

Abstract
The central theme of personalized medicine is the premise that an individual’s unique physiologic characteristics play a significant role in both disease vulnerability and in response to specific therapies. The major goals of personalized medicine are therefore to predict an individual’s susceptibility to developing an illness, achieve accurate diagnosis, and optimize the most efficient and favorable response to treatment. The goal of achieving personalized medicine in psychiatry is a laudable one, because its attainment should be associated with a marked reduction in morbidity and mortality. In this review, we summarize an illustrative selection of studies that are laying the foundation towards personalizing medicine in major depressive disorder, bipolar disorder, and schizophrenia. In addition, we present emerging applications that are likely to advance personalized medicine in psychiatry, with an emphasis on novel biomarkers and neuroimaging.
Keywords: Major depressive disorder, Schizophrenia, Personalized medicine, Psychiatric hereditability, Epigenetics, Environmental factors, Endophenotypes, Pharmacogenomics, Neuroimaging genetics
Introduction
The foundation of personalized medicine centers on the assumption that an individual’s unique characteristics play a significant role in tailoring their therapies. Such characteristics include: genetic alterations and epigenetic modifications, clinical symptomatology, observable biomarker changes, and environmental factors [1]. The goals of personalized medicine are to predict the individual’s susceptibility to disease, achieve an accurate diagnosis, and result in an efficient and favorable response to treatment (Figure 1). Although there are clearly some very successful examples of personalized medicine, especially in oncology, relatively few such examples exist in psychiatry [2]. In this review, we summarize an illustrative selection of the advancements toward personalizing medicine in major depressive disorder (MDD), bipolar disorder (BD), and schizophrenia (SZ). We also discuss some new approaches currently being used and how they are likely to affect the field in the years to come.
Figure 1.

Personalized medicine. Forefront shows the schematic of the various factors that play into developing a unique phenotypic profile: genetic alterations, epigenetic modifications, clinical diagnostics, biomarker changes, and environmental changes. Upon obtaining a unique phenotypic profile, the psychiatrist is in a better position to either predict susceptibility to disease or make an accurate diagnosis. This, is in turn, allows for therapy targeted to the individual. Background: each individual will have differences in these components, giving rise to a unique phenotypic profile.
Contributing factors to psychiatric heritability
Genetics
One major expectation of personalized medicine is the ability to determine susceptibility or protective factors imparted through genetic change. Interestingly, in the era of genome-wide association studies (GWAS), the majority of replicable findings do not pinpoint common genes underlying susceptibility or protection from disease; instead, our current understanding centers primarily on rare genetic variants, although a number of common variants have furthered understanding as well. However, both common and rare variants account for relatively small percentages of heritability, and far greater percentages are still attributed to ‘missing heritability’. Of the diseases reviewed in this report, no variation confers an autosomal dominant mendelian inheritance typical of Huntington’s disorder [3]; moreover, no single genetic change has shown an effect on heritability percentage that is in double digits. Nonetheless, there have been seminal genetic findings, each deepening our understanding of the major psychiatric illnesses. A sample of such findings in MDD, BD, and SZ is given below.
Genetics: major depressive disorder
MDD has a strong genetic component, with an estimated 40 to 70% of the risk for developing MDD thought to be genetic [4]. Prominent findings in susceptibility studies of MDD include several polymorphisms in the serotonergic system, and in various elements of the hypothalamic-pituitary-adrenocortical (HPA) axis.
Genes involved in the serotonergic neural system have been intensively scrutinized in candidate gene and linkage studies in MDD for a number of reasons: this system is a major target of several antidepressants, and patients with MDD have alterations in multiple components of the serotonin system [5,6]. Polymorphisms and variable number tandem repeat regions (VNTR) in the 5-hydroxytryptamine (5HT; serotonin) transporter (5-HTT) gene have been associated with development of MDD. In 1996, Ogilvie and colleagues identified three novel alleles of a 5-HT VNTR region, and identified an association between the nine-copy VNTR allele and risk for developing MDD [7]. In a group of 466 German patients with MDD and 836 controls, Hoefgen et al. [8] reported a significant increased frequency of a 44-base pair insertion/deletion polymorphism in the 5′ promoter region of the 5-HTT gene (5-HT transporter-linked polymorphic region; 5-HTTLPR) in patients with MDD relative to controls. Furthermore, this 5-HTTLPR polymorphism has been studied extensively both in the relationship between MDD and environmental factors and in the pharmacologic response to treatment (as discussed in the sections on ‘Environmental factors’ and ‘Prediction of treatment response,’ respectively).
A rate-limiting enzyme involved in serotonin synthesis, tryptophan hydrolylase (TPH), has been implicated in susceptibility for MDD by a number of reports, although attempts at replication have shown discordant findings [9]. Homologs 1 and 2 of the TPH gene (TPH1 and TPH2, respectively), have both been associated with MDD susceptibility. Although both TPH1 and TPH2 are involved in serotonin synthesis, their distribution is markedly different. TPH1 is found primarily in the periphery, with effects on melatonin synthesis, hemostasis, and immune system function. Conversely, TPH2 is expressed in the CNS, with central effects on sleep, aggression, food intake, and mood [10]. Two groups [11,12] analyzed TPH1 single-nucleotide polymorphism (SNP) and haplotype differences between participants with depression and control participants. In the former study, Nash et al. related quantitative phenotypes of depression in a community-based sample of 119 sibling groups to genetic alterations in TPH1, and identified a significant association between MDD susceptibility and a microsatellite downstream of TPH1[11]. In the latter study, Gizatullin et al. conducted a genetic screen of 228 patients with MDD and 253 healthy controls, and identified six haplotypes that were associated with MDD [12].
With respect to TPH2, Zill et al. [13] identified two SNPs (rs1386494 and rs1843809) associated with MDD in a sample of 300 Caucasian patients with MDD and 265 healthy controls. Subsequently, Zhang et al. searched for a novel loss-of-function mutation, G1463A, which they had identified and characterized in the previous year in sample of 87 patients with MDD and 219 controls [14,15]. The authors found that this mutation was significantly more common in patients with MDD than in controls, and suggested that defects in brain serotonin synthesis may be an important contributor to MDD susceptibility [15]. A subsequent study by Garriock et al. [16] failed to replicate these findings in a population similar in ethnicity and gender distribution to that of Zill et al. [13] Serretti et al. evaluated TPH2 SNPs in MDD, BD, and SZ in Korean psychiatric inpatients and controls; their findings suggested that TPH2 SNPs are not associated with MDD, BD, or SZ [17]. In a meta-analysis of TPH2 genetic polymorphisms and MDD, Gao et al. examined 74 TPH2 SNPs published through the end of October 2011, and using fixed-effects modeling, they found that two SNPs, rs4570625 and rs17110747, were associated with MDD susceptibility. The relationship between SNP rs4570625 and MDD was more robust, remaining significant using more conservative random-effects calculations [9]. Notably, this SNP was not one of the SNPs reported in the Zill et al. or Zhang et al. studies, although there have been later reports suggestive of a minor role for rs4570625 in MDD as well as in SZ, panic disorder, obsessive-compulsive disorder, and attention-deficit hyperactivity disorder [18-22].
For almost five decades, hyperactivity of the HPA axis has been reported, and this is suggested to be contributory to depressive symptomatology [23]. Accordingly, the components of the HPA axis provides numerous genes that might be associated with risk for MDD, including, but not limited to, a key component of the glucocorticoid receptor complex, FK506-binding protein 5 (FKBP5), corticotropin-releasing hormone receptor 1 (CRHR1) and corticotropin-releasing hormone-binding protein (CRHBP) [1]. In 2004, Binder and colleagues evaluated several genes that might be responsible for the HPA hyperactivity characteristic of depression. They identified SNPs in FKBP5 associated with increased recurrence of MDD episodes and to a more rapid therapeutic response to antidepressant therapy (discussed in the section ‘Prediction of treatment response’) [24]. A 2009 study by Tatro et al. genotyped two SNPs in FKBP5, rs3800373 and rs1360780, in 60 frozen brain samples distributed over five clinical groups: MDD, MDD with psychosis, HIV-positive with MDD, and HIV-positive and HIV-negative controls. The rs3800373-CC genotype was found significantly more frequently in the MDD and MDD with psychosis groups than would be expected based on published allelic frequencies. The authors also reported that rs1360780 allele frequencies differed significantly from the expected allelic frequencies in the MDD and MDD with psychosis groups [25]. Using a sample of 155 European adolescents with MDD from the TORDIA (Treatment of SSRI (selective serotonin reuptake inhibitor)-Resistant Depression in Adolescents) trial, Brent et al. provided preliminary reports of two FKBP5 genotypes (rs1360780-TT and rs3800373-GG) significantly associated with suicidal events [26].
CRHR1 codes for a G-protein-coupled receptor involved in the regulation of the HPA axis by mediating the effects of corticotropin-releasing hormone (CRH) [27]. Raised levels of CRH in regional brain and cerebrospinal fluid (CSF) are a consistently replicated finding in patients with depression, and is also seen in suicide victims [28-31], rendering CRHR1 an attractive candidate gene for MDD susceptibility. In 2006, Liu et al. identified three SNPs in CRHR1, which were significantly more common in a group of 206 Han Chinese patients with MDD compared with 195 controls matched for age, gender and ethnicity [32]. Subsequently, Papiol et al., compared CRHR1 SNP frequencies in 159 Spanish outpatients with MDD and 96 healthy controls, and found an association between the CRHR1 SNP rs110402 and early age of MDD onset. This SNP was also associated with increased risk for a seasonal pattern of illness [33]. Following these findings, Lekman et al. analyzed clinical data from 1,809 outpatients with MDD and a collection of 739 ‘Black’ and ‘non-Hispanic White’ ethnically matched controls enrolled in the STAR*D (Sequenced Treatment Alternatives to Relieve Depression) study. These authors found that the SNP rs1360780 was associated with MDD susceptibility in the non-Hispanic White sample, and the SNP rs4713916 was associated with disease remission to citalopram in the overall patient sample [34].
A consistent finding that clearly contributes to the HPA axis hyperactivity in MDD is hypersecretion of CRH. The CRH-binding protein (CRHBP) regulates the availability of CRH, both centrally and in the systemic circulation, which modulates HPA axis activity. SNPs in CRHPB were first associated with MDD in a case–control study conducted in a Swedish population [35]. Claes et al. examined 89 Swedish patients with recurrent MDD and 88 control samples matched for age, gender and ethnicity, and found two SNPs that were marginally associated with MDD. The authors reported one haplotype block comprised of the CRHPB SNPs s02-TT, s11-TT, and s14-T, whose presence significantly increased susceptibility to MDD [35]. In 2007, Van Den Eede et al. sought to replicate the CRHBP Swedish study findings in an extended Swedish sample, and in a larger and ethnically distinct sample (Belgian population). They analyzed 317 patients with MDD and 696 controls, but were unable to detect any statistically significant association (capable of withstanding correction for multiple testing) between the CRHBP SNPs in either the extended Swedish sample or Belgian sample [36].
Genetics: bipolar disorder
Evidence from family, twin, and adoption studies show that BD is highly heritable, with genetic variables estimated to account for 60 to 85% of risk [37]. Efforts to discover the genetic sources for BD risk have led to innumerable linkage studies, some of which have identified promising susceptibility loci. However, these linkage studies have been fraught with inconsistent replication and indeterminate genetic causes of increased linkage signal [38,39]. Hence, there have been increasing efforts toward association studies. As with MDD, genes of the HPA axis have been probed for candidates increasing susceptibility to BD. Willour et al. genotyped FKBP5 SNPs in a family sample of 317 BD pedigrees and 554 affected offspring. They found evidence for an association with BD for five SNPs (rs4713902, rs7757037, rs9296158, rs3800373, and rs9380525), with rs4713902 showing the most robust signal (P= 0.0001). Furthermore, Willour and colleagues identified four SNPs (rs1043805, rs3800373, rs9296158, and rs1360780) in covariate-based analyses that showed differential association with BD depending on the covariates of attempted suicide and/or the number of depressive episodes [40]. However, other studies were unable to replicate the findings of association of BD with FKBP5[41,42]. Subsequent GWAS in BD have contributed modest evidence of BD susceptibility attributable to SNPs in FKBP5[43,44].
Many candidate genes have been derived from the dopaminergic, serotonergic, and noradrenergic systems, based on the evidence for a role of these circuits in BD pathogenesis. In particular, the genes encoding 5-HTT, monoamine oxidase A (MAOA) and catechol-O-methyltransferase (COMT) have generated both positive and negative association findings with BD. However, there is no conclusive evidence for indisputable association of any of these genes with BD susceptibility [39]. Circadian rhythm disturbances are commonly seen in BD, which has led to multiple studies of association between circadian rhythm genes and BD [38,45,46]. The genes coding for aryl hydrocarbon receptor nuclear translocator-like BmaL1 (ARNTL) and circadian locomotor output cycles kaput (CLOCK) are two genes partially responsible for control of the internal circadian clock in the suprachiasmatic nucleus of the hypothalamus [47]. Both of these genes have been identified in association studies of BD. For example, Mansour et al. compared 234 Caucasian individuals with BD with 180 community-based controls in a BD association study. The authors genotyped 44 SNPs from eight circadian rhythm genes: ARNTL, CLOCK, Period 1, 2, and 3 (PER1, PER2, PER3), cryptochrome 1 and 2 (CRY1 and CRY2) and TIMELESS. They found modest associations with SNPs for ARNTL and TIMELESS, although they cautioned that additional studies are necessary to corroborate the findings [48]. Shi et al. conducted an association study of 10 circadian genes using the Sibling-Transmission Disequilibrium Test (sib-tdt) in an extended family collection (composed of 70 trios and 237 quads) in BD, and identified nominally significant association of three SNPs near or within the CLOCK gene; however, these associations did not survive correction for multiple testing [46].
GWAS in BD have identified four genes for further study that had SNPs of genome-wide statistical significance: calcium channel, voltage-dependent, L type alpha 1C subunit (CACNA1C), ankyrin 3 (ANK3), neurocan (NCAN) and odd Oz/ten-m homolog 4 (ODZ4) [49-51]. Although not significant at the genome-wide level, spectrin repeat containing, nuclear envelope 1 (SYNE1), has been recently associated with BD and with recurrent MDD [52]. Green et al. tested SYNE1 SNP rs9371601 in 1,527 subjects with BD and compared them with 1,579 non-psychiatrically screened controls, finding evidence for the association of the SNP with BD (P= 0.0095) [52]. Furthermore, they identified a significant association between the SNP and recurrent MDD in a sample of 1,159 subjects with recurrent MDD compared with 2,592 controls (P= 0.032). SYNE1 codes for Nesprin-1, a protein comprising part of the scaffolding that links the nucleoskeleton to the cytoskeleton (LINC) [53]. The association findings in SYNE1 relating to BD and to MDD are likely to spark subsequent genetic and functional studies.
Genetics: schizophrenia
With estimates of heritability of 50 to 80%, SZ is one of the most heritable of the disorders discussed in this paper [54,55]. Innumerable candidate gene studies and more than a handful of GWAS have contributed to the impression that SZ, despite its high heritability, is genetically complex, probably with a large polygenic component explaining a substantial amount of susceptibility [56]. In fact, the International Schizophrenia Consortium (ISC) tested a polygenic model for SZ by summarizing nominally significant associations in their GWAS data into quantitative scores. They then related these derived scores to disease states in three large, independent target samples [57]. From these analyses, the ISC concluded that one-third of genetic susceptibility for SZ lies in the collective effect of hundreds or thousands of common polygenic variants, each contributing small effects [56,57]. GWAS are poised to reveal such variants, and the results of the SZ GWAS are largely in agreement with the ISC assessment. Three comprehensive analyses of GWAS data from the ISC, Molecular Genetics of Schizophrenia (MGS) and the Schizophrenia Genetics Consortium (SGENE) have implicated a few discrete genes with odds ratios in the range of 0.73 to 1.23 [56-59]. These analyses also implicated the genome region (between 26 and 33 million bp) containing the major histocompatibility complex (MHC), a group of genes that encodes proteins necessary for the immune system to recognize foreign substances [56-59]. Clarifying the relationship between the MHC genes and SZ continues to be an area of active research.
GWAS and linkage studies have homed in on possible SZ-associated genes and genetic regions outside of the MHC region that have garnered interest, including the zinc finger 804A (ZNF804A) gene. The 2008 study of O’Donovan et al. GWAS identified 12 loci with ‘moderately strong’ (P<10-5) evidence for association in their primarily European-ancestry sample of 479 SZ and 2,937 controls. Subsequently, one SNP within the intron of the ZNF804A gene, rs1344706, remained significant in their larger replication samples (6,666 SZ subjects and 9,897 controls, including 1,782 Chinese and Japanese SZ subjects, and 1,865 controls) at the more stringent ‘strong’ threshold of P<5 × 10-7 (P = 1.61 × 10-7) although not at genome-wide significance threshold (P<7.2 × 10-8) [60]. The authors then broadened their phenotype to include BD, and found a strengthening of this association beyond the genome-wide significance threshold (P = 9.96 × 10-9) [60]. Several independent replication studies identified at least nominal associations between ZNF804A and SZ [61-63]. A 2010 case–control study by Riley et al., compared 1,021 Irish subjects with SZ or poor-outcome schizoaffective disorder with 626 Irish control subjects, and reported nominally significant association between SZ and ZNF804A rs1344706 (P = 0.0227) and two other ZNF804A SNPs (rs17508595 at P = 0.0230 and rs7597593 at P = 0.0013) [61]. Steinberg and colleagues (2011) reported an association between rs1344706 and SZ (P = 0.0029) and SZ plus BD (P = 0.00065) in their sample (European ancestry with Chinese in follow-up study). Notably, when stratified by population, only two of the thirteen ethnicities, that is, Danish and Russian, exhibited a significant association between rs1344706 and SZ [62]. Zhang et al. conducted a population case–control study and a family-based study in a Han Chinese population. They reported a significant over-representation of the T allele of rs1344706 in their SZ population relative to controls in the population-based study (P = 0.00083) and a trend toward positive association in their trios (P = 0.058) [63]. In the wake of these findings, Williams et al. performed a meta-analysis of the data from the discovery GWAS along with data from thousands of additional European-ancestry samples from in-house sources, five GWAS (ISC, MGS, SGENE-plus, the Lencz et al. GWAS, one unpublished Swedish GWAS from Sullivan and Hultman [57-59,64,65]), and a handful of collaborative groups (totaling 18,309 SZ cases and 35,739 controls). The meta-analysis supported the original findings for association between rs1344706 and SZ (P = 2.5 × 10-11; odds ratio (OR) = 1.10; 95% confidence interval (CI) 1.07 to 1.14) and with SZ plus BD (P = 4.1 × 10-13; OR = 1.11; 95% CI 1.07 to 1.14) [65]. To date, ZNF804A remains one of the most promising of the GWAS findings in SZ.
Another gene with genome-wide association with SZ is transcription factor 4 (TCF4), which is a gene important for neurodevelopment [59,66,67]. Mutations in this gene have been linked to Pitt-Hopkins syndrome, a neurodevelopmental condition characterized by mental retardation, epilepsy, hyperventilation episodes, and distinct facial features [67,68]. With respect to SZ, one TCF4 intronic SNP (rs9960767; C allele), was found to be significantly associated in a GWAS and analysis of nearly 50,000 subjects (data from SGENE-plus, follow-up samples, ISC and MGS GWAS samples) (P = 4.1 × 10-9; OR = 1.23; 95% CI 1.15 to 1.32) [59]. Steinberg et al. (2011) expanded on these findings, evaluating SNPs that had P-values of less than 1 × 10-4 in the combined sample (SGENE-plus, MGS, ISC dataset) of Stefansson et al. [69]. In the Steinberg et al. study, an additional 9,246 Caucasian SZ samples (SZ, schizoaffective disorder, or persistent delusional disorder) and 22,356 controls were assessed, followed by analysis of another 1,014 SZ and 1,144 control samples in a subsequent follow-up. These analyses implicated the C allele of the original rs9960767 SNP in SZ, in the primary, but not in the secondary follow-up (P = 2.5 × 10-4; OR = 1.16; 95% CI 1.07 to 1.25 and P = 0.5; OR = 1.09; 95% CI 0.85 to 1.41, respectively) [69]. Considered together, the Stefansson et al. combined sample and the two follow-up studies yielded a genome-wide significant association of rs9960767-C with SZ (P = 4.2 × 10-9; OR = 1.20; 95% CI 1.135 to 1.27) [69]. In 2011, Ripke et al. completed a replication study in Caucasians that also identified association of TCF4 with SZ (P = 2.29 × 10-5), albeit with a different SNP, rs12966547 (which is not in linkage disequilibrium (LD) with rs9960767) [70]. Analysis of their replication data pooled with their discovery sample (which has significant overlap with the Stefansson et al. sample), yielded an association of genome-wide significance (P = 2.6 × 10-10) [70]. Notably, a TCF4-SZ association has also been established in a non-Caucasian population. Li et al. explored the TCF4 gene in a Han Chinese population of 2,496 subjects with SZs and 5,184 controls [71]. Interestingly, the TCF4 SNP highlighted by Stefansson et al. is polymorphic in Caucasians, but is not polymorphic in the Chinese population [59,71]. Instead of using this SNP, Li et al. substituted a nearby SNP, rs2958182 (which is in complete LD with rs9960767, and is polymorphic in the Chinese population). This group found rs2958182 to be significantly associated with SZ in their Chinese population (P = 3.64 × 10-6), which provides additional support for a TCF4-SZ association [71]. Together, these findings continue to fuel genetic and functional exploration of TCF4 in SZ.
Neuregulin 1 (NRG1) is a gene that is also involved in many neurodevelopmental functions, including regulation of the expression and activation of glutamate receptors, neuronal migration, and oligodendrocyte development [72-76]. Mutant mice heterozygous for NRG1 (or for its receptor, ERBB4) have fewer NMDA receptors than their wild-type littermates. Moreover, they exhibit a SZ-like phenotype, which is reversible with the atypical antipsychotic drug, clozapine [77]. Linkage studies in the 1990s and early 2000s originally highlighted the cytogenic region containing the NRG1 gene, 8p22-21, as a possible ‘schizophrenia susceptibility locus’ (SSL) [78-81]. A more specific gene-disease association was first suggested in a 2002 Icelandic linkage study in which a NRG1 haplotype, HapICE (composed of five SNPs and two microsatellite markers), was found significantly more often in SZ samples than in control samples [77]. These initial findings spurred more than 60 additional studies of NRG1 as a putative schizophrenia risk gene, yielding mixed findings [82]. By 2011, several large case–control studies [83-88] (>1000 subjects) had found at least one positive NRG1-SZ association, whereas comparably sized studies found no association at all [89-94]. NRG1 analyses in large family-based studies (>200 families) fared similarly, with nearly equal positive [95-99] and negative [41,100-102] association findings. Meta-analyses by Munafò and colleagues in 2005 and updated in 2008 [103,104], noted a gene-level association between NRG1 and SZ, but their data did not support an association between any particular NRG1 SNP or haplotype with the disease. The results suggest that analyses at the level of the gene, rather than the level of an individual SNP, functional variant, or haplotype, might be a more reliable method of analysis [104].
Over 8,000 polymorphisms spaced over more than 1,000 candidate genes have been systematically analyzed in SZ (full results of the analyses can be found in the comprehensive SZ association study database, SZGene; http://www.szgene.org) [82]. Notably, Allen et al. highlighted SNPs in 16 genes with at least nominally significant effects (OR ranging from 0.7 to 1.52): DRD2, GRIN2B, TPH1, DTNBP1, MTHFR, DRD1, APOE, DRD4, COMT, TP53, HP, DAO, SLC6A4, GABRB2, PLXNA2, and IL1B. [82]. One of these genes receiving a great deal of attention is COMT, in part because of its known functions, and in part due to its location in the genome. The COMT gene encodes a protein responsible for the metabolic degradation of catecholamines. In addition, COMT is located on chromosome 22, within a region that when deleted causes velocardiofacial syndrome. Notably, nearly 25% of the individuals inheriting the large deletion at 22q11.21 go on to develop SZ, making the inheritance of this deletion one of the greatest risk factors for development of SZ [105]. In 2002, Shifman et al. found a significant association between a COMT haplotype and SZ in a case–control study in Ashkenazi Jews [106]. One 5′-UTR SNP involved in the haplotype (rs737865) has been implicated in a subsequent studies, and despite both positive and negative findings, maintains nominal significance by SZGene meta-analysis, with a modest OR of 1.06 (95% CI 1.01 to 1.11, P<0.05) [82].
The disrupted-in-schizophrenia (DISC) locus has also been associated with SZ after its initial characterization in a large Scottish family with high psychiatric load [107]. The DISC locus was identified at the breakpoint of a balanced translocation between chromosomes 1 and 11. The translocation significantly segregated with a range of psychiatric illness in the family, including SZ, MDD, and BD [107]. Further analysis showed that the translocation is in significant LD with SZ, outside of the effects of the other psychiatric illnesses (log of the odds ratio score being 3.6) [108]. Fine mapping focused further studies on two genes, DISC1 and DISC2, identified along the chromosome 1 component of the translocation [109]. DISC1 codes for a protein known to have diverse roles, including regulation of neuronal axonal and dendritic outgrowth, cell proliferation, and cell differentiation [110]. DISC2 is a non-protein-coding gene thought to be important in the regulation of DISC1[108,111]. The majority of DISC1 family and association studies have had positive findings of a relationship between the gene and SZ [112-130]. However, there have also been a number of studies which have not been able to show any significant findings [41,93,131]. To date, there have not been any positive association studies for DISC2 and SZ despite one large association study of 1,054 Scottish individuals (328 with SZ, 726 controls), which did not find any significant associations between DISC2 SNPs and SZ [132].
Epigenetic modifications
Although genetics clearly play an important role in personalized medicine, the role of epigenetics should not be overlooked. Epigenetics refers to the phenomenon of heritable changes to gene regulation caused by mechanisms that do not involve modification of the nucleotide sequence. Increasingly, it is being realized that these mechanisms are far more complex than originally envisioned, requiring adjustment of the current perspective on the central dogma of molecular biology; namely, that DNA transcribes RNA, which is subsequently translated into protein [133]. It is estimated that approximately 98% of the human genome is not translated into protein, and these epigenetic changes occur within the framework of DNA being transcribed into both coding and non-coding RNA, with the subsequent translation of coding RNA into protein, and the concurrent regulation of each of these steps by non-coding RNA [134,135] (Figure 2). Specific epigenetic mechanisms include DNA methylation, histone modification, alternative splicing, RNA editing, and non-transcriptional gene silencing via microRNAs [136]. Epigenetic regulation has been implicated in a myriad of neural processes, from learning and memory to seizures and neurogenesis, and recently to depression, chronic stress, and various addictions [137,138].
Figure 2.
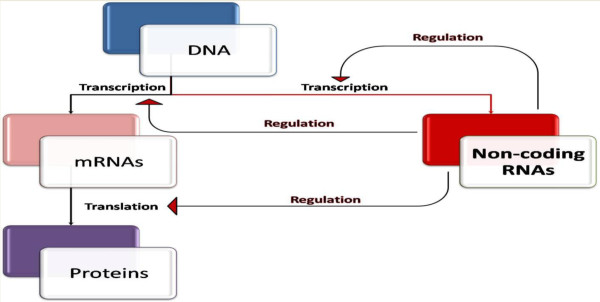
‘Modified’ central dogma. The central dogma of molecular biology states that DNA is transcribed to RNA, which is then translated into protein. In light of the emerging importance of non-coding RNAs, this diagram shows how non-coding RNAs serve to regulate each step in the central dogma, including regulating their own transcription.
Epigenetic modifications: major depressive disorder
Study of the epigenetics of MDD, particularly involving DNA methylation and histone modification, has generated some interesting findings [6]. Electroconvulsive therapy (ECT), the induction of controlled seizures to relieve depressive symptoms, can be remarkably effective in severe treatment refractory cases of depression [139]. The effectiveness of ECT requires multiple treatments, and without continued antidepressant therapy or maintenance, nearly all patients will have a relapse of their symptoms [138,139]. Epigenetic changes are thought to partially account for this phenomenon.
In animal models of depression, chromatin remodeling of brain-derived neurotrophic factor (BDNF) promoters leads to site-specific increased BDNF transcription, a change shown to mediate susceptibility to stress in humans and in mouse models [6,140]. Another notable example of epigenetic changes is seen in rats reared by mothers who exhibit especially high levels of nurturing behaviors of pup licking and grooming (LG) [141]. The authors described the adult rat offspring from high-LG mothers as being less anxious than their low-LG counterparts. The high-LG offspring also displayed corresponding measurable changes to the HPA axis (for example, increased glucocorticoid receptor mRNA and protein expression, decreased hypothalamic corticotropin-releasing factor (CRF) mRNA expression and increased sensitivity to glucocorticoid negative feedback), changes that are due, in part, to gene-expression changes that remain stable until adulthood. Of note, these gene-expression alterations are reversible with cross-fostering of rats between high-LG and low-LG mothers [138,141-143]. Building on these principal findings, McGowan et al. [144] explored the epigenetic regulation of a neuron-specific glucocorticoid receptor gene (nuclear receptor subfamily 3, group C, member 1; NR3C1) in human suicide victims subjected to childhood abuse. In their study, they compared post-mortem hippocampal tissue samples from 12 suicide victims with a history of childhood abuse, 12 suicide victims with no history of childhood abuse, and 12 controls (persons experiencing sudden, accidental death without a history of childhood abuse). They found that the expression of total glucocorticoid receptor mRNA was significantly lower in the abused suicide victims than in either the non-abused suicide victims or the controls. Furthermore, the level of methylation of the NR3C1 promoter sites was significantly higher in the abused suicide victims compared with the non-abused suicide victims or controls. Notably, in both of these findings, there were no significant differences between the non-abused suicide victims and the controls. The authors report that these findings are consistent with those in animal models showing epigenetic modifications of the glucocorticoid receptor leading to increased HPA responses to stress. Moreover, they speculate that early adverse life events, such as childhood abuse, may lead to epigenetic changes that contribute to risk for developing psychopathology [144].
