

Abstract
Comparative functional genomics studies the evolution of biological processes by analyzing functional data, such as gene expression profiles, across species. A major challenge is to compare profiles collected in a complex phylogeny. Here, we present Arboretum, a novel scalable computational algorithm that integrates expression data from multiple species with species and gene phylogenies to infer modules of coexpressed genes in extant species and their evolutionary histories. We also develop new, generally applicable measures of conservation and divergence in gene regulatory modules to assess the impact of changes in gene content and expression on module evolution. We used Arboretum to study the evolution of the transcriptional response to heat shock in eight species of Ascomycota fungi and to reconstruct modules of the ancestral environmental stress response (ESR). We found substantial conservation in the stress response across species and in the reconstructed components of the ancestral ESR modules. The greatest divergence was in the most induced stress, primarily through module expansion. The divergence of the heat stress response exceeds that observed in the response to glucose depletion in the same species. Arboretum and its associated analyses provide a comprehensive framework to systematically study regulatory evolution of condition-specific responses.
Comparative functional genomics approaches are increasingly used to study regulatory evolution in unicellular (Jensen et al. 2006; Gasch 2007; Thompson and Regev 2009; Wohlbach et al. 2009; Romero et al. 2012) and multicellular organisms (Brawand et al. 2011; Schmidt et al. 2012; Xiao et al. 2012). Such studies measure and compare genomic profiles, including mRNA levels (Bergmann et al. 2003b; Tirosh et al. 2006; Wapinski et al. 2010; Brawand et al. 2011; Fowlkes et al. 2011; Rhind et al. 2011; Tirosh et al. 2011), chromatin organization (Segal et al. 2006; Tsankov et al. 2010; Xiao et al. 2012), or protein–DNA interactions (Borneman et al. 2007; Schmidt et al. 2010, 2012; Kutter et al. 2011) across two or more species.
Although comparing genomic profiles between pairs of species is relatively straightforward, deriving evolutionary insights requires us to compare many species in a phylogeny (Brawand et al. 2011; Rhind et al. 2011). For example, one important feature of transcriptional programs is their organization into regulatory modules of coexpressed genes (Ihmels et al. 2002; Segal et al. 2003). There are many approaches to identify such modules in a single species (Eisen et al. 1998; Bergmann et al. 2003a; Segal et al. 2005; Joshi et al. 2009), but mapping genes and modules across multiple species is challenging. The few studies that compared modules across more than two species (Bergmann et al. 2003b; Stuart et al. 2003; Kuo et al. 2010b; Waltman et al. 2010) typically ignore phylogenetic relationships. Rather, they either identify modules in each species independently (Bergmann et al. 2003b; Tanay et al. 2005) or identify modules from a single merged data matrix, often requiring matched samples across species (possibly preferring orthologs to reside in the same module [Kuo et al. 2010b]). Neither strategy infers the modules in the ancestors of the extant species.
Here, we developed Arboretum, a novel algorithm that takes expression profiles from multiple species and the species' and genes' phylogenies and infers both extant and ancestral modules. By rooting the module identities at the last common ancestor (LCA) of the species, Arboretum automatically maps modules across species and allows us to trace the evolution of the module assignment of each gene. We used Arboretum to study the evolution of the transcriptional program to heat stress in eight species of Ascomycota and to reconstruct the environmental stress response (ESR) at the LCA of a subset of five species. We found substantial conservation of stress response across species, including the S. cerevisiae ESR, and highlight species- and clade-specific divergence; changes in gene content and gene duplication both contribute to this divergence.
Results
Arboretum: An algorithm to infer the evolution of expression modules
Arboretum takes as input expression profiles measured for multiple extant species in a phylogeny, the species' tree, and gene trees, and infers modules in each of the extant and ancestral species and the evolutionary transitions from the modules of an ancestral species to those of its descendant species (Fig. 1; Methods; Supplemental Methods).
Figure 1.

Arboretum. (A) Generative model. Shown are the components of the generative model for a phylogeny with three extant species (X, Y, Z, gray rectangles) and two ancestral species (A, B, white rectangles) with k = 2 modules (heatmaps). The model consists of two parts: module evolution (top) and expression generation (bottom). Module evolution is modeled by transition matrices, one for every branch of the tree (black and white matrices on branches and bottom). The observed expression (heatmaps) is modeled by a mixture of Gaussians—one mixture for each extant species, one mixture component per module. The parameters of each Gaussian are shown on top of each species-specific module. For example, Σ1X and μ1X denote the covariance and mean of module 1 in species X. (B) Modeling module evolution of a gene family with duplication. (Top) Shown is a gene tree; (star) duplication event. All species after duplication (B, X, and Y) have two copies of the ancestral gene (B1, B2, X1, X2, and Y1, Y2). (Bottom) Module evolution procedure. (CAi) Module assignments of the ith orthogroup in species A. Module assignments post-duplication are denoted as, for example, CX1i and CX2i, for genes X1, X2 in species X. The assignments CB1i and CB2i are both sampled from the transition matrix of the phylogenetic point right after the duplication (B) and evolved independently down the rest of the subtree.
Arboretum is based on a generative probabilistic model that consists of two parts (Fig. 1A): (1) evolution of “hidden” module membership of both ancestral and extant species; and (2) observed expression generation at the extant species only. Evolution of module membership is modeled by a transition matrix for every branch of the species tree, describing the conditional probability of a gene's module membership in a species, given that gene's module membership in that species' immediate ancestor (Fig. 1A, black and white matrices). A Gaussian mixture models the expression data of each module at the extant species (Fig. 1A, red and green matrices). The model's parameters are the Gaussian mixture parameters, mean 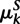 and covariance
and covariance 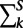 for each module k, at each extant species S, the transition matrices for each branch, and the initial module probability distribution at the root. These parameters are learned using expectation maximization (EM) (Dempster et al. 1977), with the module membership of each gene in each species (extant or ancestral) inferred based on the observed expression data. The module IDs across species are all linked to the same module in the LCA, such that module m in one species corresponds to module m in another species.
for each module k, at each extant species S, the transition matrices for each branch, and the initial module probability distribution at the root. These parameters are learned using expectation maximization (EM) (Dempster et al. 1977), with the module membership of each gene in each species (extant or ancestral) inferred based on the observed expression data. The module IDs across species are all linked to the same module in the LCA, such that module m in one species corresponds to module m in another species.
