expression proved stable in vivo because subsequent flow cytometry performed on dissociated ex vivo tumor cells (as described in Materials and Methods) analyzed on day 13 of tumor growth showed that the DNR tumors retained high levels of mutant CD119 (IFN-γ receptor) expression (Supplemental Figure S1A) and exhibited lower MHC class I expression compared to ex vivo parental tumors (Supplemental Figure S1B). We compared the growth of Colon38 and Colon38/DNR.1 tumors treated with or without radiation as described above. Both tumor lines grew similarly in untreated animals (Figure 3B). More importantly, radiation was able to control tumor growth in a similar fashion in both parental and DNR transfected tumors. Therefore, IFN-γ is not acting directly on tumor cells to mediate the antitumor effects of radiotherapy.
Figure 3.

Tumors unable to respond to IFN-γ are still susceptible to radiation treatment. Colon38 cells were stably transfected with a plasmid encoding a DNR to IFN-γ. A: IFN-γ responsiveness of cells following either 1 or 3 hours of 50 ng/mL IFN-γ stimulation in vitro was assessed by Western blot for pSTAT1, total STAT1, IRF1, and loading control β-actin. Two separate clones (0.1 and 0.2) of Colon38/DNR including parental Colon38 (Par) were tested. B: Mice were injected i.m. with either Colon38 or Colon38/DNR.1 and treated with or without 15-Gy radiation on day 7. Tumor growth was measured over time (n = 6). ∗P < 0.05 as determined by analysis of variance and the Bonferroni post hoc test.
Radiation Enhances CTL in an IFN-γ–Dependent Manner
We next focused on whether IFN-γ may modulate immune cell function, in particular, T cells. Both unirradiated and irradiated tumors were examined by whole-mount microscopy on day 11 and exhibited abundant T cells, predominantly CD8+, present within the tumor microenvironment (Figure 4, A and B). Surprisingly, there was no apparent difference in the number of T cells within the tumors between the unirradiated and irradiated groups as demonstrated by the whole-mount images (Figure 4A compared to 4B). This observation was verified using quantitative flow cytometry (described below and in Materials and Methods) (Table 2). Additionally, we FACS-sorted CD8+ cells from both unirradiated and irradiated tumors and examined four genes associated with effector status (T-bet, Prf1, FasL, and CTLA4) by quantitative real-time PCR. Unexpectedly, no differences were observed in any of the four genes between the CD8+ T cells from unirradiated and irradiated tumors (data not shown). Therefore, we examined whether IFN-γ may enhance cytolytic T-cell function. T cells were purified from treated versus untreated tumors on day 11 and immediately assayed (without further culture or stimulation) for their ability to lyse Colon38 tumor cells in a 51Cr release assay on a per tumor basis as described in Materials and Methods. T cells from unirradiated tumor exhibited modest lytic activity (∼25% at highest effector/target ratio); however, following radiation therapy, the i.t. T cells had a significantly enhanced capacity to lyse tumor cells (Figure 4C). To determine the role of IFN-γ in this response, the experiment was repeated using T cells isolated from tumors growing in IFNγKO mice. T cells from unirradiated tumors grown in IFNγKO mice exhibited little to no cytolytic activity against Colon38. More importantly, radiotherapy did not significantly enhance this response. Furthermore, the cytolytic activity of T cells from irradiated tumors in IFNγKO mice was below the lytic capacity of T cells from both unirradiated and irradiated WT samples (Figure 4C). Therefore, IFN-γ, generated as a result of radiotherapy, enhances the ability of T cells to lyse tumor cells.
Figure 4.

T cells exhibit enhanced cytotoxicity following radiation therapy. Mice were injected i.m. with 1 × 105 Colon38 cells and treated with or without 15-Gy radiation on day 7. A and B: Tumors were removed on day 11 and examined by whole-mount histology for anti-CD31 to label blood vessels (red), anti-CD8 (green), and anti-CD4 (blue) T cells. C: Thy1+ tumor-infiltrating lymphocytes were isolated from individual day 11 tumors (as described above) from either WT or IFNγKO mice and assayed for specific lysis against 51Cr-labeled Colon38 targets. Data were generated by determining the percent specific lysis from six individual tumors (tumor-infiltrating lymphocytes were not pooled) at each effector/target (E:T) ratio as described in Materials and Methods. Means ± SEM were reported (n = 6). ∗P < 0.05 versus WT 0 Gy as determined by analysis of variance and the Bonferroni post hoc test; †P < 0.05 versus IFNγKO 15 Gy as determined by analysis of variance and the Bonferroni post hoc test.
