

Abstract
Cys2-His2 zinc finger proteins (ZFPs) are the largest group of transcription factors in higher metazoans. A complete characterization of these ZFPs and their associated target sequences is pivotal to fully annotate transcriptional regulatory networks in metazoan genomes. As a first step in this process, we have characterized the DNA-binding specificities of 129 zinc finger sets from Drosophila using a bacterial one-hybrid system. This data set contains the DNA-binding specificities for at least one encoded ZFP from 70 unique genes and 23 alternate splice isoforms representing the largest set of characterized ZFPs from any organism described to date. These recognition motifs can be used to predict genomic binding sites for these factors within the fruit fly genome. Subsets of fingers from these ZFPs were characterized to define their orientation and register on their recognition sequences, thereby allowing us to define the recognition diversity within this finger set. We find that the characterized fingers can specify 47 of the 64 possible DNA triplets. To confirm the utility of our finger recognition models, we employed subsets of Drosophila fingers in combination with an existing archive of artificial zinc finger modules to create ZFPs with novel DNA-binding specificity. These hybrids of natural and artificial fingers can be used to create functional zinc finger nucleases for editing vertebrate genomes.
The deconvolution of transcriptional regulatory networks in metazoan genomes remains a problem of intense scientific interest. Analysis of transcriptional regulation in Drosophila has provided a mainstay for efforts to understand regulatory systems on an organismic level. Foundational studies focused on subsystems (both cis-regulatory elements and their collaborating trans-acting factors) controlling aspects of early developmental patterning (Hong et al. 2008; Wunderlich and DePace 2011). More recently, the advent of system-wide methodologies coupled with high-throughput sequencing technology has fueled the genome-wide analysis of nucleosome occupancy, chromatin modification states, insulator elements, transcription factor (TF) and RNA polymerase II binding sites, and tissue and temporal gene expression patterns (MacArthur et al. 2009; Schuettengruber et al. 2009; Negre et al. 2010, 2011; Roy et al. 2010; Graveley et al. 2011; Kaplan et al. 2011; Kharchenko et al. 2011; Li et al. 2011; The ENCODE Project Consortium 2012). However, for TFs in particular there is a limited (but growing) amount of genome-wide binding data (MacArthur et al. 2009; Schuettengruber et al. 2009; Roy et al. 2010; Negre et al. 2011; Neph et al. 2012; Wang et al. 2012). In its absence, knowledge of TF DNA-binding specificities within regulatory networks in concert with data sets on chromatin accessibility and modifications can be exploited by computational algorithms to predict genomic occupancy and thereby construct more elaborate transcriptional regulatory models (Elrod-Erickson et al. 1996; Noyes et al. 2008b; Segal et al. 2008; Badis et al. 2009; Jaeger et al. 2010; Kazemian et al. 2010; Negre et al. 2011; Zhu et al. 2011b; The ENCODE Project Consortium 2012; Marbach et al. 2012; Neph et al. 2012).
Cys2-His2 zinc finger proteins (ZFPs) are the largest class of TFs within the majority of metazoan genomes (Vaquerizas et al. 2009) and, as such, hold great potential for elaborating tissue/temporal-specific transcriptional regulatory programs. While many other large families of DNA-binding domains (e.g., homeodomains [Berger et al. 2008; Noyes et al. 2008a], basic helix-loop-helix (bHLH) [Grove et al. 2009], and E-twenty six [ETS] [Wei et al. 2010]) have been partially or completely characterized in a metazoan genome, ZFPs remain an outstanding group that has only seen a small fraction of its members characterized (Badis et al. 2008, 2009; Noyes et al. 2008b; Zhu et al. 2009; Jolma et al. 2010; Neph et al. 2012; Wang et al. 2012). Moreover, unlike other TF families where there is a high degree of homology between the resident factors in diverse species (Berger et al. 2008; Noyes et al. 2008a; Grove et al. 2009; Wei et al. 2010), evolutionary analysis of metazoan genomes reveals a dichotomy within the resident ZFPs: A subset displays a high degree of homology within their DNA-binding domains across species presupposing a conservation of function (Seetharam et al. 2010), whereas for other ZFPs the number and composition of fingers appear highly dynamic even over short evolutionary distances (Emerson and Thomas 2009; Groeneveld et al. 2012).
Correspondingly, ZFPs, unlike many other prominent families of DNA-binding domains, have the potential to specify a wide variety of different DNA sequences. This property is a function of the diverse DNA recognition potential of the zinc finger motif and the ability of finger units to be assembled in a tandem array to facilitate the recognition of a target sequence that represents the composite specificities of the incorporated finger modules. The recognition properties of individual zinc fingers can be influenced by their position in an array and the recognition determinants of their immediate neighbors (Desjarlais and Berg 1993; Wolfe et al. 1999; Dreier et al. 2001; Sander et al. 2009; Zhu et al. 2011a), but in some cases, in particular for subsets of specificity determinants with well-defined recognition properties, individual fingers can be assembled in novel combinations to create new recognition modalities (Desjarlais and Berg 1993; Segal et al. 1999; Dreier et al. 2000, 2001, 2005; Liu et al. 2002; Bae et al. 2003; Kim et al. 2009; Zhu et al. 2011a). Although some principles that govern the recognition properties of zinc fingers have been developed through the analysis of natural (Pavletich and Pabo 1991, 1993; Fairall et al. 1993; Laity et al. 2000; Bae et al. 2003) and artificial (Rebar and Pabo 1994; Segal et al. 1999; Dreier et al. 2000, 2001, 2005; Liu et al. 2002; Bae et al. 2003; Maeder et al. 2008; Kim et al. 2009; Sander et al. 2011; Zhu et al. 2011a; Gupta et al. 2012) ZFPs, the ability to accurately predict the DNA-binding specificity of naturally occurring zinc finger assemblies remains suboptimal.
Herein we describe a broad survey of the DNA-binding specificities of ZFPs within Drosophila. Using a bacterial one-hybrid (B1H) selection system (Noyes et al. 2008b), we have characterized the DNA-binding specificities of 93 Cys2-His2 ZFPs. This data set includes 23 alternate splice isoforms that change the finger composition within the ZFP and their resulting DNA-binding specificity, highlighting how different isoforms can increase the complexity of available trans-acting factors for gene regulation without expanding gene number. These data can be used to predict genomic targets for these TFs within the Drosophila genome. In addition, we have defined the orientation and register of individual fingers on their characterized recognition sequences for the majority of these ZFPs, which allows us to estimate the breadth of recognition potential present for fingers within the Drosophila genome. We demonstrate the utility of these data by constructing ZFPs from a combination of Drosophila and artificial fingers with adequate specificity for use in zinc finger nucleases (ZFNs).
Results
Determining the DNA-binding specificities of Drosophila ZFPs
Based on hidden Markov model (HMM) analysis of proteins in the Drosophila genome, there are at least 327 genes containing putative Cys2-His2 zinc fingers (Fig. 1A). In general, identified fingers conform to the consensus sequence: (F/Y)-X-C-X(2-5)-C-X3-(F/Y)-X5-Ψ-X2-H-X(3-5)-(H/C), where X represents any amino acid and Ψ a large hydrophobic amino acid (Klug 2010). This sequence folds into a ββα motif around a single zinc ion, where residues on the “recognition” helix make base-specifying contacts in DNA-binding fingers (Fig. 1B). However, Cys2-His2 zinc fingers can also participate in protein–RNA (Pelham and Brown 1980) and protein–protein (Brayer and Segal 2008) interactions. Two hundred eighty-two genes contain tandem finger arrays with a broad distribution of linker lengths joining neighbors (Supplemental Fig. 1A). Five amino acids is the most common linker length, and this group displays a consensus (TGE[K/R]P) (Supplemental Fig. 1B) that is a hallmark of DNA-binding fingers that dock in a “canonical” mode within the major groove (Laity et al. 2000; Wolfe et al. 2000). Thus, if we conservatively assume that any five-amino-acid linker within our data set is related to a TGE(K/R)P-type linker, a large fraction of multi-finger ZFPs (216 of 282) have DNA-recognition potential (Supplemental Fig. 1C).
Figure 1.

Distribution of Cys2-His2 zinc fingers in genes within D. melanogaster genome. (A) Distribution of the number of fingers identified within each zinc-finger-containing gene in the fruit fly genome. (B) A schematic depicting canonical DNA recognition by a Cys2-His2 zinc finger. The numbered spheres on the α-helix represent the residues that are anticipated to contact DNA in the canonical recognition mode. These residues are numbered relative to the start of the α-helix and make contact (arrows) with their respective color-coded DNA bases (boxes). Each finger (in an N-terminal to C-terminal orientation) binds its DNA subsite (labeled 5′ to 3′) in an anti-parallel arrangement. (C) Number of ZFPs attempted and the success rate of these B1H selections. (D) Comparative MatAlign analysis of ZFP motifs determined by B1H and other methods (Hallikas et al. 2006; Robasky and Bulyk 2011). B1H motifs are designated by red ovals.