Arboretum handles complex orthology relations
By considering the gene tree associated with each group of orthologs (orthogroup) (Wapinski et al. 2007b), Arboretum handles many-to-many relationships between orthologs that result from gene duplication and loss. For loss, the generative model simply does not generate expression for the species where the gene is lost. For an orthogroup with paralogs, Arboretum proceeds from the LCA down the tree generating module assignments until it reaches the phylogenetic point where duplication happened (Fig. 1B, star), as indicated by the gene tree associated with the orthogroup. At that node, Arboretum independently draws two samples from the transition probability matrix of the module, assigns each to one of the paralogs, and independently evolves it down the rest of the tree (Supplemental Methods). This allows the paralogs to subsequently evolve along different trajectories. By using the gene tree structure, Arboretum avoids iterating over all pairs of orthologs and naturally handles the many-to-many relationships across the species.
Arboretum identifies coherent and conserved expression modules compared to other methods
We compared Arboretum's performance to that of two existing methods of clustering multispecies expression profiles (Supplemental Methods) that do not explicitly model the phylogenetic relationships: the orthoseeded algorithm—similar to Waltman et al. (2010), but without biclustering—and soft k-means clustering (Kuo et al. 2010b). We used expression data from different subsets of five species from a large panel of 15 species for which we have measured expression under glucose depletion (described in D Thompson, S Roy, M Chan, M Styczynsky, J Pfiffner, unpubl.). Different subsets of species enable us to study robustness to the specific species composition in the data. We used four criteria (Supplemental Methods): (1) module stability (the proportion of gene pairs that are in the same module under different random initializations); (2) expression coherence (the average proportion of genes whose expression profiles had >0.8 correlation with the module's mean); (3) conservation of gene content in expression modules (the degree of overlap in orthologous genes between maximally overlapping pairs of modules in two species); and (4) ability to recover the ‘ground truth’ assignment of genes into modules based on simulated data generated with our module evolution model.
Arboretum performed well in all measures (Fig. 2; Supplemental Fig. 1). First, modules inferred by Arboretum were as stable as orthoseeded clustering and outperformed soft k-means clustering (Methods), for all subsets of species used. Second, the expression coherence of modules generated by all three methods was comparable, across different random initializations. Third, soft k-means clustering yielded the most conserved modules, followed by Arboretum, and then orthoseeded clustering. This is expected since soft k-means clustering explicitly favors orthologous genes to be in the same module, whereas Arboretum only imposes a prior distribution on the module assignment via the tree, to allow measured expression to uncover regulatory divergence during evolution. Fourth, for the simulated data with known module assignments, Arboretum performed significantly better than soft k-clustering and was on par (or slightly better in some cases) with the orthoseeded algorithm (Supplemental Fig. 1). The lower performance of soft k-clustering on the simulated data suggests that it likely overestimates conservation (as reflected in the third criterion above).
Figure 2.
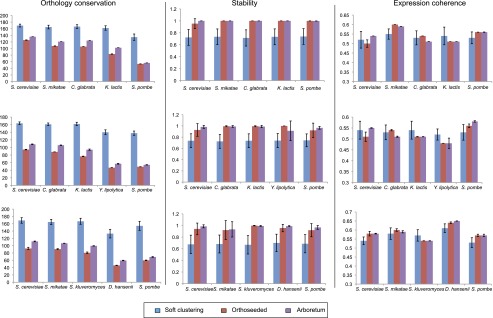
Performance of Arboretum. Shown is a comparison of Arboretum's performance (purple) to that of soft k-means clustering (blue) and orthoseeded clustering (red), based on degree of ortholog conservation measured as the average negative logarithm of the P-value of the hypergeometric test for significance of overlap across modules (left), module stability (middle), and expression coherence of modules (right), for three different sets of five species each (rows). Error bars were obtained by running each algorithm with different random initializations.
Arboretum also infers transition matrices and ancestral module assignments that provide insights into the evolutionary history of modules. To assess their quality, we compared the accuracy of the inferred modules to the ‘ground truth’ in the simulated data across different input parameters (Supplemental Figs. 2, 3). We found that Arboretum performs very well for extant species and recent ancestors, with—as expected—some diminishing performance for more ancient ancestors. The majority of errors in ancestors is due to mis-assignments between modules with close expression patterns (Supplemental Fig. 4).
Comparative transcriptional analysis of the heat shock response in eight yeast species
We used Arboretum to study the evolution of the transcriptional response to heat shock in eight Ascomycota yeasts—Saccharomyces cerevisiae, Candida glabrata, Saccharomyces castellii, Kluyveromyces lactis, Kluyveromyces waltii, Candida albicans, Schizosaccharomyces japonicus, and Schizosaccharomyces pombe (Fig. 3A, left). In each species, we measured at least four time points following heat shock (Fig. 3A; Supplemental Fig. 5; Methods).
Figure 3.

Heat shock transcriptional response in eight Ascomycota species is captured by five modules. (A) Expression modules identified by Arboretum in the transcriptional response to heat shock in eight species. Shown are the expression modules (1–5, heat maps, middle) in each of eight species (species tree, left) at denoted time points prior to and following heat shock (time axis, right). Color bar denotes expression relative to pre-stress time zero. (Red) induced; (green) repressed; (black) no change. Each heatmap shows the expression profile of all genes assigned to that module in a given species. The heatmap height is proportional to the number of genes in the module (marked on top). All modules in one column are mapped to the same ancestral module ID (1–5, top) at the LCA of these eight species. (B) Overlap of modules between species. Shown is the degree of overlap in orthologous genes between every pair of modules 1–5 (rows and columns in each matrix) in every pair of extant species. Diagonal elements (red): overlap between modules of the same ID; off-diagonal elements (blue): overlap between modules of different IDs. Red and blue intensity is proportional to −log (P-value) of the hypergeometric distribution (color scales, right).