Epigenetic modifications: bipolar disorder and schizophrenia
Because many epigenetic studies of BD also include patients or tissue samples with SZ, the epigenetic studies involving both disorders are summarized here. The role of methylation in SZ has been suggested for over 50 years, when patients with SZ who were administered a methylating agent, methionine, were noted to experience exacerbation of their psychosis [145,146]. Since that time, many post-mortem brain studies have focused on methylation patterns in SZ. For example, differential methylation patterns in the regulatory regions of COMT and reelin (RELN) have been found between individuals with SZ compared with controls. From a functional standpoint, dysregulation of either of these genes could feasibly result in psychopathology. COMT is an important gene for metabolizing catecholamines, while RELN encodes an extracellular matrix glycoprotein theorized to control cell-cell interactions during neuronal positioning and migration during brain development [147]. Abdolmaleky and colleagues analyzed 115 post-mortem brain samples from the frontal lobe of individuals with BD or SZ and controls (35 samples per group). The membrane-bound (MB)-COMT promoter was hypomethylated significantly more often in the brains of individuals with SZ or BD relative to the brains of controls [148]. Generally, methylating DNA sequences tends to decrease their transcription, and accordingly, this hypomethylation in the BD and schizophrenic brains corresponded with higher MB-COMT transcript expression than seen in the controls [148]. The authors remarked that the resulting overexpression of MB-COMT could lead to significant increases in dopamine degradation. Moreover, the resulting hypodopaminergic state and frontal-lobe hypoactivity may be amenable to COMT-inhibitor drugs or preventative strategies to modulate methylation of the COMT promoter [148]. Lower levels of RELN mRNA expression and protein concentrations were found in post-mortem brain samples from patients with schizophrenia relative to control brain samples, which led to investigations of whether promoter regions are differentially methylated in such patients relative to controls [149]. In a small (n = 10) post-mortem study of schizophrenic brains compared with controls, Abdolmaleky et al. found a significant increase in DNA methylation of CpG islands in the RELN promoter in brains from individuals with SZ compared with controls [150]. However, neither set of findings (the hypomethylation of the COMT regulatory regions and the hypermethylation of the RELN regulatory regions) have been replicated in subsequent studies [151,152].
Some studies have suggested that epigenetic dysregulation of the 5-HT2A receptor gene (HTR2A) may contribute to the pathophysiology in SZ. The focus on HTR2A is not surprising, as it has been the subject of many studies, which have provided conflicting evidence for a small, but significant, association of an HTR2A SNP, rs6313 (c.102C>T), with SZ in European populations [82,153-155]. This 102-nucleotide position is near the promoter region, and is thought to be a putative methylation site, which is consistent with the lower levels of 5HT2A mRNA seen with the C allele (in both patients and controls). Indeed, in a study on post-mortem human brain tissue, Polesskaya et al. reported that one of the key determinants of HTR2A expression was cytosine methylation at rs6313 [156]. Notably, this decrease in HTR2A mRNA is a property that is exaggerated in SZ [153,154]. Furthermore, antipsychotic treatment decreases methylation at the 102C polymorphic site, while HTR2A mRNA expression increases [157]. Interestingly, this SNP is in complete LD with a promoter SNP, rs6311 (c.-1438G>A), for which there is also some evidence in favor of an association with SZ in Caucasian samples (SZGene 2010 meta-analysis: OR = 1.20; 95% CI 1.07 to1.36]) [82,158]. Carrying the minor allele in rs6311 results in the creation of a methylation binding site for the E47 transcription activator at position −1,438, and results in the loss of a CpG binding site at position −1,439 [158]. These findings indicate how genetic alteration can trigger a chain of downstream epigenetic effects. Clearly, deciphering the effects of HTR2A gene-expression dysregulation in SZ will continue to be a rich source of study.
Histone modification is another epigenetic mechanism hypothesized to contribute to the pathogenesis of SZ and BD. Post-mortem studies have shown increased levels of histone deacetylase, type 1 (HDAC1), an enzyme generally responsible for silencing gene expression, in the pre-frontal cortices of patients with SZ [159-161]. Sharma et al. found a strong negative correlation between HDAC1 and GAD67 mRNA, which encodes an isoform of glutamate decarboxyase (GAD) [160]. Taken together, these findings provide a possible mechanism for the common observation of decreased expression of GAD, the rate-limiting enzyme involved in the synthesis of gamma-aminobutyric acid (GABA) in the brains of patients with schizophrenia or BD [162-165]. Furthermore, in an animal model that used methionine to induce SZ-like behavioral abnormalities, HDAC inhibitors (HDACi) were sufficient to attenuate the behavioral abnormalities [166]. Valproate is a mood stabilizer and a potent HDACi, whose mood-stabilizing effects in BD and SZ may be based on its epigenetic properties [167].
MicroRNAs (miRNAs) are important contributors to epigenetic modifications. Specifically, miRNAs are non-coding RNA sequences that can broadly regulate gene expression post-transcription [168]. Studies of miRNA in BD and SZ have thus far yielded somewhat inconsistent results [169]. Perkins et al. observed the expression pattern of miRNAs in the pre-frontal cortex (PFC), and identified 16 miRNAs differentially regulated in their sample of 15 patients with SZ or schizoaffective disease, relative to their 21 control samples. Of the 16 identified miRNAs, 15 were downregulated in SZ [170]. Studies conducted by Beveridge and colleagues in 2008 and 2010 showed miRNA dysregulation and altered miRNA biogenesis in the brains (specifically, the PFC and the subgenual superior temporal gyrus) of patients with SZ [171,172]. Interestingly, many of their identified miRNAs were upregulated, and those that overlapped with the Perkins study were upregulated in the Beveridge studies but downregulated in the Perkins study [170-172]. Furthermore, the identified alterations in miRNA in the Perkins study suggested a decrease in global miRNA biosynthesis whereas the 2010 Beveridge study suggested a global increase in miRNA biosynthesis in SZ [170,172]. A recent study by Miller et al. explored the expression of over 800 miRNAs in the dorsolateral PFC (dlPFC) in RNA samples from 35 patients with SZ, 31 with BD, and 34 controls [173]. The authors identified significant dysregulation in 10 miRNAs in BD and alteration of the miRNA-132 in SZ. Notably, there were no significant miRNA overlaps between BD and SZ. Further analyses, which ranked the associations of miRNA with BD or SZ by fold change and uncorrected P-values, identified six dysregulated miRNAs common to both conditions. In fact, 754 protein-coding genes are affected by two or more of these miRNAs, which is significantly higher than would be expected by chance (P<0.0001), suggesting that co-dysregulation of these targets may result in pathologic alterations in functionally overlapping molecular networks [173]. Reliably associating miRNAs with psychiatric illness is a relatively new field of study, although there are data to suggest that other individual miRNAs may be linked with SZ and or BD (for an excellent review, see Miller and Wahlestedt [169]).
Clinical diagnostics: endophenotypes/biological markers
Endophenotypes are markers of an illness regardless of the phenotypic presence or absence of the illness. Because psychiatric disorders are primarily defined based on symptoms, many of which overlap between different disorders, the concept of defining endophenotypes for the disorders is an attractive one [174]. Gottesman and Gould in 2003 discussed endophenotypes in psychiatry and outlined criteria for defining endophenotypes. The endophenotype should segregate with the illness in the population, and co-segregate with the illness within families; it should be heritable and more prevalent in affected than in unaffected families; it should not depend on whether the illness is currently clinically manifested, and finally, it should be specific to the illness and capable of being reliably measured [174,175].
Biomarkers are measureable characteristics that reflect biologic function or dysfunction, response to a therapeutic measure, or indication of the natural progression of disease [176]. Historically, psychiatric illnesses have been diagnosed on the basis of behavioral signs and symptoms, rather than on biomarkers, but the overwhelming evidence that severe psychiatric disorders are in fact brain diseases, has led to considerable efforts to identify and introduce biomarkers into clinical psychiatry [177]. Current biomarkers for psychiatric illness fall into roughly three categories: metabolite levels, molecular genetics, and imaging findings. There are subtle distinctions between endophenotypes and biomarker changes, because endophenotypes are trait markers, whereas biomarkers may be state or trait markers [175]. However, there exists substantial overlap between these two categories, and we have therefore combined discussion of each into one section.
Clinical diagnostics: major depressive disorder
Negative mood is a central feature in MDD, and there have been a number of studies evaluating this as an endophenotype for depression. Wichers et al. conducted a study in female twin pairs, assessing the relationship between daily life negative mood bias and the lifetime diagnosis of MDD [178]. They studied 259 twin pairs; 17.4% of the sample represented twin pairs of which one twin had a diagnosis of lifetime depression while the co-twin did not. The authors found that probands with co-twins meeting a diagnosis for lifetime depression exhibited greater negative affect responsiveness to daily life stressors. This relationship held true after controlling for proband continuous depression score, past depressive score, and exclusion of probands who were depressed at the time of the study. The authors concluded that increased negative affect reactivity is present in probands with high familial load for depression, regardless of past or current depression [178]. Furthermore, mood-congruent memory (MCM) bias was first reported in the 1980s and has been repeatedly demonstrated in MDD and dysphoria in a multitude of recent studies [179-183].
Similar to negative mood, anhedonia is a central feature of the MDD diagnosis and has been proposed as an endophenotype for MDD. Dryman and Eaton analyzed data from 49 individuals enrolled in the US National Institute of Mental Health Catchment Area Program (NIMH-ECA) during the period 1981 to 1985, and found that anhedonia often precedes the onset of MDD [184]. Furthermore, some researchers have found an association between family history for depression and anhedonia in unaffected relatives [185-187]. McCabe et al. used functional magnetic resonance imaging (fMRI) to compare 13 unmedicated individuals recovered from MDD with 14 healthy controls to evaluate whether deficits in the processing of reward-relevant stimuli are present in people at risk for developing MDD [188]. Compared with the healthy controls, the recovered MDD individuals showed decreased neural response in the ventral striatum to pleasant stimuli and an increased response in the caudate nucleus to aversive stimuli. The authors suggested that patients in remission from MDD may have deficits in the neural basis of reward [188].
CSF concentrations of the norepinephrine metabolite 3-methoxy-4-hydroxphenylglycol (MHPG) has long been proposed as a biomarker for suicide risk, although studies have generated conflicting findings [189,190]. In a 2009 study, Galfalvi et al. completed a prospective study of the relationship between MHPG levels and suicidal behavior in 184 subjects with MDD or BD experiencing a depressive episode. They identified that lower levels of the metabolite were predictive of suicidal behavior in this population. Additionally, lower MHPG levels correlated with higher medical lethality of the future suicide attempt. The authors suggested that CSF MHPG may be useful as a biomarker for risk for short-term suicidal behavior [190].
Neuroimaging findings are being increasingly proposed as endophenotypes in MDD. Ramel et al. used fMRI to compare 14 unmedicated remitted-depressed (RD) individuals with 14 matched, never-depressed (ND) individuals on a self-referent encoding/evaluation task completed before and after a sad mood challenge [191]. For a subset of the RD group, after priming with sad mood induction, bilateral amygdala activation predicted negative recall bias. Notably, this association was not present before the sad mood priming, nor was it present in the ND group for any mood state [191].
Clinical diagnostics: bipolar disorder
BDNF is a protein that contributes to synaptic plasticity and neuronal survival, and has been implicated in the pathophysiology of a number of psychiatric illnesses [192,193]. Because BDNF levels in serum reportedly have high correlation (r = 0.8) with CSF levels, and are purported to cross the blood–brain barrier, BDNF is an attractive candidate for real-time study of BD mood states [194,195]. Recently, two meta-analyses by Fernandes et al.[196] and Lin [197] probed whether serum or plasma BDNF levels could reliably serve as a state marker of mood episodes in BD. In an analysis that combined studies of BDNF levels in either serum or plasma, Lin [197] identified that relative to controls, patients with BD in either manic or depressive states exhibited significant decreases in peripheral blood BDNF levels, and additionally, that pharmacologic treatment of manic states was accompanied by a significant increase in BDNF blood levels (plasma or serum). The Fernandes et al. [196] meta-analysis comprised 13 case–control studies (548 patients with BD and 565 controls), and measured serum BDNF levels during each of the three BD states: mania, euthymia, and depression. The analysis showed that, relative to levels in controls, serum BDNF levels were reliably decreased during manic and depressive states, and normal in euthymia. The Fernandes group suggested that serum BDNF concentrations can be used to accurately discriminate between the depressive episodes of MDD and those of BD (the suggested cut-off of 0.26 resulted in 88% sensitivity and 90% specificity) [198]. This has significant clinical implications because the ability to distinguish between BD and MDD is extremely important, especially in view of the likelihood of antidepressant monotherapy precipitating manic episodes in patients with BD [199].
Various cognitive phenotypes have also been suggested as endophenotypes for BD, including processing speed, visual learning and memory, verbal fluency, psychomotor retardation, and response inhibition [200]. In their meta-analysis of BD endophenotype candidates, Bora et al. [200] noted that of the 18 cognitive tests reviewed, euthymic patients with BD performed significantly more poorly than controls on all but one measure, the visual copy task. Moreover, the authors found that response inhibition is the most significant endophenotype for BD, which is significantly impaired in both patients with BD and unaffected relatives of such patients, but not in controls. Other proposed endophenotypes with strong support from their meta-analysis are verbal memory (large effect size in BD versus controls; modest effect size between relatives of patients with BD versus controls) and psychomotor retardation (moderate effect size in BD versus controls, no significant difference in relatives of patients with BD versus controls). The investigators recommended longitudinal studies to determine the relationship between cognitive impairment and brain connectivity and genetics [200].
Clinical diagnostics: schizophrenia
The possibility that certain cognitive deficits should be considered as SZ endophenotypes has been supported by multiple lines of evidence [201,202]. Three meta-analyses reflect efforts to interpret which cognitive domains are most consistently deficient in the SZ population relative to controls [201,203,204]. The meta-analysis by Sitskoorn and colleagues [203] compiled data from 37 studies, and identified nine weighted variables (broadly derived from the three domains of attention, memory, and executive function) that differed significantly in SZ subjects relative to controls. The largest effect sizes found were for deficiency in verbal memory recall and executive functioning in subjects with SZ relative to healthy controls (Cohen’s d = 0.54, 95% CI 0.43 to 0.66, P<0.0001; d = 0.51, 95% CI 0.06 to 0.50, P<0.0001). Smaller effect sizes for their other variables ranged from d = 0.28 to d = 0.38 for the remainder of the variables (in decreasing effect size): visuomotor tracking, attention/verbal memory span, verbal fluency, sustained attention, visual memory, executive function, and selective attention [203]. Szöke et al. [204] reported in their meta-analysis six altered cognitive variables in first-degree relatives of subjects with SZ relative to controls, with the greatest effect sizes seen for semantic and phonological verbal fluency (Hedges’ unbiased g = 0.87, 95% CI 0.64 to 1.10; g = 0.65, 95% CI 0.48 to 0.82, respectively). The remainder of the effect sizes were smaller, ranging from g = 0.26 to g = 0.49 for visuomotor speed/executive function, selective attention, and executive function (Wisconsin Card Sorting Test categories and perseverative errors) [204]. The meta-analysis by Fioravanti et al. [205] identified five cognitive domains in which subjects with SZ performed significantly worse than controls: memory, language, attention, executive function, and IQ (with memory having the greatest standardized mean difference of −1.18 (95% CI −1.31 to−1.05, P<0.00001) [205]. In 2007, Gur et al. [202] assessed neurocognitive function in 349 members of 35 multigenerational families with SZ and in 154 medically and psychiatrically healthy controls in an attempt to characterize a neurocognitive profile for SZ. They found that probands with SZ were the most impaired with regard to certain neurocognitive domains, the control group was the least impaired, and the unaffected relatives of the probands with SZ were impaired at an intermediate level between the other two groups. In particular, the authors reported strong genetic influences on the variability of performance on memory and emotion processing accuracy and on speed of attention. Further, because these measures were associated with SZ, they could be applied to differentiate between unaffected relatives and controls, and because they appear to be heritable, the authors proposed that they could be suitable for use as endophenotypes for genetic studies [202]. Despite some heterogeneity between studies, others have identified cognitive deficits in individuals with SZ and their unaffected relatives compared with controls, consistent with generalized cognitive impairment in these patients and their genetically susceptible relatives [206-210].
Environmental factors: stress
All of the reviewed psychiatric illnesses are characterized by various degrees of symptom exacerbation upon exposure to stress. Often underlying this response is dysregulation of the HPA axis, an important modulator of adaptive responses to stress. In fact, increased secretion of cortisol, one of the downstream products of the HPA axis, has been reported for decades in MDD [211,212]. Upstream of cortisol in the HPA axis is corticotropin releasing hormone (CRH, also known as corticotropin releasing factor, CRF), which is both an important regulator of the HPA axis and a key regulator in many extra-hypothalamic brain areas, including the amygdala, septum, stria terminalis, and cerebral cortex [213]. In these areas of the brain, CRH acts as a neurotransmitter, orchestrating endocrine, immune, and behavioral responses to stress [214]. In fact, injection of CRH into the CNS of rodents produces a host of endocrine and immune changes, and results in behavioral symptoms resembling those of MDD in humans [215,216]. In view of this, special attention has been given to identifying associations in HPA axis genes, in particular those affecting CRH levels. There have been numerous studies identifying SNPs in the gene coding for the CRH receptor 1 (CRHR1), which are associated with MDD [28]. Some studies have also identified differential treatment response to antidepressant therapy with various CRHR1 haplotypes. Importantly, certain CRHR1 SNPs and haplotypes appear to modify the adult risk of MDD in individuals subjected to childhood trauma [217-219]. Bradley et al. [217] conducted a study in a group of primarily socioeconomically disadvantaged African-Americans with high rates of lifetime trauma (n = 422), to explore whether the effects of child abuse on MDD susceptibility is moderated by variation in the CRHR1 gene. Linear regression analyses identified a significant protective effect of the gene and environment (G × E) interaction between two SNP haplotypes each with three SNPs (P<0.001; rs7209436-T, rs4792887-C, rs110402-A and P<0.005; rs7209436-T, rs242924-C and rs110402-A), and with the three individual SNP components of the latter haplotype. In an independent sample of primarily Caucasian individuals [217] (n = 299), the authors found similar findings, namely significant over-representation of the protective SNP alleles and haplotypes. Polancyzk et al. [218] replicated the Bradley findings in a sample of English women, but could not replicate the sample when using a different measure of child abuse than the Childhood Trauma Questionnaire (CTQ) used in the Bradley study. The CRHR1 gene interaction with trauma in childhood and with MDD in adulthood has been scrutinized by two other groups, who used a dexamethasone/corticotropin-releasing hormone test (DEX/CRH) as a measure of HPA axis activity [219,220]. Tyrka et al. [219] found that two of the CRHR1 SNPs identified in the Bradley et al. study [217], rs110402 and rs242924, showed a significant interaction with childhood abuse and prediction of cortisol response to the DEX/CRH challenge. Similarly, Heim et al. [220] used the DEX/CRH measure, and found the same relationship, although only in men, and suggested that the nature of the type of abuse experienced is crucial because in their sample, men experienced more physical abuse than did women.
In addition to its contribution to susceptibility for MDD and to epigenetic alterations in MDD (see ‘Genetic and epigenetics of MDD’) the serotonin transporter gene 5-HTT polymorphism, 5-HTTLPR, has been implicated in G × E interactions. Specifically, Caspi et al. [221] evaluated the relationship between MDD, the 5-HTTLPR genotype, and stressful life events over five years in a group of 847 Caucasian non-Maori individuals involved in the Dunedin Multidisciplinary Health and Development Study prospective longitudinal study. The authors used a moderated regression framework with sex as a covariate, and reported that the effect of stressful life events was significantly stronger in individuals carrying the short allele than in those carrying the long allele (P = 0.02). Moreover, they identified that childhood maltreatment predicted adult depression in individuals carrying the short allele but not in individuals homozygous for the long allele (P = 0.05) [221].
Environmental factors: in utero exposure
Much of the work focusing on in utero exposure to psychiatric illness has centered on SZ, with limited study of BD. Monozygotic twins are concordant for SZ in approximately 45% of cases, which indicates a potentially large contribution from environmental factors in the development of the illness [222]. Some studies have sought data from in utero and perinatal environmental exposures for potential explanations as to how monozygotic twins can be discordant for SZ. In 1967, the NIMH completed its first study on monozygotic twins discordant for SZ, examining 14 pairs of monozygotic twins, and summarizing published reports from 100 pairs of monozygotic twins discordant for SZ [223]. Comparing available information on the twin pairs illustrated that the SZ-affected twin was more likely to have sustained birth complications (index twin: control twin (I:C) = 4.0, n = 30), including perinatal asphyxia (I:C = 4.0; n = 15), and to be more than twice as likely to be the lighter-weight twin (I:C = 2.1; n = 61; P<0.01) [223]. These data laid the groundwork for many additional examinations of prenatal and perinatal insults. Cannon et al. [224] conducted a meta-analysis of eight prospective population-based studies of obstetric complications and SZ, and identified three groups of complications that significantly increased the risk for SZ: 1) pregnancy complications, including bleeding, Rhesus factor incompatibility, diabetes, and pre-eclampsia, 2) abnormal fetal growth and development, including low birth weight, congenital malformations, and reduced head circumference, and 3) delivery complications, including uterine atony, asphyxia, and emergency Cesarean section. Although many of the independent characteristics had modest effect sizes, with OR≥2 (bleeding, Rhesus factor incompatibility, pre-eclampsia, reduced head circumference, and asphyxia), other characteristics showed sizeable ORs: diabetes in pregnancy (OR = 7.76, 95% CI 1.37 to 43.90, P<0.03), low birth weight (< 2000g; OR = 3.89, 95% CI 1.40 to 10.84, P = 0.009), emergency Cesarean section (OR = 3.24, 95% CI 1.40 to 7.50, P = 0.006), congenital malformations (OR = 2.35, 95% CI 1.21 to 4.57, P<0.02), and uterine atony (OR = 2.29, 95% CI 1.51 to 3.50, P<0.001) [224]. By contrast, another study found no robust evidence for an association between BD and obstetric complications [225].
Other in utero exposures theorized to be involved in SZ are maternal influenza and maternal stress. Following an influenza pandemic in 1957, studies in Finland [226], England and Wales [227], and the USA [228] suggested a possible association between SZ development and prenatal exposure to the influenza virus. However, subsequent studies produced conflicting findings. A 2010 meta-analysis of 13 studies of SZ and maternal influenza exposure following the 1957 pandemic could not identify an association risk for SZ and prenatal influenza exposure [229]. The relationship between prenatal exposure to maternal stress and SZ was also analyzed by meta-analysis after individual studies produced equivocal findings. The meta-analysis by Selten et al. [230] could not identify a statistically significant association, despite querying various forms of maternal stress (wars, spousal demise, natural disasters, and undesired pregnancies).
Prediction of treatment response or non-response (therapeutic and side-effect profile)
Role of genetic alteration in drug-metabolizing enzymes
Many of the SNPs reviewed above and many others have been associated with predicting treatment response (both in terms of therapeutic efficacy and side-effect profile) to a pharmacologic intervention. Beyond the effects specific to particular illnesses or drugs are the genetic changes in drug-metabolizing genes that underlie differential response to pharmacologic agents. These genetic changes will be reviewed first, followed by specific polymorphisms in non-drug-metabolizing enzymes (non-DME) and their relationship to specific psychiatric illnesses.
The most studied group of drug-metabolizing enzymes (DME) in psychiatry are the cytochrome P450 (CYP) enzyme family [231]. CYP enzymes are expressed predominantly in the liver, although they are also found in many extra-hepatic locations, including in the brain, where their levels are approximately 0.5 to 2% of those in the liver [231]. Moreover, the genes coding for the CYP enzyme family are highly polymorphic, and the effects of many of the genetic differences contribute to differential metabolism of psychotropic agents [1,231]. DME phenotypes are broadly grouped into four categories based on genotype effect on enzyme activity: 1) poor metabolizers (PM), 2) intermediate metabolizers (IM), 3) extensive metabolizers (EM), and 4) ultra-rapid metabolizers (UM) [232]. CYP2D6 is an important member of the CYP enzyme family, because it is responsible for metabolizing nearly 50% of the most commonly prescribed psychotropics, and evidence in animal models indicates its involvement in the biosynthesis of dopamine and 5-HT [232-234]. It is also highly polymorphic, with 7% of Caucasians falling into the CYP2D6-PM group [231]. Common substrates metabolized by CYPD26 are tricyclic antidepressants, many SSRIs, venlafaxine, and antipsychotics. Therefore, when presented with any of these drugs, CYP2D6-PM are generally at risk for side effects secondary to the increased bioavailability and prolonged elimination half-lives of these drugs. Theoretically, these patients are also subject to an exaggerated drug response by the same mechanisms. At the other phenotypic extreme, the CYP2D6-UM would be expected to exhibit a poorer therapeutic response to CYP2D6 substrates [235]. Depending on the enzyme affected, this simplified scheme can be used as a basis for preliminary predictions of the effect a particular genotype will have on responsiveness to pharmacologic treatments. However, despite theoretic considerations of using genetic alteration in CYP enzymes, there has thus far been insufficient evidence for clinical applicability of CYP genotyping prior to prescribing either antidepressants (in particular, SSRIs) or antipsychotics for their respective applications [236-238].
Role of genetic alteration in treatment response in major depressive disorder
The response to antidepressants in MDD has been reported to vary as a function of specific genetic alterations in certain candidate genes (comprehensively reviewed by Porcelli et al. [239]). Genes that have an effect on serotonin signaling have been a major focus of study for the pharmacogenomics of depression. The polymorphism in the 5′-promoter region of 5-HTT, 5-HTTLPR, has been shown reliably to influence not only susceptibility for developing MDD, but also response to antidepressant therapy. A number of studies have linked the 5-HTTLPR long (L) allele with greater therapeutic response to SSRIs [234,240-252]. Other studies have indicated that the 5-HTTLPR short (S) allele is associated with poorer or slower therapeutic response to SSRIs [250,251,253-258]. However, a number of discordant studies have appeared. In a population of Korean patients, Kim et al. [259] found that the 5-HTTLPR S-allele conferred a greater therapeutic response to SSRIs. Yoshida et al. [260] found this same association in a population of Japanese patients and Lotrich et al. [261] also found this association in older patients. Moreover, a number of studies failed to show any relationship between 5-HTTLPR genotypes and antidepressant response or side effects [262,263]. Such discrepant findings may be related to particular ethnic or age-specific populations, and these factors should also be considered when evaluating genetic response to antidepressants.
There is also evidence for a pharmacogenomic role for genetic polymorphisms in the serotonin receptors HTR1A and HTR2A, although there are inconsistencies in these findings as well. Some studies showed an association between the HTR1A rs6295-C/C genotype and increased therapeutic response to SSRIs in Chinese populations [246,264,265] but Kato et al. [266] found the alternate genotype, rs6295-G/G, to be associated with increased response to SSRIs. However, other groups have been unsuccessful in identifying a relationship between SSRIs and polymorphisms in HTR1A[267-269]. The data for HTR2A presents similarly, with many authors observing that particular genetic alterations, including the rs6311-G allele, the rs9316233-G allele, rs2224721, rs1923884, rs7997012, rs6313, rs799701, rs3125, rs1923882, and rs6314, are associated with increased response to SSRIs [269-273]. Murphy et al. [274] compared the SSRI paroxetine with the tetracyclic antidepressant mirtazapine in an 8-week double-blind randomized control trial, to identify potential genetic predictors of efficacy and side effects. This group characterized variation in HTR2A in 246 older patients with MDD undergoing the trial, and found that discontinuing use of paroxetine was significantly associated with the HTR2A rs6313-C allele in a dose-dependent manner. Moreover, carriers of the HTR2A rs6313-C/C genotype experienced more severe side effects than did those with the other genotypes. Notably, there was no significant influence of the HTR2A rs6313 genotypes on mirtazapine side effects [274]. Such findings stand in contrast with other groups, who did not observe an effect of HTR2A variation on antidepressant response [246,275-277].
Because HPA axis disruption has been associated with MDD susceptibility and pathogenesis, genetic targets within the HPA axis represent plausible candidates for differential antidepressant response. Notably, polymorphisms in the FKBP5 gene have been associated both with increasing susceptibility to MDD (whose conferred susceptibility is further attenuated by environmental stress) and with differential response to antidepressant therapy [24,278-280]. In fact, Binder et al. [24] characterized a significantly faster response to SSRIs, TCAs, and mirtazapine in patients with MDD who were homozygous (TT) for the FKBP5 marker rs1360780 compared with the heterozygotes (TC) and homozygote major allele carriers (CC) combined. In a pharmacogenetic association study of genetic variants in CRHBP and response to citalopram, Binder and colleagues [281] identified a SNP significantly associated with both remission and reduction of depressive symptoms in Hispanic and African-American outpatients with MDD.
Finally, the endogenous opioid system has been implicated in MDD, making the μ-opioid receptor, OPRM1, an attractive target for evaluation of antidepressant response variance secondary to genetic variance [282]. Garricock et al. [282] genotyped 53 separate SNPs in the OPRM1 gene in 1,631 STAR*D samples, and found significant associations for polymorphic variation in OPRM1 and citalopram response and remission. Moreover, the association between the commonly studied SNP rs1799971 and citalopram treatment outcome provided some evidence that this effect is influenced by ancestry groups. The authors expressed some caution in the interpretation of the findings, because even with the use of the same samples, none of the OPRM1 variants met genome-wide significance, and the associations identified could reflect a placebo effect [282].