We have employed a B1H system to determine the DNA-binding specificity of these zinc finger domains (Meng et al. 2005, 2008; Noyes et al. 2008b; Chu et al. 2012). We extracted a “cluster” of closely linked fingers (fewer than 20 amino acids between adjacent fingers) for analysis to minimize the amount of superfluous sequence expressed in the B1H system. Some proteins, such as CG4360, contain multiple well-separated finger clusters, which were characterized as independent recognition units (Supplemental Fig. 1D). Each zinc finger cluster was displayed as a C-terminal fusion to the omega subunit of Escherichia coli RNA polymerase without an accessory DNA-binding domain (Noyes et al. 2008b). Complementary binding sites for each ZFP were identified through a single round of selection from a 28-bp randomized library with the recovered sequences characterized by both Sanger and Illumina sequencing (Zhu et al. 2011a; Gupta et al. 2012). Recognition motifs were identified as overrepresented sequence motifs within the recovered sequences (Zhu et al. 2011a; Christensen et al. 2012).
To date, we have successfully characterized the DNA-binding specificity of ZFPs encoded by 70 Drosophila genes (Fig. 1C; Supplemental Fig. 2). Our success rate varied depending on the number of zinc fingers present in the cluster and the presence of canonically linked fingers (Supplemental Fig. 3). In general, our B1H motifs show a high degree of similarity to previously defined recognition motifs where these data exist, providing confidence in the quality of our data set (Fig. 1D).
Predictive value of ZFP recognition motifs
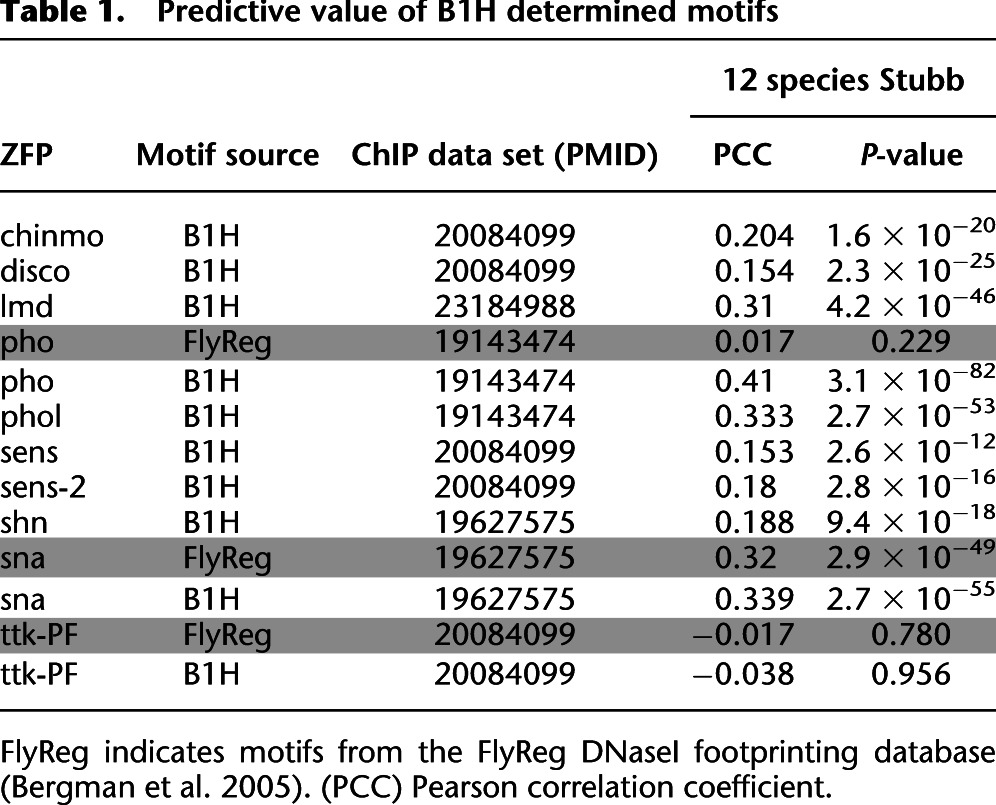
Recognition motifs for TFs within a common regulatory network can be used to computationally identify putative cis-regulatory modules and define the regulatory role of each member (Kazemian et al. 2010; Kaplan et al. 2011; Schroeder et al. 2011; Marbach et al. 2012; Neph et al. 2012). Previously, we validated B1H-defined recognition motifs for TFs involved in anterior-posterior axis segmentation by demonstrating their ability to discriminate genomic regions corresponding to ChIP-chip peaks for each factor from randomly chosen noncoding regions (Kazemian et al. 2010). These TFs spanned multiple families, including ZFPs. We performed a similar assessment of our new ZFP recognition motifs using recently published ChIP data for nine factors (Chinmo, Disco, Lmd, Pho, Phol, Sens, Shn, Sna, and Ttk) (MacArthur et al. 2009; Schuettengruber et al. 2009; Negre et al. 2010; Busser et al. 2012). We evaluated binding potential to each genomic segment using Stubb scores, which reflect motif frequency and strength within each region, phylogenetically averaged over 12 fruit fly species (Kazemian et al. 2010, 2011). For all but one factor, Ttk (Tramtrack), we find that the B1H motif provides significant discrimination between the top 1000 ChIP-bound regions and a random set of noncoding regions (Table 1). In this analysis, our B1H motifs perform similar to or better than FlyReg motifs for three of these factors (Pho, Sna, and Ttk) (Bergman et al. 2005).
Table 1.
Predictive value of B1H determined motifs
Added recognition potential from alternately spliced ZFP isoforms
Organisms can diversify the regulatory potential of a TF through the generation of alternately spliced isoforms (Nilsen and Graveley 2010). In many instances, an alteration in the composition of domains associated with a DNA-binding domain can change its regulatory potential at a common set of target sites. However, alternate splicing can also change the composition of the DNA-binding domain and thereby its DNA-recognition potential (e.g., Cf2) (Gogos et al. 1992). In Drosophila, 28 zinc finger-encoding genes have alternately spliced isoforms of this type (Supplemental Table 3). Many alterations simply change the number of fingers at the N or C terminus of an array, which should preserve the core recognition potential of common fingers between isoforms. However, 10 genes encode alternate isoforms where the insertion or substitution of one or more internal fingers within an array could radically alter recognition properties. We determined the DNA-binding specificity of 23 splice isoforms from this group to assess their recognition potential. Many of these alternately spliced ZFP isoforms, such as found in broad (Supplemental Fig. 4) and ttk (Supplemental Fig. 5), display distinct specificities that expand their regulatory potential (Supplemental Discussion).
The 23 isoforms of lola (longitudinals lacking) highlight the increased regulatory capacity realized through this mechanism. In the developing nervous system, lola directs a myriad of axon guidance decisions through the spatial and temporal expression of different isoforms (Supplemental Fig. 6; Seeger et al. 1993; Giniger et al. 1994; Madden et al. 1999; Crowner et al. 2002; Goeke et al. 2003). We determined the DNA-binding specificity of 17 Lola isoforms, which include 13 distinct sets of zinc finger clusters. The resulting family of motifs reveals the diverse recognition potential generated through alternate splicing (Fig. 2). Notably, all of the Lola isoforms contain a common BTB domain. This domain could facilitate heterodimerization between isoforms (Badenhorst et al. 2002; Bonchuk et al. 2011), which would further expand the complexity of recognition motifs recognizable by isoforms from this locus.
Figure 2.

Comparison of isoform specificities. DNA-binding specificities of 17 Lola isoforms generated through alternate splicing. MatAlign clustergram emphasizing the diversity within the recognition motifs of the various Lola isoforms. All of the characterized ZFPs utilize a pair of zinc fingers to recognize DNA. Identical fingers are present in the lola-PN and -PY isoforms and the lola-PT and -PU isoforms, and both pairs have identical specificity.
