

Abstract
The C. elegans modENCODE Consortium has defined in vivo binding sites for a large array of transcription factors by ChIP-seq. In this article, we present examples that illustrate how this compendium of ChIP-seq data can drive biological insights not possible with analysis of individual factors. First, we analyze the number of independent factors bound to the same locus, termed transcription factor complexity, and find that low-complexity sites are more likely to respond to altered expression of a single bound transcription factor. Next, we show that comparison of binding sites for the same factor across developmental stages can reveal insight into the regulatory network of that factor, as we find that the transcription factor UNC-62 has distinct binding profiles at different stages due to distinct cofactor co-association as well as tissue-specific alternative splicing. Finally, we describe an approach to infer potential regulators of gene expression changes found in profiling experiments (such as DNA microarrays) by screening these altered genes to identify significant enrichment for targets of a transcription factor identified in ChIP-seq data sets. After confirming that this approach can correctly identify the upstream regulator on expression data sets for which the regulator was previously known, we applied this approach to identify novel candidate regulators of transcriptional changes with age. The analysis revealed nine candidate aging regulators, of which three were previously known to have a role in longevity. We experimentally showed that two of the new candidate aging regulators can extend lifespan when overexpressed, indicating that this approach can identify novel functional regulators of complex processes.
The development of chromatin immunoprecipitation (ChIP) followed by quantification by microarray (ChIP-chip) or high-throughput sequencing (ChIP-seq) has enabled the identification of transcription factor binding sites in vivo (Ren et al. 2000; Johnson et al. 2007). These DNA sites bound by transcription factors can be used to characterize DNA binding motifs, identify novel regulated targets, and understand a transcription factor's biological function through analysis of its targets (Spitz and Furlong 2012; Wang et al. 2012). Identification of targets for factors that play cooperative roles during development can provide insight into the redundant and specific functions of the individual factors, as well as the molecular mechanisms through which multiple factors interact to regulate gene expression (for review, see Spitz and Furlong 2012).
The Caenorhabditis elegans modENCODE Consortium has generated 98 ChIP-seq data sets identifying directly bound targets for 57 transcription factors in one or more developmental stages (Supplemental Table 1; Zhong et al. 2010; Niu et al. 2011). Similar efforts by the ENCODE and modENCODE Consortia as well as efforts from individual laboratories have identified targets bound by hundreds of transcription factors in humans, the mouse, and fly (MacArthur et al. 2009; Negre et al. 2011; Garber et al. 2012; Gerstein et al. 2012). Compendia of ChIP-seq data sets can be used to construct regulatory networks and to find novel pairs of transcription factors with similar sets of bound targets that suggest new cases of transcription factor co-association (Niu et al. 2011; Gerstein et al. 2012). Additionally, in combination with data sets describing histone modifications and other measures of chromatin state, such compendia can be used to predict gene expression (Cheng et al. 2011, 2012; Marbach et al. 2012). These examples illustrate how combining data for many transcription factors can provide emergent insights about gene regulation that cannot be found by studying one factor by itself.
Efforts to profile transcription factor binding by the ENCODE and modENCODE Consortia, as well as by many groups, have revealed a high degree of overlap between the binding sites of different transcription factors (Gerstein et al. 2010; The modENCODE Consortium 2010; Garber et al. 2012; for review, see Biggin 2011). Studies of transcription factor complexity, defined as the total number of transcription factors bound to each genomic region, have revealed that highly occupied target (HOT) regions (bound by many transcription factors) and low-complexity regions (bound by one or only a few factors) are functionally different in many ways (Gerstein et al. 2010; The modENCODE Consortium 2010; Garber et al. 2012). Low-complexity binding sites are often enriched for DNA binding motifs of the bound transcription factor, suggesting that the transcription factor directly binds to DNA. In contrast, high-complexity sites often show weaker or no enrichment for the DNA binding motifs of bound transcription factors, suggesting that the transcription factors may instead associate with the region through protein–protein interactions (Moorman et al. 2006; Gerstein et al. 2010; The modENCODE Consortium 2010; Yip et al. 2012). HOT regions are also associated with histone modification profiles and chromatin signatures characteristic of open chromatin in human and D. melanogaster, with strong enhancer activity in D. melanogaster, and with essential genes with high levels of expression and ubiquitous expression across tissues in C. elegans (Gerstein et al. 2010; Negre et al. 2011; Kvon et al. 2012; Yip et al. 2012).