CD8+ T Cells Are the Predominant Producers of Intratumoral IFN-γ
To determine which cell type(s) were responsible for producing i.t. IFN-γ, we first identified the immune subsets that were present within the tumor. Tumors grown for 7, 9, or 11 days and untreated or treated with 15-Gy radiation on day 7 were dissociated into single-cell suspensions, stained for immune cell markers, and analyzed using multicolor flow cytometry as described in Materials and Methods. Data are presented as the number of cells standardized to tumor weight in Table 2. We identified T cells, natural killer cells, monocytes, macrophages, and dendritic cells within the tumor microenvironment. We primarily compared differences in cell number within a particular immune subset between the unirradiated and irradiated tumors at either day 9 or day 11. Surprisingly, the only changes were observed in the T-cell populations as cell numbers decreased in the irradiated tumors when compared to unirradiated tumors at day 9. This observation is most likely a result of the radiosensitivity of T cells. Otherwise, no significant changes in cell density were observed in any other immune subset, including total CD45+ cells at either time point. Interestingly, an increase of CD8+ T cells from day 9 to day 11 was noted in both the treated and untreated groups, suggesting that Colon38 promotes the infiltration of potentially tumor reactive T cells.
We next distinguished IFN-γ–producing immune cells within the tumor using two complementary methods. First, we used bicistronic IFN-γ reporter mice (Yeti) in which transcription of the IFNG gene also results in enhanced yellow fluorescent protein (eYFP) reporter expression. Therefore, immune cells producing IFN-γ mRNA will also be YFP+. Colon38 tumors were grown in either WT or Yeti mice and treated with or without radiation as described above. Tumors were dissociated and single-cell suspensions were analyzed for YFP expression by flow cytometry. A YFP+ population that was not present in WT mice was clearly evident in all tumor samples (Figure 5A). Further subset analysis revealed that most YFP+ cells (ie, positive for IFN-γ mRNA) on day 9 were CD8+ T cells and NK cells (Figure 5B). By day 11, about 70% of YFP+ cells were CD8+ T cells. No differences were observed between irradiated and unirradiated tumor samples with regard to YFP+ immune subsets.
Figure 5.

CD8+ T cells are the predominant producers of i.t. IFN-γ. WT and IFN-γ reporter mice (Yeti) were injected with 1 × 105 Colon38 cells i.m. and treated with or without 15 Gy on day 7. Tumors were removed at various time points, dissociated into a single-cell suspension, and examined for YFP expression by flow cytometry. A: Representative dot plots from either WT or Yeti+ mice analyzed for YFP+ cells. B: YFP+ cells were gated and immune subsets were determined by multicolor flow cytometry (n = 4). Intracellular staining for IFN-γ was performed on single-cell suspensions from WT mice injected with Colon38 (as described above). C: Representative dot plots of IFN-γ stained and control are provided. FMO, fluorescence minus one. D: IFN-γ+ cells were gated and immune cell subsets were determined by multicolor flow cytometry. All data were normalized to tumor burden as described in Materials and Methods (n = 6). DC, dendritic cells; Mac, macrophages; Mono, monocytes; SSC, side scatter; Tg, transgenic.
Yeti mice report for IFN-γ transcripts but not necessarily those actively producing the cytokine.24 Therefore, to examine IFN-γ protein production, we used intracellular IFN-γ staining of i.t. immune subsets from WT mice as described in Materials and Methods. IFN-γ+ cells were detected primarily in the CD45+ gated cells (Figure 5C). On further characterization by flow cytometry, IFN-γ+ cells were determined to be predominantly CD8+ T cells (Figure 5D). The number of IFN-γ–producing CD8+ T cells increased in untreated tumors from day 7 to day 11. More importantly, irradiation enhanced the number of IFN-γ+ CD8+ T cells on both day 9 and day 11 (2 and 4 days following radiation therapy, respectively). Therefore, results from both the Yeti mice and intracellular IFN-γ staining concurrently identified CD8+ T cells as the chief producers of i.t. IFN-γ.
