

Abstract
In vitro studies of hepatocytes have implicated over-activation of c-Jun N-terminal kinase (JNK) signaling as a mechanism of tumor necrosis factor-α (TNF)-induced apoptosis. However, the functional significance of JNK activation and the role of specific JNK isoforms in TNF-induced hepatic apoptosis in vivo remain unclear. JNK1 and JNK2 function was, therefore, investigated in the TNF-dependent, galactosamine/lipopolysaccharide (GalN/LPS) model of liver injury. The toxin GalN converted LPS-induced JNK signaling from a transient to prolonged activation. Liver injury and mortality from GalN/LPS was equivalent in wild-type and jnk1−/− mice but markedly decreased in jnk2−/− mice. This effect was not secondary to down-regulation of TNF receptor 1 expression or TNF production. In the absence of jnk2, the caspase-dependent, TNF death pathway was blocked, as reflected by the failure of caspase-3 and -7 and poly(ADP-ribose) polymerase cleavage to occur. JNK2 was critical for activation of the mitochondrial death pathway, as in jnk2−/− mice Bid cleavage and mitochondrial translocation and cytochrome c release were markedly decreased. This effect was secondary to the failure of jnk2−/− mice to activate caspase-8. Liver injury and caspase activation were similarly decreased in jnk2 null mice after GalN/TNF treatment. Ablation of jnk2 did not inhibit GalN/LPS-induced c-Jun kinase activity, although activity was completely blocked in jnk1−/− mice. Toxic liver injury is, therefore, associated with JNK over-activation and mediated by JNK2 promotion of caspase-8 activation and the TNF mitochondrial death pathway through a mechanism independent of c-Jun kinase activity.
Tumor necrosis factor-α (TNF)2 mediates a number of forms of organ injury through its induction of cellular apoptosis. In the liver the biological effects of TNF have been implicated as a mechanism of hepatic injury from toxins (1, 2), ischemia/reperfusion (3, 4), viral hepatitis (5, 6), and cholestasis (7, 8). Experimental toxin-induced liver injury results in large part not from the biochemical effects of the toxin but from TNF cytotoxicity (1, 2). This concept has been proven by studies in which TNF inhibition dramatically reduced liver injury from toxins such as carbon tetrachloride (1), galactosamine (9), and ethanol (2). Hepatocytes are normally resistant to TNF toxicity (10, 11) and proliferate in response to this cytokine (12), as exemplified by the mitogenic role of TNF in vivo after partial hepatectomy (13). Hepatotoxins, therefore, act by an unknown mechanism to convert the cellular outcome emanating from TNF stimulation from one of proliferation to one of cell death.
The sequential steps of the TNF-induced death receptor pathway have been well characterized. Binding of TNF to TNF receptor 1 (TNFR1) results in receptor trimerization and the recruitment of a series of intracellular proteins (14) that ultimately bind caspase-8, leading to its activation (15). Activated caspase-8 initiates a proteolytic cascade that results in release of lysosomal cathepsin B (16), cleavage of the proapoptotic Bcl-2 family member Bid (17), initiation of the mitochondrial death pathway with release of cytochrome c, and activation of downstream effector caspases that ultimately induce apoptosis (18). The inherent resistance of hepatocytes to TNF-induced cytotoxicity results from up-regulation of a protective factor(s). This conclusion is based on findings that inhibition of RNA or protein synthesis sensitizes hepatocytes to death from TNF (10, 11). NF-κB has been identified as the transcriptional regulator of the hepatocyte protective response to TNF (19, 20). Blocking TNF-induced NF-κB activation in cultured hepatocytes (20) or the liver in vivo (21) converts the hepatocellular TNF response from proliferation to apoptosis. NF-κB inactivation sensitizes hepatocytes to the classic TNF death pathway involving cathepsin B, mitochondria, and caspases (16, 19, 20).
NF-κB activation mediates resistance to TNF toxicity by down-regulating the mitogen-activated protein kinase c-Jun N-terminal kinase (JNK) in hepatocytes and nonhepatic cells (22–24). Hepatocytes like most cell types express two JNK genes, jnk1 and jnk2. Their respective gene products, JNK1 and JNK2, both consist of two alternative splicing forms, p54 and p46 JNK (25). The p54 JNK isoform is predominantly expressed by jnk2, and p46 JNK is primarily a jnk1 product (26, 27). jnk1 and jnk2 null mice are both viable (26, 27), whereas the double knock-out is an embryonic lethal (28), suggesting that JNK1 and JNK2 have redundant functions. Recent studies, however, have demonstrated that the products of jnk1 and jnk2 may have distinct functions (29–31). In cultured hepatocytes, bile acid toxicity is mediated by jnk1, whereas jnk2 expression is cytoprotective (32). One possible reason for the two gene products to have differential functions is the in vitro finding that JNK1 accounts for classical JNK activity as measured by c-Jun phosphorylation, whereas JNK2 lacks this kinase activity and may even act to decrease overall JNK activity (33, 34). Whether JNK1 and JNK2 perform differential functions in TNF-mediated cell death is uncertain, with prior in vitro studies in mouse embryonic fibroblasts demonstrating the promotion of cell death specifically by JNK1 or JNK2 (33, 35).
An established mouse model of TNF-dependent liver injury was utilized to investigate JNK function in the TNF death pathway in vivo. It was hypothesized that similar to the effect of in vitro NF-κB inhibition, hepatotoxins sensitize the liver to injury from TNF by inducing over-activation of JNK. This hypothesis was tested in the murine model of TNF-dependent hepatotoxicity resulting from the combined administration of galactosamine (GalN) and lipopolysaccharide (LPS) (36, 37). The toxin GalN converted the transient induction of JNK signaling by LPS into a sustained activation. JNK activation promoted hepatocellular injury as jnk2 null mice had significantly reduced liver damage and lethality from GalN/LPS. The effect was specific for jnk2 and involved the requirement of JNK2 for caspase-8 activation, Bid cleavage, and induction of the mitochondrial death pathway. These effects were unrelated to changes in classic c-Jun kinase activity, indicating a novel effect of JNK2 in the promotion of the TNF apoptotic death pathway in vivo.
EXPERIMENTAL PROCEDURES
Animal Model
Wild-type male and female C57BL/6, jnk1−/− (26), and jnk2−/− (27) mice (The Jackson Laboratory, Bar Harbor, ME) were maintained under 12-h light/dark cycles with unlimited access to food and water. Genotypes were confirmed by PCR with established primers (26, 27). Both knock-out mice had been backcrossed onto a C57BL/6 background for greater than six generations. Six-week-old mice were given intraperitoneal injections of 100 μg/kg of LPS (Escherichia coli 0111:B4; Sigma) alone or together with 700 mg/kg of d-galactosamine (Sigma) dissolved in phosphate-buffered saline. Alternatively, mice were pretreated with GalN for 1 h and then administered 20 μg/kg of mouse recombinant TNF (R&D Systems, Minneapolis, MN) intraperitoneally. At various intervals after injection, serum was obtained by retro-orbital bleed, and the mice were sacrificed for the removal of liver tissue. All studies were approved by the Animal Care and Use Committee of the Albert Einstein College of Medicine and followed the National Institutes of Health guidelines for animal care.