In this work, we further explore how integration of multiple ChIP-seq data sets can enable insights not possible from a single ChIP experiment. First, we address the question of how to identify the subset of ChIP-seq targets that are likely to be factor-responsive. Often, only a subset of targets that are directly bound by a transcription factor are observed to change expression upon either an increase or decrease in the activity of that transcription factor (e.g., an average of 58% of ChIP-seq targets for 37 transcription factors in yeast [Gao et al. 2004], 26% of NANOG targets and 50% of POU5F1 targets in mice [Loh et al. 2006], and ∼10% of hlh-1 targets in C. elegans [Kuntz et al. 2012; for review, see Spitz and Furlong 2012]). By analyzing ChIP-seq data in combination with data sets describing genes altered upon single transcription factor perturbation, we find that low-complexity ChIP-seq targets are more likely to be factor-responsive than are high-complexity targets.
Second, we use the large number of data sets to identify transcription factors that have different sets of targets identified in multiple developmental stages. Analysis of the low-complexity targets for one such transcription factor, UNC-62/Homothorax, enabled us to characterize two mechanisms by which it may associate with distinct targets in different tissues: tissue-specific alternative isoforms, and differential co-association with LIN-39/HOX.
Third, we show that a compendium of ChIP-seq data sets can be used to screen for candidate regulators that bind to the upstream regions of genes that change expression in an expression profiling experiment. We validated this approach by using previously published gene expression profiling data to show that HLH-1 and SKN-1 can be correctly identified as transcription factors that bind to the upstream regions of genes responsive to hlh-1 and skn-1, respectively. We then applied this approach to find candidate upstream regulators of age-regulated genes and identified nine transcription factors, five of which extend lifespan in either overexpression or knockdown experiments.
Results
Efforts to experimentally identify direct targets of transcription factors have revealed an unexpected degree of complexity among transcription factor binding sites (Gerstein et al. 2010; The modENCODE Consortium 2010; Garber et al. 2012). To explore the distinct functions of low- and high-complexity binding sites, we performed analysis on 98 ChIP-seq data sets generated by the C. elegans modENCODE Consortium that identifies direct binding sites for 57 transcription factors (Supplemental Table 1). For each ChIP-seq binding site, we defined complexity as the maximum number of transcription factors that are bound within that genomic region (The modENCODE Consortium 2010; Garber et al. 2012). We observed a wide range of binding events; for example, a DNA site in the pat-3 promoter (a previously characterized target of HLH-1) (Fukushige et al. 2006) has HLH-1 binding sites that are bound by three, eight, and 16 other transcription factors (∼5%, 14%, and 28% of the 57 factors considered). In contrast, the promoter region of translation elongation factor 1-alpha homolog eef-1A.1 and RFC (DNA replication factor) family gene rfc-4 contains a region bound by 44 different transcription factors (77%) (Fig. 1A,B). The 98 ChIP-seq data sets showed a wide range of complexity profiles, ranging from experiments in which more than half of binding sites are bound by eight or less total transcription factors to those with more than half bound by 38 or more transcription factors (Supplemental Table 1).
Figure 1.

Examples of highly occupied and low-complexity regions of transcription factor binding. Binding of 57 transcription factors in one or more developmental stages (98 ChIP-seq data sets in total) were obtained from the C. elegans modENCODE Consortium and are shown for two examples. Data sets are shown in identical order to Supplemental Table 1. (A) Binding proximal to translation elongation factor 1-alpha homolog eef-1A.1. An ∼450-nt HOT region indicated by the red dashed box is significantly enriched for binding in ChIP-seq analyses of 44 different transcription factors. Transcription factor complexity (the number of factors with ChIP-seq binding sites overlapping that position) is shown at the top. (Below) Regions of significant enrichment (Q-value ≤ 10−5) observed in each of the 98 ChIP-seq data sets are shown as black boxes. (B) Low-complexity HLH-1 binding proximal to HLH-1–activated target pat-3 is shown.