Global comparison of ZFP specificities
We constructed a pairwise alignment of the 94 ZFP B1H recognition motifs based on their similarity to assess the breadth of the recovered recognition sequences. These data were used to construct a phylogenetic tree, providing a visual framework for examining the interrelatedness of the recognition preferences of each ZFP (Fig. 3). This global perspective highlights the degree of diversity within these ZFP recognition sequences. As expected, families of ZFPs sharing similar finger arrays display similar recognition motifs (e.g., Sp/KLF, EGR, YY1, Gli/Opa, Snail/Slug, Odd, Gfi, and ZFAM4) (Seetharam et al. 2010). Interestingly, while three of the four Broad isoforms cluster together, the Lola isoforms are highly dispersed throughout the tree, demonstrating the diversity of recognition sequences that can be generated from a single locus. It is not uncommon for TFs in different families to have overlapping DNA-binding specificities, where potential competition for binding sites can create an added layer of regulatory potential (Ip et al. 1992; Kuo and Calame 2004; Reece-Hoyes et al. 2009). Likewise, some ZFP motifs overlap with the previously defined recognition motifs of other factors. For example, the recognition motifs for Shn and NF-KB are highly similar (Supplemental Fig. 7). Consistent with this observation, HIVEP1, the human homolog of Shn (Staehling-Hampton et al. 1995), can bind NF-KB recognition sequence in the HIV LTR (Maekawa et al. 1989; Baldwin et al. 1990; Fan and Maniatis 1990).
Figure 3.

Phylogenetic comparison of the B1H-determined recognition motifs for 94 Drosophila ZFPs based on the primary recognition strand. ZFPs conserved across the Drosophila and human genomes are specified with their family labels.
Assigning individual fingers to subsites within each recognition motif
We made strand-specific assignments of individual fingers to specific DNA subsites within each ZFP recognition motif to estimate the diversity of finger specificities encoded within Drosophila. In many cases, these assignments were straightforward as certain fingers within a cluster had specificity determinants with well-defined recognition preferences (Supplemental Discussion) that could be associated with a complementary DNA subsite within the recovered motif (Supplemental Fig. 8). Such a positioned finger served as an anchor, allowing the positions of neighboring fingers within the recognition sequence to be assigned assuming that fingers within the cluster docked to the DNA in a canonical geometry (with overlapping four base-pair recognition elements).
This assumption is likely valid for the majority of our characterized ZFPs since they are predominantly canonically linked (Supplemental Fig. 3). Using this anchoring approach, we associated fingers with subsites for 61 of 94 recognition motifs.
To facilitate the assignment of the remaining finger sets, we determined the DNA-binding specificity of a subset of fingers from a characterized cluster deemed likely to harbor some of its recognition potential. This strategy utilized two related approaches. In most cases, we extracted a subset of the fingers (typically three) from a larger finger array and determined their DNA-binding specificity (Supplemental Fig. 9). As an alternate assessment, we spliced subsets of one or two fingers from a cluster in question to fingers from another ZFP with well-defined DNA-binding specificity (Supplemental Fig. 10). Once determined, these subset specificities provided anchors for assigning the recognition positions of other linked fingers within the array. Using these approaches, we determined the specificity of 34 zinc finger subsets or spliced finger sets from 26 different genes (Supplemental Fig. 11). Based on this analysis, we could successfully dock 83 of the 94 zinc fingers sets (genes and alternately spliced variants) on their recognition sequences. Delineating the mode of recognition for a small number of ZFPs (e.g., CG14962) remains problematic even after this additional analysis.
Using these assignments, we deconvoluted the assigned 83 ZFPs into 238 single finger–DNA subsite combinations (Supplemental Data Set 1). Sorting these fingers based on their apparent core DNA triplet preference provides a perspective on the breadth of “recognition” space that appears to be specified by this extant zinc finger set. As expected, a high percentage of classical recognition fingers are found within this data set. For example, the RSDELXR recognition helix occurs eight times, displaying a G(c/t)G specificity. In addition, a number of novel recognition units are present, such as the second finger of Sens (QKSDMKK), which appears to specify TC(a/t) within its primary triplet sequence. Remarkably, 157 of these 238 fingers demonstrate a strong preference at the three core recognition positions. These fingers span 47 of the 64 possible triplet sequences (Fig. 4; Supplemental Table 4), demonstrating the inherent diversity of the recognition modalities within naturally occurring zinc fingers sets. For bins of recognition helices that have multiple unique members, there is typically a preference for certain determinants at the key recognition positions (Supplemental Fig. 12; Supplemental Table 5).
Figure 4.

Diversity of triplet recognition sequences. Coverage of the 64 possible triplet sequences based on the specificity of the extracted single finger–DNA subsites combinations. Each panel represents 16 different triplets, where the 5′ base is fixed (e.g., upper left is the ANN triplets). The height of the buttons at each position reflects that number of fingers that prefer this triplet within the data set, where those triplets without complementary fingers are white.
Examining specificity determinant–DNA base associations
We analyzed the specificity determinants associated with assigned finger–DNA subsite combinations to gain further insight into fundamental aspects of DNA-recognition. Assuming a canonical binding model, we assigned specificity determinants to each DNA base within the primary triplet (i.e., positions 6, 3, and −1 of the recognition helix to the 5′, middle, and 3′ base, respectively as shown in Fig. 1B). This analysis suggests complementarity between particular amino acid–base combinations (Fig. 5; Supplemental Fig. 13). We note, however, that this analysis only includes the naturally occurring diversity of our ZFP set and should not be interpreted to represent all of the possible specificities that might be observed in in vitro experiments. Nonetheless, many of these associations, such as the pairing of Arg at position −1 with Guanine and Asn at position 3 with Adenine, represent well-defined recognition preferences (Isalan et al. 1998; Wolfe et al. 2000; Dreier et al. 2001; Sera and Uranga 2002; Gupta et al. 2012). In addition, other strong associations are present, particularly for aromatic residues, that have not been broadly employed in artificial fingers or characterized across multiple naturally occurring ZFPs. Notably, a preference of Tyr at position −1 for Thymine is consistent with the specificity of artificial fingers containing Tyr at this position (Zhu et al. 2011a). Likewise, the preference of Tyr at position 3 for Adenine is consistent with the specificity of artificial fingers generated by Sangamo BioSciences (Hockemeyer et al. 2009) and us (Supplemental Fig. 14).
Figure 5.

Amino acid–base correlations. Frequency logo displaying the average base preference for each amino acid at each recognition position on the recognition helix (RH) assuming canonical recognition. The total number of recognition helices and the number of unique recognition helices (having a unique set of residues at positions −1, 2, 3, and 6) that contain the amino acid at that position are indicated above each logo. Base position nomenclature is defined in Figure 1B.
In the context of canonical recognition, position 2 of the recognition helix can influence base preference immediately 3′ to the primary recognition triplet through contact with the complementary DNA strand (Elrod-Erickson et al. 1996; Isalan et al. 1997). Assigning base preference at this position is complicated by the potential of a neighboring N-terminal finger to influence specificity at this base through position 6 of its recognition helix. Thus, associations between a particular amino acid at position 2 and a certain neighboring base should be interpreted cautiously. At minimum, any preference implies compatibility of the observed amino acid–base combination, and for some amino acids at position 2, this interaction may be the dominant determinant defining base preference (Supplemental Discussion).
Testing the recognition preference of a subset of Drosophila fingers
To demonstrate the quality of our zinc finger–DNA subsite assignments, we utilized these finger sets in the assembly of artificial zinc finger arrays (ZFAs) with new composite DNA-binding specificities. Characterized fingers from naturally occurring ZFPs have been successfully utilized as modules to assemble artificial TFs or nucleases for targeted gene disruption (Bae et al. 2003; Kim et al. 2009, 2011). While single fingers—primarily of artificial origin (Segal et al. 1999; Dreier et al. 2000, 2001, 2005; Liu et al. 2002; Zhu et al. 2011a)——have been the mainstay of archives for the assembly of ZFAs with novel DNA-binding specificity (Liu et al. 1997; Carroll et al. 2006; Mandell and Barbas 2006; Wright et al. 2006; Kim et al. 2009; Zhu et al. 2011a; Bhakta et al. 2013), more recent assembly methods have focused on archives of two-finger modules (Doyon et al. 2008; Kim et al. 2011; Sander et al. 2011; Gupta et al. 2012; Zhu et al. 2013) to reduce the number of “novel” finger–finger interfaces that are incorporated into the ZFA (Urnov et al. 2010). Consequently, we examined the utility of one and two finger Drosophila modules for the creation of ZFAs with novel specificity. Target sites were chosen to allow the construction of ZFNs from these ZFAs for six different genes (cpe, irs1, irs1b-like, nhlh2, nr3c1, and pparg) within the zebrafish genome to provide an in vivo assessment of their quality.