A conserved transcriptional program to heat stress
Arboretum identified five expression modules (Fig. 3A; Methods), ranging from strongly repressed Module 1 to strongly induced Module 5 and enriched for genes with coherent functions in most (>90%) extant and ancestral species (Supplemental Table 1). Modules of the same ID (‘orthologous’ modules) exhibit the most significant overlap (Fig. 3B, red diagonal elements), with increased conservation for more closely related species. Modules 1 and 2 (strong and milder repression, respectively) are significantly associated with growth-related processes (e.g., ribosome biogenesis, RNA processing, RNA methylation, FDR <0.05) (Supplemental Table 1), consistent with their known repression during stress. Conversely, Modules 4 (mild induction) and 5 (strong induction) are enriched with genes whose function is important in heat stress, including cellular response to heat, proteolysis, protein catabolism, and protein folding. There is also conserved enrichment of cis-regulatory elements in some modules. In most species, Module 1 is enriched for binding sites of the growth regulators SFP1 and TOD6 (Supplemental Fig. 6), and Module 5 is enriched for binding sites of stress and glucose regulators MSN2/4, RGT1, and ADR1. This suggests that basic functional features of the heat stress response are evolutionarily conserved. Indeed, the module assignment of the vast majority of individual genes (98.6%) changed in <50% of the species since the LCA (Methods).
Species- and clade-specific innovation in the response to heat stress
Arboretum also highlights species and lineage-specific innovation in the regulation of other processes. For example, Module 4 (mild induction) of all species, except the Schizosaccharomyces species, is enriched for sporulation genes. This suggests a change in the coupling of meiosis and stress response in the fission yeasts (Supplemental Table 2), possibly related to the different way in which antisense transcription of meiotic genes is responsive to stress in Schizosaccharomyces (Rhind et al. 2011). Furthermore, sexual reproduction genes are particularly enriched in Module 5 in C. albicans, where stress has been previously implicated in induction of the parasexual cycle (Berman and Hadany 2012). In another example, Module 4 in the human pathogen, C. glabrata, is enriched for genes involved in iron sulfur cluster assembly and sulfur assimilation. This may be an adaptation to the human host, where the pathogen competes on limited iron (Nevitt and Thiele 2011). C. glabrata Module 5 (strong induction) is also uniquely enriched for histidine, lysine, and arginine metabolism genes; this was not previously observed, to the best of our knowledge, and may reflect a unique lifestyle choice for this pathogen.
A pan-stress environmental stress response (ESR) is apparent and conserved across species
In all extant and ancestral species, Modules 1 and 5 significantly overlap with the repressed and induced modules of the environmental stress response (ESR), respectively, as previously defined in S. cerevisiae (Fig. 4A; Methods; Gasch et al. 2000). To test if this conservation extends to the response to other stresses, we used Arboretum to identify modules in profiles measured in five of the eight species under oxidative and salt stress (Wapinski et al. 2010). In each case, we found substantial overlap in gene content between modules with similar expression (e.g., strongly induced) across different responses within a species (Fig. 4B; Supplemental Fig. 7) and between the same response in different species (Fig. 4C; Supplemental Fig. 8), as well as to the induced and repressed modules of the S. cerevisiae ESR (Supplemental Fig. 9). The conserved, pan-stress ESR is apparent in all species, including C. albicans, in contrast to previous suggestions that C. albicans may not have a robust ESR (for review, see Gasch 2007). The repressed S. cerevisiae ESR was more conserved than the induced ESR (Supplemental Fig. 9). The salt stress response is the most conserved, and the oxidative stress is the least (Supplemental Fig. 8).
Figure 4.

Reconstructed evolutionary history of the environmental stress response. (A) Shown are the heat shock module assignments for each gene (column) in each extant and ancestral species (row) for genes previously observed as repressed in the ESR (top) and induced in the ESR (bottom) in S. cerevisiae. Rows are ordered using post-fix ordering (left: child; right: child and parent) of the species tree. Columns are ordered based on the module assignment of a gene in S. cerevisiae. (White) Gene lost in species. (B) Similarity of stress modules within a species. Shown is a comparison of modules identified by Arboretum in the transcriptional response to heat shock (Heat), oxidative stress (Ox), and salt stress (Salt) in S. cerevisiae (top) and ancestor A4 (bottom). Each matrix shows gene content conservation (F-score overlap) between all pairs of modules for one pair of conditions in one species. F-score overlap ranges from 0 (no overlap, white) to 1 (full overlap, dark blue). (C) Similarity of stress modules between species. Shown is the degree of overlap in orthologous genes between every pair of modules 1–5 (rows and columns in each matrix) for S. cerevisiae and C. glabrata (top) and C. glabrata and K. lactis (bottom) in heat, salt, and oxidative stress. Black intensity is proportional to −log (P-value) of the hypergeometric distribution. (D,E) Ancestral ESR. Shown are the expression profiles and Arboretum assignments of genes assigned to the most repressed (D) and most induced (E) modules of the LCA, A11 in a pan-stress Arboretum analysis. Genes corresponding to these panels are provided in Supplemental Table 4.
To determine the ancestral ESR and identify potential modules with unique stress- or species-specific behavior, we next applied Arboretum to the combined data set of all three stress responses across all species (three time courses with at least four time points in five species). We found that k = 7 modules explain the expression data best (Supplemental Fig. 10A). Consistent with the preceding analysis, most modules were largely conserved across species, both in gene membership (Supplemental Fig. 10B) and in expression patterns across stresses (Supplemental Fig. 10A). The modules were typically enriched with similar conserved processes: growth related in the repressed modules and stress related in the induced modules (Supplemental Table 3).
The pan-species pan-stress analysis also highlights species-specific differences. First, down-regulation of growth genes (in Module 1) and up-regulation of stress genes (in Module 7) is delayed during oxidative stress in K. lactis and C. albicans, suggesting that the ESR is initiated later in this stress in these species. A delayed kinetic may suggest that the stress as perceived as “milder” by these species. Second, comparison of Module 5 (mild induction) across species suggests that this module emerged as a pan-stress ESR module only at the LCA of S. cerevisiae and C. glabrata but was ancestrally induced only in heat shock. Genes in this module are enriched in actin cytoskeleton organization and protein targeting to the vacuole in all extant species. Third, Module 3 (mild repression, especially in heat stress) in C. albicans consists of distinct genes than in other species (Supplemental Fig. 10B), and is enriched for fatty acid oxidation genes and sulfur amino acid metabolism genes. This suggests a unique repression of these genes in this species under heat stress, which may be related to their clade-specific duplication in Candida, as we discuss below.