Of note, there are four published GWAS for SSRI antidepressant response, three of which stem from assessment of the samples involved in the STAR*D study and one from the Genome-Based Therapeutic Drugs for Depression (GENDEP). Despite the number of GWAS, only one marker in the Papilin (PAPLN) gene had a sufficiently robust association with antidepressant response (to suicidal ideation) to survive correction for multiple testing [270,283-285]. This highlights a benefit of the GWAS approach to pharmacogenomic inquiries; based on a candidate gene approach, PAPLN, which encodes a proteoglycan-sulfated glycoprotein, would probably have been overlooked. However, the paucity of significant findings also emphasizes the likely multifactorial nature of antidepressant response, not easily uncovered with current GWAS designs [286].
Role of genetic alteration in treatment response in bipolar disorder
Similar to antidepressant response in MDD, numerous candidate genes have been evaluated for the effect of their genetic variation on BD therapy, most of which have focused on lithium [1]. Lithium response to variation in candidate genes has some notable associations including 5-HTTLPR, CLOCK, BDNF, X-box binding protein 1 (XBP1), glycogen-synthase kinase 3 beta (GSK3B), breakpoint cluster region (BCR), cAMP responsive element binding proteins 1to 3 (CREB 1, 2, and 3) and neurotrophic tyrosine kinase receptor, type 2 (NTRK2) [287-294].
Role of genetic alteration in treatment response in schizophrenia
Numerous studies have begun to pinpoint associations between genetic variants in some individuals with SZ and the tolerability and therapeutic efficacy of antipsychotic medications [295-298]. Tardive dyskinesia (TD) is an often debilitating motor disorder characterized by hyperkinetic, repetitive, and involuntary movements, which is experienced by approximately 30% of individuals with chronic SZ on long-term antipsychotic treatment (usually with first-generation antipsychotics) [299]. There are several studies providing evidence for genetic variation affecting susceptibility to TD in response to antipsychotic treatment. For example, polymorphisms near the DA receptor D2 gene (Taq1A, minor allele, rs1800497) and in the DA receptor D3 gene (DRD3-Ser9Gly, rs6280) have been linked with protection against TD and increased susceptibility for developing TD, respectively [300-302]. However, Woo and colleagues [303] were unable to find any association between the DRD3 Ser9Gly in their study of 113 Korean patients with SZ. Moreover, data analysis from the NIMH Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE), a double-blind study that compared the first-generation antipsychotic perphenazine with four second-generation antipsychotics, did not find evidence for association with TD for either polymorphism [304,305]. Other genetic variations positively associated with TD have been identified in the genes for the COMT catecholamine-degrading enzyme (Met158Val, Val allele), the manganese isoform of superoxide dismutase (MnSOD), a mitochondrial enzyme involved in oxidative metabolism (homozygote Ala genotype at rs4880, MnSOD-Ala9Val, with a protective effect for Val carriers) and the serotonergic receptor gene 5-HT2A (C allele at promoter region T102C, rs6313 in older patients) [306-310]. However, conflicting reports have also been published for the MnSOD and 5-HT2A variants [311,312].
First-generation antipsychotics are associated with a risk for TD, whereas several of the second-generation antipsychotics (and first-generation antipsychotic, chlorpromazine) can increase the risk for the metabolic syndrome and or isolated weight gain in nearly one-third of patients with SZ [313-315]. Studies of association of genetic variation with these side effects include associations with polymorphisms in the gene for the serotonin receptor subtype 2C (HTR2C) and in the gene for leptin (LEP), which is important for satiety and adipose regulation. These association studies have reported positive associations with metabolic syndrome (HTR2C intragenic polymorphism rs1414334-C for risperidone and clozapine), less weight gain with HT2RC SNPs (HTR2C −759 C→T and −697 G→C promoter polymorphisms, rs3813929-T and rs518147-C [316-318], respectively), and increased weight gain with the LEP promoter SNP −2548 A→G (G allele) ([317,319]). However, contradictory findings have also been published [320,321]. Furthermore, Opgen-Rhein et al. [322] were unable to replicate the LEP association with increased weight gain.
Agranulocytosis, or severe reduction in a particular class of white blood cells, is a serious side effect occurring in approximately 1% of individuals receiving treatment with the atypical antipsychotic clozapine. This property limits clozapine’s clinical use despite its clear therapeutic efficacy in refractory patients [323,324]. Evidence for genetic variants conferring resistance or increasing susceptibility to clozapine-induced agranulocytosis (CIA) is reflected in a number of studies, although the relatively low incidence of CIA has limited study sizes and effective replication studies. The human leukocyte antigen (HLA) system is comprised of a host of genes that are important in immune system modulation, and it has thus far produced the most promising genetic associations with CIA [325]. These associations include several genetic variants involving the HLA-DQB1 locus, and have become incorporated into a commercially available test, although the clinical usefulness of this test has not been established, owing to its low sensitivity [325,326]. Additionally, a small GWAS provides preliminary confirmation of the HLA-CIA association [327].
In a double-blind study, the NIMH CATIE compared the first-generation antipsychotic perphenazine with four second-generation antipsychotics: olanzapine, risperidone, quetiapine, and ziprasidone [304]. The CATIE trial is a rich resource explored by researchers interested in determining why there were differential responses to antipsychotics. A 2009 analysis of data from the CATIE study was unable to find any significant association (following correction for multiple testing) between 2,769 potential SNPs and 21 pharmacogenetic phenotypes [328]. A GWAS, also drawing from CATIE data, analyzed 738 subjects with SZ for association between SNPs and antipsychotic response, and identified one significant locus at rs17390445 associated with improved SZ-positive symptoms to ziprasidone treatment, although the specific biological relevance of the locus is unclear [329].
Studies of the gene encoding a voltage-gated potassium channel, subtype H (KCNH2), have generated some discussion about the possibility of detecting likely responders to atypical antipsychotic therapy. Previous reports of SZ-KCNH2 SNP associations, along with knowledge that antipsychotics bind to KCNH2-encoded channels, fueled inquiry of how risk alleles in KCNH2 might predict treatment response in SZ [330,331]. Apud et al. [332] analyzed data on the relationship between various KCNH2 genotypes and atypical antipsychotic treatment response from two sources: the 364 patients of the CATIE study and an NIMH cohort of 54 partially treatment-resistant patients with SZ enrolled in a double-blind, placebo-controlled, crossover study (antipsychotics included olanzapine, quetiapine, risperidone, ziprasidone, and aripiprazole). The authors found that individuals with a TT genotype for rs1036145 (T allele previously established as an SZ-risk allele) exhibited significantly greater symptom improvement than those with either the TC or CC-genotypes (P = 0.0395) [332]. Specifically, two groups of patients homozygous for rs1036145-TT from the NIMH cohort (those receiving placebo first and those receiving active drug first), showed greater change in positive symptom ratings on the Positive and Negative Syndrome Scale (PANSS) compared with their heterozygote and CC-genotype counterparts. Similarly, in the larger CATIE group, patients with the rs1036145-TT or rs3800779-TT genotypes also showed greater improvement in positive symptoms and general psychopathology than their counterparts (rs1036145-TC/CC or rs3800779-TC/CC, respectively). Furthermore, patients carrying the rs1036145-TT homozygous genotype were far more likely to remain on their medication than the homozygous non-risk genotype (rs1036145-CC) (hazard ratio = 0.208,P = 0.033) [332]. The similar relationships of atypical antipsychotic treatment response with KCNH2 genotypes in two separate trials is encouraging evidence that genotyping may help guide therapeutic decisions in SZ.
Finally, meta-analyses on the DA D2 and D3 receptor polymorphisms have been conducted, seeking association with therapeutic response to antipsychotics [333,334]. Zhang et al. [333]carried out a meta-analysis of 10 studies to determine the relationship between the DRD2 polymorphisms, Taq1A and –141C Ins/Del, and antipsychotic response. They were unable to detect any relationship between clinical response and the Taq1A polymorphism A1/A1 genotype and A1 carriers compared with A2/A2 genotype (pooled OR = 1.39, 95% CI 0.91 to 2.13, P = 0.13 and pooled OR = 1.30, 95% CI 0.92 to 1.84, P = 0.14, respectively), despite previous association findings of clinical response to haloperidol in acute psychosis, risperidone in first-episode antipsychotic-naïve patients with SZ, and aripiprazole in patients with acutely exacerbated SZ [333,335-337]. They did report a significant relationship between the -141C Del carriers compared with patients with the 141C Ins/Ins genotype: -141C Del carriers tended to have poorer therapeutic response to antipsychotics compared with 141C Ins/Ins carriers (pooled OR = 0.65, 95% CI 0.43 to 0.97, P = 0.03) [333]. Results for association of the DRD3 polymorphism, Ser9Gly, with antipsychotic treatment response have been equivocal. An initial meta-analyses conducted by Jönsson et al. [338] found that people with the DRD3 Ser9 allele, the Ser/Ser and Ser/Gly genotypes were more likely to respond to typical antipsychotics, and less likely to respond to clozapine compared with their counterparts. However, a more recent meta-analysis by Hwang et al. [334] was unable to identify any significant association between the Ser9Gly polymorphism and antipsychotic drug response after increasing the sample size from 233 to 758 by including original analyses and studies published after the Jönsson meta-analysis.
Emerging applications
The types of applications that will greatly affect the prospect of personalized medicine in psychiatry will involve continued improvements in genomic inquiries and continued development of genomic testing protocols. Apart from increasing efficiency in genomic techniques, expanding the substrate types for study to include transcriptomics and proteomics (the study of all expressed messenger RNA (mRNA) and all proteins, respectively), will exponentially improve the knowledge base and ability to make informed decisions in personalizing psychiatry [339]. Furthermore, studies that take a multidisciplinary approach, combining the categories (for example, gene × environment interactions or endophenotypes × environmental interactions.) that generate unique phenotypic expression of illness will provide new perspectives and will increase the database from which to draw further studies. Moreover, development of tools to manage and integrate this information and the development of pharmacogenomic tests for use in clinical practice will be greatly beneficial in generating clinically relevant decision trees for use in personalized psychiatry [340]. For example, targeting the molecules responsible for epigenetic regulation may prove to be a promising means of modifying outcomes in psychiatric illnesses [134]. In addition, studies that relate genetics not only to pharmacologic therapy, but to other psychotherapeutics, including various psychotherapies (such as cognitive behavioral therapy and dialectical behavioral therapy), light therapy, deep-brain stimulation, electroconvulsive therapy, and transcranial magnetic stimulation (TMS), will serve to further refine and optimize psychiatric care. Finally, the importance of developing neuroimaging methods and their application to psychiatry will continue to have significant effects on personalizing psychiatry, and this is discussed in greater detail below.
Imaging genetics in psychiatry
A rapidly advancing area likely to have a major influence on personalized medicine in psychiatry is neuroimaging, and more specifically, neuroimaging genomics. Briefly, imaging genetics uses imaging measures as quantitative biological phenotypes. It seeks to incorporate genetics, psychiatry, and neuroscience in a way that relates genetic variation with protein function, brain structure and connectivity, and psychopathology [341]. Neuroimaging genetics allows conduction of experiments that relate genetic change to outcomes of measurable, repeatable tests of brain structure or function. Two major neuroimaging genetic approaches are 1) identification of imaging alterations in a population with a well-defined genetically determined illness; and/or 2) verification of the effects of specific genetic changes. A particularly useful aspect of defining biological phenotypes using imaging is the distancing of disease from subjective self-report and inconsistent diagnostics [341]. Furthermore, the ability to study in vivo change and change over time in psychiatric illness is paramount given the neuronal plasticity (or lack thereof) already identified in various psychopathologies. In essence, neuroimaging genetics has the potential to lead to better comprehension of the pathophysiology of common psychiatric disorders by evaluating the relationship between imaging phenotypes and genetic variation. Once interventions such as antidepressant therapy or TMS treatment (discussed below) are introduced into the equation, conclusions drawn from correlations may give rise to an understanding of causation. Explorations of concurrent intervention with real-time imaging, such as the assessment of self-regulation of frontal cortical activation (using neurofeedback) in nicotine dependence and craving, coupled with real-time fMRI, have led to this technology and its applications expanding in exciting new directions [342]. For example, trials using this type of volitional reduction with neurofeedback in the treatment of MDD are under way, building upon previous works by the same group ([343]; I. Gotlib, personal communication, 2012). One group is conducting additional trials in PTSD using concurrent TMS-EEG (technique as described by Johnson et al. [344]), which employs active therapeutic intervention with concurrent imaging (A. Etkin, personal communication, 2012) and is probably informed by the group member’s previous characterization of anxiety using fMRI [345].
Furthermore, there already exist a number of examples of imaging genetics with psychiatric implications [341]. We describe several examples that epitomize the types of studies and progress being made in imaging genetics (additional notable findings relevant to genes discussed throughout the review can be found in Table 1). Prominent examples are seen with in MDD, with the association between smaller hippocampal volumes and polymorphisms within the 5-HTTLPR region, and with the BDNF Val66Met polymorphism [346]. However, there are some limitations to this field. Neuroimaging genetics is a relatively new area, subject to initial missteps, including false positives and imprecise interpretations. There is skepticism that functional neuroimaging studies (for example, those involving fMRI) actually show the functional change being queried. The output measure of fMRI is blood oxygen level dependent (BOLD) change, which is thought to reflect neuronal activation. Of note, there is a significant lag between the supposed measured neuronal activity and fMRI change due to the cardiovascular changes necessary to increase BOLD in response to neuronal activation. Various imaging methods, each with their own strengths and weaknesses, have the potential to contribute to clarification of this process, often in complementary ways. For example, where there is some temporal disconnection between neuronal activation and BOLD response, diffusion tensor imaging (DTI), which is sensitive to directional diffusion of water along neural pathways, exhibits a more proximate relationship to neuronal activation [347]. MR spectroscopy (MRS) can provide chemical profiles capable of differentiating between brain pathologies. Although the use of positron emission tomography/single-photon emission computed tomography (PET/SPECT) is more invasive than the other methods described, it has the advantage of allowing in vivo monitoring of molecular changes such as receptor or transporter binding density in response to therapy [348-350] (a summary of select imaging findings is given in Table 1).
Table 1.
Selection of imaging findings in psychiatric illness
| Psychiatric illness | Subgroup/genotype | Imaging feature | Sample | Imaging method | Reference |
|---|---|---|---|---|---|
|
Major depressive disorder |
|
• Reduced activity in frontal lobes |
Various |
PET, fMRI, EEG, SPECT |
[351] |
| |
|
• Reduced HPC volumes |
Meta-analyses |
MRI |
[352,353] |
| |
|
• Greater reduction in HPC volume with increased duration of untreated MDD |
38 male outpatients |
MRI |
[354] |
| |
|
• Increased baseline activity in pulvinar nuclei bilaterally |
Meta-analysis |
PET and SPECT |
[343] |
| |
|
• Increased amygdala, dorsal ACC, and insular response to negative stimuli compared with healthy controls |
Meta-analysis |
Task-based fMRI |
[343] |
| |
|
• Reduced dlPFC and dorsal striatum response to negative stimuli compared with healthy controls |
Meta-analysis |
Task-based fMRI |
[343] |
| |
5-HTTLPR -S or Lg allele: |
Increased amygdala reactivity to masked emotional faces; correlated to lifetime psychiatric hospitalization in MDD |
35MDD, 32 controls |
fMRI |
[355] |
| |
5-HTTLPR -S or Lg allele: |
Increased bilateral amygdala activation after emotional stimuli (allele effects are additive) |
27 MDD on medication |
fMRI |
[356] |
| |
5-HT1A - 1019G allele: |
||||
| |
5-HTTLPR- L/L: |
Reduced HPC volumes compared with controls |
40 MDD, 40 controls |
MRI |
[357] |
| Late onset (LO) MDD |
5-HTTLPR- L/L: |
Reduced HPC volumes compared with S/L or S/S genotypes |
63 LO MDD, 72 EO MDD, 83 controls |
MRI |
[358] |
| Early onset (EO) MDD |
5-HTTLPR -S allele: |
Reduced HPC volumes compared with left allele carriers |
|
|
|
|
Bipolar disorder |
|
• Increased size of lateral ventricles (right ventricle only in [359]) |
Meta-analyses |
CT and MRI |
[359-361] |
| |
|
• Increased number of deep white-matter hyperintensities |
Meta-analyses |
CT and MRI |
[360-363] |
| |
|
• Increased number of subcortical gray-matter hyperintensities |
Meta-analysis |
MRI |
[362] |
| |
|
• Increased activity in limbic structures (left side only in [364]) |
Meta-analyses |
fMRI and PET |
[364,365] |
| |
|
• Reduced activity in frontal structures (vlPFC and dlPFC [364]) |
|||
| |
Pediatric population: |
• Reduced amygdala volume |
Meta-analyses |
MRI |
[365,366] |
|
Schizophrenia |
|
• Reduced frontal-lobe activity at rest and during task activation |
Meta-analysis |
MRI, PET |
[367,368] |
| |
|
• Increased lateral ventricular size |
Meta-analyses |
CT and MRI |
[369,370] |
| |
|
• Increased D2 dopamine receptor density |
Meta-analysis |
PET and SPECT |
[371] |
| |
|
• Reduced frontal gray matter |
Meta-analyses |
MRI |
[372,373] |
| |
SZ: |
• Reduced bilateral HPC volume relative to controls |
Meta-analyses |
MRI |
[374-376] |
| |
nonpsychotic 1st deg family: |
Meta-analysis |
MRI |
[370] |
|
| BDNF 66Val/Val carriers: | • Reduced activation of the cingulate, lateral PFC and lateral parietal regions during verbal memory task | 58 high-risk subjects (first-degree or second-degree family with SZ) | fMRI | [377] |
+/−A, alcohol abuse positive/negative subjects; ACC, anterior cingulate cortex; Cho, choline; CT, computed tomography; DA, dopamine; dlPFC, dorsolateral pre-frontal cortex; EO, early onset; fMRI, functional magnetic resonance imaging; 1H-MRS, proton MR spectroscopy; HPC, hippocampal; LO, late onset; MDD, major depressive disorder; MRI, magnetic resonance imaging; NAA, N-acetylaspartate; parahippocampal gyrus; PET, positron emission tomography; Tx, treated; vlPFC, ventral-lateral pre-frontal cortex; vmPFC, ventral-medial pre-frontal cortex.
Conclusion
In this review, we have focused on the components and tools that are proving to be instrumental in personalizing medicine in psychiatry. We have discussed the types of information that can be garnered and eventually used in tailoring psychiatric therapies to the individual. This information can be found in the form of particular genetic and epigenetic changes more characteristic in the psychiatrically ill or in observable biomarkers that are reflective of illness. Additionally, environmental influences are evaluated; in particular, how their interaction with genetic variation can lead to disease attenuation or exacerbation. Furthermore, we have discussed how the definition of the illnesses can influence the tailoring of individualized therapies. For example, many psychiatric disorders show phenotypic heterogeneity at the same time as having symptoms that overlap with other psychiatric illnesses that may presumably share some fundamental biologic underpinnings. Indeed, uncovering the biological basis of individual symptoms may prove to be as or more helpful in understanding the pathophysiology of the illness than forcing a constellation of co-occurring symptoms to fit together under one biologically plausible explanation. What we will probably experience is a refining of the diagnostic process; in some cases, recognizing spectrums of disease and in others, homing in on specific biological features of an illness.
It should be noted that these categories for defining an individual’s unique psychiatric phenotype are artificially separated to facilitate a conceptual framework, and there is substantial overlap between each category (Table 2). For example, an illustration of how genetic variation interacts with environmental factors is apparent in CRHR1 polymorphism haplotypes, which are not only associated with MDD but also interact synergistically with childhood trauma to increase the risk of MDD [217,281]. Another example is the dexamethasone-binding capacity of leukocytes; although this is not used in the diagnosis of PTSD, it can be used as a biomarker or a proxy measure for glucocorticoid receptor number. In turn, this measurement might help screen persons likely to develop PTSD, because greater glucocorticoid receptor density is predictive of risk for PTSD symptoms in military personnel returning from deployment [378].
Table 2.
Psychiatric disease susceptibilities
| Gene(s) | Variant(s) | Population(s) | EPI | EF | BM | Key findings | Outcome | Ref. |
|---|---|---|---|---|---|---|---|---|
|
Major depressive disorder | ||||||||
|
5-HTT |
5-HTTLPR and EF: stressful life events |
Caucasian |
|
+ |
|
Homozygous or heterozygous carriers of the short allele had higher frequency of depression and suicidality when exposed to stressful life events. |
SIG |
[221] |
|
5-HTT |
STin2.9 |
Caucasian |
|
|
|
Increased frequency in MDD relative to controls |
SIG |
[7] |
|
5-HTT |
5-HTTLPR |
Caucasian |
|
|
|
Increased frequency in MDD relative to controls |
SIG |
[8] |
|
TPH1 |
microsatellite at 11p15.3-p14 |
Caucasian community-based sibships |
|
|
|
Association with MDD susceptibility and microsatellite |
SIG |
[11] |
|
TPH1 |
Various |
Caucasian |
|
|
|
Six haplotypes associated with MDD risk |
SIG |
[12] |
|
TPH2 |
rs120074175 (p.R441H) |
Caucasian (90%), AA (8%), East Asian (2%) |
|
|
|
Higher frequency of SNPs in patients with MDD compared with controls or patients with BP |
SIG |
[15] |
|
TPH2 |
rs120074175 (p.R441H) |
Caucasian (84%), Hispanic (6%), East Asian (5%), AA (3%), others (2%) |
|
|
|
SNP not identified in non-treatment-resistant and treatment-resistant patients with MDD, or in treatment-resistant patients with BP, or in controls |
NS |
[16] |
|
TPH2 |
rs120074175 (p.R441H), rs1843809 (c.608 + 5263G>T) |
Caucasian |
|
|
|
Higher frequency of SNPs in MDD relative to controls |
SIG |
[13] |
|
TPH2 |
Various |
East Asian (Korean) |
|
|
|
No association of the SNPs rs4570625, rs10748185, rs11179027, rs4469933, or rs17110747 in MDD, BP, or SZ |
NS |
[17] |
|
TPH2 |
rs4570625 (c.-141-703G>T), rs17110747 (c.*479G>A) |
Meta-analysis |
|
|
|
SNPs associated with MDD susceptibility by fixed-effects modeling; rs4570625 remained significant using random-effects calculations |
SIG |
[9] |
|
TPH2 |
rs4570625-rs10748185 (G>A). |
East Asian (Korean) inpatients |
|
|
|
Haplotype significantly associated with higher MADRS endpoints in MDD |
SIG |
[19] |
|
FKBP5 |
rs3800373 (c.*1136G>T)-(CC) rs1360780 (c.106-2636A>G) |
Post-mortem brain samples, ethnicity not specified |
|
|
|
Five clinical groups were compared: MDD, MDD + psychosis, MDD + HIV, HIV-positive, and HIV-negative. Genotype frequencies in the MDD and the MDD + psychosis groups differed from published allelic frequencies |
SIG |
[25] |
|
FKBP5 |
rs1360780 (c.106-2636A>G) |
Caucasian inpatients with MDD, BD, or dysthymia |
|
|
|
Carriers of the TT genotype experienced more depressive episodes, by a factor of 2:1 compared with the CC or CT genotypes |
SIG |
[24] |
|
FKBP5 |
rs1360780 (c.106-2636A>G) (TT), rs3800373 (GG) |
Caucasian treatment-resistant adolescents |
|
|
|
Genotypes were associated with suicidal events |
SIG |
[26] |
|
FKBP5 |
rs9470080, rs9394309, rs7748266, rs1360780; BM: reduced daytime cortisol secretion |
Caucasian older people |
|
|
+ |
Minor alleles were associated with decreased daytime cortisol levels and increased likelihood of depressive symptoms |
SIG |
[279] |
|
FKBP5 |
rs9470080 (c.-19-35815A>G), rs9296158 (c.509-1901T>C) and EF: prolonged stress exposure |
East Asian (Korean) |
|
+ |
|
Two SNPs were associated with anxiety and depression after prolonged stress in patients with cancer patients |
SIG |
[280] |
|
CRHR1 |
rs110402 (GG), rs242924 (GG); and EF: childhood trauma; and BM: response to DEX/CRH test |
Healthy Caucasians with history of early life stress |
