

SUMMARY
The crystal structure of Complexin bound to a pre-fusion SNAREpin mimetic shows that the accessory helix extends away from the SNAREpin in an “open” conformation, binding another SNAREpin, and inhibiting its assembly to clamp fusion. In contrast, the accessory helix in the post-fusion complex parallels the SNARE complex in a “closed” conformation. Here we use targeted mutations, FRET spectroscopy, and a functional assay that reconstitutes Ca2+-triggered exocytosis to show that the conformational switch from open to closed in Complexin is needed for Synaptotagmin-Ca2+ to trigger fusion. Triggering fusion requires the zippering of three key Asp residues in the “switch” region (64–68) of v-SNARE. Conformational switching in Complexin is integral to clamp release and is likely triggered when its accessory helix is released from its trans-binding to the neighboring SNAREpin, allowing the v-SNARE to complete zippering and open a fusion pore.
SNARE proteins are the core machinery driving membrane fusion between cargo-carrying vesicles and their target membranes1–4 as v-SNAREs (anchored in the vesicle membrane) zipper into a coiled-coil four helix bundle5 with cognate t-SNAREs (anchored in the target membrane). In neuronal synapses, the principal SNAREs responsible for neurotransmitter release are the v-SNARE, VAMP2, localized on the synaptic vesicle, and the t-SNARE, a binary complex of SNAP25 with Syntaxin1, localized on the pre-synaptic plasma membrane. VAMP2 and Syntaxin1 each contribute one helix to the coiled-coil, and SNAP25 contributes the other two. Fusion by the isolated synaptic SNAREs is rapid and spontaneous6, implying that in the synapse, additional protein machinery is needed to arrest exocytosis until the secretion signal is provided by the entry of calcium ions.
One such protein is the calcium- and SNARE-binding protein Synaptotagmin7,8, which is the immediate sensor for synchronous vesicle fusion9–11. The SNARE complex binding protein Complexin12,13 (CPX) is equally required for synaptic transmission, functioning both positively as an activator of fusion and negatively as a clamp, to prevent fusion prior to the calcium signal14–19.
In the preceding manuscript, we established the precise nature of the clamped state using X-ray crystallography together with confirmatory solution and functional studies20. To capture this intermediate, we used a SNARE complex containing a C-terminally truncated VAMP (termed VAMP2-60) to mimic the half-zippered v-SNARE that is trapped in the clamped state14,18,21–23. In the structure, hCPX binds one partially-zippered SNARE complex via its central helix (CPXcen, residues 48–75), while its accessory helix (CPXacc, residues 26–48) extends away from this SNARE complex at ~45° to bridge to a second SNAREpin, binding its C-terminal three helix bundle so as to sterically block that SNAREpin’s own v-SNARE from completing its zippering. As a result of repeating these trans-interactions, the clamped SNAREpins are cross-linked into a rigid zig-zag array20, which is itself topologically incompatible with the opening of a fusion pore.
In this paper, we address the question: how is fusion switched “on” from the clamped state? A novel feature of the pre-fusion mimetic structure is that in the clamped state, CPXacc angles away from the SNAREpin to which its central helix is attached, a conformation that we refer to as “open” (Fig. 1a, cyan). This differs markedly from that observed in the fully-zippered post-fusion structure, where the CPXacc nearly parallels the SNARE complex24,25 which we refer to as “closed” (Fig. 1a, light cyan). Taken together, the two structures suggest that CPXacc undergoes a dramatic re-orientation as part of the mechanism (requiring calcium binding to Synaptotagmin) that switches fusion “on” from the clamped state. In this paper, we show that the movement from the open to closed state is a molecular switch required to activate fusion from the clamped state. We report that three closely clustered Aspartate residues (positions 64, 65, and 68) in the C-terminal half of VAMP2 (termed the “Asp switch region”) must zipper into the t-SNARE for CPX to move its accessory helix from the open (clamped) to the closed (post-fusion) conformation. Mutating these residues inhibits activation of fusion from the clamped state by destabilizing the open conformation.
Figure 1.

Zippering of one turn of VAMP2 helix triggers the switch in CPX position (a) Superposition of the structures of the pre- and post-fusion CPX-SNARE complexes20,24, showing the FRET label positions. Syntaxin1 (residues 190–250) is in yellow, SNAP 25 N-terminal SNARE motif (residues 10–74) is in lime, SNAP 25 C-terminal SNARE motif (residues 141–203) is in green and VAMP2 is in blue (residues 25–60 dark blue; residues 61–96 in light blue). The “switch” residues (Asp 64, Asp 65 and Asp 68) are marked in red. The Complexin (residues 26–73) in the pre-fusion complex is in cyan, while in the post-fusion complex is in light cyan. The FRET label positions, residue 193 on SNAP25 and 38 on Complexin, are marked in magenta. The sequence of the C-terminal hydrophobic layer of VAMP2 (residues 57–90) with the C-terminal truncations tested in this paper (denoted by the residue number) is also shown. The black arrow in the post-fusion structure references CPX residue 48, the demarcation line between CPXcen and CPXacc. (b) FRET experiments with C-terminal truncations of VAMP2. Fluorescence emission spectra of Stilbene and Bimane labeled CPX-SNARE complexes containing VAMP2-60 (residues 25–60, orange), VAMP2-65 (residues 25–65, green), VAMP2-69 (residues 25–69, purple), VAMP2-73 (residues 25–73, blue), VAMP2-77 (residues 25–77, red), and VAMP2 (residues 1–96, olive). A representative emission spectrum of a Stilbene (Donor)-only CPX-SNARE complex is shown in black. The donor-only spectrum was identical in all CPX-SNARE complexes
RESULTS
Zippering One Turn of VAMP2 Helix Triggers the CPX Switch
We use here a Förster Resonance Energy Transfer (FRET) with a Stilbene-Bimane donor-acceptor pair to monitor the conformational state of CPXacc in CPX-SNARE complexes20. In this assay, residue 193 of SNAP25 is labeled with the donor dye (Stilbene), and the acceptor dye (Bimane) is attached at residue 38 of CPXacc (Fig. 1a). This pair was chosen because of its sensitivity to changes in separations in the range of interest. When CPXacc adopts the open conformation, the fluorescent probes are far apart, resulting in low FRET, but when CPXacc moves to the closed conformation, the FRET probes are close together, resulting in a larger FRET signal. Distances obtained in solution from FRET analysis of VAMP2 constructs used in crystallization (VAMP2-60 or VAMP2) were in excellent agreement with the distances observed in the respective crystals (Table 1).