Eight of the constructed four-finger (4F) ZFAs incorporate one or two Drosophila fingers in combination with artificial single- and two-finger modules from our existing archives (Gupta et al. 2011; Zhu et al. 2011a, 2013). In the construction of these ZFAs, the incorporated Drosophila finger sequences were used in their entirety, whereas fingers from our artificial archive use the Zif268 or SP1C (Shi and Berg 1995) backbone (Supplemental Table 6). The DNA-binding specificity of these ZFAs were characterized using our B1H system to determine if the incorporated Drosophila modules display the anticipated DNA-binding specificity and are compatible with neighboring finger units for recognition. Five of eight ZFAs containing Drosophila fingers displayed the expected specificity and exhibited coordinated recognition with neighboring fingers within the array (Fig. 6). For two of the failed ZFAs (3p_nr3c1 and 3p_pparg), the Drosophila fingers displayed the desired DNA-binding specificity but proved incompatible with neighboring fingers. The two Lola-PW fingers in 3p_nr3c1 failed to collaborate in recognition with neighboring fingers until their recognition helices were grafted into the Zif268 backbone (3p_nr3c1_n ZFA). The Ci and Sna fingers in 3p_pparg ZFA, which are joined by a canonical linker, display a preference for an additional “C” between their subsites (GAC and CTG, respectively). This noncanonical behavior originates from the Ci finger, as the structure of the human homolog (Gli) reveals an altered docking geometry that affords recognition of an additional 3′ base pair (Pavletich and Pabo 1993). The preservation of specificity in both the Ci and Sna fingers in this artificial assembly implies that their docking geometry is driven by intrinsic features (e.g., the constellation of phosphate contacts) rather than the composition of the interfinger linker. Thus, these results demonstrate that the individual finger specificity assignments tested in these arrays were correct but that the interfaces between fingers are not always compatible.
Figure 6.

Drosophila finger sets maintain their specificity when incorporated into artificial arrays. The left column displays the B1H-determined recognition motif for each assembled ZFA. For each motif, the subsite recognized by the utilized fingers in the ZFA and Drosophila ZFP (middle column) is boxed, and where these are similar, the assembly was deemed a success (check; right column). In some cases fingers from more than one Drosophila ZFP were used in the artificial finger assembly. In the case of 3p_nr3c1, due to the initial failure (X), two additional variants were constructed (3p_nr3c1_n and 3p_nr3c1_nn) to achieve the desired DNA-binding specificity. The complements for some of these ZFN pairs are entirely artificial in construction and are thus shown in Supplemental Figure 15.
ZFNs containing Drosophila fingers are functional in vivo
Overall, pairs of ZFAs with compatible specificity for five of six ZFN target sites were successfully constructed (Fig. 6; Supplemental Fig. 15). The activity of ZFNs constructed from these ZFAs was determined in zebrafish embryos (Meng et al. 2008). Often, equal concentrations of mRNA encoding each ZFN monomer are coinjected into embryos. However, in some cases we also examined ZFN activity at different monomer ratios based on the B1H activity of individual ZFAs (Supplemental Table 7). An altered monomer ratio sometimes appeared to modestly increase activity or reduce toxicity. Three of five tested ZFN pairs generated lesions at the desired target site with efficiencies in normal embryos between 2% and 7% (Supplemental Figs. 16–18). Activity in a fourth ZFN pair (irs1b-like) was achieved by introducing Arg at position 6 within the recognition helix of the C-terminal Sens2 finger to improve its preference for G within the corresponding position of its subsite (Supplemental Figs. 15, 19). These data demonstrate that ZFAs containing Drosophila fingers in combination with artificial fingers have sufficient specificity and affinity to generate functional ZFNs in a complex vertebrate genome.
Discussion
Our B1H analysis of Cys2-His2 zinc fingers within the Drosophila genome has generated 94 recognition motifs that span 70 genes and 23 additional alternately spliced isoforms with variant specificities. To our knowledge, this represents the largest block of ZFP specificities that have been curated for any metazoan genome. Where specificity data are available for orthologous ZFPs from other species, we find that there is good concordance between the data sets. Consequently, we believe that these data are of high quality. Consistent with this assertion, we find that our motifs provide significant predictive power for the identification of bound genomic regions in existing ChIP data sets for the corresponding ZFPs (Table 1). The size of our recovered recognition motif increases as the number of fingers in the ZFP increases from two to three fingers but plateaus thereafter (Supplemental Fig. 20). Consequently, for ZFPs containing a large numbers of fingers (e.g., crol), our identified motif may represent only a portion of its full recognition potential due to limitations of our selection method.
Recognition motifs and primary data for these ZFPs are available through our web portal FlyFactorSurvey (http://pgfe.umassmed.edu/ffs/), which now harbors published and unpublished recognition motifs for more than 300 predicted Drosophila TFs (Zhu et al. 2011b). Predicted genome binding profiles for these Drosophila factors have been constructed within Genome Surveyor (http://veda.cs.uiuc.edu/gs) where combinations of these motifs can be coupled with evolutionary comparisons across 12 Drosophila species for the discovery of cis-regulatory modules (Noyes et al. 2008b; Kazemian et al. 2011). These specificity data can be combined with expression patterns of these TFs to further refine cis-regulatory module prediction (Kazemian et al. 2010).
In this study we surveyed ZFPs from 184 genes, representing 56% of the predicted ZFPs within the genome. Our success rate was lower (∼38%) than in previous studies utilizing the B1H system for TF analysis (Noyes et al. 2008a,b). Some failures likely represent true negatives, where the characterized ZFP binds to other proteins or RNA, instead of DNA. Consistent with this hypothesis, higher success rates were achieved for ZFPs that are entirely canonically linked (Supplemental Fig. 3), which is a hallmark of DNA-binding zinc fingers. However, we failed to determine the specificity of some ZFPs, such as CTCF and TRL (also known as GAGA), that have sequence-specific DNA-binding activity (Bergman et al. 2005; Holohan et al. 2007). Some failures (false negatives) may originate from biases in our library. For example, we found that the CTCF binding site when cloned into our reporter vector activated transcription of the reporter genes in the absence of CTCF, likely through the function of an endogenous factor (data not shown). Self-activating sequences are depleted from the library prior to use via counter-selection (Meng et al. 2005). In other cases, such as Cbt (a paralog to successfully characterized Sp1 family members), the gene or protein sequence may be incompatible with function in bacteria.
Where possible, we have extended our characterization of ZFPs by assigning DNA subsites to the recognition of individual fingers within each ZFA. This provides an opportunity to assess the true breadth of the recognition potential of extant ZFPs within a genome, even for this incomplete set. We find that 47 of the 64 potential DNA triplets are represented within the finger subsites recognized by 83 characterized ZFPs, where we could putatively assign the orientation and register of the fingers on the DNA. The recognition potential of these fingers is the most diverse described to date for naturally occurring ZFPs, substantially surpassing the analysis of approximately 2000 individual human fingers that generated an archive capable of recognizing 25 of the 64 potential triplets (Bae et al. 2003). Whether ZFPs within the fly genome are more diverse in their recognition potential than those found in humans will remain unclear until a comprehensive analysis of all ZFPs in both genomes is available. However, there are specificity determinant sets in the fly genome, such as the Aef1 fingers that specify a repeating ACA triplet, that are not present within the human zinc finger repertoire.
From our results, it is clear that naturally occurring ZFPs utilize a broad palette of specificities to define distinguishing recognition sequences. This is consistent with the evolutionary diversity within this family (Tadepally et al. 2008; Emerson and Thomas 2009; Thomas and Emerson 2009), and with selection-based approaches to engineer zinc fingers with novel DNA-binding specificity that have generated fingers capable of recognizing a broad variety of sequences (Carroll et al. 2006; Urnov et al. 2010). The utilization of a broad range of DNA recognition preferences by naturally occurring ZFPs is in sharp contrast to homeodomains, the second most-common family of DNA-binding domains in metazoan genomes, which appear to utilize only a small fraction of their true recognition potential in natural systems (Chu et al. 2012). In contrast to homeodomains, zinc fingers appear to function as highly malleable units that permit facile rewiring of regulatory systems by providing a wealth of new regulatory potential as trans-acting factors that can readily evolve novel recognition modalities.
The assignment of zinc finger–DNA subsite combinations within this data set allows the correlation of specificity determinants and base preferences. This information can be used in conjunction with existing data sets to train improved predictive recognition models for ZFPs. The expansive evolutionary diversity present among naturally occurring ZFPs underlies the importance of creating a robust predictive model to assess the regulatory potential of members of this family in any genome, as it is unlikely that the specificity of all extant ZFPs can be inferred by direct homology from characterized ZFPs resident in a small number of organisms.