|
+ |
+ |
In adults who had experienced maltreatment, the GG genotypes were associated with increased cortisol response to DEX/CRH test |
SIG |
[219] |
|
CRHR1 |
rs10473984 EF: childhood trauma |
|
|
+ |
|
SNP works synergistically with childhood trauma to increase risk of MDD |
SIG |
|
|
CRHR1 |
rs110402 (c.34-4338G>A); EF: childhood abuse; and BM: cortisol response to DEX/CRH test |
1: AA, 2: ethnically diverse |
|
+ |
+ |
In adult men who had experienced child abuse, the A allele was associated with reduced MDD symptoms and reduced cortisol response to DEX/CRH test |
SIG |
[220] |
|
CRHR1 |
rs110402 (c.34-4338G>A), rs7209436 (c.33 + 8207C>T) and rs7209436-rs110402-rs242924 (TAT); EF: childhood abuse |
AA, Caucasian |
|
+ |
|
Rare alleles were protective in a dose-dependent manner against MDD in the presence of child abuse |
SIG |
[217] |
|
CRHR1 |
rs7209436-rs110402-rs242924 (TAT); EF: childhood abuse |
Caucasian (>90%) |
|
+ |
|
TAT haplotype was protective against MDD in women exposed to severe maltreatment, but not in a replication study using different measure of trauma |
SIG |
[218] |
|
CRHR1 |
rs242939 (c.241 + 1631C>T), three haplotypes |
East Asian (Chinese) |
|
|
|
Allele and genotype association with MDD |
SIG |
[32] |
|
CRHR1 |
rs110402 (c.34-4348G>A) |
Caucasian |
|
|
|
Association between SNP and early onset of MDD and increased risk for a seasonal pattern |
SIG |
[33] |
|
CRHPB |
Haplotype block |
Caucasian (Swedish) |
|
|
|
In patients with recurrent MDD, haplotype block (s02-TT and s11-TT and s14-T) was significantly associated with disease compared with controls |
SIG |
[35] |
|
CRHPB |
Haplotype block |
Caucasian (Swedish and Belgian) |
|
|
|
Could not replicate findings of [35] in an extended Swedish or Belgian sample. Found higher frequency of haplotype block (s02-TT, s11-TT and s12C) in Swedish men compared with control men |
NS |
[36] |
|
HTR3A |
42 (CC); EF: early life stress (ELS); BM: frontolimbic gray-matter alterations |
Healthy Caucasian |
|
+ |
+ |
Genotype + ELS was a predictor of depressed mood. Carriers had greater frontolimbic gray-matter alterations, which were increased by ELS |
SIG |
[379] |
|
SYNE1 |
rs9371601 (c.1653 + 2159C>A) |
Caucasian |
|
|
|
Higher frequency of SNPs in recurrent MDD relative to controls |
SIG |
[52] |
|
NR3C1 |
EPI: NR3C1 promoter site methylation; and EF: history of childhood abuse |
Suicide victims |
+ |
+ |
|
In abused victims, NR3C1 promoter methylation was increased and glucocorticoid receptor mRNA reduced compared with non-abused victims or controls |
SIG |
[144] |
| -- |
BM: CSF concentration of CRF |
Various |
|
|
+ |
Increased CSF concentration of CRF is a replicable finding in MDD. Also seen in suicide victims |
SIG |
[28-31] |
| -- |
EF: birth trauma |
Monozygotic twins discordant for MDD |
|
+ |
|
Increased occurrence of birth trauma in SZ-affected twin |
SIG |
[223] |
| -- |
EF: obstetric complications, e.g. abnormal fetal growth/development, pregnancy and delivery complications |
Meta-analysis of population-based prospective studies |
|
+ |
|
Obstetric complications increased risk for SZ |
SIG |
[224] |
| -- |
BM: CSF concentration of norepinephrine metabolite MHPG |
Caucasian (81%) with MDD (85%) or BD (15%) |
|
|
+ |
Lower levels of MHPG were predictive of suicidal behavior, and correlated with higher medical lethality of suicide attempt |
SIG |
[190] |
| -- |
rs1360780 (c.106-2636A>G) |
Caucasian, Black |
|
|
|
Association of SNP with MDD risk in Caucasian sample |
SIG |
[34] |
|
Bipolar disorder | ||||||||
|
FKBP5 |
rs4713902 (c.-19-3406A>G), rs7757037 (c.841-238C>A), rs9296158 (c.509-1901T>C), rs3800373 (c.*1136G>T), rs9380525 (c.-19-22418C>G) |
Family trios and quads with BD-I, or BD-II + rMDD, or SZA-BD |
|
|
|
SNPs associated with BD in populations studied (BD-I, BD-II + rMDD, SZA/BD); rs4713902 remained significant after correction for multiple testing |
SIG |
[40] |
|
FKBP5 |
various |
Caucasian (Ashkenazi Jewish) |
|
|
|
No significant SNP or haplotype associations with BD or SZ identified |
NS |
[41] |
|
FKBP5 |
rs4713916 (c.20 + 18122T>C), rs1360780 (c.106-2636A>G), rs380037 |
Caucasian |
|
|
|
No significant association between SNPs and BD |
NS |
[42] |
|
ARNTL |
rs7107287 (c.-208 + 13499G>T), rs895682 (c.-135 + 13626T>C), rs1481892 (c.-208 + 2451G>C), rs4757142 (c.-207-5839G>A) |
Caucasian family trios |
|
|
|
SNPs rs7107287 and rs895682 showed significant transmission bias in family samples. In Pittsburg sample, genotype distribution of SNPs rs1481892, rs7107287 and rs4757142 differed from that of controls |
SIG |
[48] |
|
TIMELESS |
rs2279665 (c.114G>C), rs2291738 (c.2726-4A>G), rs774026 (c.1578 + 22T>C), rs2291739 (p.P1018L) |
Caucasian family trios |
|
|
|
SNPs (rs2279665, 2291738) showed transmission bias in family samples. Haplotype over-transmission involving SNPs rs2279665, rs774026, rs2291738, and rs2291739 |
SIG |
[48] |
|
CLOCK |
rs534654 (c.793-485A>G), rs6850524 (c.-289-5765G>C), rs4340844 (c.559 + 996T>G) |
Family trios and quads |
|
|
|
Suggestive evidence for transmission disequilibrium |
SUG |
[46] |
|
SYNE1 |
rs9371601 (c.1653 + 2159C>A) |
Caucasian |
|
|
|
Higher frequency of SNP in BD compared with controls |
SIG |
[52] |
|
COMT |
EPI: MB-COMT promoter methylation |
Post-mortem brain samples (97% Caucasian) |
+ |
|
|
Reduced methylation of COMT promoter in BD compared with controls led to higher MB-COMT expression in BD compared with controls |
SIG |
|
|
COMT |
EPI: MB-COMT promoter methylation |
Caucasian post-mortem brain samples |
+ |
|
|
Promoter methylation did not differ between BD and control brains |
NS |
[151] |
| -- |
EF: obstetric complications |
Meta-analysis |
|
+ |
|
No findings to suggest higher risk for BD relative to MDD or controls after exposure to obstetric complications |
NS |
[225] |
| -- |
BM: peripheral blood levels of BDNF |
Meta-analysis |
|
|
+ |
Relative to controls, patients with BD in manic or depressed states had reduced serum and plasma BDNF levels |
SIG |
[197] |
| -- |
BM: serum or plasma levels of BDNF |
Meta-analysis |
|
|
+ |
Relative to controls, patients with BD in manic or depressed states had reduced serum and plasma BDNF levels |
SIG |
[196] |
|
Schizophrenia | ||||||||
| GWAS |
various |
GWAS of MGS sample (Caucasian, AA) |
|
|
|
No significant finding in MGS case–control sample GWAS |
NS |
[58] |
| MHC region on chr6 |
rs3130375 (7kb from NOTCH4) and large sets of nominally associated ‘score alleles’ |
Caucasian, AA |
|
|
|
Imputed SNP rs3130375 reached genome-wide significance. Strong suggestion for a polygenic basis for SZ |
SIG |
[95,96] |
| MHC region on chr6 |
various |
Meta-analysis of MGS, ISC, and SGENE data |
|
|
|
Association between SZ and region of LD on chromosome 6p22.1 |
SIG |
[58] |
| MHC region on chr6 |
HIST1H2BJ: rs6913660, PRSS16: rs13219354, rs6932590, PGBD1: rs13211507 (c.642 + 2432T>C), NOTCH4: rs3131296 (c.2866-827A>G) |
GWAS of SGENE-plus, ISC, and MGS (Caucasian) |
|
|
|
With combined samples, MHC region SNPs showed genome-wide significance |
SIG |
[59] |
|
COMT |
rs165688 (p.V158M) |
Caucasian with velocardiofacial syndrome (VCFS) ± SZ |
|
|
|
No correlation between allelic distribution and SZ in individuals with VCFS |
NS |
[105,380] |
|
COMT |
rs165599 (c.*522G>A), rs737865 (c.-92 + 701A>G), rs165688 (p.V158M) |
Caucasian (Ashkenazi Jewish) |
|
|
|
G allele in the SNPs was associated with SZ. Haplotype rs737865-rs165599 (G-G) had most significant overall association with SZ |
SIG |
[106] |
|
COMT |
rs737865 (c.-92 + 701A>G) |
Meta-analysis (Caucasian) |
|
|
|
Nominally significant association between SNP and SZ in analyses restricted to European samples |
SIG |
[82] |
|
DISC1 |
t(1:11)(q43,q21) |
Caucasian (Scottish pedigree) |
|
|
|
Translocation found to be in significant LD with SZ |
SIG |
[107,108] |
|
DISC1 |
rs821616 (p.S704C), rs821597 (c.2042 + 7630G>A), rs7546310 (c.1982-32754A>C) BM: hippocampal structure and function |
Caucasian, replication: family trios (Caucasian and AA) |
|
|
+ |
704-Ser associated with altered hippocampal structure and formation in healthy subjects. Association between 704-Ser and SZ. Three-SNP haplotype associated with SZ in the family sample |
SIG |
[112] |
|
DISC2 |
n.9481C>T, n.11085C>A, n.11160G>A, n.11870T>C, n.11859T>C |
Caucasian (Scottish) |
|
|
|
No co-segregation with SZ or BD or significant association was detected. SNPs were not in LD |
NS |
[132] |
|
COMT |
EPI: Membrane-bound COMT (MB-COMT) promoter methylation |
Caucasian post-mortem brain samples |
+ |
|
|
COMT promoter methylation did not differ between SZ and control brains |
NS |
[151] |
|
COMT |
MB-COMT promoter EPI: COMT methylation. |
Post-mortem brain samples (97% Caucasian) |
+ |
|
|
Reduced methylation of COMT promoter in SZ compared with controls, resulting in increased MB-COMT expression in SZ compared with controls |
SIG |
[148] |
|
ZNF804A |
rs1344706 (c.256-19902A>C) |
GWAS: Caucasian (English); replication: Caucasian and East Asian (BUL, GRM, US, AUS, JPN, CHN, and ISR) |
|
|
|
Nominally significant association between SNP and SZ in samples; genome-wide association when case sample extended to include BD |
SIG |
[60] |
|
ZNF804A |
rs1344706 (c.256-19902A>C), rs7597593 (c.111 + 69783T>C), rs17508595 (c.111 + 19311C>G) |
Caucasian (Irish) |
|
|
|
Nominally significant association between SNPs and SZ + poor-outcome schizoaffective disorder |
SIG |
[61] |
|
ZNF804A |
rs12477914 and rs1366840 as surrogates for rs1344706 (c.256-19902A>C) |
Initial study: Caucasian; follow-up: Caucasian + CHN |
|
|
|
Nominally significant association between SNPs and SZ. When stratified by population, significant in 2 (RUS and DNK) of 13 (HUN, NOR, RUS, SWE, FIN, DEU, DNK, GBR, SCO, ISL, NLD, ITA, CHN) ethnic groups |
SIG |
[62] |
|
ZNF804A |
rs1344706 (c.256-19902A>C) |
East Asian (Han Chinese) |
|
|
|
Nominally significant association between SNP and SZ in a population-based sample. In a family-based trio study, trend toward significant over-transmission |
SIG/SUG |
[63] |
|
TCF4 |
rs9960767 (c.146-23634T>G) |
Caucasian (BEL, DNK, DEU, IRL, ITA, FIN, SPA, UK, USA) |
|
|
|
Association between the C allele and SZ in GWAS and in replication studies |
SIG |
[59,69] |
|
TCF4 |
rs2958182 (c.146-17653T>A) (as surrogate for rs9960767) |
East Asian (Han Chinese) |
|
|
|
SNP substituted for rs9960767 as rs9960767 is not polymorphic in CHN, is in LD with rs9960767, and is significantly associated with SZ in CHN |
SIG |
[71] |
|
TCF4 |
rs12966547 (g.542881G>A) |
Caucasian |
|
|
|
Significant association between SNP and SZ |
SIG |
[70] |
|
NRG1 |
HapICE (SNP8NRG221132, SNP8NRG221533, SNP8NRG241930, SNP8NRG243177 and SNP8NRG433E1006, & microsatellite repeats 478B14-848 and 420M9-1395) |
Caucasian |
|
|
|
Haplotype significantly associated with SZ, with a relative risk of 2.2 |
SIG |
[77] |
|
RELN |
EPI: RELN promoter methylation |
Post-mortem brain samples |
+ |
|
|
Increased methylation of RELN promoter in SZ compared with controls, leading to reduced RELN mRNA expression |
SIG |
[150] |
|
RELN |
EPI: RELN promoter methylation |
Post-mortem brain samples |
+ |
|
|
By contrast to [150], neither SZ nor control samples found promoter hypermethylation |
NS |
[152] |
|
HTR2A |
EPI: cytosine methylation at rs6313 (c.102>T) |
Post-mortem brain samples |
+ |
|
|
102C carriers have reduced 5HT2A gene expression. In SZ, there is a greater reduction in carriers than in non-SZ carriers. Antipsychotics that reduce CpG methylation lead to increased HTR2A expression |
SIG |
rev. in [153] |
|
TPH2 |
rs4570625 (c.-141-703G>T) rs4570625- rs4565946 ((c.-141-703G>T)-(c.255 + 1256C>T) (G-C)) |
Caucasian |
|
|
|
Higher frequency of SNP in patients with MDD compared with controls in discovery sample; not replicated in replication sample. Trend for rs4570625-rs4565946 G-C haplotype |
SUG |
[18] |
|
KCNH2 |
rs1036145 (c.76 + 496G>A) |
NIMH and CATIE cohorts |
|
|
|
Carriers of rs1036145-TT genotype showed greater change on the PANSS than carriers of TC and CC genotypes. rs1036145-TT and rs3800779-TT showed significant improvement in positive symptoms compared with TC/CC genotypes |
SIG |
[332] |
| -- |
EF: prenatal exposure to influenza (determined by ecologic data only) |
Caucasian (Finnish) |
|
+ |
|
Exposure to influenza during second and third trimesters increased risk of hospitalization for SZ |
SUG |
[226] |
| -- |
EF: prenatal exposure to influenza (determined by ecologic data only) |
Caucasian (English, Welsh) |
|
+ |
|
Number of births with subsequent SZ development was higher during influenza epidemic relative to corresponding time during non-epidemic years |
SUG |
[227] |
| -- |
EF: prenatal exposure to influenza (serologically documented) |
Caucasian, AA, Others (Native American, MEX, East Asian) |
|
+ |
|
Early to mid-gestational exposure to influenza increased risk for SZ |
SIG |
[228] |
| -- |
EF: prenatal exposure to influenza |
Meta-analysis |
|
+ |
|
No association between exposure and SZ identified |
NS |
[229] |
| -- | EF: prenatal exposure to maternal stress (wars, spousal demise, disasters, etc.) | Meta-analysis | + | Data show no effect of prenatal stress on risk for SZ | NS | [230] | ||
5-HTTLPR, 5-hydroxytryptophan transporter-linked polymorphic region; AA, African-American; AU, Australian; BD, bipolar disorder; BD-I, BD-II bipolar disorder types I and II; BDNF, brain-derived neurotrophic factor; BEL, Belgian; BGR, Bulgarian; BM: biomarkers; CATIE, Clinical Antipsychotic Trials of Intervention Effectiveness; CHN, Chinese; CRF, corticotropin-releasing factor; CSF, cerebrospinal fluid; DEX/CRH, Dexamethasone/corticotropin-releasing hormone; DNK, Danish; EF, environmental factors, ELS, early life stress; EPI, epigenetic factors; FIN, Finnish; GRM, German; GWAS, GWAS, Genome-wide association studies; HIV, human immunodeficiency virus; ISL, Icelandic; IRL, Irish; ISC, International Schizophrenia Consortium; ITA, Italian; ISR, Israeli; JPN, Japanese; KOR, Korean; LD, linkage disequilibrium; MB-COMT, membrane-bound catechol-O-methyltransferase; MDD, major depressive disorder; MEX, Mexican; MHPG, 3-methoxy-4-hydroxyphenylglycol;NIMH, National Institute of Mental Health; NLD, Dutch (Netherlands); NS, not significant; rMDD, recurrent MDD; RUS, Russian; SCO, Scottish; SGENE, Schizophrenia Genetics Consortium; SIG, significant; SNP, single-nucleotide polymorphism; SPA, Spanish; SUG, suggestive; SZ, schizophrenia; SZA-BD, schizoaffective disorder, manic or bipolar type; UK, United Kingdom; USA, American; VCFS, velocardiofacial syndrome.
We also discussed the use of pharmacogenomics in psychiatry. The best-studied pharmacogenomics in psychiatry are the CYP450 liver enzymes, responsible for the metabolism of many psychotropic drugs. Various polymorphisms in these enzymes predispose an individual to enhanced or poorer therapeutic and/or side-effect response to certain medications. Despite robust findings of association with the particular CYP450 genotypes and altered response to psychotropics, there remains insufficient evidence to support CYP450 genotype screening [236,381]. Additionally, many of the genetic alterations described in this review also are relevant to pharmacogenomic paradigms. Examples include the poor response to SSRIs seen in Caucasian women with MDD who are carriers of the 5-HTTLPR short allele, or the faster response to SSRIs seen in patients with MDD who are homozygous T for the FKBP5 marker rs1360780 [24,253]. Naturally, there is still work to be done in psychiatric pharmacogenomics, as causative treatment strategies for these disorders have yet to be implemented.
The prospect of personalized medicine in psychiatry more or less reflects ideals still largely unrealized. Currently, the field is at the information-gathering infancy stage. The greatest progress can be expected at the intersections of the categories described above, such as gene × environment and genes × biomarkers, which will poise psychiatry to make biological system-based evaluations. Furthermore, some of the emerging applications, including imaging genomics, strengthen our conviction that the future for personalized medicine is highly promising.
Abbreviations
5-HT: 5-hydroxytryptophan (serotonin); 5-HTTLPR: 5-HT transporter-linked polymorphic region; ACC: anterior cingulate cortex; ANK3: ankyrin 3; APOE: apolipoprotein E; ARNTL: aryl hydrocarbon receptor nuclear translocator-like BmaL1; BCR: breakpoint cluster region; BD: bipolar disorder; BDNF: brain-derived neurotrophic factor; BOLD: blood oxygen level dependent; CACNA1C: calcium channel, voltage-dependent, L type alpha 1C subunit; CATIE: Clinical Antipsychotic Trials of Intervention Effectiveness; Cho: choline; CIA: clozapine-induced agranulocytosis; CLOCK: circadian locomotor output cycles kaput protein; CNS: central nervous system; COMT: catechol-O-methyltransferase; CpG: cytosine-phosphate-guanine; CREB: cAMP responsive element binding proteins 1 to 3; CRF: corticotropin-releasing factor; CRH: corticotropin-releasing hormone; CRHBP: corticotropin-releasing hormone-binding protein; CRHR1: corticotropin-releasing hormone, receptor 1; CRY1/CRY2: cryptochrome 1 and 2; CSF: cerebrospinal fluid; CT: computed tomography; CYP: cytochrome P450; DA: dopamine; DAO: D-amino acid oxidase; DEX/CRH: dexamethasone/corticotropin-releasing hormone; DISC1: disrupted in schizophrenia, 1; dlPFC: dorsolateral pre-frontal cortex; DME: drug-metabolizing enzymes; DMNT: DNA methyltransferase; DRD1/2/4: dopamine receptor, D1/D2/D4; DTI: diffusion tensor imaging; DTNBP1: dystobrevin binding protein 1; ECT: electroconvulsive therapy; EM: extensive metabolizers; FKBP5: FK506-binding protein; fMRI: functional magnetic resonance imaging; GABA: gamma-aminobutyric acid; GABRB2: GABA A receptor, beta 2; GENDEP: Genome-Based Therapeutic Drugs for Depression; GHQ: General Health Questionnaire; GRIN2B: glutamate receptor, ionotrophic, N-methyl D-aspartate 2B; GWAS: genome-wide association studies; 1H-MRS: positron magnetic resonance spectroscopy; ISC: International Schizophrenia Consortium; HDAC1: histone deacetylase 1; HDACi: HDAC inhibitors; HLA: human leukocyte antigen; HP: haptoglobin; HPA: hypothalamic-pituitary-adrenal; HPC: hippocampus; HTR2A and HRT2C: 5-hydroxytryptamine (serotonin) receptor 2A and 2C, G-protein coupled; IL-1B: interleukin 1 beta; KCNH2: potassium voltage-gated channel, subfamily H; IM: intermediate metabolizers; ISC: International Schizophrenia Consortium; LEP: leptin; LD: linkage disequilibrium; LG: licking and grooming; LINC: links the nucleoskeleton to the cytoskeleton; MAOA: monoamine oxidase A; MB: membrane-bound; MCM: mood-congruent memory; MDD: major depressive disorder; MGS: Molecular Genetics of Schizophrenia; MHC: major histocompatibility complex; MHPG: 3-methoxy-4-hydroxphenylglycol; miRNA: microRNA; MnSOD: manganese isoform of superoxide dismutase; MRI: magnetic resonance imaging; MRS: mRNA, messenger RNA; MRS: magnetic resonance spectroscopy; MTHFR: methylenetetrahydrofolate reductase (NAD(P)H); NAA: N-acetylaspartate; NCAN: neurocan; ND: never-depressed; NIMH: National Institute of Mental Health; NIMH-ECA: National Institute of Mental Health Catchment Area; NMDA: N-methyl-D-aspartate; NR2B: NMDA receptor subunit 2B; NR3C1: neuron-specific glucocorticoid receptor; NRG1: neuregulin 1; ODZ4: odd Oz/ten-m homolog 4; PANSS: Positive and Negative Syndrome Scale; PAPLN: papilin; PER1/PER2/PER3: period 1/2/3; PET: positron emission tomography; PFC: pre-frontal cortex; PLXNA2: plexin A2; SLC6A4: dopamine transporter gene A4; PM: poor metabolizers; PTSD: post-traumatic stress disorder; RD: remitted-depressed; SGENE: Schizophrenia Genetics Consortium; sib-tbt: Sibling-Transmission Disequilibrium Test; SNP: single-nucleotide polymorphism; SSL: schizophrenia susceptibility locus; SSRI: Selective serotonin reuptake inhibitor; STAR*D: Sequenced Treatment Alternatives to Relieve Depression; SYNE: spectrin repeat containing, nuclear envelope; SZ: Schizophrenia; SPECT: single-photon emission computed tomography; TP53: tumor protein p53; TPH 1/2: tryptophan hydroxylase 1/2; TD: tardive dyskinesia; TORDIA: Treatment of SSRI-Resistant Depression in Adolescents; TMS: transcranial magnetic stimulation; UM: ultra-rapid metabolizers; vlPFC: ventrolateral pre-frontal cortex; VNTR: variable number tandem repeat regions; XBP1: X-box binding protein 1; ZNF804A: zinc finger 804A.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
UO wrote the first draft of the manuscript; UO, CW, and CN made substantial contributions to conception and design of the review, reviewed and revised the first and subsequent drafts for intellectual content. All authors have read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Uzoezi Ozomaro, Email: uozomaro@med.miami.edu.
Claes Wahlestedt, Email: cwahlestedt@med.miami.edu.
Charles B Nemeroff, Email: cnemeroff@med.miami.edu.
Acknowledgements
This work was supported in part by the following: the National Institutes of Health (NIH) under Ruth L. Kirschstein NRSA F31 MH862752 from the NIH/NIMH to UO, NIH grants MH083733 and MH084880 to CW and NIH MH094759 to CBN. Financial disclosure: At the time of this publication, CW was a consultant for Opko Health, while CBN was a consultant for Xhale, Takeda, SK Pharma, Shire, Roche, Lilly, Allergan; had received grant/support from the NIH, Agency for Healthcare Research and Quality (AHRQ); was a stock shareholder of CeNeRx BioPharma, PharmaNeuroBoost, Revaax Pharma, Xhale, NovaDel Pharma; was on the Board of Directors of the American Foundation for Suicide Prevention (AFSP), Mt. Cook Pharma (2010), NovaDel (2011), Skyland Trail, Gratitude America, ADAA; sat on the Scientific Advisory Board for AFSP, CeNeRx BioPharma, National Alliance for Research on Schizophrenia and Depression (NARSAD), Xhale, PharmaNeuroBoost, Anxiety Disorders Association of America (ADAA), Skyland Trail, AstraZeneca Pharmaceuticals (2009); holds a patent for method and devices for transdermal delivery of lithium (US 6,375,990B1), and for a method of assessing antidepressant drug therapy via transport inhibition of monoamine neurotransmitters by ex vivo assay (US 7,148,027B2) and had equity or other financial interests in AstraZeneca Pharmaceuticals, PharmaNeuroBoost, CeNeRx BioPharma, NovaDel Pharma, Reevax Pharma, American Psychiatric Publishing and Xhale.
References
- Myers AJ, Nemeroff CB. New Vistas in the Management of Treatment-Refractory Psychiatric Disorders: Genomics and Personalized Medicine. Focus. 2010;8:525–535. [Google Scholar]
- Mehta R, Jain RK, Badve S. Personalized medicine: the road ahead. Clin Breast Cancer. 2011;11:20–26. doi: 10.3816/CBC.2011.n.004. [DOI] [PubMed] [Google Scholar]
- Conneally PM. Huntington disease: genetics and epidemiology. Am J Hum Genet. 1984;36:506–526. [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, McGonagle KA, Zhao S, Nelson CB, Hughes M, Eshleman S, Wittchen HU, Kendler KS. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch Gen Psychiatry. 1994;51:8–19. doi: 10.1001/archpsyc.1994.03950010008002. [DOI] [PubMed] [Google Scholar]
- Risch SC, Nemeroff CB. Neurochemical alterations of serotonergic neuronal systems in depression. J Clin Psychiatry. 1992;53:3–7. [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogilvie AD, Battersby S, Bubb VJ, Fink G, Harmar AJ, Goodwim GM, Smith CA. Polymorphism in serotonin transporter gene associated with susceptibility to major depression. Lancet. 1996;347:731–733. doi: 10.1016/s0140-6736(96)90079-3. [DOI] [PubMed] [Google Scholar]
- Hoefgen B, Schulze TG, Ohlraun S, Von Widdern O, Höfels S, Gross M, Heidmann V, Kovalenko S, Eckermann A, Kölsch H, Metten M, Zobel A, Becker T, Nöthen MM, Propping P, Heun R, Maier W, Rietschel M. The power of sample size and homogenous sampling: association between the 5-HTTLPR serotonin transporter polymorphism and major depressive disorder. Biol Psychiatry. 2005;57:247–251. doi: 10.1016/j.biopsych.2004.11.027. [DOI] [PubMed] [Google Scholar]
- Gao J, Pan Z, Jiao Z, Li F, Zhao G, Wei Q, Pan F, Evangelou E. TPH2 Gene Polymorphisms and Major Depression - A Meta-Analysis. PLoS One. 2012;7:e36721. doi: 10.1371/journal.pone.0036721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther DJ, Bader M. A unique central tryptophan hydroxylase isoform. Biochem Pharmacol. 2003;66:1673–1680. doi: 10.1016/s0006-2952(03)00556-2. [DOI] [PubMed] [Google Scholar]
- Nash MW, Sugden K, Huezo-Diaz P, Williamson R, Sterne A, Purcell S, Sham PC, Craig IW. Association analysis of monoamine genes with measures of depression and anxiety in a selected community sample of siblings. Am J Med Genet B Neuropsychiatr Genet. 2005;135B:33–37. doi: 10.1002/ajmg.b.30063. [DOI] [PubMed] [Google Scholar]
- Gizatullin R, Zaboli G, Jönsson EG, Asberg M, Leopardi R. Haplotype analysis reveals tryptophan hydroxylase (TPH) 1 gene variants associated with major depression. Biol Psychiatry. 2006;59:295–300. doi: 10.1016/j.biopsych.2005.07.034. [DOI] [PubMed] [Google Scholar]
- Zill P, Baghai TC, Zwanzger P, Schüle C, Eser D, Rupprecht R, Möller H-J, Bondy B, Ackenheil M. SNP and haplotype analysis of a novel tryptophan hydroxylase isoform (TPH2) gene provide evidence for association with major depression. Mol Psychiatry. 2004;9:1030–1036. doi: 10.1038/sj.mp.4001525. [DOI] [PubMed] [Google Scholar]
- Zhang X, Beaulieu J-M, Sotnikova TD, Gainetdinov RR, Caron MG. Tryptophan hydroxylase-2 controls brain serotonin synthesis. Science. 2004;305:217. doi: 10.1126/science.1097540. [DOI] [PubMed] [Google Scholar]
- Zhang X, Gainetdinov RR, Beaulieu J-M, Sotnikova TD, Burch LH, Williams RB, Schwartz DA, Krishnan KRR, Caron MG. Loss-of-function mutation in tryptophan hydroxylase-2 identified in unipolar major depression. Neuron. 2005;45:11–16. doi: 10.1016/j.neuron.2004.12.014. [DOI] [PubMed] [Google Scholar]
- Garriock HA, Allen JJB, Delgado P, Nahaz Z, Kling MA, Carpenter L, Burke M, Burke W, Schwartz T, Marangell LB, Husain M, Erickson RP, Moreno FA. Lack of association of TPH2 exon XI polymorphisms with major depression and treatment resistance. Mol Psychiatry. 2005;10:976–977. doi: 10.1038/sj.mp.4001712. [DOI] [PubMed] [Google Scholar]
- Serretti A, Chiesa A, Porcelli S, Han C, Patkar AA, Lee S-J, Park MH, Pae C-U. Influence of TPH2 variants on diagnosis and response to treatment in patients with major depression, bipolar disorder and schizophrenia. Psychiatry Res. 2011;189:26–32. doi: 10.1016/j.psychres.2011.02.001. [DOI] [PubMed] [Google Scholar]