Protein Isolation and Western Blotting
For total protein isolation, portions of liver were sonicated in homogenization buffer containing 1 mm sodium bicarbonate, 0.5 mm calcium chloride, and protease and phosphatase inhibitors as previously described (38). Western blotting was performed by denaturing 50 μg of protein at 100 °C for 5 min in Laemmli sample buffer containing 62.5 mm Tris-HCl, pH 6.8, 2% SDS, 25% glycerol, 0.01% bromphenol blue, and 5% β-mercaptoethanol. Proteins were resolved by SDS-PAGE and transferred to a nitrocellulose membrane (Schleicher & Schuell) in transfer buffer containing 25 mm Tris, pH 8.3, 192 mm glycine, 0.01% SDS, and 15% methanol using a Bio-Rad Trans-blot SD semidry transfer cell to which 150 mA were applied for 90 min. Membranes were blocked in 5% nonfat dry milk, 20 mm Tris, pH 7.5, 500 mm sodium chloride, and 0.5% Tween 20 (TBS-T) for 1 h. Membranes were incubated for 18 h at 4 °C with the following primary antibodies at 1:1000 to 1:2000 dilutions in 5% bovine serum albumin or nonfat milk: rabbit anti-JNK, rabbit anti-poly(ADP-ribose) polymerase (PARP), mouse anti-Bax, rabbit anti-Bcl-XL, rabbit anti-cIAP1, rabbit anti-cIAP2, rabbit anti-TNFR1, rabbit anti-TRAF2, and rabbit anti-c-FLIP (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit anti-phospho-JNK, rabbit anti-caspase-3, rabbit anti-caspase-7, rabbit anti-Bad, and mouse anti-phospho-Bad (Cell Signaling, Beverly, MA), rat anti-caspase-8 (Axxora Life Sciences, San Diego, CA), rabbit anti-Bim (EMD Biosciences, San Diego, CA), and rabbit anti-Bid (kind gift of Xiao-Ming Yin, University of Pittsburgh, PA). Membranes were exposed to goat anti-rabbit, anti-mouse (KPL, Gaithersburg, MD) or anti-rat (SouthernBiotech, Birmingham, AL) secondary antibodies conjugated with horseradish peroxidase at a dilution of 1:10,000 in 5% nonfat milk in TBS-T for 1 h at room temperature. Signals were detected with a chemiluminescence detection system (Western Lightning Chemiluminescence Plus, PerkinElmer Life Sciences) and exposure to x-ray film. To ensure that equivalent amounts of protein were loaded among samples, membranes were stripped and immunoblotted with a mouse antibody for β-actin (Abcam, Cambridge, MA).
Mitochondrial and cytosolic protein fractions were obtained by homogenization of liver tissue in buffer A (20 mm Hepes, pH 7.5, 250 mm sucrose, 10 mm KCl, 1.5 mm MgCl2, 0.5 mm EDTA, 0.5 mm EGTA, 1 mm dithiothreitol, 1 mm phenylmethylsulfonyl fluoride) with a Dounce homogenizer and centrifugation of the homogenate at 4 °C for 10 min at 600 × g. The resultant supernatant was centrifuged a second time under the same conditions. This supernatant was centrifuged at 4 °C for 15 min at 10,000 × g. The pellet was redissolved in buffer A and formed the mitochondrial fraction. The supernatant was further centrifuged at 4 °C for 10 min at 10,000 × g. This last supernatant comprised the cytosolic protein fraction. Western blotting was performed as above with mouse anti-cytochrome c (Pharmingen), and mouse anti-cytochrome oxidase (MitoSciences, Eugene, OR) antibodies in addition to the antibodies described above.
JNK Activity Assay
JNK activity was measured in portions of liver tissue using a JNK in vitro kinase activity assay according to the manufacturer’s instructions (Cell Signaling). JNK was immunoprecipitated from 200 μg of total protein with a c-Jun fusion protein. After washing, kinase reactions were performed in the presence of 10 mm ATP using c-Jun as substrate. Samples were resolved in 10% SDS-polyacrylamide gels, and immunoblotting was performed as previously described. The relative amounts of the phosphorylated c-Jun fusion protein were detected with a phospho-c-Jun antibody. To control for the loading of equivalent amounts of protein among samples, membranes were stripped and immunoblotted with an antibody for total c-Jun (Santa Cruz Biotechnology).
Alanine Aminotransferase (ALT) Assay
Serum ALT was measured by commercial kit (TECO Diagnostics, Anaheim, CA).
Histological Analysis
Liver specimens were fixed in 10% neutral formalin, and 6-μm sections were stained with hematoxylin and eosin. Tissue sections were examined in a blinded fashion by a single pathologist and graded for the degree of liver injury and inflammation. The approximate percentage of hepatic parenchyma involved by apoptosis/necrosis or inflammation was semiquantitatively graded on a sliding scale of 0 (absent), 0.5 (minimal), 1 (mild), 1.5 (mild to moderate), 2 (moderate), 2.5 (moderate to marked), and 3 (marked).
Terminal Deoxynucleotide Transferase-mediated Deoxyuridine Triphosphate Nick End-labeling (TUNEL) Assay
TUNEL-positive cells in liver sections were detected with a commercial kit (Promega, Madison, WI). Tissue sections were deparaffinized in xylene and gradually rehydrated in decreasing concentrations of ethanol, and the TUNEL assay was performed according to the manufacturer’s instructions. Under light microscopy, the numbers of TUNEL-positive cells in 10 randomly selected high power fields (400× magnification) were counted per liver section.
TNF Levels
Serum TNF levels were measured by ELISA using a commercial kit (BD Biosciences).
Real-time Reverse Transcription (RT)-PCR
Hepatic total RNA was isolated by cesium chloride gradient centrifugation, as previously described (39). Real-time RT-PCR for TNF, interferon γ, interleukin-6, and β-actin was performed with Qiagen QuantiProbes and primers (Qiagen, Valencia, CA) in an ABI PRISM 7000 sequence detection system (Applied Biosystems, Foster City, CA). One-step RT-PCR was carried out in triplicate with 250 ng of total RNA using the QuantiTect Probe™ RT-PCR kit (Qiagen). The cycling parameters were reverse transcription at 50 °C for 30 min, HotStar Taq DNA polymerase (Qiagen) activation at 95 °C for 15 min, extension at 76 °C for 30 s, denaturation at 94 °C for 15 s, and annealing/detection at 56 °C for 30 s (for 45 cycles). Data analysis was performed using the 2−ΔΔCT method for relative quantification (40). All samples were normalized initially to β-actin, which served as an endogenous control, and then to a control sample.
Caspase-8 Activity
Total liver lysates were prepared as previously described by Zender et al. (41). Briefly, liver tissue was homogenized in buffer containing 25 mm Hepes, pH 7.5, 5 mm MgCl2, 5 mm EDTA, 2 mm dithiothreitol, 0.1% CHAPS, 0.5 mm Pefabloc (Roche Applied Science), 0.1 mg/ml leupeptin, and 0.1 mg/ml pepstatin. The homogenates were centrifuged at 12,000 × g for 15 min at 4 °C, and 50 μg of protein from the supernatant were assayed for caspase-8 activity. Activity was measured with the BD ApoAlert™ caspase-8 colorimetric assay kit (BD Biosciences) according to the manufacturer’s instructions.
Statistical Analysis
All numerical results are expressed as the mean ± S.E. and represent data from a minimum of three independent experiments. Calculations were made with Sigma Plot 2000 (SPSS Science, Chicago, IL).
RESULTS
GalN/LPS-induced Liver Injury Is Associated with Prolonged JNK Activation
The GalN/LPS model of toxin-induced liver injury was examined for the effect of the hepatotoxin GalN on JNK activation. GalN/LPS induces liver injury that is primarily apoptotic and mediated by the effects of secreted TNF on TNFR1 (36, 37). An advantage of this model is that insight into the mechanism by which the toxin GalN sensitizes to liver injury from TNF can be obtained by contrasting cell signals induced by the nontoxic dose of LPS to those that occur with the hepatotoxic GalN/LPS combination. Hepatic JNK activation induced by LPS alone or the combination of GalN and LPS was compared by Western blot analysis of levels of active, phosphorylated p54 and p46 JNK. LPS alone induced a transient increase in phosphorylated p54 and p46 JNK within 1 h, but levels returned to base line within 2 h of LPS administration (Fig. 1A). In contrast, levels of phosphorylated JNK remained markedly elevated in GalN/LPS-treated mice even at 6 h (Fig. 1A). Levels of total JNK were unchanged with either treatment (Fig. 1A). JNK over-activation preceded the onset of significant hepatocyte apoptosis that occurred at 6 h as indicated by decreased procaspase levels and appearance of the cleaved forms for the effector caspases-3 and -7 at this time point (Fig. 1A).