Methods
Discovery and clustering of Cys2-His2 ZFPs for analysis
ZFPs were identified based on the motif annotations within the SMART database (http://smart.embl.de/) (Letunic et al. 2012) and HMMER analysis using hmmsearch (Finn et al. 2011) of proteins within FlyBase (McQuilton et al. 2012) with a HMM based on the consensus Cys2-His2 zinc finger motif within PFAM (Punta et al. 2012). ZFAs within these genes were then classified into clusters, where a single cluster is any set of fingers linked by an amino acid sequence of less than 20 residues. Thus, ZFPs composed of two or more fingers could exist as a single cluster or multiple clusters of fingers (Supplemental Table 1). Boundaries for the core Drosophila melanogaster DNA-binding domain to be used in the specificity analysis were defined through TBLASTN comparisons with Drosophila pseudoobscura, Drosophila virilis, and Drosophila grimshawi, by identifying two sequential amino acid positions that were not conserved between these species.
Preparation of Drosophila genomic DNA for amplification of ZFAs
Ten anesthetized flies were collected in an Eppendorf tube, frozen at −80°C and ground in 200 μL Buffer A (100 mM Tris-HCl at pH 7.5, 100 mM EDTA, 100 mM NaCl, 0.5% SDS) with a disposable tissue grinder (Kontes). With the addition of another 200 μL aliquot of Buffer A, grinding was continued until only cuticles remained. This mixture was incubated for 30 min at 65°C, after which 800 μL LiCl/CH3COOK solution (1 part 5 M CH3COOK stock: 2.5 parts 6 M LiCl stock) was added and incubated on ice for at least 10 min. This was followed by a 15-min spin at 15000 r.p.m. in a table-top centrifuge. One milliliter of the resulting supernatant was transferred into a new tube, avoiding the floating debris. Six hundred microliters of isopropanol was added to the supernatant, mixed and further spun at 15,000 r.p.m. for 15 min. The supernatant was aspirated away, and the pelleted DNA washed gently with 70% ethanol, air-dried, and resuspended in 75 μL TE buffer. This genomic DNA was stored at −20°C.
B1H-binding site selections using the 28-bp library
In our characterization of D. melanogaster ZFPs, we truncated the coding sequence of each gene to span a “cluster” of fingers that were closely linked (less than 20 amino acids between adjacent fingers) (Supplemental Tables 1, 2). For example, for CTCF all 11 zinc fingers were assayed as a single cluster. For genes with multiple well-separated finger clusters, the clusters were characterized as independent recognition units. ZFA clusters were obtained by PCR from cDNA clones of the BDGP DGC Gold and TF collections (Stapleton et al. 2002; Lin et al. 2007) or D. melanogaster genomic DNA. Each zinc finger cluster was cloned as a C-terminal fusion to the omega subunit of E. coli RNA polymerase in the B1H system. Selections were carried out according to the method previously described (Noyes et al. 2008b) by plating 1–2 × 107 selection strain cells transformed with the 1352-omega-UV2, 1352-omega-UV5, or 1352-omega-lppC ZFA-containing expression plasmid and the 28-bp pH3U3 library plasmid on NM minimal medium selective plates. These selection plates contained 0 μM or 5 μM uracil, 10 μM IPTG, and 3-amino-1,2,4-triazole (3-AT; 2.5 mM, 5 mM, 10 mM, or 15mM) as the HIS3 competitive inhibitor and were incubated for 36–72 h at 37°C. After the number of surviving bacterial colonies were counted, ZFAs displaying threefold or greater increase in colony numbers over a no ZFA control were deemed successful selections. Sanger sequencing was initially used to characterize complementary binding sites for each successful ZFP selection with overrepresented motifs identified through MEME analysis (Bailey and Elkan 1994). Promising selections were further characterized by Illumina sequencing amplicons spanning the library region from pooled surviving colonies where the sample preparation of selected binding sites for deep sequencing was undertaken according to the method described previously (Gupta et al. 2011; Zhu et al. 2011a). Unique sequences from each selection were ranked based on the number of recovered reads. Subsequently, binding site recognition motifs were identified as overrepresented sequence motifs within these recovered sequences using MEME, where motifs constructed from the Illumina sequencing can contain thousands of unique binding sites (Christensen et al. 2012).
Clustering of determined binding site motifs
Strand-specific comparative MatAlign (Matalign-v2a) (T Wang and GD Stormo, unpubl.) analysis of ZFP motifs was used to generate neighbor joining trees (NJs), to depict the inherent diversity, similarity, and clustering of the characterized Cys2-His2 ZFP specificities.
Evaluation of the predictive value of the ZFP motifs based on existing ChIP data
TF-ChIP profiles of eight TFs from early stages of Drosophila embryonic development were downloaded from multiple sources. Data for Disco, Chinmo, Sens, and Ttk were acquired from Negre et al. (2010); Pho and Phol from Schuettengruber et al. (2009); and Shn and Sna from MacArthur et al. (2009). In the case of Disco, ChIP-seq data were used rather than ChIP-chip. For each factor, the raw TF-ChIP read scores were smoothed by averaging them over 500-bp windows with shifts of 50 bp. After this transformation, 1000 nonoverlapping windows with the highest ChIP score (“bound regions”) were selected, along with 1000 random, nonexonic windows from the remaining genome. For each selected window, we used the related DNA-binding motif from B1H (Zhu et al. 2011b) or FlyReg (Bergman et al. 2005) to calculate the STUBB scores of orthologous windows across 12 Drosophila species and then found the average based on the phylogenetic tree, according to the method previously described by Kazemian et al. (2010). This phylogenetically weighted average is called the “motif score” of the window. Finally, the predictive value of the motif was quantified using the Pearson correlation coefficient (PCC) between the motif scores and ChIP scores of the selected 2000 windows.
Assignment of the preferred triplet for each zinc finger
Three base pair submotifs were extracted for individual zinc fingers that were successfully aligned to their target site. A consensus recognition site for each finger was determined based on a refined consensus alphabet with the following probability thresholds (Mahony and Benos 2007): A/C/G/T is used if the appropriate single base frequency is greater than 0.6; M/R/W/S/Y/K is used if the sum of the appropriate two bases is greater than 0.8; and N is used otherwise. In the assessment of triplet coverage, fingers were counted toward a triplet only if they do not contain “N” at any position, and a two base code (M/R/W/S/Y/K) is allowed only at a single position.
Creation and B1H characterization of ZFAs
Four-finger ZFAs for use in ZFNs were assembled from our characterized Drosophila ZFPs and our in-house two-finger module and single-finger module archives via overlapping PCR according to the method described previously (Gupta et al. 2011; Zhu et al. 2011a). In this assembly, the Drosophila finger sequences were used in their entirety; i.e., their recognition helices were not grafted into the Zif268 backbone, which is the basis of the fingers in our artificial archive. Assembled four-finger ZFAs were cloned into the 1352-UV2 expression vector and characterized in the B1H system using the 28-bp randomized library (Noyes et al. 2008b). Selections were undertaken at 2.5–10 mM 3-AT, 10-50 μM IPTG with or without 200 μM uracil according to the method described previously (Zhu et al. 2011a). A successful selection and recovery of the binding site motif for each ZFA was determined as indicated above for the Drosophila Cys2-His2 ZFPs.
ZFN injections and analysis of somatic lesion frequency
In order to create ZFNs to target genes in zebrafish, assembled ZFA PCR amplicons were digested with Acc65I and BamHI-HF (New England Biolabs). Following gel extraction and purification, these were cloned into pCS2 vectors containing the sequence encoding the DD/RR obligate heterodimeric version of the FokI nuclease domain according to the method described previously (Gupta et al. 2011). For ZFNs targeting sites with a 7-bp spacer, an eight-amino-acid TGPGAAGS linker of nucleotide sequence ACCGGTCCTGGTGCCGCGGGATCC was used in place of the typical LRGS linker to span the ZFA and DD/RR FokI domains (Handel et al. 2009). Subsequently, the pCS2-ZFN constructs were linearized with NotI, and mRNA was transcribed using the mMessage mMachine SP6 kit (Ambion). Injections of ZFN mRNAs into the blastomere of one-cell-stage zebrafish embryos were carried out according to the method described previously (Meng et al. 2008; Gupta et al. 2011). Different ratios of 5′ and 3′ ZFNs were tested for some nucleases to improve the lesion frequencies, where these choices were guided by the relative activities of the associated ZFAs exhibited in the B1H system. After 24 h, ZFN mRNA–injected embryos with normal and deformed appearance (eight to 30 embryos) and uninjected embryos were collected and incubated in 50 mM NaOH (15 μL/embryo) for 15 min at 95°C to isolate genomic DNA. This was subsequently neutralized with 0.5 M Tris-HCl (4 μL/embryo) and centrifuged at 13,000 r.p.m. for 1 min, after which the supernatant containing genomic DNA was utilized in PCRs for lesion analysis (below).