HOT regions
Previously, 304 highly occupied target (HOT) regions were defined as those bound by 15 or more out of 23 transcription factors (Gerstein et al. 2010). We re-annotated HOT regions as 296 regions bound by 38 or more of the 57 assayed transcription factors (>65%, equivalent to the previous definition) (Fig. 2A; Supplemental Datafile 2). We observed that HOT regions tended to be positioned close to the transcriptional start site of genes, unlike regulatory enhancers that can act at a long distance (Krivega and Dean 2012); specifically, 83% of HOT regions were within 1000 bp of the transcriptional start site, whereas only 59% of low-complexity binding sites (with eight or less factors bound) were located within 1000 bp of the transcriptional start site (P < 10−10 by Fisher's exact test) (Fig. 2B).
Figure 2.

Highly occupied target (HOT) regions. (A) The histogram indicates the number of genomic regions observed for different binding complexities. In blue, thousands of low-complexity regions are bound by eight or fewer factors; in red, 296 HOT regions are bound by 38 or more transcription factors. (B) HOT regions tend to be located close to the transcription start sites (TSSs) of genes. The cumulative distribution plot shows the cumulative fraction of regions (y-axis) that have a maximal distance to the nearest annotated TSS indicated on the x-axis. HOT regions have significantly shorter distances to nearby TSSs than low-complexity regions (P = 1.7 × 10−23; Kolmogorov-Smirnov test). Results shown here are for all annotated genes (WS220); similar results were observed using only protein-coding genes (data not shown).
We found that many specific gene classes were overrepresented among HOT-associated genes, including SL2 transcripts, snoRNA transcripts, and genes encoding ribosomal subunit proteins (each P < 10−5 by Fisher's exact test). Gene ontology analysis indicated enrichment for genes involved in multiple aspects of embryonic and larval development as well as reproduction (Supplemental Table 2), consistent with the previous finding that HOT-associated genes are expressed at a high level, are expressed ubiquitously in all tissues, and have essential functions (Gerstein et al. 2010). In contrast, genes that are highly expressed at only specific points in the worm life-cycle, such as vitellogenin and collagen genes, were not associated with HOT regions (zero out of six and zero out of 159, respectively).
Low-complexity targets tend to respond to altered transcription factor expression
Next, we asked whether binding site complexity was predictive as to whether the expression of a target gene was altered in response to changes in a bound factor. For this analysis, we selected two transcription factors (HLH-1 and SKN-1) with binding targets characterized from ChIP-seq experiments and responsive genes identified from overexpression or knockdown expression experiments. The first, HLH-1, is a helix-turn-helix transcription factor that is the C. elegans ortholog of MYOD1 and is a key regulator of muscle differentiation and development (Fukushige and Krause 2005; Lei et al. 2009). Previous experiments identified 2128 genes that were significantly induced upon overexpression of HLH-1 in early embryos (Fukushige et al. 2006; Fox et al. 2007), and ChIP-seq experiments from modENCODE indicate that HLH-1 binds to 4191 genes in mixed embryos (Niu et al. 2011). The second, SKN-1, is a bZIP domain–containing transcription factor that is orthologous to NRF1/2/3. SKN-1 plays roles in specification of intestinal, muscular, and pharyngeal cell fates of the EMS blastomere and in the response to oxidative stress (Bowerman et al. 1992; Maduro et al. 2001; An and Blackwell 2003; Tullet et al. 2008). DNA microarray experiments profiling skn-1 knockdown in worms exposed to oxidative stress identified 91 SKN-1–responsive genes (Park et al. 2009), and modENCODE ChIP-seq data sets identified sites bound by SKN-1 associated with 3572 genes in L1-stage larvae and 3131 genes in L2-stage larvae (with Q-value ≤ 10−5) (Niu et al. 2011). For each, binding sites were associated with genes if they were located within the gene body or within 5 kb upstream of the annotated transcription start site (see Methods).