Table 1.
FRET distances were determined from quenching of donor fluorescence between SNAP25 Asp193 and CPX Gln38 in CPX-SNARE complexes with VAMP2 deletions and mutants. The distances measured in the pre-fusion and post-fusion crystal structures20,24 are given for comparison. Standard deviations are reported from n = 4–6 independent experiments and they reflect the reproducibility of the spectra rather than accuracy of the distance measurements.
| Constructs | Distance between SNAP25 Asp193 and CPX Gln38 (Å) |
|---|---|
| VAMP2 Deletions | |
| VAMP2-60 | 34 ± 1 Å |
| VAMP2-65 | 33 ± 2 Å |
| VAMP2-69 | 34 ± 1 Å |
| VAMP2-73 | 21 ± 2 Å |
| VAMP2-77 | 24 ± 1 Å |
| VAMP2 and VAMP2 Mutants (Residues 1-96) | |
| VAMP2 | 20 ± 1 Å |
| VAMP-4X | 33 ± 1 Å |
| VAMP-3xDA | 32 ± 3 Å |
| Measured in Crystal Structure | |
| Pre-fusion (VAMP2-60) | 28 Å |
| Post-fusion (VAMP2) | 18 Å |
This FRET assay allows us to distinguish the open conformation (complexes with VAMP2 truncated at residue 60) from the closed conformation (complexes in which the cytoplasmic domain of VAMP is nearly complete, truncated at residue 96) (Fig. 1b). To explore what happens in complexes with VAMP2 truncations in between these two extremes, we assembled full length CPX-SNARE complexes with the VAMP2 C-terminus progressively extended beyond residue 60 (VAMP2-65, VAMP2-69, VAMP2-73 and VAMP2-77) to mimic the progressive zippering of the VAMP2 C-terminus.
The FRET spectra show that CPXacc adopts the open conformation in truncated CPX-SNARE complexes when the VAMP2 C-terminus was extended to the +2 (VAMP2-65) or +3 (VAMP2-69) hydrophobic layers (Fig. 1b). In these complexes, we observe low FRET efficiency, and the distance between the probes as measured by quenching of the donor fluorescence is consistent with the distances observed in the crystal structure of the CPX-SNARE-60 complex (Fig. 1b and Table 1).
In contrast, when we extended the VAMP2 C-terminus only about one turn of the helix further, to the +4 hydrophobic layer (VAMP2-73) or beyond (VAMP2-77), we observed high FRET, quantitatively corresponding to the closed conformation, in which CPXacc runs parallel to the SNARE complex (Fig. 1b). In these complexes, the distance between donor and acceptor probes calculated from the FRET spectra is consistent with distances observed in the post-fusion CPX-SNARE structure (Table 1).
Remarkably, there is an all-or-none, discrete switch from open to closed conformation when one more turn of the helix is added between VAMP2 residues 69 and 73, without hybrid spectra of the two states. This shows that zippering of VAMP2 to at least the +4 hydrophobic layer is required for the switch in CPX conformation, indicating that a discrete ‘switch region’ is located in this stretch of VAMP2. The full-length “superclamp” CPX (scCPX) was used in these and later studies in this paper for consistency and direct comparability with the crystal structure, which also employed mutations in the CPXacc (D27L, E34F, R37A) that increase binding to the t-SNARE.
Asp residues in the Critical Region of VAMP2 Throw the Switch
In the post-fusion structure24, CPX binds the SNARE complex in the groove between VAMP2 and Syntaxin1, and the CPXcen makes three distinct contacts with VAMP2. Two of these interactions, a hydrophobic contact with VAMP2 V50,L54 and a salt-bridge with VAMP2 D57 are found in both the pre-fusion, clamped CPX-SNARE complex20 and the post-fusion fully-zippered CPX-SNARE complex24 structures. A third, distinct contact region involving VAMP2 residues Asp 64, Asp 65 and Asp 68 is present in the post-fusion complex only24 (Fig. 2a), because the VAMP C-terminus was truncated at residue 60 in the pre-fusion complex20. These three Asp residues are located within the +2 to +4 hydrophobic layers of VAMP2, which we identified (Fig. 1b) as the minimal ‘switch region’ required to switch CPXacc from the open to the closed conformation. They form hydrogen bonds and salt bridges to CPXcen which serve to anchor the accessory helix parallel to the four helix SNARE bundle in the closed, post-fusion conformation (Fig. 2a).
Figure 2.

Aspartate residues 64, 65 and 68 on VAMP2 mediate the switch in CPX position (a) Hydrogen bonding and salt bridge interactions between the switch Asp residues 64, 65, and 68 with CPXcen helix in the post-fusion complex24. (b) Complexin adopts an open conformation in CPX-SNARE complexes containing either VAMP-3xDA or VAMP-4X. Donor fluorescence at 410 nm (normalized to a Donor-only sample) for Stilbene-Bimane labeled CPX-SNARE complexes containing VAMP2-D64A, VAMP2-D65A, VAMP2-D68A, VAMP-3xDA, or VAMP-4X is shown. (The raw fluorescence emission curves are shown in Supplementary Fig. 3.) The donor fluorescence (at 410 nm) for VAMP2-60 (“open”) and VAMP2 (“closed”) are shown for comparison. Averages and standard deviations for 3–4 independent experiments are shown.