CD8+ T-Cell Depletion Abrogates the Antitumor Effects of Radiation
On the basis of the previous data, we hypothesized that depletion of CD8+ T cells should result in decreased levels of i.t. IFN-γ. Therefore, we depleted CD8+ T cells with i.p. injections of 200 μg of anti-CD8 starting 1 day before tumor inoculation and every 4 days thereafter. Rat IgG was used as a control. Anti-CD8 treatment routinely resulted in a knockdown of approximately 90% CD8+ T cells in both the tumor microenvironment and spleen (data not shown). Mice were challenged with tumor and irradiated as described above. Indeed, CD8+ T-cell depletion resulted in a substantial decrease in i.t. levels of IFN-γ at both day 9 and day 11, confirming that CD8+ T cells are the predominant producers of i.t. IFN-γ (Figure 6A). We next examined tumor growth kinetics with and without radiation between anti-CD8–treated and rat IgG–treated mice. Tumor growth was similar in unirradiated tumors between the two groups. However, the antitumor effects of radiation were completely lost in mice that were depleted of CD8+ T cells, and therefore had low levels of i.t. IFN-γ (Figure 6B). These data identify IFN-γ–producing CD8+ T cells as an essential cell type required to mediate the antitumor effects of radiotherapy.
Figure 6.
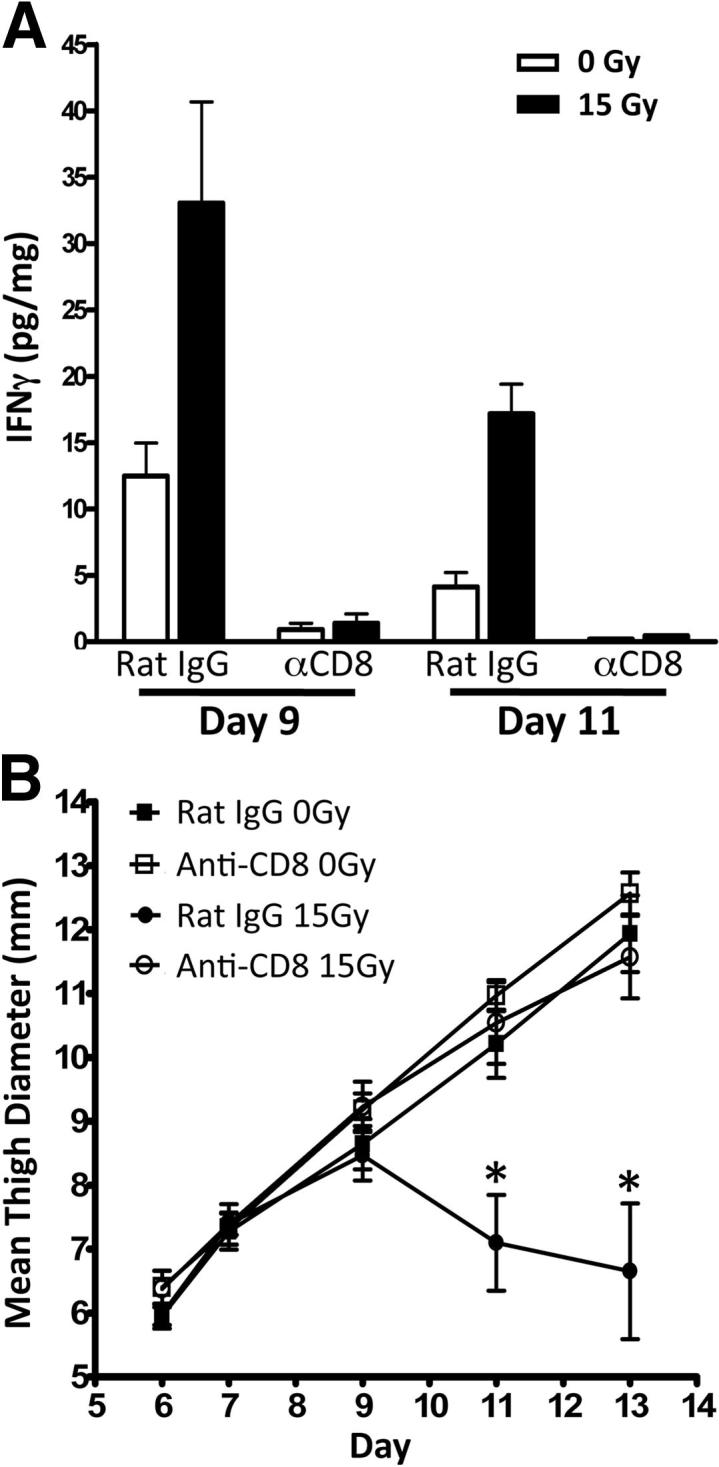
CD8+ T-cell depletion abrogates the antitumor effects of radiation. Mice were treated with 200 μg of anti-CD8 or rat IgG antibodies i.p. 1 day before tumor challenge and every 4 days after. Mice were challenged with 1 × 105 Colon38 cells i.m. and treated with or without 15 Gy radiation 7 days later. A: Tumor homogenate was assayed for IFN-γ by ELISA and normalized to total protein. B: Tumor growth was monitored and presented as mean thigh diameter (mm) (n = 4). ∗P < 0.05 as determined by analysis of variance and the Bonferroni post hoc test.