By using these data sets, we asked whether the complexity score for an HLH-1 or SKN-1 binding site correlated with factor-responsive targets. First, we determined the overlap between the list of HLH-1–activated genes and the set of genes with HLH-1 binding sites with complexity of at most n, with n ranging from one to 57 factors associating to the same genomic locus. We observed that low-complexity sites tended to be associated with genes that were HLH-1–responsive, whereas inclusion of intermediate- and high-complexity sites yielded fewer HLH-1–responsive targets (Fig. 3A; Supplemental Fig. 1A). Using the Matthews correlation coefficient to optimize the trade-off between the increased percentage of HLH-1–responsive genes at low-complexity thresholds and the increased number of targets when intermediate- and high-complexity sites are included, we found that a complexity score of eight provided the optimal threshold for HLH-1 (Fig. 3A). Using this cutoff, 39.5% (591) of the 1496 genes associated with HLH-1 binding regions with a complexity score of eight or less were activated by HLH-1 (3.1-fold enriched, χ2 = 1047). This was a 99% improvement over regions with a complexity score of nine or more, and a 49% improvement over using all HLH-1 binding sites, which were only 1.6-fold and 2.1-fold enriched (χ2 = 132 and 789), respectively (Fig. 3B).
Figure 3.

Low binding site complexity correlates with factor-responsive expression. (A) Using 4191 significant HLH-1 binding sites identified by the modENCODE Consortium (Niu et al. 2011), the set of genes with HLH-1 binding sites with complexity less than or equal to n (for n = 1–57) was identified. Each set was then compared to 2128 genes activated upon HLH-1 overexpression (Fukushige et al. 2006; Fox et al. 2007), with the percentage of directly bound targets activated indicated in red. Except for complexities of two or less, binding sites with lower complexity had higher precision in predicting HLH-1–activated genes. By use of the Matthews correlation coefficient (in blue) to weight both false-positives and false-negatives, a complexity of eight or less was identified as optimal for predicting factor-responsive targets (indicated by *). (B) Using only binding sites with complexity of eight or less significantly improves prediction of HLH-1–responsive binding. (Circles) The set of genes with any HLH-1 binding site (top) or only those with low-complexity or intermediate/high-complexity binding sites (bottom). The overlap with HLH-1–activated genes is indicated in red, with the expected overlap indicated by white hash marks. Significance of enrichment was calculated by Yates' χ2 test. (C,D) The same complexity criteria significantly delineate SKN-1 targets in L2 and L1 larvae. Circles indicate 91 SKN-1–activated genes (Park et al. 2009), with the percentage overlap with SKN-1 ChIP-seq data sets indicated in blue and expected overlap indicated by white hash marks. Low-complexity regions for SKN-1 in L2 larvae (C), and L1 larvae (D), are enriched for genes responsive to skn-1 knockdown. Enrichment significance was determined by Fisher's exact test. The set of all SKN-1 targets in L1 was not significantly enriched for SKN-1–activated genes (1.1-fold-enriched, P > 0.5), indicating that the correlation between SKN-1 binding in L1 larvae and SKN-1–responsive expression is only observed when binding site complexity is taken into account.
Next, we applied the same complexity criteria to determine whether the complexity of SKN-1 binding sites was also correlated with responsiveness to decreased activity of skn-1. The first SKN-1 ChIP-seq data set (from L2 larvae) identified 579 low-complexity targets and 1668 intermediate- and high-complexity targets bound by SKN-1. We found that genes associated with the low-complexity sites showed a 4.8-fold enrichment for activation by SKN-1 (P = 6.3 × 10−22 by χ2 test). In contrast, the intermediate- and high-complexity targets were not significantly enriched for skn-1–responsive genes (Fig. 3C).
We obtained a similar result for the second SKN-1 ChIP-seq data set (from L1 larvae), which has 344 low-complexity targets and 2301 intermediate- and high-complexity targets. Although this set showed surprisingly low overlap with the ChIP-seq performed in L2 larvae (only 16% of L1 low-complexity binding sites also bound in L2 larvae), SKN-1 (L1) low-complexity targets still showed a 4.3-fold enrichment for activation by SKN-1 (P = 7.5 × 10−7) (Fig. 3D). In contrast, neither intermediate- and high-complexity targets nor all SKN-1 (L1) targets were significantly enriched for skn-1–activated genes. Thus, without incorporating information from the 97 other ChIP-seq data sets, analysis of the SKN-1 (L1) ChIP-seq data set on its own would not show any enrichment for skn-1–responsive genes. Additionally, these results indicate that the complexity criteria trained using the HLH-1 data sets can be applied to analysis of other transcription factors. Training using the SKN-1 data sets themselves gave a different cutoff for maximal correlation, but showed the same anti-correlation between binding site complexity and factor-responsive expression (Supplemental Fig. 1B). The number of transcription factors bound to each base in the C. elegans genome is listed in Supplemental Datafile 1 to enable incorporation of binding site complexity in future analyses.