FIGURE 1. GalN/LPS co-administration leads to prolonged JNK activation before the onset of caspase cleavage.
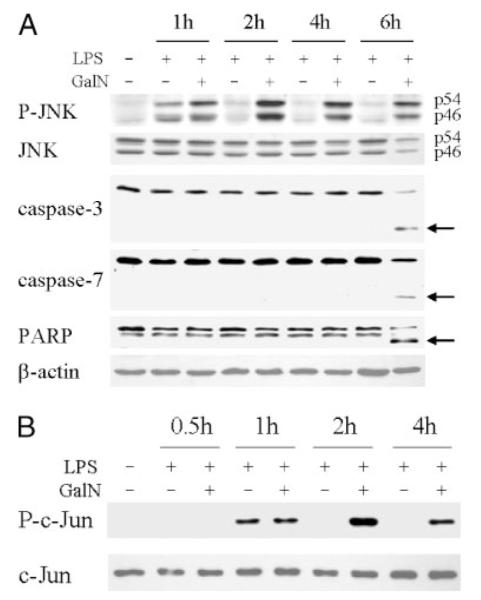
A, protein was isolated from the livers of C57BL/6 mice that were untreated or treated with LPS or GalN/LPS for the number of hours indicated. Samples were immunoblotted with antibodies against phospho-JNK (P-JNK), total JNK, caspase-3 and -7, PARP, and β-actin. The p54 and p46 JNK bands are labeled, and arrows indicate the caspase-3 and -7 and PARP cleavage products. B, hepatic JNK activity was determined in untreated and LPS- or GalN/LPS-treated livers by an in vitro kinase activity assay employing c-Jun as substrate. JNK activity was assayed by immunoblots for phosphorylated c-Jun (P-c-Jun) as described under “Experimental Procedures.” Stripped membranes were immunoblotted with an anti-total c-Jun antibody to check the equivalency of protein loading. Results are representative of three independent experiments.
As additional confirmation of the effect of GalN on JNK activation, hepatic JNK activity was assayed by an in vitro kinase assay employing c-Jun as substrate. Identical to the Western blot findings, the transient 1-h increase in JNK activity induced by LPS was converted into sustained JNK activation by GalN co-administration (Fig. 1B). TNF-dependent, GalN-induced liver injury is, therefore, associated with prolonged activation of the JNK signaling pathway similar to the JNK over-activation that occurs in cultured hepatocytes sensitized to TNF toxicity by NF-κB inhibition.
Ablation of jnk1 Does Not Reduce Liver Injury from GalN/LPS
To assess whether JNK activation has a functional role in GalN/LPS liver injury, the effect of jnk1 ablation on liver injury was examined. JNK1 was examined because in vitro studies in nonhepatic cells have suggested that only the JNK1 and not the JNK2 isoforms are important in mediating JNK kinase activity (33, 34). Additionally, in cultured hepatocytes, JNK1 but not JNK2 was shown to mediate hepatocyte injury from bile acids (32). To determine the function of JNK1, the degree of liver injury was examined in wild-type C57BL/6 and jnk1−/− mice after GalN/LPS administration. Consistent with findings in other organs in jnk1−/− mice (26), the absence of jnk1 expression markedly decreased hepatic levels of p46 JNK, whereas p54 JNK was minimally affected (Fig. 2A). After GalN/LPS treatment, levels of both p54 and p46 phospho-JNK increased significantly in jnk1 null mice, but the levels of both isoforms were decreased relative to those in wild-type mouse livers (Fig. 2A).
FIGURE 2. Liver injury from GalN/LPS is unaffected by loss of jnk1.

A, total hepatic protein was isolated from wild-type (+/+) or jnk1−/− (−/−) mice at the indicated hours after treatment with GalN/LPS and immunoblotted with phospho-JNK (P-JNK), total JNK, caspase-3 and -7, PARP, and β-actin antibodies. The p54 and p46 JNK bands are labeled, and the caspase-3 and -7 and PARP cleavage products are indicated by arrows. Findings are representative of three independent experiments. B, serum ALT levels from wild-type (WT) and jnk1−/− mice at 4 and 6 h after GalN/LPS administration. C, graded histological scoring of the degree of apoptosis (Apop) and inflammation (Inflam) in the same livers. D, numbers of TUNEL positive cells per high power field in the identical liver sections. Data in B–D are from three independent experiments and total five–eight animals per data point.
The effect of jnk1 ablation on liver injury from GalN/LPS was assessed by ALT levels, liver histology, and TUNEL staining. Serum levels of ALT, a marker of hepatocellular injury, were equivalent in wild-type and jnk1−/− mice after GalN/LPS administration with mild elevations at the 4-h onset of injury and marked increases at the 6-h peak of liver injury (Fig. 2B). Histological evidence of significant liver injury was present in both types of mice at 6 h and included hepatocyte apoptosis, inflammation, and vascular congestion (Fig. 3, A and B). Blinded grading of the liver histology confirmed an equivalent degree of apoptosis and inflammation in both groups of mice (Fig. 2C). As a final measure of liver injury, the numbers of apoptotic cells were identified by TUNEL staining. Equivalent numbers of TUNEL-positive cells were present in wild-type and jnk1−/− mice at 4 and 6 h (Figs. 2D and 3, panels d and e). Thus, by the three measures of serum ALT, liver histology, and TUNEL staining, loss of JNK1 failed to attenuate liver injury induced by GalN/LPS.
FIGURE 3. Liver histology and TUNEL staining in wild-type and jnk1 and jnk2 knock-out mice after GalN/LPS treatment.
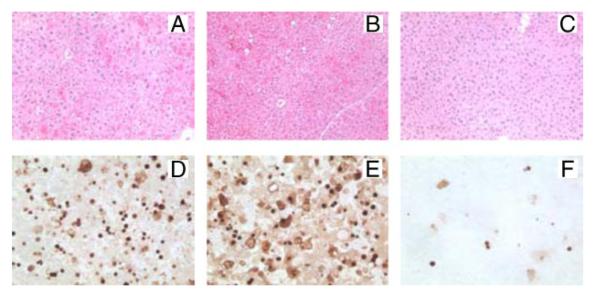
Shown are hematoxylin- and eosin-stained sections of wild-type (A), jnk1−/− (B), and jnk2−/− (C) mice at 6 h after GalN/LPS treatment. TUNEL staining at the same time point in wild-type (D), jnk1−/− (E), and jnk2−/− (F) mice (magnification 400×) is also shown.
jnk2−/− Mice Are Protected from GalN/LPS-induced Liver Injury
With the failure of the absence of jnk1 to alter injury, GalN/LPS-induced liver injury was compared in wild-type and jnk2−/− mice. Western blot analysis of hepatic JNK expression in jnk2−/− animals demonstrated a significant decrease in levels of the p54 JNK isoforms and no significant reduction in p46 JNK (Fig. 4A), consistent with findings in other tissues in these mice (27). In jnk2 knock-out mice after GalN/LPS treatment, levels of phospho-p54 JNK were markedly decreased, whereas phospho-p46 JNK levels were equivalent to those in wild-type mice, suggesting that the majority of activated p54 JNK was derived from jnk2 expression.
FIGURE 4. jnk2−/− mice are protected from GalN/LPS-induced liver injury.