ZFN activity analysis at endogenous zebrafish genes
PCR primers were designed to amplify a ∼200-bp region bordering the ZFN target site using the Phire Hot Start DNA polymerase (Finnzymes), and the PCR was run with 1 μL of the extracted genomic zebrafish DNA in a total reaction volume of 20 μL. ZFN activity was determined via restriction fragment length polymorphism analysis or T7 Endonuclease I assay (New England Biolabs). In the restriction fragment length polymorphism analysis, the 20 μL PCR product was directly digested with a restriction enzyme unique to the spacer region at the ZFN target site in a compatible NEB Buffer for 1 h at 37°C. The digestion products were run on a 3.5% 0.5× TBE UltraPure Agarose (Invitrogen) gel at 200 V for 15–20 min. Band intensities for the uncut PCR product relative to the entire product was used to estimate for the lesion at the ZFN target site using ImageJ (Schneider et al. 2012). Additionally, the restriction enzyme–resistant PCR product fragment was gel extracted and cloned into a Bluescript vector pBS2SK+ (Stratagene) via the EcoRV site. By utilizing blue-white screening, sequences harboring lesions at the ZFN site were recovered after PCR with T7 and T3 universal primers and Sanger sequencing with T3 universal primer.
When T7 Endonuclease I was used to assay for gene targeting by the ZFN constructs (Kim et al. 2009; Reyon et al. 2012), 20 μL PCR product was submitted to the following protocol on a thermocycler: 95°C for 5 min; 95°C to 85°C at −2°C/sec; 85°C to 25°C at −0.1°C/sec; hold at 4°C. Reannealed PCR products from this step were incubated with 10 U of T7 Endonuclease I in a 23 μL reaction for 45 min at 37°C in NEB Buffer 2. The digestion products were run on a 3.5% 0.5× TBE UltraPure Agarose (Invitrogen) gel at 200 V for 15–20 min. Band intensities for the cut PCR product relative to the entire PCR product was used to estimate for the lesion rate (fractional modification = fraction of cleaved bands/2) at the ZFN target site (Guschin et al. 2010) using Image J (Schneider et al. 2012). Furthermore, a set of primers were designed to clone a <100-bp region of genomic DNA bordering the target site of interest into a modified pBS2SK+ vector via the XbaI and Acc65I sites, such that it is in frame with the lacZ gene. By utilizing blue-white screening, sequences harboring out of frame lesions at the ZFN site were recovered by colony PCR of white colonies with T7 and T3 universal primers, subsequent to Sanger sequencing with T3 universal primer (JC McNulty, VL Hall, and SA Wolfe, unpubl.).
Zebrafish lines
The use of zebrafish was in accordance with established protocols (Westerfield 1993) and in conformity with Institutional Animal Care and Use Committee guidelines of the University of Massachusetts Medical School.
Data access
The sequencing data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo) under accession number GSE42709.
Acknowledgments
We thank the other members of the Wolfe and Brodsky laboratories for insightful comments and discussions. Additionally, we thank Richard Weiszmann for generating the Zn-finger TF clone set. We thank Nathan Wolfe for his assistance with constructing Figure 4. Funding for this work was supported by the National Institutes of Health (NIH) grants HG004744 (M.H.B. and S.A.W.), GM068110 (S.A.W), HG000249 (G.D.S.), and P41HG3487 (S.E.C.). Work at Lawrence Berkeley National Laboratory was conducted under Department of Energy contract DEAC02-05CH11231.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.151472.112.
References
- Badenhorst P, Finch JT, Travers AA 2002. Tramtrack co-operates to prevent inappropriate neural development in Drosophila. Mech Dev 117: 87–101 [DOI] [PubMed] [Google Scholar]
- Badis G, Chan ET, van Bakel H, Pena-Castillo L, Tillo D, Tsui K, Carlson CD, Gossett AJ, Hasinoff MJ, Warren CL, et al. 2008. A library of yeast transcription factor motifs reveals a widespread function for Rsc3 in targeting nucleosome exclusion at promoters. Mol Cell 32: 878–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, et al. 2009. Diversity and complexity in DNA recognition by transcription factors. Science 324: 1720–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae KH, Kwon YD, Shin HC, Hwang MS, Ryu EH, Park KS, Yang HY, Lee DK, Lee Y, Park J, et al. 2003. Human zinc fingers as building blocks in the construction of artificial transcription factors. Nat Biotechnol 21: 275–280 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Elkan C 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36 [PubMed] [Google Scholar]
- Baldwin AS Jr, LeClair KP, Singh H, Sharp PA 1990. A large protein containing zinc finger domains binds to related sequence elements in the enhancers of the class I major histocompatibility complex and κ immunoglobulin genes. Mol Cell Biol 10: 1406–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger MF, Badis G, Gehrke AR, Talukder S, Philippakis AA, Pena-Castillo L, Alleyne TM, Mnaimneh S, Botvinnik OB, Chan ET, et al. 2008. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133: 1266–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman CM, Carlson JW, Celniker SE 2005. Drosophila DNase I footprint database: A systematic genome annotation of transcription factor binding sites in the fruitfly, Drosophila melanogaster. Bioinformatics 21: 1747–1749 [DOI] [PubMed] [Google Scholar]
- Bhakta MS, Henry IM, Ousterout DG, Das KT, Lockwood SH, Meckler JF, Wallen MC, Zykovich A, Yu Y, Leo H, et al. 2013. Highly active zinc-finger nucleases by extended modular assembly. Genome Res 23: 530–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonchuk A, Denisov S, Georgiev P, Maksimenko O 2011. Drosophila BTB/POZ domains of “ttk group” can form multimers and selectively interact with each other. J Mol Biol 412: 423–436 [DOI] [PubMed] [Google Scholar]
- Brayer KJ, Segal DJ 2008. Keep your fingers off my DNA: Protein-protein interactions mediated by C2H2 zinc finger domains. Cell Biochem Biophys 50: 111–131 [DOI] [PubMed] [Google Scholar]
- Busser BW, Huang D, Rogacki KR, Lane EA, Shokri L, Ni T, Gamble CE, Gisselbrecht SS, Zhu J, Bulyk ML, et al. 2012. Integrative analysis of the zinc finger transcription factor Lame duck in the Drosophila myogenic gene regulatory network. Proc Natl Acad Sci 109: 20768–20773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll D, Morton JJ, Beumer KJ, Segal DJ 2006. Design, construction and in vitro testing of zinc finger nucleases. Nat Protoc 1: 1329–1341 [DOI] [PubMed] [Google Scholar]
- Christensen RG, Enuameh MS, Noyes MB, Brodsky MH, Wolfe SA, Stormo GD 2012. Recognition models to predict DNA-binding specificities of homeodomain proteins. Bioinformatics 28: i84–i89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu SW, Noyes MB, Christensen RG, Pierce BG, Zhu LJ, Weng Z, Stormo GD, Wolfe SA 2012. Exploring the DNA-recognition potential of homeodomains. Genome Res 22: 1889–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowner D, Madden K, Goeke S, Giniger E 2002. Lola regulates midline crossing of CNS axons in Drosophila. Development 129: 1317–1325 [DOI] [PubMed] [Google Scholar]
- Desjarlais JR, Berg JM 1993. Use of a zinc-finger consensus sequence framework and specificity rules to design specific DNA binding proteins. Proc Natl Acad Sci 90: 2256–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon Y, McCammon JM, Miller JC, Faraji F, Ngo C, Katibah GE, Amora R, Hocking TD, Zhang L, Rebar EJ, et al. 2008. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol 26: 702–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier B, Segal DJ, Barbas CF III 2000. Insights into the molecular recognition of the 5′-GNN-3′ family of DNA sequences by zinc finger domains. J Mol Biol 303: 489–502 [DOI] [PubMed] [Google Scholar]
- Dreier B, Beerli RR, Segal DJ, Flippin JD, Barbas CF III 2001. Development of zinc finger domains for recognition of the 5′-ANN-3′ family of DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem 276: 29466–29478 [DOI] [PubMed] [Google Scholar]
- Dreier B, Fuller RP, Segal DJ, Lund CV, Blancafort P, Huber A, Koksch B, Barbas CF III 2005. Development of zinc finger domains for recognition of the 5′-CNN-3′ family DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem 280: 35588–35597 [DOI] [PubMed] [Google Scholar]
- Elrod-Erickson M, Rould MA, Nekludova L, Pabo CO 1996. Zif268 protein-DNA complex refined at 1.6 A: A model system for understanding zinc finger-DNA interactions. Structure 4: 1171–1180 [DOI] [PubMed] [Google Scholar]
- Emerson RO, Thomas JH 2009. Adaptive evolution in zinc finger transcription factors. PLoS Genet 5: e1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairall L, Schwabe JW, Chapman L, Finch JT, Rhodes D 1993. The crystal structure of a two zinc-finger peptide reveals an extension to the rules for zinc-finger/DNA recognition. Nature 366: 483–487 [DOI] [PubMed] [Google Scholar]