Finally, we identified putative genes of the LCA ESR response. Specifically, 381 and 243 genes, respectively, belong to the most repressed (Module 1) (Fig. 4D) and the most induced modules (Module 7) (Fig. 4E) of the LCA in our pan-stress analysis (Supplemental Table 4). Another 874 and 302 genes belong to the next most repressed (Module 2) (Supplemental Fig. 11A) and induced (Module 6) (Supplemental Fig. 11B) modules. Two hundred two and 155 genes from Modules 1 and 2 are also members of the S. cerevisiae repressed (517 genes) ESR. Sixty-two and 52 genes from Modules 7 and 6 are also present in the induced (242 genes) ESR (Gasch et al. 2000), suggesting substantial conservation of the ancestral response (P < 10−12 for repressed modules to P < 10−15 for induced modules, hypergeometric test). The ancestral induced ESR is enriched for genes involved in proteolysis, carbon metabolism, glutathione metabolism, amino acid transport, sporulation, autophagy, and response to stress. The repressed ESR is enriched for growth processes, such as ribosome biogenesis, RNA processing, mitochondrial organization, purine metabolism, and chromatin silencing.
Conserved expression of orthologous genes underlies conserved expression of functional processes
Consistent with the overall conservation, some processes are associated with the same module across species (e.g., ribosome biogenesis with Module 1, protein folding with Module 5) (Supplemental Table 2). This may be due to two possible scenarios: (1) the ‘same’ (orthologous) genes from the associated process have conserved expression across species and are hence members of the ‘same’ (orthologous) modules; or (2) distinct (nonorthologous) genes from the same process are members of the ‘same’ (orthologous) modules in different species. Although the first possibility is simpler, there is support for the second possibility in cell cycle genes in S. cerevisiae and S. pombe (Jensen et al. 2006).
Supporting the first hypothesis, in ∼40% of the processes associated with the same heat shock module in two species (Supplemental Methods), >70% of the genes associated with the process in the two modules are orthologous (Supplemental Fig. 12A, blue curve, dashed lines). One notable exception is ‘response to stimulus’ (Supplemental Fig. 12; Supplemental Table 4), which is enriched in Module 4 in several species through largely distinct genes (Methods), reflecting the diverse set of processes included in this category (nutrient sensing, mating, DNA damage, etc.) and consistent with a faster evolution of the mechanisms by which species interact with their environment.
Regulatory rewiring of processes is conducted through distinct genes
In other cases, the same process is associated with distinct modules (and expression patterns) in different species. For example, DNA repair is associated with Module 2 in S. cerevisiae and C. glabrata and with Module 3 in most of the other species. As before, this could have occurred either through reassignment of orthologous genes from one module to another or due to distinct genes. Supporting the latter hypothesis, in 80% of the cases when two different (nonorthologous) modules in two species are associated with the same process, there is <50% overlap between the process' genes associated with the two modules (Supplemental Fig. 12A, purple curve, dashed lines). One of the few exceptions where process ‘reassignment’ was mediated through reassignment of orthologous genes is ‘mitochondrial translation’ (Supplemental Fig. 12C; Supplemental Table 4). This may be related to the distinction in carbon metabolism between species (Piskur et al. 2006).
Increased module divergence in heat shock compared to glucose depletion
We next compared the heat shock modules to modules learned independently by Arboretum from transcriptional profiles measured in the same eight species during gradual glucose depletion in growth in batch culture (D Thompson, S Roy, M Chan, M Styczynsky, J Pfiffner, unpubl.). By several measures, the degree of conservation in the heat shock response is lower than in the response to glucose depletion. These include: a less significant overlap in gene content between each pair of orthologous modules in the heat shock response (KS test, P < 10−4) (Fig. 5A); a lower average probability of a gene to conserve its ancestral module assignment in the heat stress response (ancestral module conservation index [AMCI]; Methods) (paired t-test P-value < 10−2) (Fig. 5B,C); more frequent reassignment between modules in heat shock than in glucose depletion (KS test, P < 10−29) (Fig. 5D); and an overall higher degree of member turnover in the heat shock response, defined as the fraction of genes that transitioned between modules at a given phylogenetic point (Fig. 5E). The notable exception is the whole genome duplication (WGD) ancestor, with lower AMCI and higher turnover in the glucose depletion response (A5) (Fig. 5C,E, arrow), consistent with the rewiring of carbon metabolism at the WGD (Piskur et al. 2006; Conant and Wolfe 2007; D Thompson, S Roy, M Chan, M Styczynsky, J Pfiffner, unpubl.).
Figure 5.

Heat shock modules diverge more than glucose depletion modules. (A) Conservation of gene content in orthologous modules (of the same ID) for a pair of species in heat shock (y-axis) versus glucose depletion (x-axis). All points below the diagonal indicate that conservation of the ‘corresponding’ module pairs is lower in heat shock than in glucose depletion. (B) Ancestral module conservation index (AMCI). Shown are transition matrices learned for C. glabrata in glucose depletion (top) and heat shock response (bottom). The matrix specifies the conditional distribution of modules in C. glabrata given modules in its immediate ancestor, A4. Element intensity is proportional to the probability value. AMCI quantifies the extent to which a species preserves its immediate ancestral module assignment and is calculated as the average of the diagonal elements. (C) Higher AMCI in glucose depletion than in heat shock. Each point in the scatter is the AMCI of all extant species (black circles) and ancestral species (gray circles) in response to heat shock (y-axis) versus glucose depletion (x-axis). (Arrow) WGD ancestor (A5). (D) Higher module reassignment of genes in heat shock than in glucose depletion. Shown is the histogram of the fraction of reassignments (out of the maximum possible) for heat shock (blue) and glucose depletion (red). (E) Module turnover is higher in heat shock than in glucose depletion. Shown is the degree of turnover at each ancestral (black) and extant (gray) species for heat shock (y-axis) versus glucose depletion (x-axis). (Arrow) WGD ancestor (A5).