A, protein was isolated from the livers of wild-type (+/+) or jnk2−/− (−/−) mice at the indicated hours after treatment with GalN/LPS and immunoblotted with phospho-JNK (P-JNK), total JNK, caspase-3 and -7, PARP, and β-actin antibodies. The p54 and p46 JNK bands are labeled, and arrows indicate the caspase-3 and -7 and PARP cleavage products. Findings are representative of three independent experiments. B, serum ALT levels from wild-type (WT) and jnk2−/− mice at 4 and 6 h after GalN/LPS treatment. Results are from 4 independent experiments representing a total of 4 mice at 4 h and 8 mice at 6 h per data point (*, p < 0.0001 compared with wild type). C, graded histological scoring of the degree of apoptosis (Apop) and inflammation (Inflam) in the same livers (p < 0.01 (*) and p < 0.001 (#) compared with wild type). D, numbers of TUNEL-positive cells per high power field in the identical liver sections (p < 0.01 (*) and p < 0.00001 (#) compared with wild-type mice).
At 6 h after GalN/LPS administration, jnk2−/− mice had ALT levels that were significantly decreased by 76% as compared with wild-type mice (Fig. 4B). Histological evidence of liver injury (Figs. 3, panel c, and 4C) and the numbers of TUNEL-positive cells (Figs. 3, panel f, and 4D) were also significantly decreased in jnk2−/− animals as compared with wild-type mice. These data combined with those from the experiments in jnk1−/− mice indicated that products of the jnk2 and not jnk1 gene promote hepatocellular injury in response to the toxin GalN.
jnk2−/− Mice Have Reduced Mortality from GalN/LPS
Although the jnk2−/− mice had dramatically decreased liver injury from GalN/LPS, it was possible that the absence of jnk2 merely delayed liver injury but had no ultimate beneficial effect on outcome. Shortly after the 6-h peak of injury, GalN/LPS-induced liver injury in mice led to liver failure and death. Long-term survival after GalN/LPS treatment was examined as an indication of whether the initial resistance of jnk2 null mice to injury from GalN/LPS affected the development of fatal liver failure. Within 8 h of GalN/LPS administration, mortality was 83% in wild-type mice and 100% in jnk1−/− mice but only 16% in jnk2−/− mice (Fig. 5A). Overall survival after GalN/LPS administration was only 5% in wild-type mice and 0% in jnk1−/− mice but 40% in jnk2−/− mice (Fig. 5A).
FIGURE 5. jnk2−/− mice have increased long-term survival after GalN/LPS treatment.
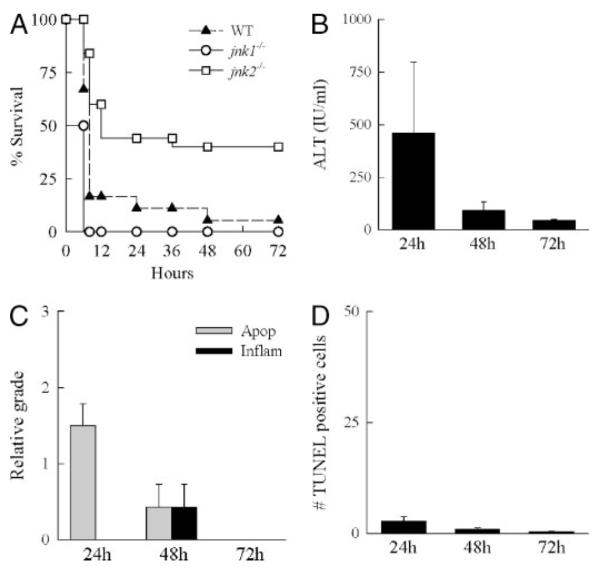
A, percentages of survival of wild-type (WT), jnk1−/−, and jnk2−/− mice at the indicated hours after administration of GalN/LPS. Data are from 3 independent experiments that include a total of 19 wild-type, 10 jnk1−/−, and 25 jnk2−/− mice (p < 0.01 for jnk2−/− as compared with wild type or jnk1−/−). B, serum ALT levels in wild-type mice surviving for the indicated hours after GalN/LPS treatment. C, graded histological scoring of the degree of apoptosis (Apop) and inflammation (Inflam) in the livers of the same animals. D, numbers of TUNEL-positive cells per high power field in the identical liver sections. Data in B–D are from four independent experiments and total three-eight animals per data point.
Although there was a marked reduction in mortality from GalN/LPS, a significant number of jnk2−/− mice still died. This mortality may reflect delayed liver injury, an alternative form of injury such as necrosis, or death from nonhepatic causes perhaps secondary to the systemic release of cytokines. To distinguish among these possibilities, mice surviving 24, 48, and 72 h after GalN/LPS treatment were examined for liver injury. At 24 h, ALT levels were variable but only moderately elevated (Fig. 5B). Serum ALTs decreased at 48 h and were essentially normal in mice surviving 72 h (Fig. 5B). Evidence of mild apoptosis and some necrosis was seen on histology of 24- and 48-h livers, but no evidence of liver injury was present by 72 h (Fig. 5C). The numbers of TUNEL-positive cells were very low in surviving animals at all three times (Fig. 5D). Knock-out of jnk2, therefore, significantly protected mice from both GalN/LPS-induced morbidity and mortality.
Absence of jnk2 Does Not Affect Cytokine Induction by GalN/LPS
GalN sensitizes the liver to injury from the cytokine TNF (36, 37). The effects of jnk2 ablation on TNF receptor expression and TNF production were investigated as a potential mechanism for the decrease in liver injury in jnk2−/− mice. Levels of TNFR1, which mediates TNF cytotoxicity in GalN/LPS injury (37), were equivalent in the livers of wild-type and jnk2−/− mice and unaffected by GalN/LPS treatment (Fig. 6A). Serum TNF levels were markedly increased to similar levels in response to GalN/LPS in wild-type and jnk2−/− mice (Fig. 6B). Finally, an equivalent increase in levels of hepatic TNF RNA occurred after GalN/LPS treatment in wild-type and jnk2−/− mice, as determined by quantitative real-time RT-PCR (Fig. 6C). Similarly, the induction of hepatic RNA expression for the cytokines interferon γ and interleukin-6, which are also induced in this model and may modulate hepatocellular injury (9, 42, 43), was equivalent in wild-type and jnk2 null mice (Fig. 6, D and E). The mechanism of the reduction in GalN/LPS liver injury by jnk2 ablation was not through the down-regulation of TNF or other cytokines.
FIGURE 6. TNFR1 expression and cytokine induction are equivalent in wild-type and jnk2−/− mice.

A, total hepatic protein was isolated from the livers of wild-type (+/+) or jnk2−/− (−/−) mice at the indicated hours after treatment with GalN/LPS and immunoblotted with TNFR1 and β-actin antibodies. B, levels of serum TNF at the times shown after GalN/LPS treatment. Serum TNF was undetectable in untreated mice. C–E, total liver RNA was isolated from wild-type and jnk2 knock-out mouse mice untreated and treated with GalN/LPS for 1, 2, and 4 h. Real-time RT-PCR was performed with primers for TNF, interferon γ (IFN), interleukin-6 (IL-6), and β-actin as described under “Experimental Procedures.” Levels of cytokine mRNA were normalized to that of β-actin. TNF mRNA was undetectable in untreated livers, and the TNF values are expressed relative to levels in 1-h-treated wild-type animals (C). Levels of interferon γ (D) and interleukin-6 (E) mRNA were normalized to the level in untreated wild-type livers. The results are from three independent experiments.