To test the hypothesis that these contacts are needed to stabilize the closed conformation, we mutated all three switch Aspartate residues to Alanines in an otherwise complete VAMP2 cytoplasmic domain (residues 1–96) containing all of the VAMP residues that assemble into the four helix SNARE bundle5, a construct referred to as VAMP-3xDA (VAMP2 with D64A, D65A, D68A mutations). As expected, VAMP-3xDA fully zippers into t-SNARE, as shown by resistance to cleavage by Tetanus and Botulinum-B neurotoxins (Supplementary Fig. 1a) and the fact that VAMP-3xDA (when produced in a full-length form, containing its membrane anchor) retains its capacity to mediate fusion with its cognate synaptic t-SNARE in the liposome fusion assay (Supplementary Fig. 1b).
As predicted by our hypothesis, CPXacc adopts the open conformation in the fully-zippered CPX-SNARE-3xDA complex, and its FRET spectrum corresponds closely to those of the pre-fusion mimetics of CPX-SNARE in which the switch region of VAMP is deleted (Fig. 2b and Table 1, raw data in Supplementary Fig. 2a). The consequences of mutating individual switch Aspartate residues reveal that all three are required for the full-switching of the CPXacc, as mutation of any of the three Asp residues (VAMP2-D64A, VAMP2-D65A and VAMP2-D68A) destabilizes the closed conformation (Fig. 2b, raw data in Supplementary Fig. 2b). Mutating residue 64 (VAMP2-D64A) has the maximum destabilizing effect, so it might act as the internal trigger for the switch (Fig. 2b).
Zippering of Asp Switch Residues Is Required for the Switch
So far, we know that CPXacc is trapped in the open conformation when the switch Asp’s are absent from VAMP (due either to point mutations or deletion). What about when the switch Asp’s are present but not able to zipper? This would correspond more closely to what occurs physiologically before these residues have zippered. To mimic this state, we employed a VAMP construct in which the normal switch Asp’s are present but which carries point mutations that prevent full zippering in the C-terminal (membrane-proximal) region of VAMP.
Specifically, we used the entire VAMP2 cytoplasmic domain (residues 1–96) and introduced mutations in its C-terminal half (L70D, A74R, A81D & L84D; termed VAMP-4X) that prevent assembly of this region with Syntaxin1 and SNAP25 and eliminate fusion activity (Supplementary Fig. 1b). However, the N-terminal half of VAMP-4X still zippers because VAMP-4X forms stable complexes with the t-SNAREs and CPX (Supplementary Fig. 3). The FRET spectrum of the CPX-SNARE-4X complex (Fig. 2b and Table 1, raw data in Supplementary Fig. 2a) is nearly identical to the open conformation, as present in CPX-SNARE complex with VAMP-60 or when the switch aspartates are mutated or deleted by truncations. (Fig. 2b and Table 1)
Importantly, this experiment establishes that the mere presence of the VAMP2 residues D64, D65 and D68 is not sufficient for switching: they must also be zippered with the t-SNARE in the helical bundle in order to throw the switch from open to closed. This strongly suggests that zippering of the switch region of the v-SNARE (i.e., progression of fusion beyond the clamped state) and movement of CPXacc from its open to its closed arrangement are thermodynamically coupled; i.e. one cannot occur without the other.
This could also explain why CPXacc adopts an open conformation in the CPX-VAMP-69 complex (Fig. 1b and Table 1). Even though the key residues required for the switch (D64, D65 and D68) are present in this complex, they are at the end of the truncated VAMP2, and they may not be properly zippered into the t-SNARE. Extending the VAMP2 C-terminus one rung on the helix to the next hydrophobic layer (VAMP2-73) could then allow the switch region to stably zipper into the t-SNARE, and switch CPXacc to the closed conformation (Fig. 1b and Table 1).
Thermodynamics of the CPX Conformational Switch
We used Isothermal Titration Calorimetry (ITC) to determine the energetics of the contributions of the switch Asp residues to the open-to-closed conformational switch. We compared the thermodynamics of the binding of CPX to SNARE complexes assembled with either VAMP2 or VAMP-3xDA (Fig. 3). Complexin binds the VAMP2 SNARE complex with 1:1 stoichiometry and high affinity (Kd = 83 nM). Mutating the switch Asp residues (VAMP-3xDA) does not alter the binding stoichiometry, but results in 88% reduction in the affinity (Kd = 670 nM) (Fig. 3 and Table 1), corresponding to a free energy difference of −1.3 kCal mole−1. The enthalpy of the interaction of CPX with the SNARE complex was greatly reduced, from −15 kCal mole−1 for the VAMP-3xDA SNARE complex to −37.5 kCal mole−1 for the wild-type VAMP2 sequence. So the difference in the enthalpy (ΔΔH = −22.5 kCal mole−1) is mainly from the interaction of CPXcen with the switch Asp residues on VAMP2.
Figure 3.

Interaction of CPXcen with Asp residues 64, 65 and 68 on VAMP2 provides thermodynamic driving force for the switch. Calorimetric titrations of superclamp CPX (scCPX; residues 1–134 with D27L, E34F, R37A mutations) into assembled SNARE complexes containing t-SNAREs and either wild-type (WT) VAMP2 (blue triangles), VAMP2-3xDA (red squares), or VAMP-3xDA with the CPXcen binding site blocked by CPX 48–134 (black circles). The solid lines represent the best fit to the corresponding data points using a nonlinear least squares fit with a one-set-of-sites model. The results of the fits are given in Table 2. All experiments were performed in triplicate at 37°C, and a representative thermogram is shown.