In addition to binding site complexity, a gene can be regulated by many transcription factors binding to multiple distinct sites. However, we found that incorporation of gene-level complexity did not improve the ability to distinguish factor-responsive from nonresponsive targets compared with incorporation of binding site-level complexity (Supplemental Fig. 1C). This result suggests that the correlation of binding site complexity with factor-responsive targets is not simply due to the number of transcription factors associated with the entire gene promoter.
Tissue specificity of transcription factor targets
Next, we explored whether the target genes bound by a transcription factor were enriched for expression in the tissue where the factor is known to be expressed. We obtained lists of genes with expression significantly enriched in a variety of tissues (e.g., intestine or neurons) and tissue subtypes (e.g., A-class neurons) (listed in Supplemental Table 3; Roy et al. 2002; Zhang et al. 2002; Colosimo et al. 2004; Fox et al. 2005; Pauli et al. 2006; Von Stetina et al. 2007; Spencer et al. 2011). For each list of tissue-enriched genes, we performed pairwise comparisons with low-complexity targets from every ChIP-seq data set, identifying many significant transcription factor target–tissue pairings (Supplemental Fig. 2). To test this approach, we identified 13 factors that had a high correlation with at least one tissue-enriched gene list and had expression patterns described in WormBase (Harris et al. 2010). In 12 of the 13 cases, the targets were enriched for expression in a tissue in which the transcription factor was expressed (Supplemental Fig. 3).
Tissue-specific gene regulatory networks controlled by UNC-62 Homothorax
Although modENCODE ChIP-seq experiments were performed using whole worms, binding of a transcription factor to distinct target genes in different developmental stages could occur if the factor was expressed in one tissue at one stage but another tissue at a later stage, or acted with different cofactors in the different stages. We identified four transcription factors (PHA-4, FOS-1, SKN-1, and UNC-62) for which targets identified in different developmental stages were enriched for expression in different tissues. We analyzed one of these transcription factors (UNC-62) as a proof-of-principle that one can use genomic analyses to uncover potential underlying molecular mechanisms responsible for binding site specificity.
UNC-62 is the ortholog of Homothorax/Meis and is a cofactor of the HOX transcription factor LIN-39. unc-62 is involved in the development of the nervous system, hypodermis, and vulva as well as in aging, and acts through both HOX-dependent and HOX-independent functions (Van Auken et al. 2002; Yang et al. 2005; Curran and Ruvkun 2007; Jiang et al. 2009; Potts et al. 2009; Van Nostrand et al. 2013). An alternative splicing event in unc-62 produces two transcripts that include either exon 7a or 7b, encoding alternative N termini of the DNA binding TALE homeodomain (Van Auken et al. 2002). Using isoform-specific fluorescent reporters UNC-62(7a) was observed to be predominantly expressed in the intestine starting at the L3 larval stage and continuing through adulthood, whereas UNC-62(7b) was expressed in neurons, the ventral nerve cord, vulval precursor cells, and hypodermis beginning in embryos and continuing through adulthood (Fig. 4A; Van Nostrand et al. 2013).
Figure 4.

Distinct sets of targets bound by UNC-62 in L2/L3 larvae and adults. (A) Strains expressing isoform-specific unc-62 translational reporters show stage- and tissue-specific expression (Van Nostrand et al. 2013). (Left) unc-62(7a):GFP is not observed in early larval stages but is highly expressed in the intestine in late larval stages and young adults (YA). (Right) unc-62(7b):GFP is not observed in the intestine at any stage but is expressed in the hypodermis (hyp), the ventral nerve cord (vnc), and other neurons (neu). Strains were imaged in a glo-4(ok623) background to limit gut autofluorescence. (B) The overlap of ChIP-seq binding sites for UNC-62 in L2 and L3 larvae, young adults (YA), and HOX transcription factor LIN-39 in L3 are shown as the percentage of binding sites in the smaller set that are also bound in the larger. (C) Targets bound by UNC-62 in L2, L3, and YA, as well as those bound by LIN-39 in L3 larvae, were compared to genes enriched for expression in various tissues (Roy et al. 2002; Zhang et al. 2002; Colosimo et al. 2004; Fox et al. 2005; Pauli et al. 2006; Von Stetina et al. 2007; Spencer et al. 2011). Colors indicate the correlation between low-complexity target genes and genes with tissue-enriched expression for the indicated tissue. Tissues are clustered according to broad tissue types, and the specific tissue for each column is listed in Supplemental Figure 2 and Supplemental Table 3. (Int) Intestine; (Hyp) hypodermis; (BWM) body wall muscle. (D) A de novo motif search with RSAT (Thomas-Chollier et al. 2012) identifies sequences significantly enriched in the 100-bp central core region of UNC-62 binding sites. A total of 200-bp flanking regions on either side of this core were used as the background sequence set. Both motifs contain the D. melonogaster Homothorax motif (TGACA) (Noyes et al. 2008).