Effector Caspase Activation Is Inhibited by the Loss of jnk2
In vitro and in vivo hepatocyte death from TNF occurs through caspase-dependent apoptosis (19, 20, 44). The failure of jnk2 ablation to affect cytokine expression suggested that JNK2 might function as a critical component in the hepatocyte, TNF-dependent, apoptotic pathway. To assess whether loss of JNK2 affected TNF-induced apoptosis, activation of the downstream effector caspases -3 and -7 was examined. Caspase-3 and -7 activation was detected in wild-type mice 6 h after GalN/LPS treatment by immunoblotting for the cleaved, active caspase forms, but activation failed to occur in GalN/LPS-treated jnk2−/− mice (Fig. 4A). In contrast, activation of caspase-3 and -7 did occur in jnk1−/− mice (Fig. 2A). The caspases were functionally active as indicated by the presence of PARP cleavage in wild-type and jnk1−/− mice at 6 h (Fig. 2A). In contrast, PARP cleavage was absent in GalN/LPS-treated jnk2−/− mice (Fig. 4A), indicating that JNK2 was required for TNF-induced activation of downstream caspases.
JNK2 Acts at the Level of Caspase-8 Activation to Promote Bid Cleavage and Cytochrome c Release
In vitro JNK-dependent TNF toxicity from NF-κB inhibition in hepatocytes results from activation of the mitochondrial death pathway with release of cytochrome c into the cytoplasm (19, 20). Critical to induction of the mitochondrial death pathway is activation of the initiator caspase-8 that cleaves the proapoptotic Bcl-2 family member Bid (17, 45). To further define the mechanism by which JNK2 promotes the TNF apoptotic death pathway, the effects of JNK2 ablation on caspase-8 activation, Bid cleavage, and hepatic mitochondrial cytochrome c release in response to GalN/LPS were examined. At 6 h after treatment, caspase-8 cleavage was detected in wild-type animals as reflected in reduced procaspase levels and the appearance of the p43 cleavage product on immunoblots (Fig. 7A). Caspase-8 activation was associated with cytosolic cleavage of Bid to tBid (Fig. 7A). Caspase-8 activation and Bid cleavage were both blocked in jnk2−/− mice (Fig. 7A). Western blot findings of inhibition of GalN/LPS-induced caspase-8 activation in jnk2−/− mice were confirmed by a caspase-8 activity assay. The 2-fold increase in caspase-8 activity 5 h after GalN/LPS treatment in wild-type mice was completely blocked in jnk2−/− mice (Fig. 7B). In contrast to findings in jnk2 null mice, caspase-8 and Bid cleavage occurred in jnk1−/− mice (Fig. 7C).
FIGURE 7. Ablation of jnk2 but not jnk1 inhibits caspase-8 activation, Bid cleavage, and mitochondrial cytochrome c release.

A, the cytosolic protein fraction was isolated from the livers of wild-type (+/+) and jnk2 knock-out (−/−) mice that were untreated or treated with GalN/LPS for 6 h. Samples were immunoblotted with antibodies against caspase-8, Bid, and β-actin. The top two panels represent different exposures of the same caspase-8 immunoblot. B, levels of capsase-8 activity in untreated control (Con) and 5-h GalN/LPS-treated wild-type (WT) and jnk2−/− animals (*, p < 0.02 compared with wild-type mice). C, immunoblots of cytosolic protein fractions from untreated or 6-h GalN/LPS-treated wild-type and jnk1−/− mice for caspase-8, Bid, and β-actin. D and E, mitochondrial protein fractions were isolated from the same livers as for the cytosolic fractions from wild-type, jnk2 (D), and jnk1 (E) knock-out mice and immunoblotted with antibodies against Bid, cytochrome c (cyto c), and cytochrome oxidase (cyto ox). F, total hepatic protein was isolated from wild-type (+/+) or jnk2 (−/−) knock-out mice that were untreated or treated with GalN/LPS for the indicated number of hours. Western blot analysis was performed with antibodies directed against TRAF-2, c-FLIP, Bax, Bad, phospho-Bad (P-Bad), Bcl-XL, cellular inhibitor of apoptosis 1 (cIAP1) and 2 (cIAP2), and β-actin. Arrows in A, C, D, and E indicate procaspase-8 and its p43 cleavage product, full size Bid and its cleaved form tBid, and the extra long (EL) and long (L) forms of Bim. Findings are representative of three independent experiments.
Bid cleavage leads to its translocation to the mitochondria where it induces release of proapoptotic factors such as cytochrome c (18). Immunoblots of mitochondrial protein isolates from wild-type mice demonstrated that GalN/LPS treatment induced tBid translocation to the mitochondria in association with loss of cytochrome c (Fig. 7D). No translocation of JNK or phospho-JNK was detected (data not shown). Translocation of tBid and cytochrome c loss did not occur in jnk2 null mice (Fig. 7D). As expected, tBid translocated to the mitochondria in jnk1−/− mice, resulting in cytochrome c release (Fig. 7E). No differential effect on the mitochondrial levels of other proapoptotic Bcl-2 family members occurred among the three types of mice. Low levels of Bax and Bim were present in the mitochondria of all mice and were unchanged with GalN/LPS treatment (Fig. 7, D and E). All mice had significant levels of Bad that decreased markedly with GalN/LPS treatment (Fig. 7, D and E). Equivalent levels of cytochrome oxidase demonstrated equal loading of mitochondrial protein among samples (Fig. 7, D and E). JNK2 but not JNK1 is, therefore, critical for caspase-8 activation, Bid cleavage, and activation of the mitochondrial death pathway.
To further validate the finding that JNK2 acts at the level of caspase-8/Bid, the effect of jnk2 expression on cytosolic modifiers of TNF-induced apoptosis was examined by immunoblotting. Loss of jnk2 had no effect on the steady-state expression of other pro- and anti-apoptotic proteins in the TNF death pathway including TRAF2, c-FLIPL, and Bcl-2 family members and the inhibitor of apoptosis proteins (IAP) (Fig. 7F). The only effect of GalN/LPS on expression of these proteins was to increase levels of phospho-Bad in both wild-type and jnk2−/− mice (Fig. 7F).
jnk2 Ablation Prevents Liver Injury and Caspase Activation from GalN/TNF
As further evidence of the proapoptotic function of JNK2 in hepatocyte death from TNF, the effect of jnk2 on liver injury from GalN/TNF was examined. Similar to the findings for GalN/LPS-induced liver injury, jnk2−/− mice had markedly reduced levels of liver injury from GalN/TNF in comparison to wild-type mice. ALT levels (Fig. 8A), the degree of liver injury and inflammation by histology (Fig. 8B), and the numbers of TUNEL-positive cells (Fig. 8C) were all significantly decreased in jnk2−/− mice. GalN/TNF treatment induced activation of caspase-8, -3, and -7 and cleavage of PARP in wild-type but not jnk2−/− mice (Fig. 8D).
FIGURE 8. jnk2−/− mice are protected from GalN/TNF-induced liver injury.

A, serum ALT levels from wild-type (WT) and jnk2−/− mice at 6 h after GalN/TNF treatment (*, p < 0.04 as compared with wild type). B, graded histological scoring of the degree of apoptosis (Apop) and inflammation (Inflam) in the same livers (p < 0.01 (*) and p < 0.02 (#) compared with wild type). C, numbers of TUNEL positive cells per high power field in the identical liver sections (*, p < 0.002 compared with wild-type mice). D, immunoblots of total protein isolated from the livers of wild-type (+/+) and jnk2 knock-out (−/−) mice that were untreated or treated with GalN/TNF for 6 h. Samples were immunoblotted with antibodies against caspase-8, -3, and -7, PARP, and β-actin. The top two panels represent different exposures of the same caspase-8 immunoblot. Results are from three independent experiments with three-five animals per data point.