Discussion
A new and exciting concept has emerged demonstrating that the immune system plays a pivotal role in mediating the antitumor effects of radiation therapy. Previous reports have highlighted the contribution of dendritic cells in the presentation of radiation-induced tumor antigen to the importance of CD8+ T cells in eliciting an antitumor response following radiotherapy.2–4,10 Our report makes a significant contribution to understanding the mechanism of how the consequential immune response following radiotherapy mediates tumor control. In particular, we demonstrated that the Th1 cytokine, IFN-γ, which was predominantly produced by i.t. CD8+ T cells, was essential to elicit the antitumor effects of radiation. This is particularly interesting because recent reports suggest that IFN-γ may be detrimental to tumor control.11 Our studies differed from these reports in that we examined the role of IFN-γ in eliciting tumor control in response to radiation therapy within a different tumor model. In other words, our data clearly show that, in the context of radiotherapy, the presence of i.t. IFN-γ in Colon38 tumors is beneficial and promotes effective antitumor cytotoxic T cells that are functionally active.
We examined IFN-γ production by i.t. immune cells using both transgenic Yeti mice and intracellular staining for protein. An attribute of transgenic Yeti mice is that they report for IFN-γ mRNA through YFP expression; however, this does not necessarily mean that IFN-γ protein is being secreted. Mayer et al24 determined that YFP+ T cells (thus cells making IFN-γ mRNA) required an additional stimulus such as antigen, anti-CD3, or phorbol 12-myristate 13-acetate (PMA)/ionomycin to induce IFN-γ protein secretion. In other words, Yeti mice, through YFP expression, report for cells that have the potential to secrete IFN-γ. This important concept can be applied to our study and is illustrated in Figure 4B where the majority of day 11 i.t. CD8+ T cells (70%) are YFP+ and therefore have the potential to secrete IFN-γ protein. Interestingly, intracellular staining confirmed that only 8% of CD8+ T cells actually stain positive for IFN-γ protein (data not shown). We were surprised to observe that YFP expression does not change in response to radiation even though we detect an increase of IFN-γ protein–producing CD8+ T cells from irradiated tumors. These data suggest that radiation, or the indirect effects of radiation therapy, may provide an additional stimulus/signal that allows cells with the potential to secrete IFN-γ (YFP+) to actually produce and release this protein. It is well established that radiotherapy results in tissue damage and the subsequent production of danger signals similar to pathogen-associated molecular patterns or damage-associated molecular patterns.8 These danger signals have the potential to induce a proinflammatory tumor microenvironment through the production of cytokines such as tumor necrosis factor α (TNF-α), IL-1, and IFN-γ.8 Also, danger signals have been shown to directly alter immune cells. In particular, specific signals such as uric acid, reactive oxygen species, and heat-shock proteins can stimulate the maturation of dendritic cells.8,25,26 Therefore, it is possible that similar danger signals may be acting on CD8+ T cells to stimulate the production of IFN-γ and secretion into the tumor microenvironment.
Our data identify CD8+ T cells as essential in mediating the antitumor effects of radiation. These cells are not only responsible for generating the increase of i.t. IFN-γ following radiation, but are likely the main effector cell type capable of destroying tumor cells via cytolytic activity. Interestingly, the numbers of i.t. CD8+ T cells were not significantly increased in irradiated tumors and, based on four genes measuring effector status (eg, T-bet, Prf1, FasL, and CTLA-4), FACS-sorted CD8+ T cells from irradiated tumors were genetically similar to CD8+ T cells isolated from unirradiated tumors as assessed by real-time PCR (data not shown). Therefore, do differences exist between CD8+ T cells from either untreated or irradiated tumors? Our data suggest that T cells from irradiated tumors exhibit high cytolytic activity against tumor targets immediately after removal without additional culture or stimulation. This implies that the cells are functionally active within the irradiated tumor microenvironment. Importantly, this functional advantage afforded to T cells from irradiated tumors is dependent on IFN-γ. We propose that i.t. CD8+ T cells are stimulated by radiation-induced danger signals (see above) within the tumor microenvironment, which promote the production of IFN-γ protein by these same cells. The IFN-γ then acts back on the T cells in an autocrine fashion, enhancing the cytolytic capacity of these cells. In essence, the CD8+ T cells are modifying a potentially inhospitable tumor microenvironment, through the release of IFN-γ, to one that is more conducive to cytolytic function.