The modENCODE Consortium performed UNC-62 ChIP-seq experiments in L1, L2, and L3 larvae (which express only unc-62(7b)) (see Supplemental Fig. 4), as well as young adults (which express both unc-62(7a) and unc-62(7b)). We identified seven low-complexity UNC-62 binding sites in L1 larvae, 47 in L2 larvae, 231 in L3 larvae, and 339 in young adults; due to the low number of targets, we removed the L1 data set from additional analyses. We observed that UNC-62 showed a dramatic shift in binding between L2/L3 larvae and adults; 62% of UNC-62 L2 peaks overlapped regions enriched in L3, but only 13% of L2 peaks and 5% of L3 peaks were enriched in young adults (Fig. 4B).
We performed three comparisons of the binding targets of UNC-62 in the L2/L3 stages to the young adult stage: overlap with binding sites of cofactor LIN-39, tissue-specific expression of target genes, and differential motif enrichment. First, we found that 45% of low-complexity UNC-62 binding sites in L2 larvae and 42% of low-complexity L3 binding sites overlapped low-complexity LIN-39 binding sites, reflecting the known shared functions of UNC-62 and LIN-39 during these larval stages. In contrast, only 2% of low-complexity UNC-62 young adult binding sites were also bound by LIN-39, indicating that these transcription factors likely have divergent roles at this stage (Fig. 4B). Second, we found that UNC-62 low-complexity targets in the L2 and L3 larval stages were enriched for genes with neuronal expression, similar to HOX LIN-39 targets in the L3 larval stages (Fig. 4C). In contrast, the UNC-62 young adult targets were enriched for genes expressed predominantly in the intestine. These profiles match the expression of the isoforms of unc-62, as unc-62(7a) is expressed in the intestine in young adults, and both unc-62(7b) and lin-39 are expressed in neuronal tissues (as well as other tissues) in larvae (Fig. 4A; Wagmaister et al. 2006).
Third, we performed a de novo search to identify DNA motifs contained within the UNC-62 peaks using the central 100-nt region of low-complexity UNC-62 binding sites in L3 and young adults. We identified two similar motifs: (1) a motif with consensus sequence GTGACA that is enriched in both the L3 and the young adult stages (2.8-fold, P = 0.0007 for the L3 stage and 3.4-fold, P = 1.3 × 10−7 for the young adult stage), and (2) a motif with consensus sequence TTGACA motif that was significantly enriched in young adult (3.3-fold, P = 1.5 × 10−22) but not in the L3 larval stage (1.7-fold enriched relative to flanking regions, P > 1 after Bonferroni correction; 2.0-fold enriched, P = 5.6 × 10−5 in young adult binding sites relative to L3 binding sites) (Fig. 4D). Both motifs contain the core TGACA sequence previously described as the binding site for Drosophila Homothorax (Noyes et al. 2008).
In summary, these results suggest that multiple mechanisms may direct stage-specific binding of UNC-62 to its targets: (1) differential use of the LIN-39 cofactor in specifying binding targets in neurons compared with the intestine, and (2) binding to distinct DNA motifs by the UNC-62(7a) versus the UNC-62(7b) isoform. Similar analysis could suggest mechanisms for tissue specificity for additional factors that could be dissected with further experimental exploration.
Identifying upstream transcription factors involved in regulating gene expression changes from genome-wide profiling experiments
A commonly arising question in high-throughput gene expression profiling experiments is to identify upstream transcriptional regulators that may be responsible for causing the observed transcriptional differences. Given the increasing availability of ChIP-seq data sets describing transcription factor targets, one approach to find such candidate regulators is to search for transcription factors that bind to the upstream regions of differentially expressed genes (Lachmann et al. 2010; Zambelli et al. 2012). Rather than test all of the targets from the ChIP-seq experiments, we use only the low-complexity targets as this subset is enriched for factor-responsive targets.