- Schuhmacher A, Becker T, Rujescu D, Quednow BB, Lennertz L, Wagner M, Benninghoff J, Rietschel M, Häfner H, Franke P, Wölwer W, Gaebel W, Maier W, Mössner R. Investigation of tryptophan hydroxylase 2 (TPH2) in schizophrenia and in the response to antipsychotics. J Psychiatric Res. 2012;46:1073–1080. doi: 10.1016/j.jpsychires.2012.04.021. [DOI] [PubMed] [Google Scholar]
- Zhang C, Li Z, Shao Y, Xie B, Du Y, Fang Y, Yu S. Association study of tryptophan hydroxylase-2 gene in schizophrenia and its clinical features in Chinese Han population. J Mol Neurosci. 2011;43:406–411. doi: 10.1007/s12031-010-9458-2. [DOI] [PubMed] [Google Scholar]
- Kim Y-K, Lee H-J, Yang J-C, Hwang J-A, Yoon H-K. A tryptophan hydroxylase 2 gene polymorphism is associated with panic disorder. Behav Genet. 2009;39:170–175. doi: 10.1007/s10519-008-9254-8. [DOI] [PubMed] [Google Scholar]
- Mössner R, Walitza S, Geller F, Scherag A, Gutknecht L, Jacob C, Bogusch L, Remschmidt H, Simons M, Herpertz-Dahlmann B, Fleischhaker C, Schulz E, Warnke A, Hinney A, Wewetzer C, Lesch K-P. Transmission disequilibrium of polymorphic variants in the tryptophan hydroxylase-2 gene in children and adolescents with obsessive-compulsive disorder. Int J Neuropsychopharmacol. 2006;9:437–442. doi: 10.1017/S1461145705005997. [DOI] [PubMed] [Google Scholar]
- Walitza S, Renner TJ, Dempfle A, Konrad K, Wewetzer C, Halbach A, Herpertz-Dahlmann B, Remschmidt H, Smidt J, Linder M, Flierl L, Knölker U, Friedel S, Schäfer H, Gross C, Hebebrand J, Warnke A, Lesch KP. Transmission disequilibrium of polymorphic variants in the tryptophan hydroxylase-2 gene in attention-deficit/hyperactivity disorder. Mol Psychiatry. 2005;10:1126–1132. doi: 10.1038/sj.mp.4001734. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB. Clinical significance of psychoneuroendocrinology in psychiatry: focus on the thyroid and adrenal. J Clin Psychiatry. 1989;50:13–20. discussion 21–22. [PubMed] [Google Scholar]
- Binder EB, Salyakina D, Lichtner P, Wochnik GM, Ising M, Pütz B, Papiol S, Seaman S, Lucae S, Kohli MA, Nickel T, Künzel HE, Fuchs B, Majer M, Pfennig A, Kern N, Brunner J, Modell S, Baghai T, Deiml T, Zill P, Bondy B, Rupprecht R, Messer T, Köhnlein O, Dabitz H, Brückl T, Müller N, Pfister H, Lieb R. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat Genet. 2004;36:1319–1325. doi: 10.1038/ng1479. [DOI] [PubMed] [Google Scholar]
- Tatro ET, Everall IP, Masliah E, Hult BJ, Lucero G, Chana G, Soontornniyomkij V, Achim CL. Differential expression of immunophilins FKBP51 and FKBP52 in the frontal cortex of HIV-infected patients with major depressive disorder. J Neuroimmune Pharmacol. 2009;4:218–226. doi: 10.1007/s11481-009-9146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brent D, Melhem N, Ferrell R, Emslie G, Wagner KD, Ryan N, Vitiello B, Birmaher B, Mayes T, Zelazny J, Onorato M, Devlin B, Clarke G, DeBar L, Keller M. Association of FKBP5 polymorphisms with suicidal events in the Treatment of Resistant Depression in Adolescents (TORDIA) study. Am J Psychiatry. 2010;167:190–197. doi: 10.1176/appi.ajp.2009.09040576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timpl P, Spanagel R, Sillaber I, Kresse A, Reul JM, Stalla GK, Blanquet V, Steckler T, Holsboer F, Wurst W. Impaired stress response and reduced anxiety in mice lacking a functional corticotropin-releasing hormone receptor 1. Nat Genet. 1998;19:162–166. doi: 10.1038/520. [DOI] [PubMed] [Google Scholar]
- Gillespie CF, Binder EB, Holtzheimer PE, Nemeroff CB. Integrative Neuroscience and Personalized Medicine. Oxford: Oxford University Press; 2010. Stress and the impact of personalized medicine; pp. 73–92. [Google Scholar]
- Hartline KM, Owens MJ, Nemeroff CB. Postmortem and cerebrospinal fluid studies of corticotropin-releasing factor in humans. Ann N Y Acad Sci. 1996;780:96–105. doi: 10.1111/j.1749-6632.1996.tb15114.x. [DOI] [PubMed] [Google Scholar]
- Arató M, Bánki CM, Bissette G, Nemeroff CB. Elevated CSF CRF in suicide victims. Biol Psychiatry. 1989;25:355–359. doi: 10.1016/0006-3223(89)90183-2. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB, Widerlöv E, Bissette G, Walléus H, Karlsson I, Eklund K, Kilts CD, Loosen PT, Vale W. Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science. 1984;226:1342–1344. doi: 10.1126/science.6334362. [DOI] [PubMed] [Google Scholar]
- Liu Z, Zhu F, Wang G, Xiao Z, Wang H, Tang J, Wang X, Qiu D, Liu W, Cao Z, Li W. Association of corticotropin-releasing hormone receptor1 gene SNP and haplotype with major depression. Neurosci Lett. 2006;404:358–362. doi: 10.1016/j.neulet.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Papiol S, Arias B, Gastó C, Gutiérrez B, Catalán R, Fañanás L. Genetic variability at HPA axis in major depression and clinical response to antidepressant treatment. J Affect Disord. 2007;104:83–90. doi: 10.1016/j.jad.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Lekman M, Laje G, Charney D, Rush AJ, Wilson AF, Sorant AJM, Lipsky R, Wisniewski SR, Manji H, McMahon FJ, Paddock S. The FKBP5-gene in depression and treatment response–an association study in the Sequenced Treatment Alternatives to Relieve Depression (STAR*D) Cohort. Biol Psychiatry. 2008;63:1103–1110. doi: 10.1016/j.biopsych.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes S, Villafuerte S, Forsgren T, Sluijs S, Del-Favero J, Adolfsson R, Van Broeckhoven C. The corticotropin-releasing hormone binding protein is associated with major depression in a population from Northern Sweden. Biol Psychiatry. 2003;54:867–872. doi: 10.1016/s0006-3223(03)00425-6. [DOI] [PubMed] [Google Scholar]
- Van Den Eede F, Venken T, Del-Favero J, Norrback K-F, Souery D, Nilsson LG, Van den Bossche B, Hulstijn W, Sabbe BGC, Cosyns P, Mendlewicz J, Adolfsson R, Van Broeckhoven C, Claes SJ. Single nucleotide polymorphism analysis of corticotropin-releasing factor-binding protein gene in recurrent major depressive disorder. Psychiatry Res. 2007;153:17–25. doi: 10.1016/j.psychres.2006.12.018. [DOI] [PubMed] [Google Scholar]
- Smoller JW, Finn CT. Family, twin, and adoption studies of bipolar disorder. Am J Med Genet C Semin Med Genet. 2003;123C:48–58. doi: 10.1002/ajmg.c.20013. [DOI] [PubMed] [Google Scholar]
- Barnett JH, Smoller JW. The genetics of bipolar disorder. Neuroscience. 2009;164:331–343. doi: 10.1016/j.neuroscience.2009.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock N, Sklar P. Genetics of bipolar disorder: successful start to a long journey. Trends Genet. 2009;25:99–105. doi: 10.1016/j.tig.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Willour VL, Chen H, Toolan J, Belmonte P, Cutler DJ, Goes FS, Zandi PP, Lee RS, MacKinnon DF, Mondimore FM, Schweizer B, DePaulo JR Jr, Gershon ES, McMahon FJ, Potash JB. Family-based association of FKBP5 in bipolar disorder. Mol Psychiatry. 2009;14:261–268. doi: 10.1038/sj.mp.4002141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallin MD, Lasseter VK, Avramopoulos D, Nicodemus KK, Wolyniec PS, McGrath JA, Steel G, Nestadt G, Liang K-Y, Huganir RL, Valle D, Pulver AE. Bipolar I disorder and schizophrenia: a 440-single-nucleotide polymorphism screen of 64 candidate genes among Ashkenazi Jewish case-parent trios. Am J Hum Genet. 2005;77:918–936. doi: 10.1086/497703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawlik M, Moller-Ehrlich K, Mende M, Jovnerovski M, Jung S, Jabs B, Knapp M, Stoeber G. Is FKBP5 a genetic marker of affective psychosis? A case control study and analysis of disease related traits. BMC Psychiatry. 2006;6:52. doi: 10.1186/1471-244X-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum AE, Akula N, Cabanero M, Cardona I, Corona W, Klemens B, Schulze TG, Cichon S, Rietschel M, Nöthen MM, Georgi A, Schumacher J, Schwarz M, Abou Jamra R, Höfels S, Propping P, Satagopan J, Detera-Wadleigh SD, Hardy J, McMahon FJ. A genome-wide association study implicates diacylglycerol kinase eta (DGKH) and several other genes in the etiology of bipolar disorder. Mol Psychiatry. 2008;13:197–207. doi: 10.1038/sj.mp.4002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti F, Serretti A, Colombo C, Barbini B, Lorenzi C, Campori E, Smeraldi E. Influence of CLOCK gene polymorphism on circadian mood fluctuation and illness recurrence in bipolar depression. Am J Med Genet B Neuropsychiatr Genet. 2003;123B:23–26. doi: 10.1002/ajmg.b.20038. [DOI] [PubMed] [Google Scholar]
- Shi J, Wittke-Thompson JK, Badner JA, Hattori E, Potash JB, Willour VL, McMahon FJ, Gershon ES, Liu C. Clock genes may influence bipolar disorder susceptibility and dysfunctional circadian rhythm. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1047–1055. doi: 10.1002/ajmg.b.30714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager-Smith K, Kay SA. Circadian rhythm genetics: from flies to mice to humans. Nat Genet. 2000;26:23–27. doi: 10.1038/79134. [DOI] [PubMed] [Google Scholar]
- Mansour HA, Wood J, Logue T, Chowdari KV, Dayal M, Kupfer DJ, Monk TH, Devlin B, Nimgaonkar VL. Association study of eight circadian genes with bipolar I disorder, schizoaffective disorder and schizophrenia. Genes Brain Behav. 2006;5:150–157. doi: 10.1111/j.1601-183X.2005.00147.x. [DOI] [PubMed] [Google Scholar]
- Ferreira MAR, O’Donovan MC, Meng YA, Jones IR, Ruderfer DM, Jones L, Fan J, Kirov G, Perlis RH, Green EK, Smoller JW, Grozeva D, Stone J, Nikolov I, Chambert K, Hamshere ML, Nimgaonkar VL, Moskvina V, Thase ME, Caesar S, Sachs GS, Franklin J, Gordon-Smith K, Ardlie KG, Gabriel SB, Fraser C, Blumenstiel B, Defelice M, Breen G, Gill M. Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet. 2008;40:1056–1058. doi: 10.1038/ng.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichon S, Mühleisen TW, Degenhardt FA, Mattheisen M, Miró X, Strohmaier J, Steffens M, Meesters C, Herms S, Weingarten M, Priebe L, Haenisch B, Alexander M, Vollmer J, Breuer R, Schmäl C, Tessmann P, Moebus S, Wichmann H-E, Schreiber S, Müller-Myhsok B, Lucae S, Jamain S, Leboyer M, Bellivier F, Etain B, Henry C, Kahn J-P, Heath S, Hamshere M. Genome-wide association study identifies genetic variation in neurocan as a susceptibility factor for bipolar disorder. Am J Hum Genet. 2011;88:372–381. doi: 10.1016/j.ajhg.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklar P, Ripke S, Scott LJ. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet. 2011;43:977–983. doi: 10.1038/ng.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green EK, Grozeva D, Forty L, Gordon-Smith K, Russell E, Farmer A, Hamshere M, Jones IR, Jones L, McGuffin P, Moran JL, Purcell S, Sklar P, Owen MJ, O’Donovan MC, Craddock N. Association at SYNE1 in both bipolar disorder and recurrent major depression. Mol Psychiatry. 2012;18:614–617. doi: 10.1038/mp.2012.48. [DOI] [PubMed] [Google Scholar]
- Warren DT, Zhang Q, Weissberg PL, Shanahan CM. Nesprins: intracellular scaffolds that maintain cell architecture and coordinate cell function? Expert Rev Mol Med. 2005;7:1–15. doi: 10.1017/S1462399405009294. [DOI] [PubMed] [Google Scholar]
- Cardno AG, Marshall EJ, Coid B, Macdonald AM, Ribchester TR, Davies NJ, Venturi P, Jones LA, Lewis SW, Sham PC, Gottesman II, Farmer AE, McGuffin P, Reveley AM, Murray RM. Heritability estimates for psychotic disorders: the Maudsley twin psychosis series. Arch Gen Psychiatry. 1999;56:162–168. doi: 10.1001/archpsyc.56.2.162. [DOI] [PubMed] [Google Scholar]
- Cardno AG, Gottesman II. Twin studies of schizophrenia: from bow-and-arrow concordances to star wars Mx and functional genomics. Am J Med Genet. 2000;97:12–17. [PubMed] [Google Scholar]
- Gejman PV, Sanders AR, Kendler KS. Genetics of schizophrenia: new findings and challenges. Annu Rev Genomics Hum Genet. 2011;12:121–144. doi: 10.1146/annurev-genom-082410-101459. [DOI] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, Sklar P. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe’er I, Dudbridge F, Holmans PA, Whittemore AS, Mowry BJ, Olincy A, Amin F, Cloninger CR, Silverman JM, Buccola NG, Byerley WF, Black DW, Crowe RR, Oksenberg JR, Mirel DB, Kendler KS, Freedman R, Gejman PV. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009;460:753–757. doi: 10.1038/nature08192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, Werge T, Pietiläinen OPH, Mors O, Mortensen PB, Sigurdsson E, Gustafsson O, Nyegaard M, Tuulio-Henriksson A, Ingason A, Hansen T, Suvisaari J, Lonnqvist J, Paunio T, Børglum AD, Hartmann A, Fink-Jensen A, Nordentoft M, Hougaard D, Norgaard-Pedersen B, Böttcher Y, Olesen J, Breuer R, Möller H-J, Giegling I. Common variants conferring risk of schizophrenia. Nature. 2009;460:744–747. doi: 10.1038/nature08186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, Nikolov I, Hamshere M, Carroll L, Georgieva L, Dwyer S, Holmans P, Marchini JL, Spencer CCA, Howie B, Leung H-T, Hartmann AM, Möller H-J, Morris DW, Shi Y, Feng G, Hoffmann P, Propping P, Vasilescu C, Maier W, Rietschel M, Zammit S, Schumacher J, Quinn EM, Schulze TG, Williams NM, Giegling I, Iwata N, Ikeda M, Darvasi A, Shifman S, He L, Duan J, Sanders AR, Levinson DF, Gejman PV, Cichon S, Nöthen MM, Gill M, Corvin A, Rujescu D, Kirov G, Owen MJ, Buccola NG, Mowry BJ, Freedman R, Amin F, Black DW, Silverman JM, Byerley WF, Cloninger CR. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40:1053–1055. doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- Riley B, Thiselton D, Maher BS, Bigdeli T, Wormley B, McMichael GO, Fanous AH, Vladimirov V, O’Neill FA, Walsh D, Kendler KS. Replication of association between schizophrenia and ZNF804A in the Irish Case–control Study of Schizophrenia sample. Mol Psychiatry. 2010;15:29–37. doi: 10.1038/mp.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg S, Mors O, Børglum AD, Gustafsson O, Werge T, Mortensen PB, Andreassen OA, Sigurdsson E, Thorgeirsson TE, Böttcher Y, Olason P, Ophoff RA, Cichon S, Gudjonsdottir IH, Pietiläinen OPH, Nyegaard M, Tuulio-Henriksson A, Ingason A, Hansen T, Athanasiu L, Suvisaari J, Lonnqvist J, Paunio T, Hartmann A, Jürgens G, Nordentoft M, Hougaard D, Norgaard-Pedersen B, Breuer R, Möller H-J, Giegling I, Glenthøj B, Rasmussen HB, Mattheisen M, Bitter I, Réthelyi JM, Sigmundsson T, Fossdal R, Thorsteinsdottir U, Ruggeri M, Tosato S, Strengman E, Kiemeney LA, Melle I, Djurovic S, Abramova L, Kaleda V, Walshe M, Bramon E, Vassos E, Li T, Fraser G, Walker N, Toulopoulou T, Yoon J, Freimer NB, Cantor RM, Murray R, Kong A, Golimbet V, Jönsson EG, Terenius L, Agartz I, Petursson H, Nöthen MM, Rietschel M, Peltonen L, Rujescu D, Collier DA, Stefansson H, St Clair D, Stefansson K. Expanding the range of ZNF804A variants conferring risk of psychosis. Mol Psychiatry. 2011;16:59–66. doi: 10.1038/mp.2009.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Lu SM, Qiu C, Liu XG, Gao CG, Guo TW, Valenzuela RK, Deng HW, Ma J. Population-based and family-based association studies of ZNF804A locus and schizophrenia. Mol Psychiatry. 2011;16:360–361. doi: 10.1038/mp.2010.55. [DOI] [PubMed] [Google Scholar]
- Lencz T, Morgan TV, Athanasiou M, Dain B, Reed CR, Kane JM, Kucherlapati R, Malhotra AK. Converging evidence for a pseudoautosomal cytokine receptor gene locus in schizophrenia. Mol Psychiatry. 2007;12:572–580. doi: 10.1038/sj.mp.4001983. [DOI] [PubMed] [Google Scholar]
- Williams HJ, Norton N, Dwyer S, Moskvina V, Nikolov I, Carroll L, Georgieva L, Williams NM, Morris DW, Quinn EM, Giegling I, Ikeda M, Wood J, Lencz T, Hultman C, Lichtenstein P, Thiselton D, Maher BS, Malhotra AK, Riley B, Kendler KS, Gill M, Sullivan P, Sklar P, Purcell S, Nimgaonkar VL, Kirov G, Holmans P, Corvin A, Rujescu D, Craddock N, Owen MJ, O’Donovan MC. Fine mapping of ZNF804A and genome-wide significant evidence for its involvement in schizophrenia and bipolar disorder. Mol Psychiatry. 2011;16:429–441. doi: 10.1038/mp.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flora A, Garcia JJ, Thaller C, Zoghbi HY. The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc Natl Acad Sci USA. 2007;104:15382–15387. doi: 10.1073/pnas.0707456104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pontual L, Mathieu Y, Golzio C, Rio M, Malan V, Boddaert N, Soufflet C, Picard C, Durandy A, Dobbie A, Heron D, Isidor B, Motte J, Newburry-Ecob R, Pasquier L, Tardieu M, Viot G, Jaubert F, Munnich A, Colleaux L, Vekemans M, Etchevers H, Lyonnet S, Amiel J. Mutational, functional, and expression studies of the TCF4 gene in Pitt-Hopkins syndrome. Hum Mutat. 2009;30:669–676. doi: 10.1002/humu.20935. [DOI] [PubMed] [Google Scholar]
- Pitt D, Hopkins I. A syndrome of mental retardation, wide mouth and intermittent overbreathing. Aust Paediatr J. 1978;14:182–184. doi: 10.1111/jpc.1978.14.3.182. [DOI] [PubMed] [Google Scholar]
- Steinberg S, De Jong S, Andreassen OA, Werge T, Børglum AD, Mors O, Mortensen PB, Gustafsson O, Costas J, Pietiläinen OPH, Demontis D, Papiol S, Huttenlocher J, Mattheisen M, Breuer R, Vassos E, Giegling I, Fraser G, Walker N, Tuulio-Henriksson A, Suvisaari J, Lönnqvist J, Paunio T, Agartz I, Melle I, Djurovic S, Strengman E, Jürgens G, Glenthøj B, Terenius L. Common variants at VRK2 and TCF4 conferring risk of schizophrenia. Hum Mol Genet. 2011;20:4076–4081. doi: 10.1093/hmg/ddr325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Sanders AR, Kendler KS, Levinson DF, Sklar P, Holmans PA, Lin D, Duan J, Ophoff RA, Andreassen OA, Scolnick E, Cichon S, St Clair D, Corvin A, Gurling H, Werge T, Rujescu D, Blackwood D, Pato CN, Malhotra AK, Purcell SM, Dudbridge F, Neale BM, Rossin L, Visscher PM, Posthuma D, Ruderfer DM, Fanous AH, Stefansson H, Steinberg S. Genome-wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969–976. doi: 10.1038/ng.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Li Z, Chen P, Zhao Q, Wang T, Huang K, Li J, Li Y, Liu J, Zeng Z, Feng G, He L, Shi Y. Common Variants in Major Histocompatibility Complex Region and TCF4 Gene Are Significantly Associated with Schizophrenia in Han Chinese. Biol Psychiatry. 2010;68:671–673. doi: 10.1016/j.biopsych.2010.06.014. [DOI] [PubMed] [Google Scholar]
- Canoll PD, Musacchio JM, Hardy R, Reynolds R, Marchionni MA, Salzer JL. GGF/neuregulin is a neuronal signal that promotes the proliferation and survival and inhibits the differentiation of oligodendrocyte progenitors. Neuron. 1996;17:229–243. doi: 10.1016/s0896-6273(00)80155-5. [DOI] [PubMed] [Google Scholar]
- Anton ES, Marchionni MA, Lee KF, Rakic P. Role of GGF/neuregulin signaling in interactions between migrating neurons and radial glia in the developing cerebral cortex. Development. 1997;124:3501–3510. doi: 10.1242/dev.124.18.3501. [DOI] [PubMed] [Google Scholar]
- Huang YZ, Won S, Ali DW, Wang Q, Tanowitz M, Du QS, Pelkey KA, Yang DJ, Xiong WC, Salter MW, Mei L. Regulation of neuregulin signaling by PSD-95 interacting with ErbB4 at CNS synapses. Neuron. 2000;26:443–455. doi: 10.1016/s0896-6273(00)81176-9. [DOI] [PubMed] [Google Scholar]
- Liu Y, Ford B, Mann MA, Fischbach GD. Neuregulins increase alpha7 nicotinic acetylcholine receptors and enhance excitatory synaptic transmission in GABAergic interneurons of the hippocampus. J Neurosci. 2001;21:5660–5669. doi: 10.1523/JNEUROSCI.21-15-05660.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corfas G, Roy K, Buxbaum JD. Neuregulin 1-erbB signaling and the molecular/cellular basis of schizophrenia. Nat Neurosci. 2004;7:575–580. doi: 10.1038/nn1258. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Sigurdsson E, Steinthorsdottir V, Bjornsdottir S, Sigmundsson T, Ghosh S, Brynjolfsson J, Gunnarsdottir S, Ivarsson O, Chou TT, Hjaltason O, Birgisdottir B, Jonsson H, Gudnadottir VG, Gudmundsdottir E, Bjornsson A, Ingvarsson B, Ingason A, Sigfusson S, Hardardottir H, Harvey RP, Lai D, Zhou M, Brunner D, Mutel V, Gonzalo A, Lemke G, Sainz J, Johannesson G, Andresson T, Gudbjartsson D, Manolescu A, Frigge ML, Gurney ME, Kong A, Gulcher JR, Petursson H, Stefansson K. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71:877–892. doi: 10.1086/342734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulver AE, Wolyniec PS, Housman D, Kazazian HH, Antonarakis SE, Nestadt G, Lasseter VK, McGrath JA, Dombroski B, Karayiorgou M, Ton C, Blouin JL, Kempf L. The Johns Hopkins University Collaborative Schizophrenia Study: an epidemiologic-genetic approach to test the heterogeneity hypothesis and identify schizophrenia susceptibility genes. Cold Spring Harb Symp Quant Biol. 1996;61:797–814. [PubMed] [Google Scholar]
- Kendler KS, MacLean CJ, O’Neill FA, Burke J, Murphy B, Duke F, Shinkwin R, Easter SM, Webb BT, Zhang J, Walsh D, Straub RE. Evidence for a schizophrenia vulnerability locus on chromosome 8p in the Irish Study of High-Density Schizophrenia Families. Am J Psychiatry. 1996;153:1534–1540. doi: 10.1176/ajp.153.12.1534. [DOI] [PubMed] [Google Scholar]
- Blouin JL, Dombroski BA, Nath SK, Lasseter VK, Wolyniec PS, Nestadt G, Thornquist M, Ullrich G, McGrath J, Kasch L, Lamacz M, Thomas MG, Gehrig C, Radhakrishna U, Snyder SE, Balk KG, Neufeld K, Swartz KL, DeMarchi N, Papadimitriou GN, Dikeos DG, Stefanis CN, Chakravarti A, Childs B, Housman DE, Kazazian HH, Antonarakis S, Pulver AE. Schizophrenia susceptibility loci on chromosomes 13q32 and 8p21. Nat Genet. 1998;20:70–73. doi: 10.1038/1734. [DOI] [PubMed] [Google Scholar]
- Kaufmann CA, Suarez B, Malaspina D, Pepple J, Svrakic D, Markel PD, Meyer J, Zambuto CT, Schmitt K, Matise TC, Harkavy Friedman JM, Hampe C, Lee H, Shore D, Wynne D, Faraone SV, Tsuang MT, Cloninger CR. NIMH Genetics Initiative Millenium Schizophrenia Consortium: linkage analysis of African-American pedigrees. Am J Med Genet. 1998;81:282–289. [PubMed] [Google Scholar]
- Allen NC, Bagade S, McQueen MB, Ioannidis JPA, Kavvoura FK, Khoury MJ, Tanzi RE, Bertram L. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40:827–834. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- Stefansson H, Sarginson J, Kong A, Yates P, Steinthorsdottir V, Gudfinnsson E, Gunnarsdottir S, Walker N, Petursson H, Crombie C, Ingason A, Gulcher JR, Stefansson K, St Clair D. Association of neuregulin 1 with schizophrenia confirmed in a Scottish population. Am J Hum Genet. 2003;72:83–87. doi: 10.1086/345442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams NM, Preece A, Spurlock G, Norton N, Williams HJ, Zammit S, O’Donovan MC, Owen MJ. Support for genetic variation in neuregulin 1 and susceptibility to schizophrenia. Mol Psychiatry. 2003;8:485–487. doi: 10.1038/sj.mp.4001348. [DOI] [PubMed] [Google Scholar]
- Benzel I, Bansal A, Browning BL, Galwey NW, Maycox PR, McGinnis R, Smart D, St Clair D, Yates P, Purvis I. Interactions among genes in the ErbB-Neuregulin signalling network are associated with increased susceptibility to schizophrenia. Behav Brain Funct. 2007;3:31. doi: 10.1186/1744-9081-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M, Takahashi N, Saito S, Aleksic B, Watanabe Y, Nunokawa A, Yamanouchi Y, Kitajima T, Kinoshita Y, Kishi T, Kawashima K, Hashimoto R, Ujike H, Inada T, Someya T, Takeda M, Ozaki N, Iwata N. Failure to replicate the association between NRG1 and schizophrenia using Japanese large sample. Schizophr Res. 2008;101:1–8. doi: 10.1016/j.schres.2008.01.010. [DOI] [PubMed] [Google Scholar]
- Alaerts M, Ceulemans S, Forero D, Moens LN, De Zutter S, Heyrman L, Lenaerts A-S, Norrback K-F, De Rijk P, Nilsson L-G, Goossens D, Adolfsson R, Del-Favero J. Support for NRG1 as a susceptibility factor for schizophrenia in a northern Swedish isolated population. Arch Gen Psychiatry. 2009;66:828–837. doi: 10.1001/archgenpsychiatry.2009.82. [DOI] [PubMed] [Google Scholar]
- So H-C, Fong PY, Chen RYL, Hui TCK, Ng MYM, Cherny SS, Mak WWM, Cheung EFC, Chan RCK, Chen EYH, Li T, Sham PC. Identification of neuroglycan C and interacting partners as potential susceptibility genes for schizophrenia in a Southern Chinese population. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:103–113. doi: 10.1002/ajmg.b.30961. [DOI] [PubMed] [Google Scholar]
- Iwata N, Suzuki T, Ikeda M, Kitajima T, Yamanouchi Y, Inada T, Ozaki N. No association with the neuregulin 1 haplotype to Japanese schizophrenia. Mol Psychiatry. 2004;9:126–127. doi: 10.1038/sj.mp.4001456. [DOI] [PubMed] [Google Scholar]
- Vilella E, Costas J, Sanjuan J, Guitart M, De Diego Y, Carracedo A, Martorell L, Valero J, Labad A, De Frutos R, Nájera C, Moltó MD, Toirac I, Guillamat R, Brunet A, Vallès V, Pérez L, Leon M, De Fonseca FR, Phillips C, Torres M. Association of schizophrenia with DTNBP1 but not with DAO, DAOA, NRG1 and RGS4 nor their genetic interaction. J Psychiatr Res. 2008;42:278–288. doi: 10.1016/j.jpsychires.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Jungerius BJ, Hoogendoorn MLC, Bakker SC, Van’t Slot R, Bardoel AF, Ophoff RA, Wijmenga C, Kahn RS, Sinke RJ. An association screen of myelin-related genes implicates the chromosome 22q11 PIK4CA gene in schizophrenia. Mol Psychiatry. 2008;13:1060–1068. doi: 10.1038/sj.mp.4002080. [DOI] [PubMed] [Google Scholar]
- Crowley JJ, Keefe RSE, Perkins DO, Stroup TS, Lieberman JA, Sullivan PF. The neuregulin 1 promoter polymorphism rs6994992 is not associated with chronic schizophrenia or neurocognition. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1298–1300. doi: 10.1002/ajmg.b.30727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders AR, Duan J, Levinson DF, Shi J, He D, Hou C, Burrell GJ, Rice JP, Nertney DA, Olincy A, Rozic P, Vinogradov S, Buccola NG, Mowry BJ, Freedman R, Amin F, Black DW, Silverman JM, Byerley WF, Crowe RR, Cloninger CR, Martinez M, Gejman PV. No significant association of 14 candidate genes with schizophrenia in a large European ancestry sample: implications for psychiatric genetics. Am J Psychiatry. 2008;165:497–506. doi: 10.1176/appi.ajp.2007.07101573. [DOI] [PubMed] [Google Scholar]
- Jönsson EG, Saetre P, Vares M, Andreou D, Larsson K, Timm S, Rasmussen HB, Djurovic S, Melle I, Andreassen OA, Agartz I, Werge T, Hall H, Terenius L. DTNBP1, NRG1, DAOA, DAO and GRM3 polymorphisms and schizophrenia: an association study. Neuropsychobiology. 2009;59:142–150. doi: 10.1159/000218076. [DOI] [PubMed] [Google Scholar]
- Yang JZ, Si TM, Ruan Y, Ling YS, Han YH, Wang XL, Zhou M, Zhang HY, Kong QM, Liu C, Zhang DR, Yu YQ, Liu SZ, Ju GZ, Shu L, Ma DL, Zhang D. Association study of neuregulin 1 gene with schizophrenia. Mol Psychiatry. 2003;8:706–709. doi: 10.1038/sj.mp.4001377. [DOI] [PubMed] [Google Scholar]