Loss of jnk1 but Not jnk2 Inhibits Hepatic c-Jun Kinase Activity
A potential explanation for the distinct functions of the JNK1 and JNK2 isoforms in liver injury could be their differences in c-Jun kinase activity. Cell culture studies have suggested that JNK1 isoforms exhibit classic JNK kinase activity as defined by phosphorylation of c-Jun, whereas the JNK2 forms lack this activity (33, 34). However, it is unknown whether JNK1 and JNK2 function similarly in hepatocytes or in the liver in vivo. To assess whether JNK1 and JNK2 had differential effects on GalN/LPS-induced c-Jun kinase activation, JNK activity was determined by in vitro kinase assay in the two knock-out animals. GalN/LPS-induced c-Jun kinase activity was completely blocked in jnk1−/− mice (Fig. 9A). In contrast, the increase in c-Jun kinase activity induced by GalN/LPS was equivalent in wild-type and jnk2 null mice (Fig. 9B), indicating that hepatic JNK2 had no c-Jun kinase activity. Thus, consistent with previous findings in nonhepatic, in vitro systems, c-Jun kinase activity in the liver was exclusively dependent on JNK1 and independent of JNK2. Both the inability of jnk1 ablation to prevent liver injury despite effective inhibition of c-Jun kinase activity and the effectiveness of loss of jnk2 in blocking liver injury in the absence of any decrease in JNK activity support the conclusion that hepatocellular injury is promoted by JNK2 through a mechanism unrelated to the classical c-Jun kinase activity of this protein.
FIGURE 9. GalN/LPS-induced JNK c-Jun kinase activity is jnk1- but not jnk2-dependent.

Hepatic JNK activity was determined in wild-type (+/+) and jnk1 (A) or jnk2 (B) knock-out (−/−) mice that were untreated or treated with GalN/LPS for the number of hours shown. JNK activity is reflected in the levels of phosphorylated c-Jun (P-c-Jun) on immunoblots. Stripped membranes were immunoblotted for total c-Jun to indicate equal protein loading.
DISCUSSION
Recent in vitro studies in several cell types including hepatocytes have implicated sustained activation of the JNK signaling pathway as a mechanism of TNF-induced apoptosis (22–24, 46). However, the relevance of this finding to TNF-mediated injury in vivo, the JNK isoform(s) involved, and the mechanism of this proapoptotic JNK effect remain unclear. The present investigations employed the in vivo model of TNF-dependent liver injury induced by GalN/LPS to examine these questions. In this model the toxin GalN sensitizes the liver to LPS-induced injury that results from TNF-dependent apoptosis mediated by TNFR1 (36, 37). Administration of a nontoxic dose of LPS alone induced a transient hepatic activation of JNK as determined by both Western blot analysis for levels of phosphorylated JNK and an in vitro activity assay using c-Jun as substrate. In contrast, the toxic combination of GalN and LPS was associated with a sustained activation of JNK. These results mirror those from studies in cultured hepatocytes in which NF-κB inhibition similarly led to over-activation of JNK (23, 47). JNK over-activation in vivo may be secondary to GalN-induced inhibition of the expression of NF-κB-dependent genes, although this possibility was not examined in the present study. Recent investigations in non-hepatic cells have indicated that NF-κB down-regulates JNK activation by limiting oxidative stress that otherwise inhibits the activity of phosphatases (48). A similar mechanism may promote the over-activation of JNK in GalN/LPS liver injury.
To test the mechanistic involvement of JNK in toxic liver injury, the extent of hepatic injury from GalN/LPS was examined in jnk1−/− and jnk2−/− mice. Loss of jnk1 failed to alter liver injury from GalN/LPS. However, hepatic injury was markedly decreased in jnk2−/− mice as indicated by reductions in serum ALT and the number of TUNEL-positive cells, improved histology, and increased survival. These results demonstrate a dramatic, differential function of the JNK1 and JNK2 isoforms in toxic liver injury. Findings of isoform-specific JNK effects are consistent with recent in vitro investigations, suggesting that the jnk1 and jnk2 gene products have distinct functions (29–32), although the findings of these studies have been contradictory. In vivo studies in non-hepatic tissues have demonstrated specific effects of JNK2 in promoting inflammatory injury in the pancreas yet protecting against arthritis in joints (30, 49). For TNF toxicity, prior investigations in murine embryonic fibroblasts have supported the promotion of TNF toxicity by both selective jnk1 and jnk2 expression (33, 35). In immune-mediated liver injury induced by the T cell activator concanavalin A, Maeda et al. (50) demonstrated that liver injury was decreased in both jnk1 and jnk2 null mice. These findings together with those in organs other than liver suggest that the functions of JNK1 and JNK2 in cell death pathways are complex and likely to differ depending on the cell type and nature of the death stimulus. The complexity of JNK signaling particularly during in vivo injury is illustrated by the recent findings of Kaiser et al. (51), which show that, surprisingly, the activation or suppression of either the JNK1 or JNK2 isoforms induced protection against ischemic cardiac damage.
The mechanism of JNK2 promotion of GalN-induced liver injury could not be explained by a pro-inflammatory effect as the absence of jnk2 failed to affect the expression of TNF or other cytokines. Injury induced by GalN and TNF directly rather than LPS was also blocked in jnk2−/− mice. These data suggested a possible direct effect of JNK2 in promotion of the hepatocyte TNF death pathway. Despite normal hepatic TNFR1 expression and TNF production in GalN/LPS-treated jnk2−/− mice, the apoptotic TNF death receptor signaling cascade was blocked, as indicated by the failure of caspase-3 and -7 activation or PARP cleavage to occur. JNK2 performed an essential proapoptotic function above the level of Bid activation, as cleavage of Bid was markedly reduced in jnk2 knock-out mice. As a result, mitochondrial release of cytochrome c failed to occur in these animals. These findings are consistent with prior investigations in non-hepatic cell lines that have implicated JNK in the promotion of the mitochondrial death pathway by several apoptotic stimuli (52–56) including TNF (23, 46). However, prior investigations attributed the mechanism of this JNK effect to phosphorylation events that activated the proapoptotic Bcl-2 family members Bad or Bax (55, 56) or to the ability of JNK to translocate to the mitochondria (53). In contrast, the present studies demonstrate a specific effect of hepatic JNK2 on Bid cleavage and not other Bcl-2 family members. Mitochondrial levels of Bax, Bim, and Bad were equivalent in wild-type and jnk2−/− mice. In contrast to a previous report of Bax mitochondrial translocation after GalN/LPS treatment (18), an increase in mitochondrial Bax was not detected in our studies. However, Bax does not appear to regulate TNF-dependent liver injury, as the same study demonstrated no decrease in GalN/LPS injury in bax null mice (18). Mitochondrial Bad levels fell in parallel with increased levels of cytosolic phospho-Bad, consistent with previous in vitro studies in which Bad phosphorylation led to its translocation to the cytoplasm and inactivation (57, 58). JNK2 had no effect on Bad translocation, as mitochondrial levels of Bad were equivalent in wild-type and jnk2−/− mice.
The findings that JNK2 acts above the level of Bid cleavage and apparently functions in the cytosol, as it failed to translocate to mitochondria, suggested that JNK2 affected caspase-8 activation. Western blots and caspase-8 activity assay confirmed that in the absence of jnk2, caspase-8 activation was blocked. This finding suggests for the first time that JNK2 regulates the activation of this initiator caspase. JNK2 could function by activating a protein that promotes caspase-8 autoactivation or by down-regulating an inhibitor of caspase-8 activation. JNK2 failed to affect two likely candidate proteins, TRAF2 and c-FLIPL, and the JNK2 target remains to be identified. Alternatively, but seemingly less likely, caspase-8 inhibition may have been the indirect result of a lack of amplification by the mitochondrial release of proapoptotic factors.