- Fan CM, Maniatis T 1990. A DNA-binding protein containing two widely separated zinc finger motifs that recognize the same DNA sequence. Genes Dev 4: 29–42 [DOI] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR 2011. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res 39: W29–W37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giniger E, Tietje K, Jan LY, Jan YN 1994. lola encodes a putative transcription factor required for axon growth and guidance in Drosophila. Development 120: 1385–1398 [DOI] [PubMed] [Google Scholar]
- Goeke S, Greene EA, Grant PK, Gates MA, Crowner D, Aigaki T, Giniger E 2003. Alternative splicing of lola generates 19 transcription factors controlling axon guidance in Drosophila. Nat Neurosci 6: 917–924 [DOI] [PubMed] [Google Scholar]
- Gogos JA, Hsu T, Bolton J, Kafatos FC 1992. Sequence discrimination by alternatively spliced isoforms of a DNA binding zinc finger domain. Science 257: 1951–1955 [DOI] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. 2011. The developmental transcriptome of Drosophila melanogaster. Nature 471: 473–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groeneveld LF, Atencia R, Garriga RM, Vigilant L 2012. High diversity at PRDM9 in chimpanzees and bonobos. PLoS ONE 7: e39064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove CA, De Masi F, Barrasa MI, Newburger DE, Alkema MJ, Bulyk ML, Walhout AJ 2009. A multiparameter network reveals extensive divergence between C. elegans bHLH transcription factors. Cell 138: 314–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Meng X, Zhu LJ, Lawson ND, Wolfe SA 2011. Zinc finger protein-dependent and -independent contributions to the in vivo off-target activity of zinc finger nucleases. Nucleic Acids Res 39: 381–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Christensen RG, Rayla AL, Lakshmanan A, Stormo GD, Wolfe SA 2012. An optimized two-finger archive for ZFN-mediated gene targeting. Nat Methods 9: 588–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guschin DY, Waite AJ, Katibah GE, Miller JC, Holmes MC, Rebar EJ 2010. A Rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol 649: 247–256 [DOI] [PubMed] [Google Scholar]
- Hallikas O, Palin K, Sinjushina N, Rautiainen R, Partanen J, Ukkonen E, Taipale J 2006. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell 124: 47–59 [DOI] [PubMed] [Google Scholar]
- Handel EM, Alwin S, Cathomen T 2009. Expanding or restricting the target site repertoire of zinc-finger nucleases: The inter-domain linker as a major determinant of target site selectivity. Mol Ther 17: 104–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, et al. 2009. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol 27: 851–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holohan EE, Kwong C, Adryan B, Bartkuhn M, Herold M, Renkawitz R, Russell S, White R 2007. CTCF genomic binding sites in Drosophila and the organisation of the bithorax complex. PLoS Genet 3: e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JW, Hendrix DA, Papatsenko D, Levine MS 2008. How the dorsal gradient works: Insights from postgenome technologies. Proc Natl Acad Sci 105: 20072–20076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip YT, Park RE, Kosman D, Bier E, Levine M 1992. The dorsal gradient morphogen regulates stripes of rhomboid expression in the presumptive neuroectoderm of the Drosophila embryo. Genes Dev 6: 1728–1739 [DOI] [PubMed] [Google Scholar]
- Isalan M, Choo Y, Klug A 1997. Synergy between adjacent zinc fingers in sequence-specific DNA recognition. Proc Natl Acad Sci 94: 5617–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isalan M, Klug A, Choo Y 1998. Comprehensive DNA recognition through concerted interactions from adjacent zinc fingers. Biochemistry 37: 12026–12033 [DOI] [PubMed] [Google Scholar]
- Jaeger SA, Chan ET, Berger MF, Stottmann R, Hughes TR, Bulyk ML 2010. Conservation and regulatory associations of a wide affinity range of mouse transcription factor binding sites. Genomics 95: 185–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolma A, Kivioja T, Toivonen J, Cheng L, Wei G, Enge M, Taipale M, Vaquerizas JM, Yan J, Sillanpaa MJ, et al. 2010. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res 20: 861–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan T, Li XY, Sabo PJ, Thomas S, Stamatoyannopoulos JA, Biggin MD, Eisen MB 2011. Quantitative models of the mechanisms that control genome-wide patterns of transcription factor binding during early Drosophila development. PLoS Genet 7: e1001290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemian M, Blatti C, Richards A, McCutchan M, Wakabayashi-Ito N, Hammonds AS, Celniker SE, Kumar S, Wolfe SA, Brodsky MH, et al. 2010. Quantitative analysis of the Drosophila segmentation regulatory network using pattern generating potentials. PLoS Biol 8: e1000456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemian M, Brodsky MH, Sinha S 2011. Genome Surveyor 2.0: cis-regulatory analysis in Drosophila. Nucleic Acids Res 39: W79–W85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, et al. 2011. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 471: 480–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Lee HJ, Kim H, Cho SW, Kim JS 2009. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res 19: 1279–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee MJ, Kim H, Kang M, Kim J-S 2011. Preassembled zinc-finger arrays for rapid construction of ZFNs. Nat Methods 8: 7. [DOI] [PubMed] [Google Scholar]
- Klug A 2010. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu Rev Biochem 79: 213–231 [DOI] [PubMed] [Google Scholar]
- Kuo TC, Calame KL 2004. B lymphocyte-induced maturation protein (Blimp)-1, IFN regulatory factor (IRF)-1, and IRF-2 can bind to the same regulatory sites. J Immunol 173: 5556–5563 [DOI] [PubMed] [Google Scholar]
- Laity JH, Dyson HJ, Wright PE 2000. DNA-induced α-helix capping in conserved linker sequences is a determinant of binding affinity in Cys2-His2 zinc fingers. J Mol Biol 295: 719–727 [DOI] [PubMed] [Google Scholar]
- Letunic I, Doerks T, Bork P 2012. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res 40: D302–D305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XY, Thomas S, Sabo PJ, Eisen MB, Stamatoyannopoulos JA, Biggin MD 2011. The role of chromatin accessibility in directing the widespread, overlapping patterns of Drosophila transcription factor binding. Genome Biol 12: R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MF, Carlson JW, Crosby MA, Matthews BB, Yu C, Park S, Wan KH, Schroeder AJ, Gramates LS, St Pierre SE, et al. 2007. Revisiting the protein-coding gene catalog of Drosophila melanogaster using 12 fly genomes. Genome Res 17: 1823–1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Segal DJ, Ghiara JB, Barbas CF III 1997. Design of polydactyl zinc-finger proteins for unique addressing within complex genomes. Proc Natl Acad Sci 94: 5525–5530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Xia Z, Zhong X, Case CC 2002. Validated zinc finger protein designs for all 16 GNN DNA triplet targets. J Biol Chem 277: 3850–3856 [DOI] [PubMed] [Google Scholar]
- MacArthur S, Li XY, Li J, Brown JB, Chu HC, Zeng L, Grondona BP, Hechmer A, Simirenko L, Keranen SV, et al. 2009. Developmental roles of 21 Drosophila transcription factors are determined by quantitative differences in binding to an overlapping set of thousands of genomic regions. Genome Biol 10: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden K, Crowner D, Giniger E 1999. lola has the properties of a master regulator of axon-target interaction for SNb motor axons of Drosophila. Dev Biol 213: 301–313 [DOI] [PubMed] [Google Scholar]
- Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, et al. 2008. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell 31: 294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa T, Sakura H, Sudo T, Ishii S 1989. Putative metal finger structure of the human immunodeficiency virus type 1 enhancer binding protein HIV-EP1. J Biol Chem 264: 14591–14593 [PubMed] [Google Scholar]