- Zhao X, Shi Y, Tang J, Tang R, Yu L, Gu N, Feng G, Zhu S, Liu H, Xing Y, Zhao S, Sang H, Guan Y, St Clair D, He L. A case control and family based association study of the neuregulin1 gene and schizophrenia. J Med Genet. 2004;41:31–34. doi: 10.1136/jmg.2003.014977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong C-J, Huo S-J, Liao D-L, Lee K, Wu J-Y, Tsai S-J. Case–control and family-based association studies between the neuregulin 1 (Arg38Gln) polymorphism and schizophrenia. Neurosci Lett. 2004;366:158–161. doi: 10.1016/j.neulet.2004.05.027. [DOI] [PubMed] [Google Scholar]
- Li T, Stefansson H, Gudfinnsson E, Cai G, Liu X, Murray RM, Steinthorsdottir V, Januel D, Gudnadottir VG, Petursson H, Ingason A, Gulcher JR, Stefansson K, Collier DA. Identification of a novel neuregulin 1 at-risk haplotype in Han schizophrenia Chinese patients, but no association with the Icelandic/Scottish risk haplotype. Mol Psychiatry. 2004;9:698–704. doi: 10.1038/sj.mp.4001485. [DOI] [PubMed] [Google Scholar]
- Georgieva L, Dimitrova A, Ivanov D, Nikolov I, Williams NM, Grozeva D, Zaharieva I, Toncheva D, Owen MJ, Kirov G, O’Donovan MC. Support for neuregulin 1 as a susceptibility gene for bipolar disorder and schizophrenia. Biol Psychiatry. 2008;64:419–427. doi: 10.1016/j.biopsych.2008.03.025. [DOI] [PubMed] [Google Scholar]
- Hall D, Gogos JA, Karayiorgou M. The contribution of three strong candidate schizophrenia susceptibility genes in demographically distinct populations. Genes Brain Behav. 2004;3:240–248. doi: 10.1111/j.1601-183X.2004.00078.x. [DOI] [PubMed] [Google Scholar]
- Thiselton DL, Webb BT, Neale BM, Ribble RC, O’Neill FA, Walsh D, Riley BP, Kendler KS. No evidence for linkage or association of neuregulin-1 (NRG1) with disease in the Irish study of high-density schizophrenia families (ISHDSF) Mol Psychiatry. 2004;9:777–783. doi: 10.1038/sj.mp.4001530. image 729. [DOI] [PubMed] [Google Scholar]
- Turunen JA, Peltonen JO, Pietiläinen OPH, Hennah W, Loukola A, Paunio T, Silander K, Ekelund J, Varilo T, Partonen T, Lönnqvist J, Peltonen L. The role of DTNBP1, NRG1, and AKT1 in the genetics of schizophrenia in Finland. Schizophr Res. 2007;91:27–36. doi: 10.1016/j.schres.2006.11.028. [DOI] [PubMed] [Google Scholar]
- Munafò MR, Thiselton DL, Clark TG, Flint J. Association of the NRG1 gene and schizophrenia: a meta-analysis. Mol Psychiatry. 2006;11:539–546. doi: 10.1038/sj.mp.4001817. [DOI] [PubMed] [Google Scholar]
- Munafò MR, Attwood AS, Flint J. Neuregulin 1 genotype and schizophrenia. Schizophr Bull. 2008;34:9–12. doi: 10.1093/schbul/sbm129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KC. Schizophrenia and velo-cardio-facial syndrome. Lancet. 2002;359:426–430. doi: 10.1016/S0140-6736(02)07604-3. [DOI] [PubMed] [Google Scholar]
- Shifman S, Bronstein M, Sternfeld M, Pisanté-Shalom A, Lev-Lehman E, Weizman A, Reznik I, Spivak B, Grisaru N, Karp L, Schiffer R, Kotler M, Strous RD, Swartz-Vanetik M, Knobler HY, Shinar E, Beckmann JS, Yakir B, Risch N, Zak NB, Darvasi A. A highly significant association between a COMT haplotype and schizophrenia. Am J Hum Genet. 2002;71:1296–1302. doi: 10.1086/344514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Clair D, Blackwood D, Muir W, Carothers A, Walker M, Spowart G, Gosden C, Evans HJ. Association within a family of a balanced autosomal translocation with major mental illness. Lancet. 1990;336:13–16. doi: 10.1016/0140-6736(90)91520-k. [DOI] [PubMed] [Google Scholar]
- Blackwood DH, Fordyce A, Walker MT, St Clair DM, Porteous DJ, Muir WJ. Schizophrenia and affective disorders–cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet. 2001;69:428–433. doi: 10.1086/321969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar JK, Christie S, Anderson S, Lawson D, Hsiao-Wei Loh D, Devon RS, Arveiler B, Muir WJ, Blackwood DH, Porteous DJ. Genomic structure and localisation within a linkage hotspot of Disrupted In Schizophrenia 1, a gene disrupted by a translocation segregating with schizophrenia. Mol Psychiatry. 2001;6:173–178. doi: 10.1038/sj.mp.4000784. [DOI] [PubMed] [Google Scholar]
- Kim JY, Duan X, Liu CY, Jang M-H, Guo JU, Pow-anpongkul N, Kang E, Song H, Ming G. DISC1 regulates new neuron development in the adult brain via modulation of AKT-mTOR signaling through KIAA1212. Neuron. 2009;63:761–773. doi: 10.1016/j.neuron.2009.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T. Molecular genetics of bipolar disorder and depression. Psychiatry Clin Neurosci. 2007;61:3–19. doi: 10.1111/j.1440-1819.2007.01604.x. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Straub RE, Pezawas L, Egan MF, Mattay VS, Hariri AR, Verchinski BA, Meyer-Lindenberg A, Balkissoon R, Kolachana B, Goldberg TE, Weinberger DR. Variation in DISC1 affects hippocampal structure and function and increases risk for schizophrenia. Proc Natl Acad Sci USA. 2005;102:8627–8632. doi: 10.1073/pnas.0500515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon TD, Hennah W, Van Erp TGM, Thompson PM, Lonnqvist J, Huttunen M, Gasperoni T, Tuulio-Henriksson A, Pirkola T, Toga AW, Kaprio J, Mazziotta J, Peltonen L. Association of DISC1/TRAX haplotypes with schizophrenia, reduced prefrontal gray matter, and impaired short- and long-term memory. Arch Gen Psychiatry. 2005;62:1205–1213. doi: 10.1001/archpsyc.62.11.1205. [DOI] [PubMed] [Google Scholar]
- Ekelund J, Hennah W, Hiekkalinna T, Parker A, Meyer J, Lönnqvist J, Peltonen L. Replication of 1q42 linkage in Finnish schizophrenia pedigrees. Mol Psychiatry. 2004;9:1037–1041. doi: 10.1038/sj.mp.4001536. [DOI] [PubMed] [Google Scholar]
- Hennah W, Varilo T, Kestilä M, Paunio T, Arajärvi R, Haukka J, Parker A, Martin R, Levitzky S, Partonen T, Meyer J, Lönnqvist J, Peltonen L, Ekelund J. Haplotype transmission analysis provides evidence of association for DISC1 to schizophrenia and suggests sex-dependent effects. Hum Mol Genet. 2003;12:3151–3159. doi: 10.1093/hmg/ddg341. [DOI] [PubMed] [Google Scholar]
- Lepagnol-Bestel A-M, Dubertret C, Benmessaoud D, Simonneau M, Adès J, Kacha F, Hamdani N, Gorwood P, Ramoz N. Association of DISC1 gene with schizophrenia in families from two distinct French and Algerian populations. Psychiatr Genet. 2010;20:298–303. doi: 10.1097/YPG.0b013e32833aa5c4. [DOI] [PubMed] [Google Scholar]
- Rastogi A, Zai C, Likhodi O, Kennedy JL, Wong AH. Genetic association and post-mortem brain mRNA analysis of DISC1 and related genes in schizophrenia. Schizophr Res. 2009;114:39–49. doi: 10.1016/j.schres.2009.06.019. [DOI] [PubMed] [Google Scholar]
- Liu Y-L, Fann CS-J, Liu C-M, Chen WJ, Wu J-Y, Hung S-I, Chen C-H, Jou Y-S, Liu S-K, Hwang T-J, Hsieh MH, Ouyang W-C, Chan H-Y, Chen J-J, Yang W-C, Lin C-Y, Lee SFC, Hwu H-G. A single nucleotide polymorphism fine mapping study of chromosome 1q42.1 reveals the vulnerability genes for schizophrenia, GNPAT and DISC1: Association with impairment of sustained attention. Biol Psychiatry. 2006;60:554–562. doi: 10.1016/j.biopsych.2006.04.024. [DOI] [PubMed] [Google Scholar]
- Hennah W, Thomson P, McQuillin A, Bass N, Loukola A, Anjorin A, Blackwood D, Curtis D, Deary IJ, Harris SE, Isometsä ET, Lawrence J, Lönnqvist J, Muir W, Palotie A, Partonen T, Paunio T, Pylkkö E, Robinson M, Soronen P, Suominen K, Suvisaari J, Thirumalai S, St Clair D, Gurling H, Peltonen L, Porteous D. DISC1 association, heterogeneity and interplay in schizophrenia and bipolar disorder. Mol Psychiatry. 2009;14:865–873. doi: 10.1038/mp.2008.22. [DOI] [PubMed] [Google Scholar]
- Hodgkinson CA, Goldman D, Jaeger J, Persaud S, Kane JM, Lipsky RH, Malhotra AK. Disrupted in schizophrenia 1 (DISC1): association with schizophrenia, schizoaffective disorder, and bipolar disorder. Am J Hum Genet. 2004;75:862–872. doi: 10.1086/425586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata I, Perez-Iglesias R, Roiz-Santiañez R, Tordesillas-Gutierrez D, Gonzalez-Mandly A, Berja A, Vazquez-Barquero JL, Crespo-Facorro B. Additive effect of NRG1 and DISC1 genes on lateral ventricle enlargement in first episode schizophrenia. Neuroimage. 2010;53:1016–1022. doi: 10.1016/j.neuroimage.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Mouaffak F, Kebir O, Chayet M, Tordjman S, Vacheron MN, Millet B, Jaafari N, Bellon A, Olié JP, Krebs M-O. Association of Disrupted in Schizophrenia 1 (DISC1) missense variants with ultra-resistant schizophrenia. Pharmacogenomics J. 2011;11:267–273. doi: 10.1038/tpj.2010.40. [DOI] [PubMed] [Google Scholar]
- Nicodemus KK, Kolachana BS, Vakkalanka R, Straub RE, Giegling I, Egan MF, Rujescu D, Weinberger DR. Evidence for statistical epistasis between catechol-O-methyltransferase (COMT) and polymorphisms in RGS4, G72 (DAOA), GRM3, and DISC1: influence on risk of schizophrenia. Hum Genet. 2007;120:889–906. doi: 10.1007/s00439-006-0257-3. [DOI] [PubMed] [Google Scholar]
- Saetre P, Agartz I, De Franciscis A, Lundmark P, Djurovic S, Kähler A, Andreassen OA, Jakobsen KD, Rasmussen HB, Werge T, Hall H, Terenius L, Jönsson EG. Association between a disrupted-in-schizophrenia 1 (DISC1) single nucleotide polymorphism and schizophrenia in a combined Scandinavian case–control sample. Schizophr Res. 2008;106:237–241. doi: 10.1016/j.schres.2008.08.024. [DOI] [PubMed] [Google Scholar]
- Schumacher J, Laje G, Abou Jamra R, Becker T, Mühleisen TW, Vasilescu C, Mattheisen M, Herms S, Hoffmann P, Hillmer AM, Georgi A, Herold C, Schulze TG, Propping P, Rietschel M, McMahon FJ, Nöthen MM, Cichon S. The DISC locus and schizophrenia: evidence from an association study in a central European sample and from a meta-analysis across different European populations. Hum Mol Genet. 2009;18:2719–2727. doi: 10.1093/hmg/ddp204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Li W, Feng J, Heston LL, Scaringe WA, Sommer SS. Identification of high risk DISC1 structural variants with a 2% attributable risk for schizophrenia. Biochem Biophys Res Commun. 2008;367:700–706. doi: 10.1016/j.bbrc.2007.12.117. [DOI] [PubMed] [Google Scholar]
- Szeszko PR, Hodgkinson CA, Robinson DG, Derosse P, Bilder RM, Lencz T, Burdick KE, Napolitano B, Betensky JD, Kane JM, Goldman D, Malhotra AK. DISC1 is associated with prefrontal cortical gray matter and positive symptoms in schizophrenia. Biol Psychol. 2008;79:103–110. doi: 10.1016/j.biopsycho.2007.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson PA, Wray NR, Millar JK, Evans KL, Hellard SL, Condie A, Muir WJ, Blackwood DHR, Porteous DJ. Association between the TRAX/DISC locus and both bipolar disorder and schizophrenia in the Scottish population. Mol Psychiatry. 2005;10:657–668. doi: 10.1038/sj.mp.4001669. 616. [DOI] [PubMed] [Google Scholar]
- Wood LS, Pickering EH, Dechairo BM. Significant support for DAO as a schizophrenia susceptibility locus: examination of five genes putatively associated with schizophrenia. Biol Psychiatry. 2007;61:1195–1199. doi: 10.1016/j.biopsych.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Zhang F, Sarginson J, Crombie C, Walker N, St Clair D, Shaw D. Genetic association between schizophrenia and the DISC1 gene in the Scottish population. Am J Med Genet B Neuropsychiatr Genet. 2006;141B:155–159. doi: 10.1002/ajmg.b.30274. [DOI] [PubMed] [Google Scholar]
- Betcheva ET, Mushiroda T, Takahashi A, Kubo M, Karachanak SK, Zaharieva IT, Vazharova RV, Dimova II, Milanova VK, Tolev T, Kirov G, Owen MJ, O’Donovan MC, Kamatani N, Nakamura Y, Toncheva DI. Case–control association study of 59 candidate genes reveals the DRD2 SNP rs6277 (C957T) as the only susceptibility factor for schizophrenia in the Bulgarian population. J Hum Genet. 2009;54:98–107. doi: 10.1038/jhg.2008.14. [DOI] [PubMed] [Google Scholar]
- Devon RS, Anderson S, Teague PW, Burgess P, Kipari TM, Semple CA, Millar JK, Muir WJ, Murray V, Pelosi AJ, Blackwood DH, Porteous DJ. Identification of polymorphisms within Disrupted in Schizophrenia 1 and Disrupted in Schizophrenia 2, and an investigation of their association with schizophrenia and bipolar affective disorder. Psychiatr Genet. 2001;11:71–78. doi: 10.1097/00041444-200106000-00003. [DOI] [PubMed] [Google Scholar]
- Crick F. Central dogma of molecular biology. Nature. 1970;227:561–563. doi: 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- Pastori C, Wahlestedt C. Involvement of long noncoding RNAs in diseases affecting the central nervous system. RNA Biol. 2012;9:860–870. doi: 10.4161/rna.20482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgar G, Vavouri T. Tuning in to the signals: noncoding sequence conservation in vertebrate genomes. Trends Genet. 2008;24:344–352. doi: 10.1016/j.tig.2008.04.005. [DOI] [PubMed] [Google Scholar]
- Bird A. Perceptions of epigenetics. Nature. 2007;447:396–398. doi: 10.1038/nature05913. [DOI] [PubMed] [Google Scholar]
- Sartor GC, St Laurent G 3rd, Wahlestedt C. The Emerging Role of Non-Coding RNAs in Drug Addiction. Front Genet. 2012;3:106. doi: 10.3389/fgene.2012.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB. The burden of severe depression: A review of diagnostic challenges and treatment alternatives. J Psychiatr Res. 2007;41:189–206. doi: 10.1016/j.jpsychires.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Meaney MJ, Szyf M. Maternal care as a model for experience-dependent chromatin plasticity? Trends Neurosci. 2005;28:456–463. doi: 10.1016/j.tins.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Champagne FA, Francis DD, Mar A, Meaney MJ. Variations in maternal care in the rat as a mediating influence for the effects of environment on development. Physiol Behav. 2003;79:359–371. doi: 10.1016/s0031-9384(03)00149-5. [DOI] [PubMed] [Google Scholar]
- Schroeder M, Krebs MO, Bleich S, Frieling H. Epigenetics and depression: current challenges and new therapeutic options. Curr Opin Psychiatry. 2010;23:588–592. doi: 10.1097/YCO.0b013e32833d16c1. [DOI] [PubMed] [Google Scholar]
- McGowan PO, Sasaki A, D’Alessio AC, Dymov S, Labonté B, Szyf M, Turecki G, Meaney MJ. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci. 2009;12:342–348. doi: 10.1038/nn.2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollin W, Cardon PV Jr, Kety SS. Effects of amino acid feedings in schizophrenic patients treated with iproniazid. Science. 1961;133:104–105. doi: 10.1126/science.133.3446.104. [DOI] [PubMed] [Google Scholar]
- Brune GG, Himwich HE. Effects of methionine loading on the behavior of schizophrenic patients. J Nerv Ment Dis. 1962;134:447–450. doi: 10.1097/00005053-196205000-00007. [DOI] [PubMed] [Google Scholar]
- D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- Abdolmaleky HM, Cheng K-H, Faraone SV, Wilcox M, Glatt SJ, Gao F, Smith CL, Shafa R, Aeali B, Carnevale J, Pan H, Papageorgis P, Ponte JF, Sivaraman V, Tsuang MT, Thiagalingam S. Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum Mol Genet. 2006;15:3132–3145. doi: 10.1093/hmg/ddl253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson DR, Chen Y, Costa E, Dong E, Guidotti A, Kundakovic M, Sharma RP. The human reelin gene: transcription factors (+), repressors (−) and the methylation switch (+/−) in schizophrenia. Pharmacol Ther. 2006;111:272–286. doi: 10.1016/j.pharmthera.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Abdolmaleky HM, Cheng K, Russo A, Smith CL, Faraone SV, Wilcox M, Shafa R, Glatt SJ, Nguyen G, Ponte JF, Thiagalingam S, Tsuang MT. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet. 2005;134B:60–66. doi: 10.1002/ajmg.b.30140. [DOI] [PubMed] [Google Scholar]
- Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, Jia P, Assadzadeh A, Flanagan J, Schumacher A, Wang S-C, Petronis A. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82:696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tochigi M, Iwamoto K, Bundo M, Komori A, Sasaki T, Kato N, Kato T. Methylation status of the reelin promoter region in the brain of schizophrenic patients. Biol Psychiatry. 2008;63:530–533. doi: 10.1016/j.biopsych.2007.07.003. [DOI] [PubMed] [Google Scholar]
- Abdolmaleky HM, Faraone SV, Glatt SJ, Tsuang MT. Meta-analysis of association between the T102C polymorphism of the 5HT2a receptor gene and schizophrenia. Schizophr Res. 2004;67:53–62. doi: 10.1016/s0920-9964(03)00183-x. [DOI] [PubMed] [Google Scholar]
- Abdolmaleky HM, Smith CL, Faraone SV, Shafa R, Stone W, Glatt SJ, Tsuang MT. Methylomics in psychiatry: Modulation of gene-environment interactions may be through DNA methylation. Am J Med Genet B Neuropsychiatr Genet. 2004;127B:51–59. doi: 10.1002/ajmg.b.20142. [DOI] [PubMed] [Google Scholar]
- Levinson DF. Meta-analysis in psychiatric genetics. Curr Psychiatry Rep. 2005;7:143–151. doi: 10.1007/s11920-005-0012-9. [DOI] [PubMed] [Google Scholar]
- Polesskaya OO, Aston C, Sokolov BP. Allele C-specific methylation of the 5-HT2A receptor gene: evidence for correlation with its expression and expression of DNA methylase DNMT1. J Neurosci Res. 2006;83:362–373. doi: 10.1002/jnr.20732. [DOI] [PubMed] [Google Scholar]
- Polesskaya OO, Sokolov BP. Differential expression of the “C” and “T” alleles of the 5-HT2A receptor gene in the temporal cortex of normal individuals and schizophrenics. J Neurosci Res. 2002;67:812–822. doi: 10.1002/jnr.10173. [DOI] [PubMed] [Google Scholar]
- Falkenberg VR, Gurbaxani BM, Unger ER, Rajeevan MS. Functional genomics of serotonin receptor 2A (HTR2A): interaction of polymorphism, methylation, expression and disease association. Neuromolecular Med. 2011;13:66–76. doi: 10.1007/s12017-010-8138-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagalingam S, Cheng K-H, Lee HJ, Mineva N, Thiagalingam A, Ponte JF. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci. 2003;983:84–100. doi: 10.1111/j.1749-6632.2003.tb05964.x. [DOI] [PubMed] [Google Scholar]
- Sharma RP, Grayson DR, Gavin DP. Histone deactylase 1 expression is increased in the prefrontal cortex of schizophrenia subjects: analysis of the National Brain Databank microarray collection. Schizophr Res. 2008;98:111–117. doi: 10.1016/j.schres.2007.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM, Lim B, Matzilevich D, Subburaju S, Walsh JP. Circuitry-based gene expression profiles in GABA cells of the trisynaptic pathway in schizophrenics versus bipolars. Proc Natl Acad Sci USA. 2008;105:20935–20940. doi: 10.1073/pnas.0810153105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti A, Auta J, Davis JM, Di-Giorgi-Gerevini V, Dwivedi Y, Grayson DR, Impagnatiello F, Pandey G, Pesold C, Sharma R, Uzunov D, Costa E, DiGiorgi GV. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry. 2000;57:1061–1069. doi: 10.1001/archpsyc.57.11.1061. [DOI] [PubMed] [Google Scholar]
- Heckers S, Stone D, Walsh J, Shick J, Koul P, Benes FM. Differential hippocampal expression of glutamic acid decarboxylase 65 and 67 messenger RNA in bipolar disorder and schizophrenia. Arch Gen Psychiatry. 2002;59:521–529. doi: 10.1001/archpsyc.59.6.521. [DOI] [PubMed] [Google Scholar]
- Woo T-UW, Walsh JP, Benes FM. Density of glutamic acid decarboxylase 67 messenger RNA-containing neurons that express the N-methyl-D-aspartate receptor subunit NR2A in the anterior cingulate cortex in schizophrenia and bipolar disorder. Arch Gen Psychiatry. 2004;61:649–657. doi: 10.1001/archpsyc.61.7.649. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Hossein Fatemi S, Stary JM, Earle JA, Araghi-Niknam M, Eagan E. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr Res. 2005;72:109–122. doi: 10.1016/j.schres.2004.02.017. [DOI] [PubMed] [Google Scholar]
- Tremolizzo L, Doueiri M-S, Dong E, Grayson DR, Davis J, Pinna G, Tueting P, Rodriguez-Menendez V, Costa E, Guidotti A. Valproate corrects the schizophrenia-like epigenetic behavioral modifications induced by methionine in mice. Biol Psychiatry. 2005;57:500–509. doi: 10.1016/j.biopsych.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Göttlicher M. Valproic acid: an old drug newly discovered as inhibitor of histone deacetylases. Ann Hematol. 2004;83:S91–92. doi: 10.1007/s00277-004-0850-2. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Miller BH, Wahlestedt C. MicroRNA dysregulation in psychiatric disease. Brain Res. 2010;1338:89–99. doi: 10.1016/j.brainres.2010.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins DO, Jeffries CD, Jarskog LF, Thomson JM, Woods K, Newman MA, Parker JS, Jin J, Hammond SM. microRNA expression in the prefrontal cortex of individuals with schizophrenia and schizoaffective disorder. Genome Biol. 2007;8:R27. doi: 10.1186/gb-2007-8-2-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beveridge NJ, Tooney PA, Carroll AP, Gardiner E, Bowden N, Scott RJ, Tran N, Dedova I, Cairns MJ. Dysregulation of miRNA 181b in the temporal cortex in schizophrenia. Hum Mol Genet. 2008;17:1156–1168. doi: 10.1093/hmg/ddn005. [DOI] [PubMed] [Google Scholar]
- Beveridge NJ, Gardiner E, Carroll AP, Tooney PA, Cairns MJ. Schizophrenia is associated with an increase in cortical microRNA biogenesis. Mol Psychiatry. 2010;15:1176–1189. doi: 10.1038/mp.2009.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BH, Zeier Z, Xi L, Lanz TA, Deng S, Strathmann J, Willoughby D, Kenny PJ, Elsworth JD, Lawrence MS, Roth RH, Edbauer D, Kleiman RJ, Wahlestedt C. MicroRNA-132 dysregulation in schizophrenia has implications for both neurodevelopment and adult brain function. Proc Natl Acad Sci USA. 2012;109:3125–3130. doi: 10.1073/pnas.1113793109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Beauchaine TP. Role of biomarkers and endophenotypes in prevention and treatment of psychopathological disorders. Biomark Med. 2009;3:1–3. doi: 10.2217/17520363.3.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- Kraemer HC, Schultz SK, Arndt S. Biomarkers in psychiatry: methodological issues. Am J Geriatr Psychiatry. 2002;10:653–659. [PubMed] [Google Scholar]
- Wichers M, Myin-Germeys I, Jacobs N, Peeters F, Kenis G, Derom C, Vlietinck R, Delespaul P, Van Os J. Genetic risk of depression and stress-induced negative affect in daily life. Br J Psychiatry. 2007;191:218–223. doi: 10.1192/bjp.bp.106.032201. [DOI] [PubMed] [Google Scholar]
- Ellwart T, Rinck M, Becker ES. Selective memory and memory deficits in depressed inpatients. Depress Anxiety. 2003;17:197–206. doi: 10.1002/da.10102. [DOI] [PubMed] [Google Scholar]
- Tarsia M, Power MJ, Sanavio E. Implicit and explicit memory biases in mixed anxiety-depression. J Affect Disord. 2003;77:213–225. doi: 10.1016/s0165-0327(02)00119-2. [DOI] [PubMed] [Google Scholar]
- Erickson K, Drevets WC, Clark L, Cannon DM, Bain EE, Zarate CA Jr, Charney DS, Sahakian BJ. Mood-congruent bias in affective go/no-go performance of unmedicated patients with major depressive disorder. Am J Psychiatry. 2005;162:2171–2173. doi: 10.1176/appi.ajp.162.11.2171. [DOI] [PubMed] [Google Scholar]
- Koster EHW, De Raedt R, Leyman L, De Lissnyder E. Mood-congruent attention and memory bias in dysphoria: Exploring the coherence among information-processing biases. Behav Res Ther. 2010;48:219–225. doi: 10.1016/j.brat.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Van Wingen GA, Van Eijndhoven P, Cremers HR, Tendolkar I, Verkes RJ, Buitelaar JK, Fernández G. Neural state and trait bases of mood-incongruent memory formation and retrieval in first-episode major depression. J Psychiatr Res. 2010;44:527–534. doi: 10.1016/j.jpsychires.2009.11.009. [DOI] [PubMed] [Google Scholar]
- Dryman A, Eaton WW. Affective symptoms associated with the onset of major depression in the community: findings from the US National Institute of Mental Health Epidemiologic Catchment Area Program. Acta Psychiatr Scand. 1991;84:1–5. doi: 10.1111/j.1600-0447.1991.tb01410.x. [DOI] [PubMed] [Google Scholar]
- Hirschfeld RM, Klerman GL, Clayton PJ, Keller MB. Personality and depression. Empirical findings. Arch Gen Psychiatry. 1983;40:993–998. doi: 10.1001/archpsyc.1983.01790080075010. [DOI] [PubMed] [Google Scholar]
- Schrader GD. Does anhedonia correlate with depression severity in chronic depression? Compr Psychiatry. 1997;38:260–263. doi: 10.1016/s0010-440x(97)90057-2. [DOI] [PubMed] [Google Scholar]
- Hecht H, Van Calker D, Berger M, Von Zerssen D. Personality in patients with affective disorders and their relatives. J Affect Disord. 1998;51:33–43. doi: 10.1016/s0165-0327(98)00154-2. [DOI] [PubMed] [Google Scholar]
- McCabe C, Cowen PJ, Harmer CJ. Neural representation of reward in recovered depressed patients. Psychopharmacology (Berl) 2009;205:667–677. doi: 10.1007/s00213-009-1573-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester D. The concentration of neurotransmitter metabolites in the cerebrospinal fluid of suicidal individuals: a meta-analysis. Pharmacopsychiatry. 1995;28:45–50. doi: 10.1055/s-2007-979587. [DOI] [PubMed] [Google Scholar]
- Galfalvy H, Currier D, Oquendo MA, Sullivan G, Huang Y-Y, John Mann J. Lower CSF MHPG predicts short-term risk for suicide attempt. Int J Neuropsychopharmacol. 2009;12:1327–1335. doi: 10.1017/S1461145709990228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramel W, Goldin PR, Eyler LT, Brown GG, Gotlib IH, McQuaid JR. Amygdala reactivity and mood-congruent memory in individuals at risk for depressive relapse. Biol Psychiatry. 2007;61:231–239. doi: 10.1016/j.biopsych.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Autry AE, Monteggia LM. Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol Rev. 2012;64:238–258. doi: 10.1124/pr.111.005108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karege F, Bondolfi G, Gervasoni N, Schwald M, Aubry J-M, Bertschy G. Low brain-derived neurotrophic factor (BDNF) levels in serum of depressed patients probably results from lowered platelet BDNF release unrelated to platelet reactivity. Biol Psychiatry. 2005;57:1068–1072. doi: 10.1016/j.biopsych.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Pan W, Banks WA, Fasold MB, Bluth J, Kastin AJ. Transport of brain-derived neurotrophic factor across the blood–brain barrier. Neuropharmacology. 1998;37:1553–1561. doi: 10.1016/s0028-3908(98)00141-5. [DOI] [PubMed] [Google Scholar]
- Fernandes BS, Gama CS, Ceresér KM, Yatham LN, Fries GR, Colpo G, De Lucena D, Kunz M, Gomes FA, Kapczinski F. Brain-derived neurotrophic factor as a state-marker of mood episodes in bipolar disorders: a systematic review and meta-regression analysis. J Psychiatr Res. 2011;45:995–1004. doi: 10.1016/j.jpsychires.2011.03.002. [DOI] [PubMed] [Google Scholar]