To test the validity of this approach, we analyzed four expression profiling experiments: (1) genes induced following overexpression of HLH-1, (2) genes decreased upon knockdown of SKN-1, (3) genes decreased upon knockdown of UNC-62, or (4) genes that have altered expression during aging. We chose the first three data sets as positive controls, as the upstream regulator was present in the modENCODE database. For the fourth data set, we chose aging as a complex process for which transcriptional changes are likely the effect of altered activity of multiple regulators.
For HLH-1, we compared the set of 2128 HLH-1–activated genes to low-complexity target genes from each of the 98 ChIP-seq data sets, and we found that HLH-1 ChIP-seq data set was the most significantly enriched for genes that change expression upon HLH-1 overexpression (3.1-fold enriched, P < 10−100, χ2 = 1047) (Fig. 5A). For SKN-1, we performed a similar analysis on the 91 genes that decrease expression following skn-1 knockdown (Park et al. 2009). Out of 98 ChIP-seq data sets, the top two were SKN-1 ChIP-seq data sets (4.8-fold enriched, P = 6.3 × 10−22 in the L2 larval stage and 4.3-fold enriched, P = 7.5 − 10−7 in the L1 stage, respectively) (Fig. 5B). For the SKN-1 ChIP-seq experiment using L1 worms, analysis using all binding sites does not show enrichment for genes responsive to skn-1 knockdown, indicating that SKN-1 is only identified as an upstream regulator of genes responsive to SKN-1 when low binding site complexity is incorporated (Supplemental Fig. 5C; as discussed in Fig. 3D).
Figure 5.

Identifying candidate regulators of expression profiling data sets. To predict candidate regulators of genes altered in expression profiling experiments, low-complexity targets from each of the 98 ChIP-seq data sets were compared against (A) 2128 genes activated by hlh-1 (Fukushige et al. 2006; Fox et al. 2007), (B) 91 genes activated by skn-1 (Park et al. 2009), or (C) 115 genes decreased upon knockdown of unc-62 (Van Nostrand et al. 2013). For each ChIP-seq data set (y-axis), the x-axis indicates the overlap between low-complexity targets and genes altered in the transcriptome profiling experiment, with enrichment indicated by positive values and depletion by negative values. (A) HLH-1 low-complexity ChIP-seq targets in mixed embryos (MxE) showed the greatest enrichment for hlh-1–activated genes ([*] P < 10−100, χ2 = 1047 by Yates' χ2 test). (B) SKN-1 targets in L2 larvae (P = 6.3 × 10−22), followed by SKN-1 targets in L1 larvae (P = 7.5 × 10−7) showed the greatest enrichment for skn-1–activated genes. (C) UNC-62 targets did not correlate with unc-62-activated genes, potentially indicating that most genes with decreased expression upon unc-62 knockdown are secondary targets of UNC-62. (D) To control for the different number of targets between ChIP-seq data sets, we developed a score (based on a naïve Bayes classifier) for each binding site that reflects both the binding site significance (Q-value) as well as binding site complexity. This classifier was trained on the set of HLH-1 ChIP-seq targets and hlh-1–activated genes, and the 500 binding sites for each ChIP-seq data set with the highest scores were then tested on skn-1–activated genes. By use of this method, SKN-1 targets showed the greatest correlation (3.6-fold enriched, P = 7.9 × 10−9 in L1 larvae and 5.1-fold enriched, P = 9.8 × 10−9 in L2 larvae) for skn-1–activated genes.
However, when we compared the ChIP-seq data sets against 115 genes that are activated by and 67 genes repressed by UNC-62 in young adults (Van Nostrand et al. 2013), neither stage-matched UNC-62 ChIP-seq targets from young adult worms nor UNC-62 targets in the L2 or L3 stage showed significant overlap (Fig. 5C; Supplemental Fig. 5D). This false-negative result may reflect that knockdown of unc-62 activity results in expression changes of a small number of direct targets, which subsequently leads to a cascade of gene expression changes of secondary indirect targets. Thus, the degree to which transcriptional changes are composed of primary, direct targets instead of secondary, indirect ones represents a limitation for this approach.