The tumor microenvironment is likely to elicit profound changes, both stimulatory and inhibitory, on the cytolytic capacity of effector T cells. Therefore, in the 51Cr release assay, we made an effort to preserve these effects by purifying T cells using anti-Thy1.1, which will isolate both CD8+ T cells along with CD4+ T cells, some of which could be regulatory cells that may function to suppress CTL function. However, our data in Table 2 demonstrate that although we could detect the presence of FoxP3+, CD4+ Tregs in both the unirradiated and irradiated tumors at each time point, the number of Tregs is decreased as a result of radiation at day 9 (possibly due to the inherent radiosensitive nature of lymphocytes), and remains down in irradiated tumors at day 11 when compared to unirradiated controls. More importantly, the CD8 to Treg ratio at day 11 is 8:1 in unirradiated tumors and further increases to 20:1 in irradiated samples. Therefore, our data suggest that radiotherapy may modify (or alleviate) immunosuppressive component(s) (in this example, Tregs) within the Colon38 tumor microenvironment, thereby promoting CTL lytic function. Additional studies would need to be performed to address this specific question; however, regardless of the mechanism, the data presented here highlight IFN-γ as the main factor in promoting the antitumor effects of radiotherapy.
IFN-γ has been shown to elicit antiproliferative or cytotoxic effects directly on tumor cells. Moreover, the proliferative capacity of Colon38 is greatly diminished in a dose-dependent fashion when these cells are incubated with IFN-γ in vitro (data not shown). However, our DNR data argue against IFN-γ acting directly on Colon38 in an antiproliferative capacity, because tumor growth was not altered in either unirradiated or irradiated DNR-expressing tumors when compared to parental Colon38 (Figure 3B). This, however, should not detract from the importance of the potential direct effects of IFN-γ in other models. We have previously shown that i.t. IFN-γ was required to up-regulate MHC class I molecules on tumor cells following radiation in a B16 tumor model.10 This increase of MHC class I likely plays an important role in providing tumor antigen targets to effector CD8+ T cells, thereby facilitating immune recognition. This is in contrast to our current model in which we demonstrated that IFN-γ does not act directly on tumor cells to promote tumor control, rather its effects may be directed toward enhancing cytotoxic cells. Interestingly, the ex vivo Colon38/DNR tumor cells (unresponsive to IFN-γ) did express lower levels of MHC class I when compared to parental tumors following radiation. However, the expression of MHC class I is inherently abundant on this cell line, and further increases of this molecule appear to have little effect on the ability of radiation to control tumor growth. Overall, these observations highlight important differences in the mode of action of IFN-γ following radiotherapy between tumor models.