The increased divergence in the heat shock program is most prominent in the highly induced Module 5 and is primarily due to module expansion
To test whether the increased divergence in the heat shock response affects all modules equally, we measured the degree of conservation of each module in each response by the average fraction of genes that were shared between each pair of species (Methods). Module 5 conservation is lower in heat shock than in glucose depletion (KS test P-value < 10−19; mean 0.43 ± 0.199 STDEV in heat shock; mean 0.59 ± 0.159 STDEV in glucose depletion). Module 1 conservation is more comparable in the two responses, albeit still significantly lower in heat shock (KS test P-value < 10−8; 0.67 ± 0.142 STDEV in heat shock; 0.75 ± 0.122 in glucose depletion). The higher conservation of Module 1 in both responses reflects the known repression of growth processes in both heat shock and nutrient limitation. The degree of divergence in the other three modules is much more comparable in the two responses. The different species had a similarly robust response to stress by several independent measures, including the effect on growth and changes in expression of ESR genes (above and Wapinski et al. [2010]) (except Schizosaccharomyces [Rhind et al. 2011]), suggesting that this increased divergence is likely not due to an experimental limitation.
The divergence in the gene content of a given module could result either from member genes ‘moving out’ of that module’s ancestor (‘module contraction’) or from new members ‘moving into’ this module (‘module expansion’), or both. We quantified these using: (1) a module contraction index (MCI) that measures the overall extent to which genes leave their ancestral module; and (2) a module expansion index (MEI) that measures the overall extent to which new genes join a module (Methods). Both Modules 1 and 5 have the lowest MCI in both responses (Fig. 6A), but Module 5 has a relatively high MEI in heat shock compared to glucose depletion (Fig. 6B), suggesting that its increased divergence is likely a result of enhanced expansion. Computing these metrics at each phylogenetic point identified the WGD ancestor (A5) to be one of the most substantial expansion points in Module 5 (Fig. 6C,D).
Figure 6.

Module contraction and expansion in heat shock and glucose depletion. (A,B) Module contraction index (A) and module expansion index (B) in heat shock (gray) and glucose depletion (black) for each module M1–M5 (x-axis). Higher bars indicate a greater expansion (contraction) of a module. (C,D) MCI and MEI for the most induced heat shock module (C) and glucose depletion module (D) at individual phylogenetic points (x-axis).
Similar genes are stationary, but distinct processes are mobile between modules in the two responses
We next examined if module reassignment of genes is recapitulated in the two responses, by testing whether genes that are reassigned between modules in one response (at a certain phylogenetic point) are as likely to be reassigned in the other response (possibly at a different phylogenetic point). The stationary genes in each response significantly overlap (189 in heat, 340 in carbon, and 102 in both; hypergeometric P-value < 10−20), and are enriched for similar processes (RNA metabolism and ribosome biogenesis). Indeed, in both responses, growth-related processes have a relatively low number of reassignments (KS test P-value < 0.05) (Supplemental Table 5). This is expected given the similar functional role of growth repression in both stress and nutrient limitation responses.
Although mobile genes also overlapped significantly between the responses (187 in heat, 110 in carbon, and 37 in both; hypergeometric P-value < 10−20), they were not enriched for the same processes (carbon metabolism and mitochondrial processes in glucose depletion and amino acid metabolism in heat shock; KS test P-value < 0.05) (Supplemental Table 6). Thus, although some high-mobility genes may have ‘intrinsic’ regulatory flexibility, this does not necessarily contribute to the regulatory rewiring at the level of functional processes. Such rewiring is instead mediated through regulatory changes in distinct genes in different responses.
Gene duplication is a major source of module divergence
To assess the role of gene duplication in module divergence, we compared modules reconstructed by Arboretum in either (1) orthogroups with no duplication events (but allowing losses); (2) orthogroups with at most one duplication event; or (3) all orthogroups, including those with many duplication events (Methods). Module conservation, as reflected by AMCI, decreases as the number of duplications increases (Supplemental Fig. 13A), suggesting that paralogous genes allow increased regulatory divergence. This increased divergence is specifically due to orthogroups with duplication: there are no significant differences between the runs in the reassignment frequencies of orthogroups with no duplications (Supplemental Fig. 13B).
Reassignment of paralogs between modules explains some of the species-specific divergence patterns we observed. For example, as noted above, in our pan-stress analysis (Supplemental Fig. 10), the C. albicans Module 3 (mild repression, especially in heat stress) is uniquely enriched for fatty acid oxidation genes, belonging to orthogroups that were specifically duplicated in the Candida clade. This duplication was accompanied by functional and regulatory divergence. Those in Module 3, uniquely repressed in C. albicans, are associated with induction of morphological changes (filamentation and the white-to-opaque transition) related to pathogenicity (Lan 2002; Shea and Del Poeta 2006; Shareck et al. 2011). Their paralogs reside in the heat induced Module 6 and are involved in peroxisomal fatty acid oxidation and iron homeostasis (Singh et al. 2011). Thus, the neofunctionalization of these genes was accompanied by regulatory divergence, reflected as species-specific module reassignment of one member of each group of paralogs.
Discussion
A major challenge in comparative functional genomics is to develop methods that can relate complex functional data in meaningful ways across a phylogeny. Unlike sequence data, studies of the evolution of genomic responses still lack specific models. Here, we addressed this challenge within the context of modular transcriptional responses. Typical approaches that compare modules in pairs of species attempt to enumerate all possible mappings between extant species, do not directly incorporate the tree structure of species and genes, and hence do not scale well to dozens of species. In contrast, Arboretum handles this mapping efficiently by associating the inferred modules through their ancestry, explicitly modeling the transition of genes between modules. This solves the mapping problem, is scalable to large numbers of species, and can be easily applied to data sets with a different number of measurements per species. To interpret Arboretum's rich output, we developed several generally applicable statistical measures. In this and a companion study (D Thompson, S Roy, M Chan, M Styczynsky, J Pfiffner, unpubl.), we showed how these can be applied to study an individual response across species (heat stress here and glucose depletion in D Thompson, S Roy, M Chan, M Styczynsky, J Pfiffner, unpubl.), as well as to compare the global evolutionary characteristics of two complex responses.