- Mahony S, Benos PV 2007. STAMP: A web tool for exploring DNA-binding motif similarities. Nucleic Acids Res 35: W253–W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell JG, Barbas CF III 2006. Zinc Finger Tools: Custom DNA-binding domains for transcription factors and nucleases. Nucleic Acids Res 34: W516–W523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbach D, Roy S, Ay F, Meyer PE, Candeias R, Kahveci T, Bristow CA, Kellis M 2012. Predictive regulatory models in Drosophila melanogaster by integrative inference of transcriptional networks. Genome Res 22: 1334–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuilton P, St Pierre SE, Thurmond J 2012. FlyBase 101: The basics of navigating FlyBase. Nucleic Acids Res 40: D706–D714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Brodsky MH, Wolfe SA 2005. A bacterial one-hybrid system for determining the DNA-binding specificity of transcription factors. Nat Biotechnol 23: 988–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Noyes MB, Zhu LJ, Lawson ND, Wolfe SA 2008. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nat Biotechnol 26: 695–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre N, Brown CD, Shah PK, Kheradpour P, Morrison CA, Henikoff JG, Feng X, Ahmad K, Russell S, White RA, et al. 2010. A comprehensive map of insulator elements for the Drosophila genome. PLoS Genet 6: e1000814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre N, Brown CD, Ma L, Bristow CA, Miller SW, Wagner U, Kheradpour P, Eaton ML, Loriaux P, Sealfon R, et al. 2011. A cis-regulatory map of the Drosophila genome. Nature 471: 527–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Stergachis AB, Reynolds A, Sandstrom R, Borenstein E, Stamatoyannopoulos JA 2012. Circuitry and dynamics of human transcription factor regulatory networks. Cell 150: 1274–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR 2010. Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyes MB, Christensen RG, Wakabayashi A, Stormo GD, Brodsky MH, Wolfe SA 2008a. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell 133: 1277–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyes MB, Meng X, Wakabayashi A, Sinha S, Brodsky MH, Wolfe SA 2008b. A systematic characterization of factors that regulate Drosophila segmentation via a bacterial one-hybrid system. Nucleic Acids Res 36: 2547–2560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavletich NP, Pabo CO 1991. Zinc finger-DNA recognition: Crystal structure of a Zif268-DNA complex at 2.1 Å. Science 252: 809–817 [DOI] [PubMed] [Google Scholar]
- Pavletich NP, Pabo CO 1993. Crystal structure of a five-finger GLI-DNA complex: New perspectives on zinc fingers. Science 261: 1701–1707 [DOI] [PubMed] [Google Scholar]
- Pelham HR, Brown DD 1980. A specific transcription factor that can bind either the 5S RNA gene or 5S RNA. Proc Natl Acad Sci 77: 4170–4174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, et al. 2012. The Pfam protein families database. Nucleic Acids Res 40: D290–D301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebar EJ, Pabo CO 1994. Zinc finger phage: Affinity selection of fingers with new DNA-binding specificities. Science 263: 671–673 [DOI] [PubMed] [Google Scholar]
- Reece-Hoyes JS, Deplancke B, Barrasa MI, Hatzold J, Smit RB, Arda HE, Pope PA, Gaudet J, Conradt B, Walhout AJ 2009. The C. elegans Snail homolog CES-1 can activate gene expression in vivo and share targets with bHLH transcription factors. Nucleic Acids Res 37: 3689–3698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK 2012. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30: 460–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robasky K, Bulyk ML 2011. UniPROBE, update 2011: Expanded content and search tools in the online database of protein-binding microarray data on protein-DNA interactions. Nucleic Acids Res 39: D124–D128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, Lin MF, et al. 2010. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330: 1787–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Zaback P, Joung JK, Voytas DF, Dobbs D 2009. An affinity-based scoring scheme for predicting DNA-binding activities of modularly assembled zinc-finger proteins. Nucleic Acids Res 37: 506–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Dahlborg EJ, Goodwin MJ, Cade L, Zhang F, Cifuentes D, Curtin SJ, Blackburn JS, Thibodeau-Beganny S, Qi Y, et al. 2011. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat Methods 8: 67–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder MD, Greer C, Gaul U 2011. How to make stripes: Deciphering the transition from non-periodic to periodic patterns in Drosophila segmentation. Development 138: 3067–3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuettengruber B, Ganapathi M, Leblanc B, Portoso M, Jaschek R, Tolhuis B, van Lohuizen M, Tanay A, Cavalli G 2009. Functional anatomy of polycomb and trithorax chromatin landscapes in Drosophila embryos. PLoS Biol 7: e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger M, Tear G, Ferres-Marco D, Goodman CS 1993. Mutations affecting growth cone guidance in Drosophila: Genes necessary for guidance toward or away from the midline. Neuron 10: 409–426 [DOI] [PubMed] [Google Scholar]
- Seetharam A, Bai Y, Stuart GW 2010. A survey of well conserved families of C2H2 zinc-finger genes in Daphnia. BMC Genomics 11: 276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal DJ, Dreier B, Beerli RR, Barbas CF III 1999. Toward controlling gene expression at will: Selection and design of zinc finger domains recognizing each of the 5′-GNN-3′ DNA target sequences. Proc Natl Acad Sci 96: 2758–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, Raveh-Sadka T, Schroeder M, Unnerstall U, Gaul U 2008. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature 451: 535–540 [DOI] [PubMed] [Google Scholar]
- Sera T, Uranga C 2002. Rational design of artificial zinc-finger proteins using a nondegenerate recognition code table. Biochemistry 41: 7074–7081 [DOI] [PubMed] [Google Scholar]
- Shi Y, Berg JM 1995. A direct comparison of the properties of natural and designed zinc-finger proteins. Chem Biol 2: 83–89 [DOI] [PubMed] [Google Scholar]
- Staehling-Hampton K, Laughon AS, Hoffmann FM 1995. A Drosophila protein related to the human zinc finger transcription factor PRDII/MBPI/HIV-EP1 is required for dpp signaling. Development 121: 3393–3403 [DOI] [PubMed] [Google Scholar]
- Stapleton M, Carlson J, Brokstein P, Yu C, Champe M, George R, Guarin H, Kronmiller B, Pacleb J, Park S et al. 2002. A Drosophila full-length cDNA resource. Genome Biol 3: RESEARCH0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadepally HD, Burger G, Aubry M 2008. Evolution of C2H2-zinc finger genes and subfamilies in mammals: Species-specific duplication and loss of clusters, genes and effector domains. BMC Evol Biol 8: 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JH, Emerson RO 2009. Evolution of C2H2-zinc finger genes revisited. BMC Evol Biol 9: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD 2010. Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11: 636–646 [DOI] [PubMed] [Google Scholar]
- Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM 2009. A census of human transcription factors: Function, expression and evolution. Nat Rev Genet 10: 252–263 [DOI] [PubMed] [Google Scholar]
- Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, et al. 2012. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res 22: 1798–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei GH, Badis G, Berger MF, Kivioja T, Palin K, Enge M, Bonke M, Jolma A, Varjosalo M, Gehrke AR, et al. 2010. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J 29: 2147–2160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. 1993. The zebrafish book. University of Oregon Press, Eugene, OR. [Google Scholar]
- Wolfe SA, Greisman HA, Ramm EI, Pabo CO 1999. Analysis of zinc fingers optimized via phage display: Evaluating the utility of a recognition code. J Mol Biol 285: 1917–1934 [DOI] [PubMed] [Google Scholar]
- Wolfe SA, Nekludova L, Pabo CO 2000. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct 29: 183–212 [DOI] [PubMed] [Google Scholar]
- Wright DA, Thibodeau-Beganny S, Sander JD, Winfrey RJ, Hirsh AS, Eichtinger M, Fu F, Porteus MH, Dobbs D, Voytas DF, et al. 2006. Standardized reagents and protocols for engineering zinc finger nucleases by modular assembly. Nat Protoc 1: 1637–1652 [DOI] [PubMed] [Google Scholar]
- Wunderlich Z, DePace AH 2011. Modeling transcriptional networks in Drosophila development at multiple scales. Curr Opin Genet Dev 21: 711–718 [DOI] [PubMed] [Google Scholar]
- Zhu C, Byers KJ, McCord RP, Shi Z, Berger MF, Newburger DE, Saulrieta K, Smith Z, Shah MV, Radhakrishnan M, et al. 2009. High-resolution DNA-binding specificity analysis of yeast transcription factors. Genome Res 19: 556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Smith T, McNulty J, Rayla AL, Lakshmanan A, Siekmann AF, Buffardi M, Meng X, Shin J, Padmanabhan A, et al. 2011a. Evaluation and application of modularly assembled zinc-finger nucleases in zebrafish. Development 138: 4555–4564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ, Christensen RG, Kazemian M, Hull CJ, Enuameh MS, Basciotta MD, Brasefield JA, Zhu C, Asriyan Y, Lapointe DS, et al. 2011b. FlyFactorSurvey: A database of Drosophila transcription factor binding specificities determined using the bacterial one-hybrid system. Nucleic Acids Res 39: D111–D117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Gupta A, Hall VL, Rayla AL, Christensen RG, Dake B, Lakshmanan A, Kuperwasser C, Stormo GD, Wolfe SA 2013. Using defined finger-finger interfaces as units of assembly for constructing zinc-finger nucleases. Nucleic Acids Res 41: 2455–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]