The tumor microenvironment contains a variety of cell types, including immune, endothelial, fibroblasts, and other stromal cells, as well as malignant cells.27–29 In aggressive malignancies, complex interactions between each cell type may promote a tumorigenic microenvironment resulting in tumor progression.27 However, certain factors can be introduced intratumorally that could skew an otherwise tumorigenic microenvironment to an immunogenic one. In our study, radiation-induced IFN-γ is an essential factor that skews the response toward tumor destruction, thereby reducing tumor burden. This factor was specifically induced within the tumor microenvironment and not detectable [as assayed by Luminex (Millipore, Billerica, MA) and ELISA] systemically within the serum of these mice (data not shown). Furthermore, our data highlight the complex nature of i.t. IFN-γ within the tumor microenvironment as this cytokine was necessary to stimulate T cells to destroy tumor cells, but was not required to act directly on tumor cells to promote tumor control. Other cell types that contain abundant IFN-γ receptors are present within the tumor stroma and could also be targets for this cytokine.30 In particular, IFN-γ can act directly on endothelial cells to induce cell senescence.31 Additionally, our previous work has demonstrated that IFN-γ up-regulates the adhesion molecule vascular cell adhesion molecule 1 (VCAM-1) on tumor endothelium10 and promotes vessel normalization,22 consequences that may promote antitumor activity by facilitating immune cell infiltration.32,33 Similarly, Lu et al34 demonstrated that stromal fibroblasts are abundant in tumors and enhance tumor growth by stimulating angiogenesis. Interestingly, IFN-γ acted directly on fibroblasts to down-regulate the production of the angiogenic molecule VEGF-A, resulting in angiostasis and tumor control.34
Although 15-Gy radiation treatment resulted in significant tumor control in our model, many of these tumors eventually grew (Figure 1). Therefore, there is considerable room for improvement. Our data clearly warrant further investigation into treatment options that increase the levels of i.t. IFN-γ during radiation treatment. Delivery of high doses of cytokines has been shown to elicit serious side effects when administered systemically.35,36 Therefore, an effective, yet safe, treatment would involve the delivery of this cytokine directly to the tumor microenvironment. Certain technologies such as cytokine-conjugated microspheres or fusion protein technology may be a possible means of targeting IFN-γ to the tumor.37,38 We speculate that the efficacy of radiotherapy can be enhanced by increasing the i.t. levels of IFN-γ at the time of, or shortly after, radiation treatment. Current studies are examining the therapeutic potential of combining radiotherapy with tumor-targeted IFN-γ delivery in the Colon38 tumor as well as other murine tumors to ensure that the effectiveness of the treatment is generalizable. Additional testing will obviously be necessary, but this innovative option has the potential to redefine how radiation oncologists treat malignancies in the clinic.
Acknowledgments
We thank Meghan E. Bushway, Ryan J. Cummings, and Joanne Y.H. Lim for critical discussions in preparation of the manuscript and Dr. Edward Brown for providing Colon38 tumor cells.
Footnotes
Supported by NIH grant RO1CA28332 (E.M.L.).
Supplemental Data
Ex vivo Colon38/DNR cells exhibit continued high level of mutant IFN-γ receptor and reduced MHC class I surface expression. Mice were injected i.m. with 1 × 105 Colon38 or Colon38/DNR.1 cells. Tumors were removed and dissociated into a single-cell suspension 13 days after injection. IFN-γ receptor levels (A) and MHC class I expression (B) on gated ex vivo tumor cells was assessed by flow cytometry and presented as histograms (n = 3).
References
- 1.Lugade A.A., Moran J.P., Gerber S.A., Rose R.C., Frelinger J.G., Lord E.M. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. 2005;174:7516–7523. doi: 10.4049/jimmunol.174.12.7516. [DOI] [PubMed] [Google Scholar]