Despite the profound effect of jnk2 ablation on early liver injury and hepatocellular apoptosis, the absence of jnk2 did not completely block GalN/LPS-induced mortality. A simple explanation for this finding may be that proinflammatory cytokine induction was unaffected, and the profound systemic effects of these factors and not liver injury led to death in some animals. Consistent with this possibility is the finding that jnk2−/− mice surviving for 24–72 h after GalN/LPS treatment had amounts of liver injury that were incompatible with death from liver failure. Alternatively, JNK2 may be essential for the Bid-mitochondrial death pathway, but in its absence, TNF-induced death occurs by other mechanisms. However, GalN/LPS-induced lethality was not prevented but, rather, was only delayed several hours in bid−/− mice on a C57BL/6 background, whereas long-term survival similar to our jnk2−/− mice was obtained in knock-out mice on a 129/SvJ background (18). The greater reduction in mortality in our study raises the possibility that JNK2 has other proapoptotic effects independent of the Bid-mitochondrial death pathway. Alternatively the difference between the findings in bid−/− and jnk2−/− mice is that in the jnk2−/− mice prevention of caspase-8 activation led to a more profound inhibition of the TNF death pathway rather than simple interference with the Bid-mitochondrial amplification of TNF death signaling. The final question raised by the incomplete prevention of GalN/LPS-induced mortality by JNK2 ablation is whether combined inhibition of both JNK2 and JNK1 would be more effective in preventing death. It is inconceivable that JNK1 plays a role in the early phase of liver injury, as its loss had no effect on injury over the initial 6 h. However, jnk1 may promote delayed liver injury by inducing alternative TNF death pathways or mediating lethal, systemic effects. Because the double jnk1/jnk2 knock-out is lethal, alternative, non-genetic approaches will have to address this question.
The findings suggest that JNK2 might provide a unique therapeutic target in the prevention and treatment of toxic liver injury. TNF-dependent liver injury has been implicated in a number of forms of toxic liver injury including that from hepatotoxins relevant to human disease such as alcohol (59). Despite strong evidence of the mechanistic involvement of TNF, attempts at mitigating human alcoholic liver disease with TNF inhibition have been disappointing (60). This failure may represent the fact that TNF exerts beneficial effects on liver regeneration and immune function, and global TNF inhibition eliminates these beneficial functions as well as apoptotic signaling cascades. Selective targeting of signaling factors such as JNK2 that specifically mediate only the toxic effects of TNF may provide a more effective approach to the treatment of these diseases.
Acknowledgment
The anti-Bid antibody was kindly provided by Xiao-Ming Yin.
Footnotes
This work was supported by National Institutes of Health Grant DK044234.
The abbreviations used are: TNF, tumor necrosis factor; TNFR1, TNF receptor 1; ALT, alanine aminotransferase; GalN, galactosamine; JNK, c-Jun N-terminal kinase; LPS, lipopolysaccharide; PARP, poly(ADP-ribose) polymerase; RT, reverse transcription; TUNEL, terminal deoxynucleotide transferase-mediated deoxyuridine triphosphate nick end-labeling; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid; TRAF, TNF receptor-associated factor; cIAP, cellular inhibitor of apoptosis.
REFERENCES
- 1.Czaja MJ, Xu J, Alt E. Gastroenterology. 1995;108:1849–1854. doi: 10.1016/0016-5085(95)90149-3. [DOI] [PubMed] [Google Scholar]
- 2.Yin M, Wheeler MD, Kono H, Bradford BU, Gallucci RM, Luster MI, Thurman RG. Gastroenterology. 1999;117:942–952. doi: 10.1016/s0016-5085(99)70354-9. [DOI] [PubMed] [Google Scholar]
- 3.Teoh N, Field J, Sutton J, Farrell G. Hepatology. 2004;39:412–421. doi: 10.1002/hep.20035. [DOI] [PubMed] [Google Scholar]
- 4.Zhou W, Zhang Y, Hosch MS, Lang A, Zwacka RM, Engelhardt JF. Hepatology. 2001;33:902–914. doi: 10.1053/jhep.2001.23073. [DOI] [PubMed] [Google Scholar]
- 5.Su F, Schneider RJ. Proc. Natl. Acad. Sci. U. S. A. 1997;94:8744–8749. doi: 10.1073/pnas.94.16.8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kallinowski B, Haseroth K, Marinos G, Hanck C, Stremmel W, Theilmann L, Singer MV, Rossol S. Clin. Exp. Immunol. 1998;111:269–277. doi: 10.1046/j.1365-2249.1998.00469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lesage G, Glaser S, Ueno Y, Alvaro D, Baiocchi L, Kanno N, Phinizy JL, Francis H, Alpini G. Am. J. Physiol. Gastrointest. Liver Physiol. 2001;281:182–190. doi: 10.1152/ajpgi.2001.281.1.G182. [DOI] [PubMed] [Google Scholar]
- 8.Yerushalmi B, Dahl R, Devereaux MW, Gumpricht E, Sokol RJ. Hepatology. 2001;33:616–626. doi: 10.1053/jhep.2001.22702. [DOI] [PubMed] [Google Scholar]
- 9.Morikawa A, Sugiyama T, Kato Y, Koide N, Jiang GZ, Takahashi K, Tamada Y, Yokochi T. Infect. Immun. 1996;64:734–738. doi: 10.1128/iai.64.3.734-738.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leist M, Gantner F, Bohlinger I, Germann PG, Tiegs G, Wendel A. J. Immunol. 1994;153:1778–1788. [PubMed] [Google Scholar]
- 11.Xu Y, Jones BE, Neufeld DS, Czaja MJ. Gastroenterology. 1998;115:1229–1237. doi: 10.1016/s0016-5085(98)70095-2. [DOI] [PubMed] [Google Scholar]
- 12.Iocca HA, Isom HC. Am. J. Pathol. 2003;163:465–476. doi: 10.1016/s0002-9440(10)63676-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akerman P, Cote P, Yang SQ, McClain C, Nelson S, Bagby GJ, Diehl AM. Am. J. Physiol. 1992;263:G579–G585. doi: 10.1152/ajpgi.1992.263.4.G579. [DOI] [PubMed] [Google Scholar]
- 14.Wajant H, Pfizenmaier K, Scheurich P. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 15.Micheau O, Tschopp J. Cell. 2003;114:181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 16.Guicciardi ME, Miyoshi H, Bronk SF, Gores GJ. Am. J. Pathol. 2001;159:2045–2054. doi: 10.1016/s0002-9440(10)63056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yin XM. J. Mol. Med. 2000;78:203–211. doi: 10.1007/s001090000099. [DOI] [PubMed] [Google Scholar]
- 18.Zhao Y, Li S, Childs EE, Kuharsky DK, Yin XM. J. Biol. Chem. 2001;276:27432–27440. doi: 10.1074/jbc.M102465200. [DOI] [PubMed] [Google Scholar]
- 19.Bradham CA, Qian T, Streetz K, Trautwein C, Brenner DA, Lemasters JJ. Mol. Cell. Biol. 1998;18:6353–6364. doi: 10.1128/mcb.18.11.6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Bialik S, Jones BE, Iimuro Y, Kitsis RN, Srinivasan A, Brenner DA, Czaja MJ. Am. J. Physiol. 1998;275:C1058–C1066. doi: 10.1152/ajpcell.1998.275.4.C1058. [DOI] [PubMed] [Google Scholar]