In a control experiment, when we block the CPXcen binding site on VAMP-3xDA SNARE complex by pre-binding CPXcen (residues 48–134), we see no further interaction with CPX (Fig. 3). We conclude that the interaction of CPX with the SNARE-3xDA complex is mediated solely by CPXcen. This observation confirms that SNARE-3xDA complex is fully zippered, since the additional binding site for CPXacc; i.e., the C-terminal t-SNARE groove, is not available.
The Switch in CPX is Required for Synaptotagmin to Trigger Fusion
To the extent that the open-to-closed conformational switch in CPX is needed to activate fusion from the clamped state, locking CPXacc in the open state will prevent activation of fusion and result in a persistent clamped state, which should inhibit activation of fusion by Synaptotagmin and calcium ions.
To test this, we used the ‘flipped’ SNARE system in which cells expressing either VAMP2 or Syntaxin1-SNAP25 proteins on their surface are mixed and the rate of cell-to-cell fusion is scored using light microscopy2. In this system, fusion occurs spontaneously unless CPX is added either as an exogenous pure protein or by endogenous gene expression and secretion. In the presence of CPX, fusion is blocked when the SNAREs are approximately half-zippered, as judged by the pattern of Botulinum and Tetanus neurotoxin resistance14. When Synaptotagmin is either added back to the medium (cytoplasmic domain only) or endogenously expressed as a flipped protein, fusion is then re-activated upon addition of Ca2+ ions14. The physiological relevance of this minimal system was established by several criteria14. For example, mutations in Synaptotagmin that alter calcium sensitivity in mice, correspondingly alter sensitivity in this reconstituted system, toxin sensitivity in the clamped state reproduces the pattern found at the neuromuscular junction and most recently, super-clamp mutations of CPXacc that increase clamping potency in neurons 26 also do so in this in vitro system15.
We tested the activation of fusion mediated by VAMP-3xDA (VAMP2 with D64A, 65A, 68A mutations) from the clamped state by Synaptotagmin and calcium and confirmed the prediction that clamp release would be impaired when the v-SNARE lacked the switch Asp residues (Fig. 4a, VAMP-3xDA, green bar). The limited extent of activation that was observed with VAMP-3xDA and wild-type CPX was similar to the essentially permanent clamped state that results when wild-type SNAREs are frozen with super-clamp CPX (Fig. 4a, scCPX, green bar). In the latter case, the open state is stabilized by stronger binding of CPXacc to the trans-t-SNARE, whereas in the former case it is the closed state that is de-stabilized by weaker binding of CPXcen to the SNARE bundle due to switch Asp mutations. As expected, mutating individual Asp residues in the switch region partially compromised activation in a manner reflecting their relative contributions to stabilizing the closed conformation measured by FRET (Fig. 2b).
Figure 4.

The switch in CPXacc position is necessary for Synaptotagmin-Ca2+ to trigger fusion. (a) Clamping of SNARE-mediated fusion by CPX and the reversal of the clamp by Synaptotagmin-Ca2+ in the presence of Phosphoinositol-Phospholipase C (PI-PLC) in wild-type (WT) VAMP2, VAMP2-D64A, VAMP2-D65A and VAMP-3xDA as measured in a cell-cell fusion assay. The effect of the superclamp CPX (scCPX; CPX D27L, E34F, R37A) on wild-type VAMP2 is shown for comparison. (b) Kinetics of the reversal of the CPX clamp by Syanptotagmin-Ca2+. The cell fusion recovery was carried out at 1mM free Ca2+ and the samples were fixed at the indicated time point after the addition of Ca2+. Averages and standard deviation of 2–3 independent experiments are shown
As a control, we tested VAMP-3xDA in the absence of any CPX to confirm that it is intrinsically fusion competent (Fig. 4a, blue bars). Furthermore, we found that the clamping of fusion by wild-type CPX was identical for cells expressing VAMP2 or VAMP-3xDA (Fig. 4a, red bars). Because the mutations of the switch Asp’s lock CPXacc in the open state, the fact that we see functional clamping in VAMP-3xDA is consistent with the prediction that the accessory helix exerts clamping in the open conformation20.
To further characterize the VAMP-3xDA mutation, we analyzed the kinetics of the activation of fusion by Synaptotagmin and calcium from the clamped state. We found the overall kinetics of clamp release was similar in cells expressing VAMP-3xDA or VAMP2 (Fig. 4b), but the extent of activation of fusion by Ca2+-Synaptotagmin was limited at all time points (Fig. 4b). These activation curves have the same calcium requirement (EC50 ~ 100 μM, Supplementary Fig. 4), indicating that the effect is mainly due to an intrinsic property of the VAMP-3xDA mutation rather an impairment in Synaptotagmin. These results clearly demonstrate that the conformational switch in CPX position is essential for Synaptotagmin to trigger fusion upon arrival of the Ca2+ signal.
DISCUSSION
The data presented establish that a region near the middle of the VAMP2 SNARE motif must be folded with the t-SNARE in order for CPX to assume the closed conformation. This positions the key Aspartate residues (Asp 64, Asp 65, Asp 68) to interact with the CPX central helix (Fig. 2a), driving CPX from its open into its closed conformation. The data also reveal that accessing the closed conformation is not required for clamping, but it is required for calcium-bound Synaptotagmin to release the clamp.