- 2.Apetoh L., Ghiringhelli F., Tesniere A., Obeid M., Ortiz C., Criollo A., Mignot G., Maiuri M.C., Ullrich E., Saulnier P., Yang H., Amigorena S., Ryffel B., Barrat F.J., Saftig P., Levi F., Lidereau R., Nogues C., Mira J.P., Chompret A., Joulin V., Clavel-Chapelon F., Bourhis J., Andre F., Delaloge S., Tursz T., Kroemer G., Zitvogel L. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 3.Lee Y., Auh S.L., Wang Y., Burnette B., Meng Y., Beckett M., Sharma R., Chin R., Tu T., Weichselbaum R.R., Fu Y.X. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114:589–595. doi: 10.1182/blood-2009-02-206870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takeshima T., Chamoto K., Wakita D., Ohkuri T., Togashi Y., Shirato H., Kitamura H., Nishimura T. Local radiation therapy inhibits tumor growth through the generation of tumor-specific CTL: its potentiation by combination with Th1 cell therapy. Cancer Res. 2010;70:2697–2706. doi: 10.1158/0008-5472.CAN-09-2982. [DOI] [PubMed] [Google Scholar]
- 5.Meng Y., Beckett M.A., Liang H., Mauceri H.J., van Rooijen N., Cohen K.S., Weichselbaum R.R. Blockade of tumor necrosis factor alpha signaling in tumor-associated macrophages as a radiosensitizing strategy. Cancer Res. 2010;70:1534–1543. doi: 10.1158/0008-5472.CAN-09-2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim K., McBride W.H. Modifying radiation damage. Curr Drug Targets. 2010;11:1352–1365. doi: 10.2174/1389450111009011352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Demaria S., Bhardwaj N., McBride W.H., Formenti S.C. Combining radiotherapy and immunotherapy: a revived partnership. Int J Radiat Oncol Biol Phys. 2005;63:655–666. doi: 10.1016/j.ijrobp.2005.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McBride W.H., Chiang C.S., Olson J.L., Wang C.C., Hong J.H., Pajonk F., Dougherty G.J., Iwamoto K.S., Pervan M., Liao Y.P. A sense of danger from radiation. Radiat Res. 2004;162:1–19. doi: 10.1667/rr3196. [DOI] [PubMed] [Google Scholar]
- 9.Formenti S.C., Demaria S. Systemic effects of local radiotherapy. Lancet Oncol. 2009;10:718–726. doi: 10.1016/S1470-2045(09)70082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lugade A.A., Sorensen E.W., Gerber S.A., Moran J.P., Frelinger J.G., Lord E.M. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J Immunol. 2008;180:3132–3139. doi: 10.4049/jimmunol.180.5.3132. [DOI] [PubMed] [Google Scholar]
- 11.Zaidi M.R., Merlino G. The two faces of interferon-gamma in cancer. Clin Cancer Res. 2011;17:6118–6124. doi: 10.1158/1078-0432.CCR-11-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taniguchi K., Petersson M., Hoglund P., Kiessling R., Klein G., Karre K. Interferon gamma induces lung colonization by intravenously inoculated B16 melanoma cells in parallel with enhanced expression of class I major histocompatibility complex antigens. Proc Natl Acad Sci U S A. 1987;84:3405–3409. doi: 10.1073/pnas.84.10.3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gorbacheva V.Y., Lindner D., Sen G.C., Vestal D.J. The interferon (IFN)-induced GTPase, mGBP-2. Role in IFN-gamma-induced murine fibroblast proliferation. J Biol Chem. 2002;277:6080–6087. doi: 10.1074/jbc.M110542200. [DOI] [PubMed] [Google Scholar]
- 14.Brody J.R., Costantino C.L., Berger A.C., Sato T., Lisanti M.P., Yeo C.J., Emmons R.V., Witkiewicz A.K. Expression of indoleamine 2,3-dioxygenase in metastatic malignant melanoma recruits regulatory T cells to avoid immune detection and affects survival. Cell Cycle. 2009;8:1930–1934. doi: 10.4161/cc.8.12.8745. [DOI] [PubMed] [Google Scholar]
- 15.Katz J.B., Muller A.J., Prendergast G.C. Indoleamine 2,3-dioxygenase in T-cell tolerance and tumoral immune escape. Immunol Rev. 2008;222:206–221. doi: 10.1111/j.1600-065X.2008.00610.x. [DOI] [PubMed] [Google Scholar]
- 16.Prendergast G.C. Immune escape as a fundamental trait of cancer: focus on IDO. Oncogene. 2008;27:3889–3900. doi: 10.1038/onc.2008.35. [DOI] [PubMed] [Google Scholar]
- 17.Ostrand-Rosenberg S., Sinha P. Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol. 2009;182:4499–4506. doi: 10.4049/jimmunol.0802740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corbett T.H., Griswold D.P., Jr., Roberts B.J., Peckham J.C., Schabel F.M., Jr. Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res. 1975;35:2434–2439. [PubMed] [Google Scholar]
- 19.Stetson D.B., Mohrs M., Reinhardt R.L., Baron J.L., Wang Z.E., Gapin L., Kronenberg M., Locksley R.M. Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J Exp Med. 2003;198:1069–1076. doi: 10.1084/jem.20030630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerber S.A., Pober J.S. IFN-alpha induces transcription of hypoxia-inducible factor-1alpha to inhibit proliferation of human endothelial cells. J Immunol. 2008;181:1052–1062. doi: 10.4049/jimmunol.181.2.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blieden T.M., McAdam A.J., Frelinger J.G., Lord E.M. Mechanism of cytolytic T lymphocyte killing of a low class I-expressing tumor. J Immunol. 1991;147:1433–1438. [PubMed] [Google Scholar]