- Lin P-Y. State-dependent decrease in levels of brain-derived neurotrophic factor in bipolar disorder: a meta-analytic study. Neurosci Lett. 2009;466:139–143. doi: 10.1016/j.neulet.2009.09.044. [DOI] [PubMed] [Google Scholar]
- Fernandes BS, Gama CS, Kauer-Sant’Anna M, Lobato MI, Belmonte-de-Abreu P, Kapczinski F. Serum brain-derived neurotrophic factor in bipolar and unipolar depression: a potential adjunctive tool for differential diagnosis. J Psychiatr Res. 2009;43:1200–1204. doi: 10.1016/j.jpsychires.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Wehr TA, Goodwin FK. Can antidepressants cause mania and worsen the course of affective illness? Am J Psychiatry. 1987;144:1403–1411. doi: 10.1176/ajp.144.11.1403. [DOI] [PubMed] [Google Scholar]
- Bora E, Yucel M, Pantelis C. Cognitive endophenotypes of bipolar disorder: a meta-analysis of neuropsychological deficits in euthymic patients and their first-degree relatives. J Affect Disord. 2009;113:1–20. doi: 10.1016/j.jad.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Snitz BE, Macdonald AW 3rd, Carter CS. Cognitive deficits in unaffected first-degree relatives of schizophrenia patients: a meta-analytic review of putative endophenotypes. Schizophr Bull. 2006;32:179–194. doi: 10.1093/schbul/sbi048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur RE, Nimgaonkar VL, Almasy L, Calkins ME, Ragland JD, Pogue-Geile MF, Kanes S, Blangero J, Gur RC. Neurocognitive endophenotypes in a multiplex multigenerational family study of schizophrenia. Am J Psychiatry. 2007;164:813–819. doi: 10.1176/ajp.2007.164.5.813. [DOI] [PubMed] [Google Scholar]
- Sitskoorn MM, Aleman A, Ebisch SJH, Appels MCM, Kahn RS. Cognitive deficits in relatives of patients with schizophrenia: a meta-analysis. Schizophr Res. 2004;71:285–295. doi: 10.1016/j.schres.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Szöke A, Schürhoff F, Mathieu F, Meary A, Ionescu S, Leboyer M. Tests of executive functions in first-degree relatives of schizophrenic patients: a meta-analysis. Psychol Med. 2005;35:771–782. doi: 10.1017/s0033291704003460. [DOI] [PubMed] [Google Scholar]
- Fioravanti M, Carlone O, Vitale B, Cinti ME, Clare L. A meta-analysis of cognitive deficits in adults with a diagnosis of schizophrenia. Neuropsychol Rev. 2005;15:73–95. doi: 10.1007/s11065-005-6254-9. [DOI] [PubMed] [Google Scholar]
- Saykin AJ, Gur RC, Gur RE, Mozley PD, Mozley LH, Resnick SM, Kester DB, Stafiniak P. Neuropsychological function in schizophrenia, Selective impairment in memory and learning. Arch Gen Psychiatry. 1991;48:618–624. doi: 10.1001/archpsyc.1991.01810310036007. [DOI] [PubMed] [Google Scholar]
- Egan MF, Goldberg TE, Gscheidle T, Weirich M, Rawlings R, Hyde TM, Bigelow L, Weinberger DR. Relative risk for cognitive impairments in siblings of patients with schizophrenia. Biol Psychiatry. 2001;50:98–107. doi: 10.1016/s0006-3223(01)01133-7. [DOI] [PubMed] [Google Scholar]
- Cannon TD, Zorrilla LE, Shtasel D, Gur RE, Gur RC, Marco EJ, Moberg P, Price RA. Neuropsychological functioning in siblings discordant for schizophrenia and healthy volunteers. Arch Gen Psychiatry. 1994;51:651–661. doi: 10.1001/archpsyc.1994.03950080063009. [DOI] [PubMed] [Google Scholar]
- Faraone SV, Seidman LJ, Kremen WS, Pepple JR, Lyons MJ, Tsuang MT. Neuropsychological functioning among the nonpsychotic relatives of schizophrenic patients: a diagnostic efficiency analysis. J Abnorm Psychol. 1995;104:286–304. doi: 10.1037//0021-843x.104.2.286. [DOI] [PubMed] [Google Scholar]
- Conklin HM, Curtis CE, Calkins ME, Iacono WG. Working memory functioning in schizophrenia patients and their first-degree relatives: cognitive functioning shedding light on etiology. Neuropsychologia. 2005;43:930–942. doi: 10.1016/j.neuropsychologia.2004.09.013. [DOI] [PubMed] [Google Scholar]
- Gibbons JL, McHugh PR. Plasma cortisol in depressive illness. J Psychiatr Res. 1962;1:162–171. doi: 10.1016/0022-3956(62)90006-7. [DOI] [PubMed] [Google Scholar]
- Carpenter WT Jr, Bunney WE Jr. Adrenal cortical activity in depressive illness. Am J Psychiatry. 1971;128:31–40. doi: 10.1176/ajp.128.1.31. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Sawchenko PE, Rivier J, Vale WW. Organization of ovine corticotropin-releasing factor immunoreactive cells and fibers in the rat brain: an immunohistochemical study. Neuroendocrinology. 1983;36:165–186. doi: 10.1159/000123454. [DOI] [PubMed] [Google Scholar]
- Arborelius L, Owens MJ, Plotsky PM, Nemeroff CB. The role of corticotropin-releasing factor in depression and anxiety disorders. J Endocrinol. 1999;160:1–12. doi: 10.1677/joe.0.1600001. [DOI] [PubMed] [Google Scholar]
- Dunn AJ, Berridge CW. Physiological and behavioral responses to corticotropin-releasing factor administration: is CRF a mediator of anxiety or stress responses? Brain Res Brain Res Rev. 1990;15:71–100. doi: 10.1016/0165-0173(90)90012-d. [DOI] [PubMed] [Google Scholar]
- Owens MJ, Nemeroff CB. Physiology and pharmacology of corticotropin-releasing factor. Pharmacol Rev. 1991;43:425–473. [PubMed] [Google Scholar]
- Bradley RG, Binder EB, Epstein MP, Tang Y, Nair HP, Liu W, Gillespie CF, Berg T, Evces M, Newport DJ, Stowe ZN, Heim CM, Nemeroff CB, Schwartz A, Cubells JF, Ressler KJ. Influence of child abuse on adult depression: moderation by the corticotropin-releasing hormone receptor gene. Arch Gen Psychiatry. 2008;65:190–200. doi: 10.1001/archgenpsychiatry.2007.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanczyk G, Caspi A, Williams B, Price TS, Danese A, Sugden K, Uher R, Poulton R, Moffitt TE. Protective effect of CRHR1 gene variants on the development of adult depression following childhood maltreatment: replication and extension. Arch Gen Psychiatry. 2009;66:978–985. doi: 10.1001/archgenpsychiatry.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrka AR, Price LH, Gelernter J, Schepker C, Anderson GM, Carpenter LL. Interaction of childhood maltreatment with the corticotropin-releasing hormone receptor gene: effects on hypothalamic-pituitary-adrenal axis reactivity. Biol Psychiatry. 2009;66:681–685. doi: 10.1016/j.biopsych.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim C, Bradley B, Mletzko TC, Deveau TC, Musselman DL, Nemeroff CB, Ressler KJ, Binder EB. Effect of Childhood Trauma on Adult Depression and Neuroendocrine Function: Sex-Specific Moderation by CRH Receptor 1 Gene. Front Behav Neurosci. 2009;3:41. doi: 10.3389/neuro.08.041.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- McGuffin P, Asherson P, Owen M, Farmer A. The strength of the genetic effect. Is there room for an environmental influence in the aetiology of schizophrenia? Br J Psychiatry. 1994;164:593–599. doi: 10.1192/bjp.164.5.593. [DOI] [PubMed] [Google Scholar]
- Stabenau JR, Pollin W. Early characteristics of monozygotic twins discordant for schizophrenia. Arch Gen Psychiatry. 1967;17:723–734. doi: 10.1001/archpsyc.1967.01730300083011. [DOI] [PubMed] [Google Scholar]
- Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry. 2002;159:1080–1092. doi: 10.1176/appi.ajp.159.7.1080. [DOI] [PubMed] [Google Scholar]
- Scott J, McNeill Y, Cavanagh J, Cannon M, Murray R. Exposure to obstetric complications and subsequent development of bipolar disorder: Systematic review. Br J Psychiatry. 2006;189:3–11. doi: 10.1192/bjp.bp.105.010579. [DOI] [PubMed] [Google Scholar]
- Mednick SA, Machon RA, Huttunen MO, Bonett D. Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psychiatry. 1988;45:189–192. doi: 10.1001/archpsyc.1988.01800260109013. [DOI] [PubMed] [Google Scholar]
- O’Callaghan E, Sham P, Takei N, Glover G, Murray RM. Schizophrenia after prenatal exposure to 1957 A2 influenza epidemic. Lancet. 1991;337:1248–1250. doi: 10.1016/0140-6736(91)92919-s. [DOI] [PubMed] [Google Scholar]
- Brown AS. Prenatal infection as a risk factor for schizophrenia. Schizophr Bull. 2006;32:200–202. doi: 10.1093/schbul/sbj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selten J-P, Frissen A, Lensvelt-Mulders G, Morgan VA. Schizophrenia and 1957 pandemic of influenza: meta-analysis. Schizophr Bull. 2010;36:219–228. doi: 10.1093/schbul/sbp147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selten J-P, Cantor-Graae E, Nahon D, Levav I, Aleman A, Kahn RS. No relationship between risk of schizophrenia and prenatal exposure to stress during the Six-Day War or Yom Kippur War in Israel. Schizophr Res. 2003;63:131–135. doi: 10.1016/s0920-9964(02)00375-4. [DOI] [PubMed] [Google Scholar]
- Ferguson CS, Tyndale RF. Cytochrome P450 enzymes in the brain: emerging evidence of biological significance. Trends Pharmacol Sci. 2011;32:708–714. doi: 10.1016/j.tips.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingl JC, Brockmöller J, Viviani R. Genetic variability of drug-metabolizing enzymes: the dual impact on psychiatric therapy and regulation of brain function. Mol Psychiatry. 2013;18:273–287. doi: 10.1038/mp.2012.42. [DOI] [PubMed] [Google Scholar]
- Bromek E, Haduch A, Gołembiowska K, Daniel WA. Cytochrome P450 mediates dopamine formation in the brain in vivo. J Neurochem. 2011;118:806–815. doi: 10.1111/j.1471-4159.2011.07339.x. [DOI] [PubMed] [Google Scholar]
- Yu A-M, Idle JR, Byrd LG, Krausz KW, Küpfer A, Gonzalez FJ. Regeneration of serotonin from 5-methoxytryptamine by polymorphic human CYP2D6. Pharmacogenetics. 2003;13:173–181. doi: 10.1097/01.fpc.0000054066.98065.7b. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB, DeVane CL, Pollock BG. Newer antidepressants and the cytochrome P450 system. Am J Psychiatry. 1996;153:311–320. doi: 10.1176/ajp.153.3.311. [DOI] [PubMed] [Google Scholar]
- Thakur M, Grossman I, McCrory DC, Orlando LA, Steffens DC, Cline KE, Gray RN, Farmer J, DeJesus G, O’Brien C, Samsa G, Goldstein DB, Matchar DB. Review of evidence for genetic testing for CYP450 polymorphisms in management of patients with nonpsychotic depression with selective serotonin reuptake inhibitors. Genet Med. 2007;9:826–835. doi: 10.1097/gim.0b013e31815bf98f. [DOI] [PubMed] [Google Scholar]
- Fleeman N, McLeod C, Bagust A, Beale S, Boland A, Dundar Y, Jorgensen A, Payne K, Pirmohamed M, Pushpakom S, Walley T, De Warren-Penny P, Dickson R. The clinical effectiveness and cost-effectiveness of testing for cytochrome P450 polymorphisms in patients with schizophrenia treated with antipsychotics: a systematic review and economic evaluation. Health Technol Assess. 2010;14:1–157. doi: 10.3310/hta14030. iii. [DOI] [PubMed] [Google Scholar]
- Fleeman N, Dundar Y, Dickson R, Jorgensen A, Pushpakom S, McLeod C, Pirmohamed M, Walley T. Cytochrome P450 testing for prescribing antipsychotics in adults with schizophrenia: systematic review and meta-analyses. Pharmacogenomics J. 2011;11:1–14. doi: 10.1038/tpj.2010.73. [DOI] [PubMed] [Google Scholar]
- Porcelli S, Drago A, Fabbri C, Gibiino S, Calati R, Serretti A. Pharmacogenetics of antidepressant response. J Psychiatry Neurosci. 2011;36:87–113. doi: 10.1503/jpn.100059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrazek DA, Rush AJ, Biernacka JM, O’Kane DJ, Cunningham JM, Wieben ED, Schaid DJ, Drews MS, Courson VL, Snyder KA, Black JL 3rd, Weinshilboum RM. SLC6A4 variation and citalopram response. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:341–351. doi: 10.1002/ajmg.b.30816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min W, Li T, Ma X, Li Z, Yu T, Gao D, Zhang B, Yun Y, Sun X. Monoamine transporter gene polymorphisms affect susceptibility to depression and predict antidepressant response. Psychopharmacology (Berl) 2009;205:409–417. doi: 10.1007/s00213-009-1550-3. [DOI] [PubMed] [Google Scholar]
- Huezo-Diaz P, Uher R, Smith R, Rietschel M, Henigsberg N, Marusic A, Mors O, Maier W, Hauser J, Souery D, Placentino A, Zobel A, Larsen ER, Czerski PM, Gupta B, Hoda F, Perroud N, Farmer A, Craig I, Aitchison KJ, McGuffin P. Moderation of antidepressant response by the serotonin transporter gene. Br J Psychiatry. 2009;195:30–38. doi: 10.1192/bjp.bp.108.062521. [DOI] [PubMed] [Google Scholar]
- Bozina N, Peles AM, Sagud M, Bilusic H, Jakovljevic M. Association study of paroxetine therapeutic response with SERT gene polymorphisms in patients with major depressive disorder. World J Biol Psychiatry. 2008;9:190–197. doi: 10.1080/15622970701308397. [DOI] [PubMed] [Google Scholar]
- Smeraldi E, Serretti A, Artioli P, Lorenzi C, Catalano M. Serotonin transporter gene-linked polymorphic region: possible pharmacogenetic implications of rare variants. Psychiatr Genet. 2006;16:153–158. doi: 10.1097/01.ypg.0000218611.53064.a0. [DOI] [PubMed] [Google Scholar]
- Smeraldi E, Zanardi R, Benedetti F, Di Bella D, Perez J, Catalano M. Polymorphism within the promoter of the serotonin transporter gene and antidepressant efficacy of fluvoxamine. Mol Psychiatry. 1998;3:508–511. doi: 10.1038/sj.mp.4000425. [DOI] [PubMed] [Google Scholar]
- Hong C-J, Chen T-J, Yu YW-Y, Tsai S-J. Response to fluoxetine and serotonin 1A receptor (C-1019G) polymorphism in Taiwan Chinese major depressive disorder. Pharmacogenomics J. 2006;6:27–33. doi: 10.1038/sj.tpj.6500340. [DOI] [PubMed] [Google Scholar]
- Kato M, Fukuda T, Wakeno M, Fukuda K, Okugawa G, Ikenaga Y, Yamashita M, Takekita Y, Nobuhara K, Azuma J, Kinoshita T. Effects of the serotonin type 2A, 3A and 3B receptor and the serotonin transporter genes on paroxetine and fluvoxamine efficacy and adverse drug reactions in depressed Japanese patients. Neuropsychobiology. 2006;53:186–195. doi: 10.1159/000094727. [DOI] [PubMed] [Google Scholar]
- Kato M, Ikenaga Y, Wakeno M, Okugawa G, Nobuhara K, Fukuda T, Fukuda K, Azuma J, Kinoshita T. Controlled clinical comparison of paroxetine and fluvoxamine considering the serotonin transporter promoter polymorphism. Int Clin Psychopharmacol. 2005;20:151–156. doi: 10.1097/00004850-200505000-00005. [DOI] [PubMed] [Google Scholar]
- Rausch JL, Johnson ME, Fei Y-J, Li JQ, Shendarkar N, Hobby HM, Ganapathy V, Leibach FH. Initial conditions of serotonin transporter kinetics and genotype: influence on SSRI treatment trial outcome. Biol Psychiatry. 2002;51:723–732. doi: 10.1016/s0006-3223(01)01283-5. [DOI] [PubMed] [Google Scholar]
- Zanardi R, Serretti A, Rossini D, Franchini L, Cusin C, Lattuada E, Dotoli D, Smeraldi E. Factors affecting fluvoxamine antidepressant activity: influence of pindolol and 5-HTTLPR in delusional and nondelusional depression. Biol Psychiatry. 2001;50:323–330. doi: 10.1016/s0006-3223(01)01118-0. [DOI] [PubMed] [Google Scholar]
- Serretti A, Cusin C, Rossini D, Artioli P, Dotoli D, Zanardi R. Further evidence of a combined effect of SERTPR and TPH on SSRIs response in mood disorders. Am J Med Genet B Neuropsychiatr Genet. 2004;129B:36–40. doi: 10.1002/ajmg.b.30027. [DOI] [PubMed] [Google Scholar]
- Murphy GM Jr, Hollander SB, Rodrigues HE, Kremer C, Schatzberg AF. Effects of the serotonin transporter gene promoter polymorphism on mirtazapine and paroxetine efficacy and adverse events in geriatric major depression. Arch Gen Psychiatry. 2004;61:1163–1169. doi: 10.1001/archpsyc.61.11.1163. [DOI] [PubMed] [Google Scholar]
- Smits K, Smits L, Peeters F, Schouten J, Janssen R, Smeets H, Van Os J, Prins M. Serotonin transporter polymorphisms and the occurrence of adverse events during treatment with selective serotonin reuptake inhibitors. Int Clin Psychopharmacol. 2007;22:137–143. doi: 10.1097/YIC.0b013e328014822a. [DOI] [PubMed] [Google Scholar]
- Smits KM, Smits LJM, Peeters FPML, Schouten JSAG, Janssen RGJH, Smeets HJM, Van Os J, Prins MH. The influence of 5-HTTLPR and STin2 polymorphisms in the serotonin transporter gene on treatment effect of selective serotonin reuptake inhibitors in depressive patients. Psychiatr Genet. 2008;18:184–190. doi: 10.1097/YPG.0b013e3283050aca. [DOI] [PubMed] [Google Scholar]
- Durham LK, Webb SM, Milos PM, Clary CM, Seymour AB. The serotonin transporter polymorphism, 5HTTLPR, is associated with a faster response time to sertraline in an elderly population with major depressive disorder. Psychopharmacology (Berl) 2004;174:525–529. doi: 10.1007/s00213-003-1562-3. [DOI] [PubMed] [Google Scholar]
- Arias B, Catalán R, Gastó C, Gutiérrez B, Fañanás L. 5-HTTLPR polymorphism of the serotonin transporter gene predicts non-remission in major depression patients treated with citalopram in a 12-weeks follow up study. J Clin Psychopharmacol. 2003;23:563–567. doi: 10.1097/01.jcp.0000095350.32154.73. [DOI] [PubMed] [Google Scholar]
- Joyce PR, Mulder RT, Luty SE, McKenzie JM, Miller AL, Rogers GR, Kennedy MA. Age-dependent antidepressant pharmacogenomics: polymorphisms of the serotonin transporter and G protein beta3 subunit as predictors of response to fluoxetine and nortriptyline. Int J Neuropsychopharmacol. 2003;6:339–346. doi: 10.1017/S1461145703003663. [DOI] [PubMed] [Google Scholar]
- Pollock BG, Ferrell RE, Mulsant BH, Mazumdar S, Miller M, Sweet RA, Davis S, Kirshner MA, Houck PR, Stack JA, Reynolds CF, Kupfer DJ. Allelic variation in the serotonin transporter promoter affects onset of paroxetine treatment response in late-life depression. Neuropsychopharmacology. 2000;23:587–590. doi: 10.1016/S0893-133X(00)00132-9. [DOI] [PubMed] [Google Scholar]
- Kim H, Lim S-W, Kim S, Kim J-W, Chang YH, Carroll BJ, Kim DK. Monoamine transporter gene polymorphisms and antidepressant response in koreans with late-life depression. JAMA. 2006;296:1609–1618. doi: 10.1001/jama.296.13.1609. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Takahashi H, Higuchi H, Kamata M, Ito K, Sato K, Naito S, Shimizu T, Itoh K, Inoue K, Suzuki T, Nemeroff CB. Prediction of antidepressant response to milnacipran by norepinephrine transporter gene polymorphisms. Am J Psychiatry. 2004;161:1575–1580. doi: 10.1176/appi.ajp.161.9.1575. [DOI] [PubMed] [Google Scholar]
- Lotrich FE, Pollock BG, Kirshner M, Ferrell RF. Reynolds Iii CF: Serotonin transporter genotype interacts with paroxetine plasma levels to influence depression treatment response in geriatric patients. J Psychiatry Neurosci. 2008;33:123–130. [PMC free article] [PubMed] [Google Scholar]
- Yoshimura R, Umene-Nakano W, Suzuki A, Ueda N, Miyamoto K, Ikenouchi-Sugita A, Hori H, Otani K, Nakamura J. Rapid response to paroxetine is associated with plasma paroxetine levels at 4 but not 8 weeks of treatment, and is independent of serotonin transporter promoter polymorphism in Japanese depressed patients. Hum Psychopharmacol. 2009;24:489–494. doi: 10.1002/hup.1043. [DOI] [PubMed] [Google Scholar]
- Ng CH, Easteal S, Tan S, Schweitzer I, Ho BKW, Aziz S. Serotonin transporter polymorphisms and clinical response to sertraline across ethnicities. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:953–957. doi: 10.1016/j.pnpbp.2006.02.015. [DOI] [PubMed] [Google Scholar]
- Yu YW-Y, Tsai S-J, Liou Y-J, Hong C-J, Chen T-J. Association study of two serotonin 1A receptor gene polymorphisms and fluoxetine treatment response in Chinese major depressive disorders. Eur Neuropsychopharmacol. 2006;16:498–503. doi: 10.1016/j.euroneuro.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Serretti A, Artioli P, Lorenzi C, Pirovano A, Tubazio V, Zanardi R. The C(−1019)G polymorphism of the 5-HT1A gene promoter and antidepressant response in mood disorders: preliminary findings. Int J Neuropsychopharmacol. 2004;7:453–460. doi: 10.1017/S1461145704004687. [DOI] [PubMed] [Google Scholar]
- Kato M, Fukuda T, Wakeno M, Okugawa G, Takekita Y, Watanabe S, Yamashita M, Hosoi Y, Azuma J, Kinoshita T, Serretti A. Effect of 5-HT1A gene polymorphisms on antidepressant response in major depressive disorder. Am J Med Genet B Neuropsychiatr Genet. 2009;150B:115–123. doi: 10.1002/ajmg.b.30783. [DOI] [PubMed] [Google Scholar]
- Levin GM, Bowles TM, Ehret MJ, Langaee T, Tan JY, Johnson JA, Millard WJ. Assessment of human serotonin 1A receptor polymorphisms and SSRI responsiveness. Mol Diagn Ther. 2007;11:155–160. doi: 10.1007/BF03256237. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Sawamura K, Someya T. Polymorphisms in the 5-hydroxytryptamine 2A receptor and CytochromeP4502D6 genes synergistically predict fluvoxamine-induced side effects in japanese depressed patients. Neuropsychopharmacology. 2006;31:825–831. doi: 10.1038/sj.npp.1300919. [DOI] [PubMed] [Google Scholar]
- Peters EJ, Slager SL, McGrath PJ, Knowles JA, Hamilton SP. Investigation of serotonin-related genes in antidepressant response. Mol Psychiatry. 2004;9:879–889. doi: 10.1038/sj.mp.4001502. [DOI] [PubMed] [Google Scholar]
- Uher R, Huezo-Diaz P, Perroud N, Smith R, Rietschel M, Mors O, Hauser J, Maier W, Kozel D, Henigsberg N, Barreto M, Placentino A, Dernovsek MZ, Schulze TG, Kalember P, Zobel A, Czerski PM, Larsen ER, Souery D, Giovannini C, Gray JM, Lewis CM, Farmer A, Aitchison KJ, McGuffin P, Craig I. Genetic predictors of response to antidepressants in the GENDEP project. Pharmacogenomics J. 2009;9:225–233. doi: 10.1038/tpj.2009.12. [DOI] [PubMed] [Google Scholar]
- Peters EJ, Slager SL, Jenkins GD, Reinalda MS, Garriock HA, Shyn SI, Kraft JB, McGrath PJ, Hamilton SP. Resequencing of serotonin-related genes and association of tagging SNPs to citalopram response. Pharmacogenet Genomics. 2009;19:1–10. doi: 10.1097/FPC.0b013e3283163ecd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie MJV, Smith G, Day RK, Matthews K, Smith D, Blackwood D, Reid IC, Wolf CR. Polymorphisms in the SLC6A4 and HTR2A genes influence treatment outcome following antidepressant therapy. Pharmacogenomics J. 2009;9:61–70. doi: 10.1038/sj.tpj.6500491. [DOI] [PubMed] [Google Scholar]
- McMahon FJ, Buervenich S, Charney D, Lipsky R, Rush AJ, Wilson AF, Sorant AJM, Papanicolaou GJ, Laje G, Fava M, Trivedi MH, Wisniewski SR, Manji H. Variation in the gene encoding the serotonin 2A receptor is associated with outcome of antidepressant treatment. Am J Hum Genet. 2006;78:804–814. doi: 10.1086/503820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy GM Jr, Kremer C, Rodrigues HE, Schatzberg AF. Pharmacogenetics of antidepressant medication intolerance. Am J Psychiatry. 2003;160:1830–1835. doi: 10.1176/appi.ajp.160.10.1830. [DOI] [PubMed] [Google Scholar]
- Illi A, Setälä-Soikkeli E, Viikki M, Poutanen O, Huhtala H, Mononen N, Lehtimäki T, Leinonen E, Kampman O. 5-HTR1A, 5-HTR2A, 5-HTR6, TPH1 and TPH2 polymorphisms and major depression. Neuroreport. 2009;20:1125–1128. doi: 10.1097/WNR.0b013e32832eb708. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Kobayashi D, Murakami Y, Ozaki N, Suzuki T, Iwata N, Haraguchi K, Ieiri I, Kinukawa N, Hosoi M, Ohtani H, Sawada Y, Mine K. Genetic polymorphisms in the 5-hydroxytryptamine type 3B receptor gene and paroxetine-induced nausea. Int J Neuropsychopharmacol. 2008;11:261–267. doi: 10.1017/S1461145707007985. [DOI] [PubMed] [Google Scholar]
- Sato K, Yoshida K, Takahashi H, Ito K, Kamata M, Higuchi H, Shimizu T, Itoh K, Inoue K, Tezuka T, Suzuki T, Ohkubo T, Sugawara K, Otani K. Association between -1438G/A promoter polymorphism in the 5-HT(2A) receptor gene and fluvoxamine response in Japanese patients with major depressive disorder. Neuropsychobiology. 2002;46:136–140. doi: 10.1159/000066394. [DOI] [PubMed] [Google Scholar]
- Binder EB, Bradley RG, Liu W, Epstein MP, Deveau TC, Mercer KB, Tang Y, Gillespie CF, Heim CM, Nemeroff CB, Schwartz AC, Cubells JF, Ressler KJ. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA. 2008;299:1291–1305. doi: 10.1001/jama.299.11.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velders FP, Kuningas M, Kumari M, Dekker MJ, Uitterlinden AG, Kirschbaum C, Hek K, Hofman A, Verhulst FC, Kivimaki M, Van Duijn CM, Walker BR, Tiemeier H. Genetics of cortisol secretion and depressive symptoms: a candidate gene and genome wide association approach. Psychoneuroendocrinology. 2011;36:1053–1061. doi: 10.1016/j.psyneuen.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JI, Chung HC, Jeung H-C, Kim SJ, An SK, Namkoong K. FKBP5 polymorphisms as vulnerability to anxiety and depression in patients with advanced gastric cancer: A controlled and prospective study. Psychoneuroendocrinology. 2012;37:1569–1576. doi: 10.1016/j.psyneuen.2012.02.017. [DOI] [PubMed] [Google Scholar]
- Binder EB, Owens MJ, Liu W, Deveau TC, Rush AJ, Trivedi MH, Fava M, Bradley B, Ressler KJ, Nemeroff CB. Association of polymorphisms in genes regulating the corticotropin-releasing factor system with antidepressant treatment response. Arch Gen Psychiatry. 2010;67:369–379. doi: 10.1001/archgenpsychiatry.2010.18. [DOI] [PubMed] [Google Scholar]
- Garriock HA, Tanowitz M, Kraft JB, Dang VC, Peters EJ, Jenkins GD, Reinalda MS, McGrath PJ, Von Zastrow M, Slager SL, Hamilton SP. Association of mu-opioid receptor variants and response to citalopram treatment in major depressive disorder. Am J Psychiatry. 2010;167:565–573. doi: 10.1176/appi.ajp.2009.08081167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laje G, Allen AS, Akula N, Manji H, John Rush A, McMahon FJ. Genome-wide association study of suicidal ideation emerging during citalopram treatment of depressed outpatients. Pharmacogenet Genomics. 2009;19:666–674. doi: 10.1097/FPC.0b013e32832e4bcd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriock HA, Kraft JB, Shyn SI, Peters EJ, Yokoyama JS, Jenkins GD, Reinalda MS, Slager SL, McGrath PJ, Hamilton SP. A genomewide association study of citalopram response in major depressive disorder. Biol Psychiatry. 2010;67:133–138. doi: 10.1016/j.biopsych.2009.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ising M, Lucae S, Binder EB, Bettecken T, Uhr M, Ripke S, Kohli MA, Hennings JM, Horstmann S, Kloiber S, Menke A, Bondy B, Rupprecht R, Domschke K, Baune BT, Arolt V, Rush AJ, Holsboer F, Müller-Myhsok B. A genomewide association study points to multiple loci that predict antidepressant drug treatment outcome in depression. Arch Gen Psychiatry. 2009;66:966–975. doi: 10.1001/archgenpsychiatry.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra AK. The Pharmacogenetics of Depression: Enter the GWAS. Am J Psychiatry. 2010;167:493–495. doi: 10.1176/appi.ajp.2010.10020244. [DOI] [PubMed] [Google Scholar]
- Rybakowski JK, Suwalska A, Czerski PM, Dmitrzak-Weglarz M, Leszczynska-Rodziewicz A, Hauser J. Prophylactic effect of lithium in bipolar affective illness may be related to serotonin transporter genotype. Pharmacol Rep. 2005;57:124–127. [PubMed] [Google Scholar]