Clamp release entails removing CPXacc from its trans-t-SNARE binding site so that the trans-SNAREpin can complete zippering and thereby fuse the bilayers. In light of the network of interactions constituting the zig-zag array20 (Fig. 5), it is hard to imagine how this could occur as an isolated event. For example, if any one CPX in the array (example, the CPX emanating from SNAREpin 3 in Fig. 5) were to flip from open to closed, in so doing, it would necessarily pull its CPXacc out of the binding pocket of its trans-SNAREpin across the midline of the array (number 2), where it had up until then been bound (Fig. 5). But, in actuality this could not happen unless the switch region of the v-SNARE within SNAREpin 3 had somehow zippered up to create the binding site needed to anchor its emanating CPXacc. However, this in turn, is prohibited by yet another CPXacc (emanating from SNAREpin 4) which plugs the path of the v-SNARE within SNAREpin 3 (Fig. 5). This suggests that the zippering of the VAMP2 C-terminus and the conformational switch in CPX must both occur in the short interval corresponding to clamp release, probably concomitantly. In essence, the zig-zag array seems designed either to remain as it is or to disassemble in a nearly simultaneous cascade, ideally suited for the synchronous activation of synaptic transmission. How could this be triggered?
Figure 5.
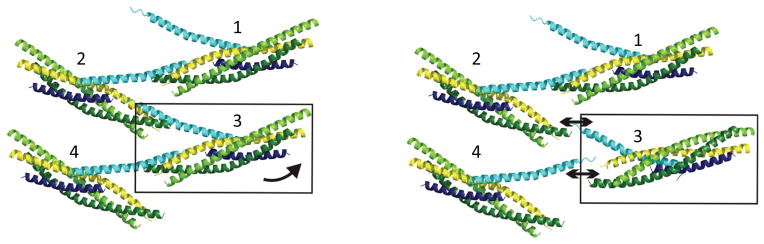
Perturbation of a single SNARE complex in the zig-zag array should be sufficient to rapidly disassemble the clamp in response to neuronal stimulus. A small perturbation of one CPX-SNARE complex in the clamped zig-zag array would eliminate interactions with both of its neighbors in the array. For example, a disruption of complex 3 would eliminate interaction with complexes 2 and 4. So, if one set of the CPXacc-SNAREpin interactions were to be perturbed by Syaptotagmin-Ca2+, then VAMP2 could zipper up, and the entire zig-zag array would disassemble very rapidly, releasing the clamp and triggering fusion.
The structure suggests an appealingly simple hypothesis. Removing any individual SNAREpin from the zig-zag entails breaking contacts with two other SNAREpins, one before and the other after it in the array (SNAREpin 3 in Fig. 5). This fracture will almost always break the CPXacc bonds rather than the CPXcen bonds, because the former (ΔG = −6.8 kCal mole−1) are much weaker than the latter (ΔG = −9.1 kCal mole−1)20. When this occurs, the cascade outlined in the last paragraph could be spontaneously triggered (see discussion below), and the SNAREpins located across the midline of the zig-zag would zipper away from each other to form the fusion pore and release neurotransmitter.
If our speculative hypothesis that fusion is triggered by perturbing a single SNAREpin in the array is correct, the activation energy that needs to be provided to enable this triggered fusion will be the energy required to disrupt the two CPXacc-t-SNARE binding sites in which that SNAREpin is engaged, totaling ~14 kCal mole−1 (corresponding to ~20 kBT)20. The source of this energy must be from Synaptotagmin binding to calcium, the event that triggers release from the clamped state8. When Synaptotagmin binds calcium, it undergoes a conformational change27 and as a result, ~21 kCal mole−1 of free energy (corresponding to ~33 kBT) is made available to do work beyond that which is needed for the conformational change itself28 as the sum of the individual ΔG values for sites 1–3 in C2A & C2B excluding site 4 because it binds calcium well above the physiological Ca2+ concentration range). In addition, the calcium-Synaptotagmin complex binds acidic phospholipid-containing bilayers with a Keq of ~2 μM28, corresponding to ~12 kBT. The total energy potentially available to perturb the zig-zag array when a single Synaptotagmin binds its complement of calcium ions is thus ~45 kBT, greatly exceeding the energy needed to remove its attached SNAREpin (~ 20 kBT in our model).
Of course, it is not known how much of Ca2+-Synaptotagmin’s energy resource is usefully funneled into disrupting the array, but these considerations make it tenable to suggest that activation of a single Synaptotagmin molecule (by its bound complement of calcium ions) could be sufficient to dislodge its single bound SNAREpin, and that this in turn is sufficient to trigger synchronous release of a quantum of neurotransmitter. Twisting, pulling, or pushing on a SNAREpin with sufficient (~20 kBT) energy will remove it from the array, as is illustrated by SNAREpin 3 in Fig. 5. Though the structural details are still missing, it is easy to imagine how a conformational change in Synaptotagmin (perhaps driven by stabilization of its compact conformation when it binds calcium27,29,30) could perturb an attached SNAREpin in this manner. In addition, Synaptotagmin is expected to rapidly adhere to the acidic phospholipids (mainly PS and PIP2) in the nearby synaptic vesicle and/or plasma membrane cytoplasmic leaflets,7,8,31–33 which would also be expected to perturb an attached SNAREpin out of planarity in the array. But, it is important to point out that lipid binding alone (~12 kBT)28 does not appear to be sufficient to activate fusion (~20 kBT) though it may well make an important contribution. The N-terminal domain of CPX (residues 1–25, which precede the CPXacc) also contributes to the activation process in vivo34, but is not needed in our minimal cell-cell fusion system15. Also, recent data suggests that Ca2+ alone could perturb the CPX-SNARE complexes even in the absence of Synaptotagmin, in certain cases35.
Neurotransmitters can be released within as little as 200 μsec after calcium ions enter the nerve terminal36. Can activation in our single Synaptotagmin-SNAREpin hypothesis keep pace with this? The rate of spontaneous fusion from the CPX-clamped state in the flipped SNARE fusion assay, toff ~ 1 hour14, indicates an energy barrier of ≥ 30 kBT (based on the well-established Kramers-Evans relationship between activation energy and dissociation rate37). Starting with the ~ 30 kBT value for the spontaneous activation energy barrier from the array, removing a single SNAREpin (by Ca2+-Synaptotagmin or any other means) is predicted to reduce the activation energy barrier by ~20 kBT. The remaining barrier of ~10 kBT will be transited37 in 2–20 μsec, in no way limiting for the overall time required to release the first quanta of transmitter.