- 22.Gerber S.A., Moran J.P., Frelinger J.G., Frelinger J.A., Fenton B.M., Lord E.M. Mechanism of IL-12 mediated alterations in tumour blood vessel morphology: analysis using whole-tissue mounts. Br J Cancer. 2003;88:1453–1461. doi: 10.1038/sj.bjc.6600907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerber S.A., Rybalko V.Y., Bigelow C.E., Lugade A.A., Foster T.H., Frelinger J.G., Lord E.M. Preferential attachment of peritoneal tumor metastases to omental immune aggregates and possible role of a unique vascular microenvironment in metastatic survival and growth. Am J Pathol. 2006;169:1739–1752. doi: 10.2353/ajpath.2006.051222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayer K.D., Mohrs K., Crowe S.R., Johnson L.L., Rhyne P., Woodland D.L., Mohrs M. The functional heterogeneity of type 1 effector T cells in response to infection is related to the potential for IFN-gamma production. J Immunol. 2005;174:7732–7739. doi: 10.4049/jimmunol.174.12.7732. [DOI] [PubMed] [Google Scholar]
- 25.Gallucci S., Matzinger P. Danger signals: sOS to the immune system. Curr Opin Immunol. 2001;13:114–119. doi: 10.1016/s0952-7915(00)00191-6. [DOI] [PubMed] [Google Scholar]
- 26.Shi Y., Evans J.E., Rock K.L. Molecular identification of a danger signal that alerts the immune system to dying cells. Nature. 2003;425:516–521. doi: 10.1038/nature01991. [DOI] [PubMed] [Google Scholar]
- 27.Hanahan D., Coussens L.M. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–322. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 28.Li H., Fan X., Houghton J. Tumor microenvironment: the role of the tumor stroma in cancer. J Cell Biochem. 2007;101:805–815. doi: 10.1002/jcb.21159. [DOI] [PubMed] [Google Scholar]
- 29.Mueller M.M., Fusenig N.E. Friends or foes—bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 30.Valente G., Ozmen L., Novelli F., Geuna M., Palestro G., Forni G., Garotta G. Distribution of interferon-gamma receptor in human tissues. Eur J Immunol. 1992;22:2403–2412. doi: 10.1002/eji.1830220933. [DOI] [PubMed] [Google Scholar]
- 31.Kim K.S., Kang K.W., Seu Y.B., Baek S.H., Kim J.R. Interferon-gamma induces cellular senescence through p53-dependent DNA damage signaling in human endothelial cells. Mech Ageing Dev. 2009;130:179–188. doi: 10.1016/j.mad.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 32.Goel S., Duda D.G., Xu L., Munn L.L., Boucher Y., Fukumura D., Jain R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91:1071–1121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jain R.K. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 34.Lu Y., Yang W., Qin C., Zhang L., Deng J., Liu S., Qin Z. Responsiveness of stromal fibroblasts to IFN-gamma blocks tumor growth via angiostasis. J Immunol. 2009;183:6413–6421. doi: 10.4049/jimmunol.0901073. [DOI] [PubMed] [Google Scholar]
- 35.Car B.D., Eng V.M., Lipman J.M., Anderson T.D. The toxicology of interleukin-12: a review. Toxicol Pathol. 1999;27:58–63. doi: 10.1177/019262339902700112. [DOI] [PubMed] [Google Scholar]
- 36.Mier J.W., Aronson F.R., Numerof R.P., Vachino G., Atkins M.B. Toxicity of immunotherapy with interleukin-2 and lymphokine-activated killer cells. Pathol Immunopathol Res. 1988;7:459–476. doi: 10.1159/000157075. [DOI] [PubMed] [Google Scholar]
- 37.Egilmez N.K., Jong Y.S., Sabel M.S., Jacob J.S., Mathiowitz E., Bankert R.B. In situ tumor vaccination with interleukin-12-encapsulated biodegradable microspheres: induction of tumor regression and potent antitumor immunity. Cancer Res. 2000;60:3832–3837. [PubMed] [Google Scholar]
- 38.Puskas J., Skrombolas D., Sedlacek A., Lord E., Sullivan M., Frelinger J. Development of an attenuated interleukin-2 fusion protein that can be activated by tumour-expressed proteases. Immunology. 2011;133:206–220. doi: 10.1111/j.1365-2567.2011.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Ex vivo Colon38/DNR cells exhibit continued high level of mutant IFN-γ receptor and reduced MHC class I surface expression. Mice were injected i.m. with 1 × 105 Colon38 or Colon38/DNR.1 cells. Tumors were removed and dissociated into a single-cell suspension 13 days after injection. IFN-γ receptor levels (A) and MHC class I expression (B) on gated ex vivo tumor cells was assessed by flow cytometry and presented as histograms (n = 3).