- Masui T, Hashimoto R, Kusumi I, Suzuki K, Tanaka T, Nakagawa S, Kunugi H, Koyama T. A possible association between the -116C/G single nucleotide polymorphism of the XBP1 gene and lithium prophylaxis in bipolar disorder. Int J Neuropsychopharmacol. 2006;9:83–88. doi: 10.1017/S1461145705005523. [DOI] [PubMed] [Google Scholar]
- Benedetti F, Dallaspezia S, Fulgosi MC, Lorenzi C, Serretti A, Barbini B, Colombo C, Smeraldi E. Actimetric evidence that CLOCK 3111 T/C SNP influences sleep and activity patterns in patients affected by bipolar depression. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:631–635. doi: 10.1002/ajmg.b.30475. [DOI] [PubMed] [Google Scholar]
- Rybakowski JK, Suwalska A, Skibinska M, Dmitrzak-Weglarz M, Leszczynska-Rodziewicz A, Hauser J. Response to lithium prophylaxis: interaction between serotonin transporter and BDNF genes. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:820–823. doi: 10.1002/ajmg.b.30420. [DOI] [PubMed] [Google Scholar]
- Adli M, Hollinde DL, Stamm T, Wiethoff K, Tsahuridu M, Kirchheiner J, Heinz A, Bauer M. Response to lithium augmentation in depression is associated with the glycogen synthase kinase 3-beta -50T/C single nucleotide polymorphism. Biol Psychiatry. 2007;62:1295–1302. doi: 10.1016/j.biopsych.2007.03.023. [DOI] [PubMed] [Google Scholar]
- Masui T, Hashimoto R, Kusumi I, Suzuki K, Tanaka T, Nakagawa S, Suzuki T, Iwata N, Ozaki N, Kato T, Takeda M, Kunugi H, Koyama T. A possible association between missense polymorphism of the breakpoint cluster region gene and lithium prophylaxis in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:204–208. doi: 10.1016/j.pnpbp.2007.08.010. [DOI] [PubMed] [Google Scholar]
- Mamdani F, Alda M, Grof P, Young LT, Rouleau G, Turecki G. Lithium response and genetic variation in the CREB family of genes. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:500–504. doi: 10.1002/ajmg.b.30617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dmitrzak-Weglarz M, Rybakowski JK, Suwalska A, Skibinska M, Leszczynska-Rodziewicz A, Szczepankiewicz A, Hauser J. Association studies of the BDNF and the NTRK2 gene polymorphisms with prophylactic lithium response in bipolar patients. Pharmacogenomics. 2008;9:1595–1603. doi: 10.2217/14622416.9.11.1595. [DOI] [PubMed] [Google Scholar]
- Arranz MJ, De Leon J. Pharmacogenetics and pharmacogenomics of schizophrenia: a review of last decade of research. Mol Psychiatry. 2007;12:707–747. doi: 10.1038/sj.mp.4002009. [DOI] [PubMed] [Google Scholar]
- Foster A, Miller DD, Buckley PF. Pharmacogenetics and schizophrenia. Psychiatr Clin North Am. 2007;30:417–435. doi: 10.1016/j.psc.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Nnadi CU, Malhotra AK. Individualizing antipsychotic drug therapy in schizophrenia: the promise of pharmacogenetics. Curr Psychiatry Rep. 2007;9:313–318. doi: 10.1007/s11920-007-0038-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds GP. The impact of pharmacogenetics on the development and use of antipsychotic drugs. Drug Discov Today. 2007;12:953–959. doi: 10.1016/j.drudis.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Thelma B, Srivastava V, Tiwari AK. Genetic underpinnings of tardive dyskinesia: passing the baton to pharmacogenetics. Pharmacogenomics. 2008;9:1285–1306. doi: 10.2217/14622416.9.9.1285. [DOI] [PubMed] [Google Scholar]
- Zai CC, Romano-Silva MA, Hwang R, Zai GC, Deluca V, Müller DJ, King N, Voineskos AN, Meltzer HY, Lieberman JA, Potkin SG, Remington G, Kennedy JL. Genetic study of eight AKT1 gene polymorphisms and their interaction with DRD2 gene polymorphisms in tardive dyskinesia. Schizophr Res. 2008;106:248–252. doi: 10.1016/j.schres.2008.08.036. [DOI] [PubMed] [Google Scholar]
- Steen VM, Løvlie R, MacEwan T, McCreadie RG. Dopamine D3-receptor gene variant and susceptibility to tardive dyskinesia in schizophrenic patients. Mol Psychiatry. 1997;2:139–145. doi: 10.1038/sj.mp.4000249. [DOI] [PubMed] [Google Scholar]
- Lerer B, Segman RH, Fangerau H, Daly AK, Basile VS, Cavallaro R, Aschauer HN, McCreadie RG, Ohlraun S, Ferrier N, Masellis M, Verga M, Scharfetter J, Rietschel M, Lovlie R, Levy UH, Meltzer HY, Kennedy JL, Steen VM, Macciardi F. Pharmacogenetics of tardive dyskinesia: combined analysis of 780 patients supports association with dopamine D3 receptor gene Ser9Gly polymorphism. Neuropsychopharmacology. 2002;27:105–119. doi: 10.1016/S0893-133X(02)00293-2. [DOI] [PubMed] [Google Scholar]
- Woo S, Kim JW, Rha E, Han S, Hahn K, Park C, Sohn J. Association of the Ser9Gly polymorphism in the dopamine D3 receptor gene with tardive dyskinesia in Korean schizophrenics. Psychiatry Clin Neurosci. 2002;56:469–474. doi: 10.1046/j.1440-1819.2002.01038.x. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, Keefe RSE, Davis SM, Davis CE, Lebowitz BD, Severe J, Hsiao JK. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–1223. doi: 10.1056/NEJMoa051688. [DOI] [PubMed] [Google Scholar]
- Tsai H-T, North KE, West SL, Poole C. The DRD3 rs6280 polymorphism and prevalence of tardive dyskinesia: a meta-analysis. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:57–66. doi: 10.1002/ajmg.b.30946. [DOI] [PubMed] [Google Scholar]
- Bakker PR, Van Harten PN, Van Os J. Antipsychotic-induced tardive dyskinesia and polymorphic variations in COMT, DRD2, CYP1A2 and MnSOD genes: a meta-analysis of pharmacogenetic interactions. Mol Psychiatry. 2008;13:544–556. doi: 10.1038/sj.mp.4002142. [DOI] [PubMed] [Google Scholar]
- Hori H, Ohmori O, Shinkai T, Kojima H, Okano C, Suzuki T, Nakamura J. Manganese superoxide dismutase gene polymorphism and schizophrenia: relation to tardive dyskinesia. Neuropsychopharmacology. 2000;23:170–177. doi: 10.1016/S0893-133X(99)00156-6. [DOI] [PubMed] [Google Scholar]
- Hitzeroth A, Niehaus DJH, Koen L, Botes WC, Deleuze JF, Warnich L. Association between the MnSOD Ala-9Val polymorphism and development of schizophrenia and abnormal involuntary movements in the Xhosa population. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:664–672. doi: 10.1016/j.pnpbp.2006.12.019. [DOI] [PubMed] [Google Scholar]
- Kang S-G, Choi J-E, An H, Park Y-M, Lee H-J, Han C, Kim Y-K, Kim S-H, Cho SN, Joe S-H, Jung I-K, Kim L, Lee M-S. Manganese superoxide dismutase gene Ala-9Val polymorphism might be related to the severity of abnormal involuntary movements in Korean schizophrenic patients. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:1844–1847. doi: 10.1016/j.pnpbp.2008.08.013. [DOI] [PubMed] [Google Scholar]
- Lerer B, Segman RH, Tan E-C, Basile VS, Cavallaro R, Aschauer HN, Strous R, Chong S-A, Heresco-Levy U, Verga M, Scharfetter J, Meltzer HY, Kennedy JL, Macciardi F. Combined analysis of 635 patients confirms an age-related association of the serotonin 2A receptor gene with tardive dyskinesia and specificity for the non-orofacial subtype. Int J Neuropsychopharmacol. 2005;8:411–425. doi: 10.1017/S1461145705005389. [DOI] [PubMed] [Google Scholar]
- Zai CC, Tiwari AK, Basile V, De Luca V, Müller DJ, Voineskos AN, Remington G, Meltzer HY, Lieberman JA, Potkin SG, Kennedy JL. Oxidative stress in tardive dyskinesia: genetic association study and meta-analysis of NADPH quinine oxidoreductase 1 (NQO1) and Superoxide dismutase 2 (SOD2, MnSOD) genes. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:50–56. doi: 10.1016/j.pnpbp.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Boke O, Gunes S, Kara N, Aker S, Sahin AR, Basar Y, Bagci H. Association of serotonin 2A receptor and lack of association of CYP1A2 gene polymorphism with tardive dyskinesia in a Turkish population. DNA Cell Biol. 2007;26:527–531. doi: 10.1089/dna.2007.0605. [DOI] [PubMed] [Google Scholar]
- Allison DB, Mentore JL, Heo M, Chandler LP, Cappelleri JC, Infante MC, Weiden PJ. Antipsychotic-induced weight gain: a comprehensive research synthesis. Am J Psychiatry. 1999;156:1686–1696. doi: 10.1176/ajp.156.11.1686. [DOI] [PubMed] [Google Scholar]
- Henderson DC, Cagliero E, Gray C, Nasrallah RA, Hayden DL, Schoenfeld DA, Goff DC. Clozapine, diabetes mellitus, weight gain, and lipid abnormalities: A five-year naturalistic study. Am J Psychiatry. 2000;157:975–981. doi: 10.1176/appi.ajp.157.6.975. [DOI] [PubMed] [Google Scholar]
- Müller DJ, Kennedy JL. Genetics of antipsychotic treatment emergent weight gain in schizophrenia. Pharmacogenomics. 2006;7:863–887. doi: 10.2217/14622416.7.6.863. [DOI] [PubMed] [Google Scholar]
- Ryu S, Cho EY, Park T, Oh S, Jang W-S, Kim S-K, Lee D, Hong KS. 759 C/T polymorphism of 5-HT2C receptor gene and early phase weight gain associated with antipsychotic drug treatment. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:673–677. doi: 10.1016/j.pnpbp.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Templeman LA, Reynolds GP, Arranz B, San L. Polymorphisms of the 5-HT2C receptor and leptin genes are associated with antipsychotic drug-induced weight gain in Caucasian subjects with a first-episode psychosis. Pharmacogenet Genomics. 2005;15:195–200. doi: 10.1097/01213011-200504000-00002. [DOI] [PubMed] [Google Scholar]
- Reynolds GP. The pharmacogenetics of symptom response to antipsychotic drugs. Psychiatry Investig. 2012;9:1–7. doi: 10.4306/pi.2012.9.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XY, Tan YL, Zhou DF, Haile CN, Cao LY, Xu Q, Shen Y, Kosten TA, Kosten TR. Association of clozapine-induced weight gain with a polymorphism in the leptin promoter region in patients with chronic schizophrenia in a Chinese population. J Clin Psychopharmacol. 2007;27:246–251. doi: 10.1097/jcp.0b013e3180582412. [DOI] [PubMed] [Google Scholar]
- De Luca V, Mueller DJ, De Bartolomeis A, Kennedy JL. Association of the HTR2C gene and antipsychotic induced weight gain: a meta-analysis. Int J Neuropsychopharmacol. 2007;10:697–704. doi: 10.1017/S1461145707007547. [DOI] [PubMed] [Google Scholar]
- Sicard MN, Zai CC, Tiwari AK, Souza RP, Meltzer HY, Lieberman JA, Kennedy JL, Müller DJ. Polymorphisms of the HTR2C gene and antipsychotic-induced weight gain: an update and meta-analysis. Pharmacogenomics. 2010;11:1561–1571. doi: 10.2217/pgs.10.123. [DOI] [PubMed] [Google Scholar]
- Opgen-Rhein C, Brandl EJ, Müller DJ, Neuhaus AH, Tiwari AK, Sander T, Dettling M. Association of HTR2C, but not LEP or INSIG2, genes with antipsychotic-induced weight gain in a German sample. Pharmacogenomics. 2010;11:773–780. doi: 10.2217/pgs.10.50. [DOI] [PubMed] [Google Scholar]
- Kane J, Honigfeld G, Singer J, Meltzer H. Clozapine for the treatment-resistant schizophrenic, A double-blind comparison with chlorpromazine. Arch Gen Psychiatry. 1988;45:789–796. doi: 10.1001/archpsyc.1988.01800330013001. [DOI] [PubMed] [Google Scholar]
- Alvir JM, Lieberman JA. A reevaluation of the clinical characteristics of clozapine-induced agranulocytosis in light of the United States experience. J Clin Psychopharmacol. 1994;14:87–89. [PubMed] [Google Scholar]
- Chowdhury NI, Remington G, Kennedy JL. Genetics of antipsychotic-induced side effects and agranulocytosis. Curr Psychiatry Rep. 2011;13:156–165. doi: 10.1007/s11920-011-0185-3. [DOI] [PubMed] [Google Scholar]
- Athanasiou MC, Dettling M, Cascorbi I, Mosyagin I, Salisbury BA, Pierz KA, Zou W, Whalen H, Malhotra AK, Lencz T, Gerson SL, Kane JM, Reed CR. Candidate gene analysis identifies a polymorphism in HLA-DQB1 associated with clozapine-induced agranulocytosis. J Clin Psychiatry. 2011;72:458–463. doi: 10.4088/JCP.09m05527yel. [DOI] [PubMed] [Google Scholar]
- Dettling M, Cascorbi I, Opgen-Rhein C, Schaub R. Clozapine-induced agranulocytosis in schizophrenic Caucasians: confirming clues for associations with human leukocyte class I and II antigens. Pharmacogenomics J. 2007;7:325–332. doi: 10.1038/sj.tpj.6500423. [DOI] [PubMed] [Google Scholar]
- Need AC, Keefe RSE, Ge D, Grossman I, Dickson S, McEvoy JP, Goldstein DB. Pharmacogenetics of antipsychotic response in the CATIE trial: a candidate gene analysis. Eur J Hum Genet. 2009;17:946–957. doi: 10.1038/ejhg.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClay JL, Adkins DE, Aberg K, Stroup S, Perkins DO, Vladimirov VI, Lieberman JA, Sullivan PF, Van den Oord EJCG. Genome-wide pharmacogenomic analysis of response to treatment with antipsychotics. Mol Psychiatry. 2011;16:76–85. doi: 10.1038/mp.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffaker SJ, Chen J, Nicodemus KK, Sambataro F, Yang F, Mattay V, Lipska BK, Hyde TM, Song J, Rujescu D, Giegling I, Mayilyan K, Proust MJ, Soghoyan A, Caforio G, Callicott JH, Bertolino A, Meyer-Lindenberg A, Chang J, Ji Y, Egan MF, Goldberg TE, Kleinman JE, Lu B, Weinberger DR. A primate-specific, brain isoform of KCNH2 affects cortical physiology, cognition, neuronal repolarization and risk of schizophrenia. Nat Med. 2009;15:509–518. doi: 10.1038/nm.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kongsamut S, Kang J, Chen X-L, Roehr J, Rampe D. A comparison of the receptor binding and HERG channel affinities for a series of antipsychotic drugs. Eur J Pharmacol. 2002;450:37–41. doi: 10.1016/s0014-2999(02)02074-5. [DOI] [PubMed] [Google Scholar]
- Apud JA, Zhang F, Decot H, Bigos KL, Weinberger DR. Genetic variation in KCNH2 associated with expression in the brain of a unique hERG isoform modulates treatment response in patients with schizophrenia. Am J Psychiatry. 2012;169:725–734. doi: 10.1176/appi.ajp.2012.11081214. [DOI] [PubMed] [Google Scholar]
- Zhang J-P, Lencz T, Malhotra AK. D2 receptor genetic variation and clinical response to antipsychotic drug treatment: a meta-analysis. Am J Psychiatry. 2010;167:763–772. doi: 10.1176/appi.ajp.2009.09040598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang R, Zai C, Tiwari A, Müller DJ, Arranz MJ, Morris AG, McKenna PJ, Munro J, Potkin SG, Lieberman JA, Meltzer HY, Kennedy JL. Effect of dopamine D3 receptor gene polymorphisms and clozapine treatment response: exploratory analysis of nine polymorphisms and meta-analysis of the Ser9Gly variant. Pharmacogenomics J. 2010;10:200–218. doi: 10.1038/tpj.2009.65. [DOI] [PubMed] [Google Scholar]
- Schäfer M, Rujescu D, Giegling I, Guntermann A, Erfurth A, Bondy B, Möller HJ. Association of short-term response to haloperidol treatment with a polymorphism in the dopamine D(2) receptor gene. Am J Psychiatry. 2001;158:802–804. doi: 10.1176/appi.ajp.158.5.802. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Yamanouchi Y, Kinoshita Y, Kitajima T, Yoshimura R, Hashimoto S, O’Donovan MC, Nakamura J, Ozaki N, Iwata N. Variants of dopamine and serotonin candidate genes as predictors of response to risperidone treatment in first-episode schizophrenia. Pharmacogenomics. 2008;9:1437–1443. doi: 10.2217/14622416.9.10.1437. [DOI] [PubMed] [Google Scholar]
- Shen Y-C, Chen S-F, Chen C-H, Lin CCH, Chen S-J, Chen Y-J, Luu S-U. Effects of DRD2/ANKK1 gene variations and clinical factors on aripiprazole efficacy in schizophrenic patients. J Psychiatr Res. 2009;43:600–606. doi: 10.1016/j.jpsychires.2008.09.005. [DOI] [PubMed] [Google Scholar]
- Jönsson EG, Flyckt L, Burgert E, Crocq M-A, Forslund K, Mattila-Evenden M, Rylander G, Asberg M, Nimgaonkar VL, Edman G, Bjerkenstedt L, Wiesel F-A, Sedvall GC. Dopamine D3 receptor gene Ser9Gly variant and schizophrenia: association study and meta-analysis. Psychiatr Genet. 2003;13:1–12. doi: 10.1097/00041444-200303000-00001. [DOI] [PubMed] [Google Scholar]
- Binder EB, Nemeroff CB. Implications for the practice of psychiatry. Progress in Brain Research. 2006;158:275–293. doi: 10.1016/S0079-6123(06)58013-6. [DOI] [PubMed] [Google Scholar]
- De Leon J. Pharmacogenomics: The Promise of Personalized Medicine for CNS Disorders. Neuropsychopharmacology. 2008;34:159–172. doi: 10.1038/npp.2008.147. [DOI] [PubMed] [Google Scholar]
- Bogdan R, Hyde LW, Hariri AR. A neurogenetics approach to understanding individual differences in brain, behavior, and risk for psychopathology. Mol Psychiatry. 2012;18:288–299. doi: 10.1038/mp.2012.35. [DOI] [PubMed] [Google Scholar]
- Hanlon CA, Jones EM, Li X, Hartwell KJ, Brady KT, George MS. Individual variability in the locus of prefrontal craving for nicotine: Implications for brain stimulation studies and treatments. Drug Alcohol Depend. 2012;125:239–243. doi: 10.1016/j.drugalcdep.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JP, Etkin A, Furman DJ, Lemus MG, Johnson RF, Gotlib IH. Functional Neuroimaging of Major Depressive Disorder: A Meta-Analysis and New Integration of Baseline Activation and Neural Response Data. Am J Psychiatry. 2012;169:693–703. doi: 10.1176/appi.ajp.2012.11071105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JS, Hamidi M, Postle BR. Using EEG to explore how rTMS produces its effects on behavior. Brain Topogr. 2010;22:281–293. doi: 10.1007/s10548-009-0118-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etkin A, Wager TD. Functional neuroimaging of anxiety: a meta-analysis of emotional processing in PTSD, social anxiety disorder, and specific phobia. Am J Psychiatry. 2007;164:1476–1488. doi: 10.1176/appi.ajp.2007.07030504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frodl T, Möller H-J, Meisenzahl E. Neuroimaging genetics: new perspectives in research on major depression? Acta Psychiatr Scand. 2008;118:363–372. doi: 10.1111/j.1600-0447.2008.01225.x. [DOI] [PubMed] [Google Scholar]
- Thompson PM, Martin NG, Wright MJ. Imaging genomics. Curr Opin Neurol. 2010;23:368–373. doi: 10.1097/WCO.0b013e32833b764c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marano CM, Workman CI, Kramer E, Hermann CR, Ma Y, Dhawan V, Chaly T, Eidelberg D, Smith GS. Longitudinal studies of cerebral glucose metabolism in late-life depression and normal aging. Int J Geriatr Psychiatry. 2013;28:417–423. doi: 10.1002/gps.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumeister A, Normandin MD, Murrough JW, Henry S, Bailey CR, Luckenbaugh DA, Tuit K, Zheng M-Q, Galatzer-Levy IR, Sinha R, Carson RE, Potenza MN, Huang Y. Positron Emission Tomography Shows Elevated Cannabinoid CB (1) Receptor Binding in Men with Alcohol Dependence. Alcohol Clin Exp Res. 2012;36:2104–2109. doi: 10.1111/j.1530-0277.2012.01815.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- Nord M, Farde L. Antipsychotic occupancy of dopamine receptors in schizophrenia. CNS Neurosci Ther. 2011;17:97–103. doi: 10.1111/j.1755-5949.2010.00222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele JD, Currie J, Lawrie SM, Reid I. Prefrontal cortical functional abnormality in major depressive disorder: a stereotactic meta-analysis. J Affect Disord. 2007;101:1–11. doi: 10.1016/j.jad.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Campbell S, Marriott M, Nahmias C, MacQueen GM. Lower hippocampal volume in patients suffering from depression: a meta-analysis. Am J Psychiatry. 2004;161:598–607. doi: 10.1176/appi.ajp.161.4.598. [DOI] [PubMed] [Google Scholar]
- Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161:1957–1966. doi: 10.1176/appi.ajp.161.11.1957. [DOI] [PubMed] [Google Scholar]
- Sheline YI, Gado MH, Kraemer HC. Untreated depression and hippocampal volume loss. Am J Psychiatry. 2003;160:1516–1518. doi: 10.1176/appi.ajp.160.8.1516. [DOI] [PubMed] [Google Scholar]
- Dannlowski U, Ohrmann P, Bauer J, Deckert J, Hohoff C, Kugel H, Arolt V, Heindel W, Kersting A, Baune BT, Suslow T. 5-HTTLPR biases amygdala activity in response to masked facial expressions in major depression. Neuropsychopharmacology. 2008;33:418–424. doi: 10.1038/sj.npp.1301411. [DOI] [PubMed] [Google Scholar]
- Dannlowski U, Ohrmann P, Bauer J, Kugel H, Baune BT, Hohoff C, Kersting A, Arolt V, Heindel W, Deckert J, Suslow T. Serotonergic genes modulate amygdala activity in major depression. Genes Brain Behav. 2007;6:672–676. doi: 10.1111/j.1601-183X.2006.00297.x. [DOI] [PubMed] [Google Scholar]
- Frodl T, Meisenzahl EM, Zill P, Baghai T, Rujescu D, Leinsinger G, Bottlender R, Schüle C, Zwanzger P, Engel RR, Rupprecht R, Bondy B, Reiser M, Möller H-J. Reduced hippocampal volumes associated with the long variant of the serotonin transporter polymorphism in major depression. Arch Gen Psychiatry. 2004;61:177–183. doi: 10.1001/archpsyc.61.2.177. [DOI] [PubMed] [Google Scholar]
- Taylor WD, Steffens DC, Payne ME, MacFall JR, Marchuk DA, Svenson IK, Krishnan KRR. Influence of serotonin transporter promoter region polymorphisms on hippocampal volumes in late-life depression. Arch Gen Psychiatry. 2005;62:537–544. doi: 10.1001/archpsyc.62.5.537. [DOI] [PubMed] [Google Scholar]
- McDonald C, Zanelli J, Rabe-Hesketh S, Ellison-Wright I, Sham P, Kalidindi S, Murray RM, Kennedy N. Meta-analysis of magnetic resonance imaging brain morphometry studies in bipolar disorder. Biol Psychiatry. 2004;56:411–417. doi: 10.1016/j.biopsych.2004.06.021. [DOI] [PubMed] [Google Scholar]
- Kempton MJ, Geddes JR, Ettinger U, Williams SCR, Grasby PM. Meta-analysis, database, and meta-regression of 98 structural imaging studies in bipolar disorder. Arch Gen Psychiatry. 2008;65:1017–1032. doi: 10.1001/archpsyc.65.9.1017. [DOI] [PubMed] [Google Scholar]
- Vita A, De Peri L, Sacchetti E. Gray matter, white matter, brain, and intracranial volumes in first-episode bipolar disorder: a meta-analysis of magnetic resonance imaging studies. Bipolar Disord. 2009;11:807–814. doi: 10.1111/j.1399-5618.2009.00759.x. [DOI] [PubMed] [Google Scholar]
- Beyer JL, Young R, Kuchibhatla M, Krishnan KRR. Hyperintense MRI lesions in bipolar disorder: A meta-analysis and review. Int Rev Psychiatry. 2009;21:394–409. doi: 10.1080/09540260902962198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempton MJ, Salvador Z, Munafò MR, Geddes JR, Simmons A, Frangou S, Williams SCR. Structural neuroimaging studies in major depressive disorder. Meta-analysis and comparison with bipolar disorder. Arch Gen Psychiatry. 2011;68:675–690. doi: 10.1001/archgenpsychiatry.2011.60. [DOI] [PubMed] [Google Scholar]
- Houenou J, Frommberger J, Carde S, Glasbrenner M, Diener C, Leboyer M, Wessa M. Neuroimaging-based markers of bipolar disorder: evidence from two meta-analyses. J Affect Disord. 2011;132:344–355. doi: 10.1016/j.jad.2011.03.016. [DOI] [PubMed] [Google Scholar]
- Hajek T, Kopecek M, Kozeny J, Gunde E, Alda M, Höschl C. Amygdala volumes in mood disorders–meta-analysis of magnetic resonance volumetry studies. J Affect Disord. 2009;115:395–410. doi: 10.1016/j.jad.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Pfeifer JC, Welge J, Strakowski SM, Adler CM, DelBello MP. Meta-analysis of amygdala volumes in children and adolescents with bipolar disorder. J Am Acad Child Adolesc Psychiatry. 2008;47:1289–1298. doi: 10.1097/CHI.0b013e318185d299. [DOI] [PubMed] [Google Scholar]
- Davidson LL, Heinrichs RW. Quantification of frontal and temporal lobe brain-imaging findings in schizophrenia: a meta-analysis. Psychiatry Res. 2003;122:69–87. doi: 10.1016/s0925-4927(02)00118-x. [DOI] [PubMed] [Google Scholar]
- Hill K, Mann L, Laws KR, Stephenson CME, Nimmo-Smith I, McKenna PJ. Hypofrontality in schizophrenia: a meta-analysis of functional imaging studies. Acta Psychiatr Scand. 2004;110:243–256. doi: 10.1111/j.1600-0447.2004.00376.x. [DOI] [PubMed] [Google Scholar]
- Raz S, Raz N. Structural brain abnormalities in the major psychoses: a quantitative review of the evidence from computerized imaging. Psychol Bull. 1990;108:93–108. doi: 10.1037/0033-2909.108.1.93. [DOI] [PubMed] [Google Scholar]
- Boos HBM, Aleman A, Cahn W, Hulshoff Pol H, Kahn RS. Brain volumes in relatives of patients with schizophrenia: a meta-analysis. Arch Gen Psychiatry. 2007;64:297–304. doi: 10.1001/archpsyc.64.3.297. [DOI] [PubMed] [Google Scholar]
- Zakzanis KK, Hansen KT. Dopamine D2 densities and the schizophrenic brain. Schizophr Res. 1998;32:201–206. doi: 10.1016/s0920-9964(98)00041-3. [DOI] [PubMed] [Google Scholar]
- Olabi B, Ellison-Wright I, McIntosh AM, Wood SJ, Bullmore E, Lawrie SM. Are there progressive brain changes in schizophrenia? A meta-analysis of structural magnetic resonance imaging studies. Biol Psychiatry. 2011;70:88–96. doi: 10.1016/j.biopsych.2011.01.032. [DOI] [PubMed] [Google Scholar]
- Shepherd AM, Laurens KR, Matheson SL, Carr VJ, Green MJ. Systematic meta-review and quality assessment of the structural brain alterations in schizophrenia. Neurosci Biobehav Rev. 2012;36:1342–1356. doi: 10.1016/j.neubiorev.2011.12.015. [DOI] [PubMed] [Google Scholar]
- Nelson MD, Saykin AJ, Flashman LA, Riordan HJ. Hippocampal volume reduction in schizophrenia as assessed by magnetic resonance imaging: a meta-analytic study. Arch Gen Psychiatry. 1998;55:433–440. doi: 10.1001/archpsyc.55.5.433. [DOI] [PubMed] [Google Scholar]
- Steen RG, Mull C, McClure R, Hamer RM, Lieberman JA. Brain volume in first-episode schizophrenia: systematic review and meta-analysis of magnetic resonance imaging studies. Br J Psychiatry. 2006;188:510–518. doi: 10.1192/bjp.188.6.510. [DOI] [PubMed] [Google Scholar]
- Adriano F, Caltagirone C, Spalletta G. Hippocampal volume reduction in first-episode and chronic schizophrenia: a review and meta-analysis. Neuroscientist. 2012;18:180–200. doi: 10.1177/1073858410395147. [DOI] [PubMed] [Google Scholar]
- Baig BJ, Whalley HC, Hall J, McIntosh AM, Job DE, Cunningham-Owens DG, Johnstone EC, Lawrie SM. Functional magnetic resonance imaging of BDNF val66met polymorphism in unmedicated subjects at high genetic risk of schizophrenia performing a verbal memory task. Psychiatry Res. 2010;183:195–201. doi: 10.1016/j.pscychresns.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Van Zuiden M, Geuze E, Willemen HLDM, Vermetten E, Maas M, Heijnen CJ, Kavelaars A. Pre-existing high glucocorticoid receptor number predicting development of posttraumatic stress symptoms after military deployment. Am J Psychiatry. 2011;168:89–96. doi: 10.1176/appi.ajp.2010.10050706. [DOI] [PubMed] [Google Scholar]
- Gatt JM, Williams LM, Schofield PR, Dobson-Stone C, Paul RH, Grieve SM, Clark CR, Gordon E, Nemeroff CB. Impact of the HTR3A gene with early life trauma on emotional brain networks and depressed mood. Depress Anxiety. 2010;27:752–759. doi: 10.1002/da.20726. [DOI] [PubMed] [Google Scholar]
- Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch Gen Psychiatry. 1999;56:940–945. doi: 10.1001/archpsyc.56.10.940. [DOI] [PubMed] [Google Scholar]
- Dunbar L, Butler R, Wheeler A, Pulford J, Miles W, Sheridan J. Clinician experiences of employing the AmpliChip® CYP450 test in routine psychiatric practice. J Psychopharmacol (Oxford) 2012;26:390–397. doi: 10.1177/0269881109106957. [DOI] [PubMed] [Google Scholar]