Put differently, the activation energy of ~10 kBT for unbinding CPXacc in the clamped array implies that single CPXacc dissociation events will occur spontaneously every 2–20 μsec, but these will fruitlessly snap back into the array and not result in spontaneous fusion. The activation barrier for spontaneous fusion from the clamped state of ~30 kBT suggests that it is only when three such contacts are broken (presumably in neighboring SNAREpins) that the fusion pore can successfully opens with high probability. In our model, perturbation by Ca2+-Synaptotagmin serves to remove two of these three accessory helices, and when the third spontaneously dissociates (2–20 μsec) the fusion pore can now open. The lifetime of the Ca2+-Synaptotagmin complex, whose lower limit is set by the duration of the rise in local calcium concentration, is far longer than this, and so the third and final CPXacc will have many chances to dissociate while calcium is still bound.
METHODS
Plasmid Constructs
The constructs used in this study are pET28a oligohistidine-Thrombin-syntaxin1A (containing rSyntaxin1a residues 191–253), pET28a oligohistidine-MBP-Thrombin-SNAP25N (containing hSNAP25A residues 7-82 and a C-terminal tryptophan), pET28a oligohistidine-Thrombin-SNAP25C (containing hSNAP25A residues 141–203) and pET15b oligohistidine-Complexin (containing hComplexin1 residues 1–134 with the following “super clamp” mutations: D27L, E34F, R37A). The VAMP2 C-terminal truncations were pET28a-oligohistidine-SUMO-VAMP2-60 (hVAMP2 residues 25–60) -VAMP2-65 (hVAMP2 residues 25–65); -VAMP-69 (hVAMP2 residues 25–69); -VAMP-73 (hVAMP2 residues 25–73); -VAMP-77 (hVAMP2 residues 25–77). VAMP-4X and VAMP-3xDA were generated by introducing L70D, A74R, A81D & L84D and D64A, D65A, & D68A mutation, respectively into pET15b-oligohistidine-Thrombin-VAMP2 (human VAMP2 residues 1–96) using QuickChange mutagenesis Kit (Stratagene). VAMP-D64A; -D65A and -D68A were also generated in the same fashion. In these constructs, ‘Thrombin’ and ‘SUMO’ refer to the protease cleavage site.
Protein Expression and Purification
All constructs were expressed and purified as described previously20. Briefly, recombinant fusion proteins were expressed in E. coli BL21 (DE3) cells by induction with 1 mM IPTG for 3 h at 37°C. Cells were harvested and re-suspended in Breaking Buffer (50mM Tris, 150 mM NaCl, 1 mM TCEP, pH 7.4) supplemented with protease inhibitors (Sigma), then lysed using a cell disruptor (Avestin). A cleared lysate obtained by centrifugation was incubated for 3–4 hours with Ni-NTA-agarose (Qiagen) beads. The beads were washed with 25 column volumes of Wash buffer (50mM Tris, 150 mM NaCl, 1 mM TCEP, pH 7.4), followed by 10 column volumes of Wash buffer supplemented with 50mM Imidazole. v- and t-SNARE proteins were cleaved with Thrombin or SUMO protease (as appropriate for the cleavage site). Complexin was eluted from the beads using 400mM Imidazole in Wash buffer and excess Imidazole was removed using a NAP25 desalting column (GE Healthcare). If needed, the proteins were further purified by size exclusion chromatography (Hi-Load Superdex 75, GE Healthcare).
FRET Analysis
Positions Asp193 on SNAP25C and Gln38 on CPX were mutated into cysteines using the Stratagene QuikChange Kit. SNAP25 D193C was labeled with the donor probe, Stilbene (4-acetamido-4′-((iodoacetyl)amino)-stilbene-2,2′-disulfonic acid, disodium salt, Invitrogen) and CPX Q38C was labeled with the acceptor Bimane (Monochlorobimane, Invitrogen) as described previously20. The double-labeled CPX-SNARE complexes were assembled overnight at 4°C and purified by gel-filtration on a Superdex 75 gel filtration column. All fluorescence data were obtained on a Perkin-Elmer LS55 luminescence spectrometer operating at 25°C and the conditions are similar to those used previously20. FRET distances were calculated as described previously using a R0 for the Stilbene-Bimane FRET pair to be 27.5 Å20
Toxin Accessibility Assay
To test the accessibility of VAMP-3xDA to the neurotoxins, 5μM of either free VAMP-3xDA or CPX-SNARE-3xDA complex were incubated with the neurotoxins at 1:20 toxin:protein ratio in a Tris Buffer pH 7.4, 150 mM NaCl containing 100 μM Zn2+ at 37°C for 2 hours and were analyzed by SDS PAGE-Coomassie Stain. Botulinum-B and Tetanus light chains were purified as described previously14.
Liposome Fusion Assay
VAMP2 and t-SNARE proteins were incorporated in liposome at 1:400 protein:lipid ratio and liposome fusion assay was carried out as described previously4,38,39. Briefly, 45 μl unlabeled t-SNARE liposomes were mixed with 5 μl labeled v-SNARE liposomes in a 96-well plate and fusion was followed by measuring the increase in NBD uorescence at 538 nm (excitation 460 nm) every 2 min at 37°C. At the end of the 2 hr reaction, 10 ml of 2.5% dodecyl-maltoside was added to the liposomes and the fusion is plotted as the percentage of the maximal NBD fluorescence38.