Our analysis indicates a higher degree of conservation of stress responses than that suggested in a recent study of a similar set of species and conditions (Tirosh et al. 2011). First, there is a somewhat higher degree of global correlation in expression profiles between matching conditions in different species in our study (Supplemental Fig. 14). This may be due to the fact that the rich medium we used was optimized to minimize differences in growth between species (D Thompson, S Roy, M Chan, M Styczynsky, J Pfiffner, unpubl.), as compared to YPD previously used (Tirosh et al. 2011). Second, our analysis relies on data collected along multiple time points (Wapinski et al. 2010), thus reducing differences that may manifest at a single time point (Tirosh et al. 2011). Indeed, the delay in the onset of the oxidative stress response in two of the species would be missed by a single early time point. Third, we carefully monitored growth curves (Wapinski et al. 2010; D Thompson, S Roy, M Chan, M Styczynsky, J Pfiffner, unpubl.) to ensure that all species were at a comparable physiological state. Finally, most of our conclusions are drawn from Arboretum's module analysis, likely increasing the robustness of our analysis and reducing our sensitivity to fluctuations in gene expression within species.
Arboretum and its associated analyses provide a promising direction for comparative functional genomics and for other cases when samples are related through a tree (e.g., a cell lineage) (Liu et al. 2009; Novershtern et al. 2011). An important future direction is to model gene expression at ancestral species (Gu et al. 2005). One possibility is to assume a mean expression profile in the ancestral species and use a random effects model capturing how the expression evolves. Other future developments can include explicit modeling of module birth and death, and direct association with changes in regulatory mechanisms. Together, these can lead to mechanistic and adaptive models of the evolution of regulatory programs.
Methods
Overview of Arboretum
The full algorithmic details of Arboretum are given in the Supplemental Methods. Briefly, Arboretum is a model-based clustering approach that uses a probabilistic generative model to cluster multiple expression data sets, one for each extant species. The generative model generates values for the ‘hidden’ module assignments and the observed expression values for each gene in a species. The generative process for each orthogroup starts with a module assignment drawn from the prior distribution at the LCA, propagating it down through the branches of the species tree for uniform orthogroups and gene trees for nonuniform orthogroups until it reaches a leaf node. We use a Gaussian mixture to generate the expression level of the gene at each leaf. The model parameters are the Gaussian mixture parameters, the module prior probability, and the transition probabilities along each branch, which are learned by expectation maximization. When the algorithm converges, we have a discrete probability distribution over module assignments for each gene-species pair. A gene is finally assigned to a module in a species s that has the highest probability of generating the gene's expression profile in the species s (if extant) or its descendant species (if ancestral).
Assessing Arboretum's performance
We compared Arboretum to two algorithms, Orthoseeded species-specific clustering (Waltman et al. 2010) and soft k-means clustering (Kuo et al. 2010a), which are also detailed in the Supplemental Methods. We used four comparison measures, estimating these from 20 different random initializations of each algorithm: (1) Module stability, defined as the proportion of gene pairs that coclustered; (2) Expression coherence, measured as the average proportion of module genes whose expression profiles had a >0.8 correlation with the module's mean; (3) Conservation of gene content, first identifying best matching modules in each pair of species (the hypergeometric P-value), and then calculating conservation for two species as the average of the maximal overlap scores; (4) Performance on (simulated) ground truth to assess how well other algorithms infer modules in extant species. We also used the simulated data for an accuracy and sensitivity analysis of initial parameter settings of Arboretum. The simulated data and all performance measures are detailed in the Supplemental Methods.
Analysis of heat shock response in eight species
We ran Arboretum on expression data measuring the heat shock response of eight species using orthology mappings from the Synergy algorithm (http://www.broadinstitute.org/regev/orthogroups/) (Wapinski et al. 2007a). The strains, growth conditions, microarray hybridization, and data preprocessing are described in detail in the Supplemental Methods, and microrray data are available at GSE38478. The majority of our analysis is on 3499 orthogroups with at most one duplication event (1069 orthogroups with one duplication event; 2430 are either uniform [no duplication or loss] or have a loss). To assess the role of gene duplication in module divergence, we included all 4215 orthogroups that had at least one gene member in S. cerevisiae and in at least one other species (with no limit on the number of duplications).
We selected the number of modules using a combination of penalized log-likelihood of data per species and manual inspection (Supplemental Methods). Based on penalized log likelihood of separate clustering of each species as well as Arboretum-based clustering of all species, the maximum number of modules for any species was k = 11 (Supplemental Fig. 15A,B). However, the k = 11 case did not produce significantly different expression modules, and were prone to seemingly arbitrary reassignment of module genes between species, given the very similar expression patterns in ‘adjacent’ modules. We therefore picked k manually (Supplemental Fig. 15C), choosing a number where different modules had clearly distinguishable expression patterns (k = 5 for heat stress and k = 7 for pan-stress).
Module conservation and divergence scores
To compare module conservation in extant species, we use a hypergeometric test–based overlap (Supplemental Methods). For comparisons that include the ancestral module assignments, we defined:
Ancestral module conservation index (AMCI)
AMCI for each species with an ancestor measures the tendency of a species to conserve the modules' assignment of its immediate ancestor. AMCI for a species t is the average of the diagonal elements of t's transition matrix. Because each element is a probability value, it is bounded between 0 and 1. The closer it is to 1, the more likely is the species to preserve the module assignments of its immediate ancestor; and the closer it is to 0, the more likely it is to diverge from the module assignments of its immediate ancestor.
Module contraction and expansion index
Module contraction index (MCI) for module m at a phylogenetic point s, is the ratio of the number of contractions (Supplemental Methods) divided by the number of genes in module m in s's ancestor t. Module expansion index (MEI) at s for m is the number of expansions (Supplemental Methods) divided by total number of genes in module m in s. We also define a global MCI of a module m as the sum of contractions for that module across all species with a parent (that is, except the LCA) divided by a normalization term, Zmc, defined as follows: 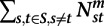 , where S is the set of all species other than the LCA; t is s's immediate parent; and
, where S is the set of all species other than the LCA; t is s's immediate parent; and 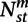 is the number of genes for which we have a module assignment in both s and t and the module assignment of the gene is m in the ancestor t. Similarly, we define a global MEI as the sum of all expansions divided by a corresponding normalization term (Supplemental Methods).
is the number of genes for which we have a module assignment in both s and t and the module assignment of the gene is m in the ancestor t. Similarly, we define a global MEI as the sum of all expansions divided by a corresponding normalization term (Supplemental Methods).