- 21.Iimuro Y, Nishiura T, Hellerbrand C, Behrns KE, Schoonhoven R, Grisham JW, Brenner DA. J. Clin. Investig. 1998;101:802–811. doi: 10.1172/JCI483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, Franzoso G. Nature. 2001;414:308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- 23.Liu H, Lo CR, Czaja MJ. Hepatology. 2002;35:772–778. doi: 10.1053/jhep.2002.32534. [DOI] [PubMed] [Google Scholar]
- 24.Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M, Lin A. Nature. 2001;414:313–317. doi: 10.1038/35104568. [DOI] [PubMed] [Google Scholar]
- 25.Davis RJ. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 26.Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA. Science. 1998;282:2092–2095. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- 27.Yang DD, Conze D, Whitmarsh AJ, Barrett T, Davis RJ, Rincon M, Flavell RA. Immunity. 1998;9:575–585. doi: 10.1016/s1074-7613(00)80640-8. [DOI] [PubMed] [Google Scholar]
- 28.Sabapathy K, Jochum W, Hochedlinger K, Chang L, Karin M, Wagner EF. Mech. Dev. 1999;89:115–124. doi: 10.1016/s0925-4773(99)00213-0. [DOI] [PubMed] [Google Scholar]
- 29.Conze D, Krahl T, Kennedy N, Weiss L, Lumsden J, Hess P, Flavell RA, Le Gros G, Davis RJ, Rincon M. J. Exp. Med. 2002;195:811–823. doi: 10.1084/jem.20011508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaeschke A, Rincon M, Doran B, Reilly J, Neuberg D, Greiner DL, Shultz LD, Rossini AA, Flavell RA, Davis RJ. Proc. Natl. Acad. Sci. U. S. A. 2005;102:6931–6935. doi: 10.1073/pnas.0502143102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.She QB, Chen N, Bode AM, Flavell RA, Dong Z. Cancer Res. 2002;62:1343–1348. [PubMed] [Google Scholar]
- 32.Qiao L, Han SI, Fang Y, Park JS, Gupta S, Gilfor D, Amorino G, Valerie K, Sealy L, Engelhardt JF, Grant S, Hylemon PB, Dent P. Mol. Cell. Biol. 2003;23:3052–3066. doi: 10.1128/MCB.23.9.3052-3066.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu J, Minemoto Y, Lin A. Mol. Cell. Biol. 2004;24:10844–10856. doi: 10.1128/MCB.24.24.10844-10856.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sabapathy K, Hochedlinger K, Nam SY, Bauer A, Karin M, Wagner EF. Mol. Cell. 2004;15:713–725. doi: 10.1016/j.molcel.2004.08.028. [DOI] [PubMed] [Google Scholar]
- 35.Dietrich N, Thastrup J, Holmberg C, Gyrd-Hansen M, Fehrenbacher N, Lademann U, Lerdrup M, Herdegen T, Jaattela M, Kallunki T. Cell Death Differ. 2004;11:301–313. doi: 10.1038/sj.cdd.4401353. [DOI] [PubMed] [Google Scholar]
- 36.Leist M, Gantner F, Bohlinger I, Tiegs G, Germann PG, Wendel A. Am. J. Pathol. 1995;146:1220–1234. [PMC free article] [PubMed] [Google Scholar]
- 37.Nowak M, Gaines GC, Rosenberg J, Minter R, Bahjat FR, Rectenwald J, MacKay SL, Edwards CK, III, Moldawer LL. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000;278:1202–1209. doi: 10.1152/ajpregu.2000.278.5.R1202. [DOI] [PubMed] [Google Scholar]
- 38.Schattenberg JM, Wang Y, Rigoli RM, Koop DR, Czaja MJ. Hepatology. 2004;39:444–455. doi: 10.1002/hep.20067. [DOI] [PubMed] [Google Scholar]
- 39.Liu H, Lo CR, Jones BE, Pradhan Z, Srinivasan A, Valentino KL, Stockert RJ, Czaja MJ. J. Biol. Chem. 2000;275:40155–40162. doi: 10.1074/jbc.M001565200. [DOI] [PubMed] [Google Scholar]
- 40.Livak KJ, Schmittgen TD. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 41.Zender L, Hutker S, Liedtke C, Tillmann HL, Zender S, Mundt B, Waltemathe M, Gosling T, Flemming P, Malek NP, Trautwein C, Manns MP, Kuhnel F, Kubicka S. Proc. Natl. Acad. Sci. U. S. A. 2003;100:7797–7802. doi: 10.1073/pnas.1330920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim WH, Hong F, Radaeva S, Jaruga B, Fan S, Gao B. Am. J. Physiol. Gastrointest. Liver Physiol. 2003;285:761–768. doi: 10.1152/ajpgi.00224.2003. [DOI] [PubMed] [Google Scholar]
- 43.Malagarie-Cazenave S, Segui B, Leveque S, Garcia V, Carpentier S, Altie MF, Brouchet A, Gouaze V, Andrieu-Abadie N, Barreira Y, Benoist H, Levade T. J. Biol. Chem. 2004;279:18648–18655. doi: 10.1074/jbc.M314294200. [DOI] [PubMed] [Google Scholar]
- 44.Jaeschke H, Fisher MA, Lawson JA, Simmons CA, Farhood A, Jones DA. J. Immunol. 1998;160:3480–3486. [PubMed] [Google Scholar]
- 45.Werneburg NW, Guicciardi ME, Bronk SF, Gores GJ. Am. J. Physiol. Gastrointest. Liver Physiol. 2002;283:947–956. doi: 10.1152/ajpgi.00151.2002. [DOI] [PubMed] [Google Scholar]
- 46.Deng Y, Ren X, Yang L, Lin Y, Wu X. Cell. 2003;115:61–70. doi: 10.1016/s0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 47.Schwabe RF, Uchinami H, Qian T, Bennett BL, Lemasters JJ, Brenner DA. FASEB J. 2004;18:720–722. doi: 10.1096/fj.03-0771fje. [DOI] [PubMed] [Google Scholar]
- 48.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 49.Han Z, Chang L, Yamanishi Y, Karin M, Firestein GS. Arthritis Rheum. 2002;46:818–823. doi: 10.1002/art.10104. [DOI] [PubMed] [Google Scholar]
- 50.Maeda S, Chang L, Li ZW, Luo JL, Leffert H, Karin M. Immunity. 2003;19:725–737. doi: 10.1016/s1074-7613(03)00301-7. [DOI] [PubMed] [Google Scholar]
- 51.Kaiser RA, Liang Q, Bueno O, Huang Y, Lackey T, Klevitsky R, Hewett TE, Molkentin JD. J. Biol. Chem. 2005;280:32602–32608. doi: 10.1074/jbc.M500684200. [DOI] [PubMed] [Google Scholar]
- 52.Chauhan D, Li G, Hideshima T, Podar K, Mitsiades C, Mitsiades N, Munshi N, Kharbanda S, Anderson KC. J. Biol. Chem. 2003;278:17593–17596. doi: 10.1074/jbc.C300076200. [DOI] [PubMed] [Google Scholar]
- 53.Eminel S, Klettner A, Roemer L, Herdegen T, Waetzig V. J. Biol. Chem. 2004;279:55385–55392. doi: 10.1074/jbc.M405858200. [DOI] [PubMed] [Google Scholar]
- 54.Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ. Science. 2000;288:870–874. doi: 10.1126/science.288.5467.870. [DOI] [PubMed] [Google Scholar]
- 55.Tsuruta F, Sunayama J, Mori Y, Hattori S, Shimizu S, Tsujimoto Y, Yoshioka K, Masuyama N, Gotoh Y. EMBO J. 2004;23:1889–1899. doi: 10.1038/sj.emboj.7600194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu C, Minemoto Y, Zhang J, Liu J, Tang F, Bui TN, Xiang J, Lin A. Mol. Cell. 2004;13:329–340. doi: 10.1016/s1097-2765(04)00028-0. [DOI] [PubMed] [Google Scholar]
- 57.Harada H, Andersen JS, Mann M, Terada N, Korsmeyer SJ. Proc. Natl. Acad. Sci. U. S. A. 2001;98:9666–9670. doi: 10.1073/pnas.171301998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 59.Schattenberg JM, Czaja MJ. Signaling Pathways in Liver Diseases. Springer-Verlag; Berlin: 2005. pp. 115–128. [Google Scholar]
- 60.Naveau S, Chollet-Martin S, Dharancy S, Mathurin P, Jouet P, Piquet MA, Davion T, Oberti F, Broet P, Emilie D. Hepatology. 2004;39:1390–1397. doi: 10.1002/hep.20206. [DOI] [PubMed] [Google Scholar]