Cell-Cell Fusion Assay
The flipped SNARE cell-cell fusion assay was performed essentially as described before2,14,15,40. In brief, HeLa cell lines were transiently transfected with flipped VAMP2 (WT or 3xDA), DsRed2-NES and either with or without CPX and Synaptotagmin. After one day, transfected v-cells were seeded onto glass coverslips containing cells stably co-expressing flipped syntaxin1, flipped SNAP-25 and CFP-NLS (t-cells). The following day, cells were fixed with 4% paraformaldehyde directly or after treatment with recovery solution (1 U ml−1 Phosphatidylinositol Specific Phospholipase-C (PI-PLC, Molecular Probes), 20 μg ml−1 laminin, with or without 1.8 mM EGTA), washed and mounted with Prolong Antifade Gold mounting medium (Molecular Probes). Confocal images were acquired on a Zeiss 510-Meta confocal microscope and processed using Adobe Photoshop software (Adobe Inc). Kinetics of the reversal of the CPX clamp by Syanptotagmin-Ca2+ was essentially carried out as described, wherein 5 minutes after the addition of PI-PLC-EGTA, the free Ca2+ concentration raised to 1mM and the samples were fixed at the noted time intervals. For the calcium sensitivity experiment, free Ca2+ was raised to the indicated concentration (ranging from 5 μM to 5000 μM) and the cells were incubated at 37°C for 30 min before fixing the cells for quantitation.
Isothermal Titration Calorimetry (ITC) analysis
ITC experiments were carried out as described previously20. Typically, ~200μM of scCPX was titrated into ~10 μM of assembled SNARE complexes in the sample cells and thermodynamic parameters were calculated using Microcal Origin ITC200 (Mircocal, GE Healthcare) software package assuming a “one-set-of-sites” binding model.
Supplementary Material
Table 2.
Thermodynamic parameters of CPX binding to SNAREs measured by Isothermal Titration Calorimetry. Superclamp CPX (scCPX, residues 1–134 carrying superclamp mutation D27L, E34F, R37A, scCPX) or CPX48–134 were titrated into assembled SNARE complexes containing VAMP2 (residues 1–96) or VAMP-3xDA (residues 1–96 with mutations D64A, D65A, & D68A). The thermodynamic parameters, dissociation constant (Kd); enthalpy (ΔH); entropy (ΔS) and free energy (ΔG) were calculated by nonlinear least squares fit with a one-set-of-sites model from the binding isotherms shown in Fig. 3. Average and standard deviations of a minimum of three independent experiments are shown.
| Titrant | In sample cell | Stoichiometric Coefficient (N) | Kd (nM) | ΔH (kCal mol−1) | ΔS (Cal mol−1 K−1) | ΔG (kCal mol−1) |
|---|---|---|---|---|---|---|
| scCPX | SNARE complex with VAMP2-WT | 0.95 ± 0.01 | 83 ± 17 | −37.5 ± 0.7 | −88.6 ± 2.7 | −10.1 ± 0.2 |
| scCPX | SNARE complex with VAMP-3xDA | 0.99 ± 0.04 | 670 ± 90 | −15.0 ± 1.0 | −20.1 ± 3.6 | −8.8 ± 0.2 |
| CPX48–134 | SNARE complex with VAMP-3xDA | 0.98 ± 0.01 | 620 ± 110 | −14.0 ± 1.1 | −16.6 ± 3.8 | −8.8 ± 0.1 |
Acknowledgments
We wish to thank Dr. Tom Melia (Yale University) for critical reading of the manuscript. This work was supported by NIH grants to JER and KMR, ANR PCV grant to FP, and a grant from the DFG to DK.
Footnotes
AUTHOR CONTRIBUTIONS
SSK, DTR and DK performed the mutagenesis and protein purification, SSK and DTR carried out the fluorescence measurements, FL did the ITC measurements and CGG contributed the cell-cell fusion assay. LK, SB and JC provided technical assistance. Results were analyzed and discussed by all authors. The manuscript was prepared by SSK, DTR, KR, FP & JER
References
- 1.Sollner T, et al. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–24. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- 2.Hu C, et al. Fusion of cells by flipped SNAREs. Science. 2003;300:1745–9. doi: 10.1126/science.1084909. [DOI] [PubMed] [Google Scholar]
- 3.McNew JA, et al. Compartmental specificity of cellular membrane fusion encoded in SNARE proteins. Nature. 2000;407:153–9. doi: 10.1038/35025000. [DOI] [PubMed] [Google Scholar]
- 4.Weber T, et al. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–72. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- 5.Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–53. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 6.Karatekin E, et al. A fast, single-vesicle fusion assay mimics physiological SNARE requirements. Proc Natl Acad Sci U S A. 2010;107:3517–21. doi: 10.1073/pnas.0914723107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perin MS, Fried VA, Mignery GA, Jahn R, Sudhof TC. Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature. 1990;345:260–3. doi: 10.1038/345260a0. [DOI] [PubMed] [Google Scholar]
- 8.Brose N, Petrenko AG, Sudhof TC, Jahn R. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. 1992;256:1021–5. doi: 10.1126/science.1589771. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez-Chacon R, et al. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 2001;410:41–9. doi: 10.1038/35065004. [DOI] [PubMed] [Google Scholar]
- 10.Geppert M, et al. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–27. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- 11.Pang ZP, Shin OH, Meyer AC, Rosenmund C, Sudhof TC. A gain-of-function mutation in synaptotagmin-1 reveals a critical role of Ca2+-dependent soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex binding in synaptic exocytosis. J Neurosci. 2006;26:12556–65. doi: 10.1523/JNEUROSCI.3804-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishizuka T, Saisu H, Odani S, Abe T. Synaphin: a protein associated with the docking/fusion complex in presynaptic terminals. Biochem Biophys Res Commun. 1995;213:1107–14. doi: 10.1006/bbrc.1995.2241. [DOI] [PubMed] [Google Scholar]