Gene ontology (GO) processes and cis-regulatory element enrichment in modules
We use the FDR-corrected hypergeometric P-value to assess enrichment of GO processes and cis-regulatory elements in a given gene set (Supplemental Methods). GO terms for S. cerevisiae were downloaded from the Saccharomyces Genome Database (SGD) Release version 1.1556. For all other species, we use orthology to transfer the gene ontology annotations, as previously described (Wapinski et al. 2007b). For cis-regulatory elements, we used a similar hypergeometric-based enrichment using a recently generated collection of species-specific motifs (Habib et al. 2012).
Assessing GO process conservation and divergence
To assess conservation in gene content for a process enriched in orthologous modules (same IDs), we use F-score overlap of gene members annotated with the process for each pair of modules (Supplemental Methods). Briefly, for each process, p, enriched in module m in at least two species, we take an average of F-scores first over each pair of such species, and then over any modules enriched in p in more than one species. Gene content conservation for processes enriched in nonorthologous modules (different IDs) are computed also using F-score, averaged between all pairs of enriched nonorthologous modules (Supplemental Methods).
Comparing the reassignment tendency of genes under different responses
Reassignment tendency measures how often a gene is reassigned at any phylogenetic point starting from the LCA to any of the leaf nodes (see Supplemental Methods for details). For orthogroups without duplications, the reassignment fraction is the number of reassignments divided by the number of phylogenetic points at which the gene is not lost. For orthogroups with duplications, we compute the reassignment fraction pre- and post-duplication separately, and take an average of these quantities (Supplemental Methods). A gene is called “high mobility” if it has a reassignment score of ≥0.5 and “low mobility” or “stationary” if it has a reassignment score of <0.05.
Source code availability
Source code and usage instructions can be downloaded from http://www.broadinstitute.org/∼sroy/arboretum or http://pages.discovery.wisc.edu/∼sroy/arboretum.
Data access
The expression data sets associated with this study have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE38478.
Acknowledgments
We thank Itay Tirosh for help with the stress RNA-seq data presented in the Discussion; Guy Zinman for sharing code for soft-clustering; Leslie Gaffney and Pouya Kheradpour for assistance with artwork; and Michelle Chan, Tal Shay, and Moran Cabili-Kalmar for helpful discussions. S.R. was supported by a Computing Innovation Fellow (CIF) grant. I.W. is the HHMI fellow at the Damon Runyon Cancer Research Foundation. Work was supported by HHMI, the Human Frontiers Science Program, the Broad Institute, an NIH PIONEER award, an NIH R01 2R01CA119176-01, a Burroughs-Wellcome Fund Career Award at the Scientific Interface, and the Sloan Foundation (A.R.).
Author contributions: A.R., S.R., and D.T. designed the research. S.R. implemented the module inference algorithm and measures for module evolution analysis. I.W., J.P., C.F., A.S., J.K., and D.T. generated microarray expression data and preprocessed data. I.W. and N.H. provided the cis-regulatory elements database. S.R., D.T., M.K., and A.R. analyzed and interpreted results and wrote the manuscript with input from all authors.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.146233.112.
References
- Bergmann S, Ihmels J, Barkai N 2003a. Iterative signature algorithm for the analysis of large-scale gene expression data. Phys Rev E Stat Nonlin Soft Matter Phys 67: 031902. [DOI] [PubMed] [Google Scholar]
- Bergmann S, Ihmels J, Barkai N 2003b. Similarities and differences in genome-wide expression data of six organisms. PLoS Biol 2: e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman J, Hadany L 2012. Does stress induce (para)sex? Implications for Candida albicans evolution. Trends Genet 28: 197–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borneman AR, Gianoulis TA, Zhang ZD, Yu H, Rozowsky J, Seringhaus MR, Wang LY, Gerstein M, Snyder M 2007. Divergence of transcription factor binding sites across related yeast species. Science 317: 815–819 [DOI] [PubMed] [Google Scholar]
- Brawand D, Soumillon M, Necsulea A, Julien P, Csárdi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M, et al. 2011. The evolution of gene expression levels in mammalian organs. Nature 478: 343–348 [DOI] [PubMed] [Google Scholar]
- Conant GC, Wolfe KH 2007. Increased glycolytic flux as an outcome of whole-genome duplication in yeast. Mol Syst Biol 3: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempster A, Laird N, Rubin D 1977. Maximum likelihood from incomplete data via the EM algorithm. J R Stat Soc Ser B Methodol 39: 1–38 [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D 1998. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci 95: 14863–14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowlkes CC, Eckenrode KB, Bragdon MD, Meyer M, Wunderlich Z, Simirenko L, Luengo Hendriks CL, Keränen SV, Henriquez C, Knowles DW, et al. 2011. A conserved developmental patterning network produces quantitatively different output in multiple species of Drosophila. PLoS Genet 7: e1002346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP 2007. Comparative genomics of the environmental stress response in ascomycete fungi. Yeast 24: 961–976 [DOI] [PubMed] [Google Scholar]
- Gasch AP, Spellman PT, Kao CM, Carmel-Harel O, Eisen MB, Storz G, Botstein D, Brown PO 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol Biol Cell 11: 4241–4257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X, Zhang Z, Huang W 2005. Rapid evolution of expression and regulatory divergences after yeast gene duplication. Proc Natl Acad Sci 102: 707–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N, Wapinski I, Margalit H, Regev A, Friedman N 2012. A functional selection model explains evolutionary robustness despite plasticity in regulatory networks. Mol Syst Biol 8: 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihmels J, Friedlander G, Bergmann S, Sarig O, Ziv Y, Barkai N 2002. Revealing modular organization in the yeast transcriptional network. Nat Genet 31: 370–377 [DOI] [PubMed] [Google Scholar]
- Jensen LJ, Jensen TS, de Lichtenberg U, Brunak S, Bork P 2006. Co-evolution of transcriptional and post-translational cell-cycle regulation. Nature 443: 594–597 [DOI] [PubMed] [Google Scholar]