- 13.McMahon HT, Missler M, Li C, Sudhof TC. Complexins: cytosolic proteins that regulate SNAP receptor function. Cell. 1995;83:111–9. doi: 10.1016/0092-8674(95)90239-2. [DOI] [PubMed] [Google Scholar]
- 14.Giraudo CG, Eng WS, Melia TJ, Rothman JE. A clamping mechanism involved in SNARE-dependent exocytosis. Science. 2006;313:676–80. doi: 10.1126/science.1129450. [DOI] [PubMed] [Google Scholar]
- 15.Giraudo CG, et al. Alternative zippering as an on-off switch for SNARE-mediated fusion. Science. 2009;323:512–6. doi: 10.1126/science.1166500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maximov A, Tang J, Yang X, Pang ZP, Sudhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–21. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xue M, et al. Distinct domains of complexin I differentially regulate neurotransmitter release. Nat Struct Mol Biol. 2007;14:949–58. doi: 10.1038/nsmb1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xue M, et al. Tilting the balance between facilitatory and inhibitory functions of mammalian and Drosophila Complexins orchestrates synaptic vesicle exocytosis. Neuron. 2009;64:367–80. doi: 10.1016/j.neuron.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–7. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kümmel D, et al. Complexin Crosslinks Pre-fusion SNAREs into Zig-Zag Array: A Structure Based Model for Complexin Clamping. Nat Struct Mol Biol. 2011 Same Issue. [Google Scholar]
- 21.Hua SY, Charlton MP. Activity-dependent changes in partial VAMP complexes during neurotransmitter release. Nat Neurosci. 1999;2:1078–83. doi: 10.1038/16005. [DOI] [PubMed] [Google Scholar]
- 22.Reim K, et al. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 2001;104:71–81. doi: 10.1016/s0092-8674(01)00192-1. [DOI] [PubMed] [Google Scholar]
- 23.Tang J, et al. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–87. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- 24.Chen X, et al. Three-dimensional structure of the complexin/SNARE complex. Neuron. 2002;33:397–409. doi: 10.1016/s0896-6273(02)00583-4. [DOI] [PubMed] [Google Scholar]
- 25.Bracher A, Kadlec J, Betz H, Weissenhorn W. X-ray structure of a neuronal complexin-SNARE complex from squid. J Biol Chem. 2002;277:26517–23. doi: 10.1074/jbc.M203460200. [DOI] [PubMed] [Google Scholar]
- 26.Yang X, Kaeser-Woo YJ, Pang ZP, Xu W, Sudhof TC. Complexin clamps asynchronous release by blocking a secondary Ca(2+) sensor via its accessory alpha helix. Neuron. 2010;68:907–20. doi: 10.1016/j.neuron.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shao X, Fernandez I, Sudhof TC, Rizo J. Solution structures of the Ca2+-free and Ca2+-bound C2A domain of synaptotagmin I: does Ca2+ induce a conformational change? Biochemistry. 1998;37:16106–15. doi: 10.1021/bi981789h. [DOI] [PubMed] [Google Scholar]
- 28.Radhakrishnan A, Stein A, Jahn R, Fasshauer D. The Ca2+ affinity of synaptotagmin 1 is markedly increased by a specific interaction of its C2B domain with phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2009;284:25749–60. doi: 10.1074/jbc.M109.042499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vrljic M, et al. Molecular mechanism of the synaptotagmin-SNARE interaction in Ca2+-triggered vesicle fusion. Nat Struct Mol Biol. 2010;17:325–31. doi: 10.1038/nsmb.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ubach J, Zhang X, Shao X, Sudhof TC, Rizo J. Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? EMBO J. 1998;17:3921–30. doi: 10.1093/emboj/17.14.3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bai J, Tucker WC, Chapman ER. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat Struct Mol Biol. 2004;11:36–44. doi: 10.1038/nsmb709. [DOI] [PubMed] [Google Scholar]
- 32.Chapman ER, Davis AF. Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. J Biol Chem. 1998;273:13995–4001. doi: 10.1074/jbc.273.22.13995. [DOI] [PubMed] [Google Scholar]
- 33.McMahon HT, Kozlov MM, Martens S. Membrane curvature in synaptic vesicle fusion and beyond. Cell. 2010;140:601–5. doi: 10.1016/j.cell.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 34.Xue M, et al. Binding of the complexin N terminus to the SNARE complex potentiates synaptic-vesicle fusogenicity. Nat Struct Mol Biol. 2010;17:568–75. doi: 10.1038/nsmb.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoon TY, et al. Complexin and Ca2+ stimulate SNARE-mediated membrane fusion. Nat Struct Mol Biol. 2008;15:707–13. doi: 10.1038/nsmb.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sabatini BL, Regehr WG. Timing of neurotransmission at fast synapses in the mammalian brain. Nature. 1996;384:170–2. doi: 10.1038/384170a0. [DOI] [PubMed] [Google Scholar]
- 37.Evans E. Probing the relation between force--lifetime--and chemistry in single molecular bonds. Annu Rev Biophys Biomol Struct. 2001;30:105–28. doi: 10.1146/annurev.biophys.30.1.105. [DOI] [PubMed] [Google Scholar]
- 38.Scott BL, et al. Liposome fusion assay to monitor intracellular membrane fusion machines. Methods Enzymol. 2003;372:274–300. doi: 10.1016/S0076-6879(03)72016-3. [DOI] [PubMed] [Google Scholar]
- 39.Ji H, et al. Protein determinants of SNARE-mediated lipid mixing. Biophys J. 2010;99:553–60. doi: 10.1016/j.bpj.2010.04.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giraudo CG, et al. Distinct domains of complexins bind SNARE complexes and clamp fusion in vitro. J Biol Chem. 2008;283:21211–9. doi: 10.1074/jbc.M803478200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.