- Joshi A, De Smet R, Marchal K, Van de Peer Y, Michoel T 2009. Module networks revisited: Computational assessment and prioritization of model predictions. Bioinformatics 25: 490–496 [DOI] [PubMed] [Google Scholar]
- Kuo D, Licon K, Bandyopadhyay S, Chuang R, Luo C, Catalana J, Ravasi T, Tan K, Ideker T 2010a. Coevolution within a transcriptional network by compensatory trans and cis mutations. Genome Res 20: 1672–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo D, Tan K, Zinman G, Ravasi T, Bar-Joseph Z, Ideker T 2010b. Evolutionary divergence in the fungal response to fluconazole revealed by soft clustering. Genome Biol 11: R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutter C, Brown GD, Gonçalves Â, Wilson MD, Watt S, Brazma A, White RJ, Odom DT 2011. Pol III binding in six mammals shows conservation among amino acid isotypes despite divergence among tRNA genes. Nat Genet 43: 948–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan CY 2002. Metabolic specialization associated with phenotypic switching in Candida albicans. Proc Natl Acad Sci 99: 14907–14912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Long F, Peng H, Aerni SJ, Jiang M, Sánchez-Blanco A, Murray JI, Preston E, Mericle B, Batzoglou S, et al. 2009. Analysis of cell fate from single-cell gene expression profiles in C. elegans. Cell 139: 623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevitt T, Thiele DJ 2011. Host iron withholding demands siderophore utilization for Candida glabrata to survive macrophage killing. PLoS Pathog 7: e1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novershtern N, Subramanian A, Lawton LN, Mak RH, Haining WN, McConkey ME, Habib N, Yosef N, Chang CY, Shay T, et al. 2011. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 144: 296–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskur J, Rozpedowska E, Polakova S, Merico A, Compagno C 2006. How did Saccharomyces evolve to become a good brewer? Trends Genet 22: 183–186 [DOI] [PubMed] [Google Scholar]
- Rhind N, Chen Z, Yassour M, Thompson DA, Haas BJ, Habib N, Wapinski I, Roy S, Lin MF, Heiman DI, et al. 2011. Comparative functional genomics of the fission yeasts. Science 332: 930–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero IG, Ruvinsky I, Gilad Y 2012. Comparative studies of gene expression and the evolution of gene regulation. Sci Transl Med 13: 505–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Wilson MD, Ballester B, Schwalie PC, Brown GD, Marshall A, Kutter C, Watt S, Martinez-Jimenez CP, Mackay S, et al. 2010. Five-vertebrate ChIP-seq reveals the evolutionary dynamics of transcription factor binding. Science 328: 1036–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt D, Schwalie PC, Wilson MD, Ballester B, Gonçalves Â, Kutter C, Brown GD, Marshall A, Flicek P, Odom DT 2012. Waves of retrotransposon expansion remodel genome organization and CTCF binding in multiple mammalian lineages. Mol Cell 148: 335–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Shapira M, Regev A, Pe'er D, Botstein D, Koller D, Friedman N 2003. Module networks: Identifying regulatory modules and their condition-specific regulators from gene expression data. Nat Genet 34: 166–176 [DOI] [PubMed] [Google Scholar]
- Segal E, Pe'er D, Regev A, Koller D, Friedman N 2005. Learning module networks. J Mach Learn Res 6: 557–588 [Google Scholar]
- Segal E, Fondufe-Mittendorf Y, Chen L, Thastrom A, Field Y, Moore IK, Wang J-PZ, Widom J 2006. A genomic code for nucleosome positioning. Nature 442: 772–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shareck J, Nantel A, Belhumeur P 2011. Conjugated linoleic acid inhibits hyphal growth in Candida albicans by modulating Ras1p cellular levels and downregulating TEC1 expression. Eukaryot Cell 10: 565–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea JM, Del Poeta M 2006. Lipid signaling in pathogenic fungi. Genomics 9: 352–358 [DOI] [PubMed] [Google Scholar]
- Singh RP, Prasad HK, Sinha I, Agarwal N, Natarajan K 2011. Cap2-HAP complex is a critical transcriptional regulator that has dual but contrasting roles in regulation of iron homeostasis in Candida albicans. J Biol Chem 286: 25154–25170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JM, Segal E, Koller D, Kim SK 2003. A gene-coexpression network for global discovery of conserved genetic modules. Science 302: 249–255 [DOI] [PubMed] [Google Scholar]
- Tanay A, Regev A, Shamir R 2005. Conservation and evolvability in regulatory networks: The evolution of ribosomal regulation in yeast. Proc Natl Acad Sci 102: 7203–7208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DAA, Regev A 2009. Fungal regulatory evolution: cis and trans in the balance. FEBS Lett 583: 3959–3965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Weinberger A, Carmi M, Barkai N 2006. A genetic signature of interspecies variations in gene expression. Nat Genet 38: 830–834 [DOI] [PubMed] [Google Scholar]
- Tirosh I, Wong KH, Barkai N, Struhl K 2011. Extensive divergence of yeast stress responses through transitions between induced and constitutive activation. Proc Natl Acad Sci 108: 16693–16698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankov AM, Thompson DA, Socha A, Regev A, Rando OJ 2010. The role of nucleosome positioning in the evolution of gene regulation. PLoS Biol 8: e1000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltman P, Kacmarczyk T, Bate A, Kearns D, Reiss D, Eichenberger P, Bonneau R 2010. Multi-species integrative biclustering. Genome Biol 11: R96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski I, Pfeffer A, Friedman N, Regev A 2007a. Automatic genome-wide reconstruction of phylogenetic gene trees. Bioinformatics 23: i549–i558 [DOI] [PubMed] [Google Scholar]
- Wapinski I, Pfeffer A, Friedman N, Regev A 2007b. Natural history and evolutionary principles of gene duplication in fungi. Nature 449: 54–61 [DOI] [PubMed] [Google Scholar]
- Wapinski I, Pfiffner J, French C, Socha A, Thompson DA, Regev A 2010. Gene duplication and the evolution of ribosomal protein gene regulation in yeast. Proc Natl Acad Sci 107: 5505–5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlbach DJ, Thompson DAA, Gasch AP, Regev A 2009. From elements to modules: Regulatory evolution in Ascomycota fungi. Curr Opin Genet Dev 19: 571–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Xie D, Cao X, Yu P, Xing X, Chen C-C, Musselman M, Xie M, West FD, Lewin HA, et al. 2012. Comparative epigenomic annotation of regulatory DNA. Mol Cell 149: 1381–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]