A potential confounding factor is that the significance level depends on the number of genes identified in the ChIP-seq experiments, which would undesirably favor ChIP-seq experiments with a large number of targets. To address this concern, we developed a method using a naïve Bayes classifier, in which each binding site is given a RP (responsiveness-predictor) score as a function of both its complexity as well as its significance Q-value (see Methods). We then selected the top-scoring 500 sites in order to compare an equal number of binding sites from each ChIP-seq data set against each other as described above.
First, we independently trained a model to predict targets that are induced by HLH-1 overexpression for each ChIP-seq data set. Similar to before, we observed that the HLH-1 ChIP-seq data set was the best of all of the modENCODE ChIP-seq data sets at predicting hlh-1–induced genes; specifically, the top 500 HLH-1 binding sites were 4.5-fold enriched for induction by hlh-1 (P < 10−100, χ2 = 761) (Supplemental Fig. 5A,B). Next, we asked whether RP scores (trained to weight ChIP-seq parameters using HLH-1 data) could be used to infer regulators for the SKN-1 data set. Consistent with the earlier results, we observed that the only data sets showing significant enrichment were the two SKN-1 ChIP-seq data sets; specifically, SKN-1 targets in L1 larvae were 3.6-fold enriched (P = 7.9 × 10−9), and those in L2 larvae were 5.1-fold enriched (P = 9.8 × 10−9) for skn-1–activated genes (Fig. 5D). This method did not, however, improve the correlation between UNC-62 binding and unc-62–responsive expression (Supplemental Fig. 5E).
In summary, our analysis correctly identified the hlh-1 and skn-1 transcription factors as upstream regulators of genes that change expression following hlh-1 overexpression or skn-1 knockdown, respectively. These results indicate that it is possible to identify upstream regulators in silico from a gene expression profiling experiment by screening ChIP-seq data sets. In one case (SKN-1 in L1 larvae), the upstream regulator is only identified once binding site complexity is incorporated.
Identification of novel aging regulators
Encouraged by the success of using ChIP-seq data to infer upstream transcriptional regulators for gene signatures in two out of three cases, we turned to analysis of genes that change expression during aging. To screen for putative aging regulators, we compared each set of transcription factor targets against 1106 genes with altered expression during aging (Budovskaya et al. 2008). We performed this screen using both scoring methods: (1) using only those binding sites with low complexity (Fig. 6A), and (2) using the 500 binding sites with the highest RP scores for each ChIP-seq data set (Fig. 6B). The two approaches are complementary, as the first uses only significant binding peaks (even if they are few in number), whereas the second uses an equal number of binding sites across data sets in order to improve sensitivity for data sets with fewer binding sites (but may include sites bound weakly by the transcription factor). We identified nine transcription factors that are significantly enriched for binding to age-regulated genes with at least one of the two methods; six of the nine were significantly enriched using both methods.
Figure 6.

Identification of candidate regulators of aging. (A,B) The ChIP-seq–based screening approach described in Figure 5 was applied to the set of genes with altered expression during aging (Budovskaya et al. 2008). For each ChIP-seq data set, the correlation was calculated twice: first, using all low-complexity binding sites (A), and second, using the 500 binding sites with the highest naïve Bayes–derived score as described in Figure 5D (B). (Bars) The P-value (log10) between age-regulated genes and the indicated ChIP-seq targets, with enrichment indicated by positive values and depletion indicated by negative values. For nine transcription factors, significant overlap (P < 10−5) was observed using at least one of the two approaches; six of nine were significant with both. (Black checkmarks) Three factors (ELT-3, UNC-62, and SKN-1) for which modulation has been shown to increase lifespan (Curran and Ruvkun 2007; Budovskaya et al. 2008; Tullet et al. 2008). (Red checkmarks) Factors for which the modENCODE strain (which contains an integrated multi-copy fosmid containing the listed transcription factor) has an extended lifespan. (C,D) Lifespan of modENCODE-generated strains in which a strain containing a fosmid with C-terminal GFP-tagged nhr-28 or nhr-76 was compared with controls. Days of adulthood are indicated on the x-axis, and the percentage of worms remaining alive is indicated on the y-axis. (C) Strain OP371 (containing a fosmid with GFP-tagged nhr-28) was compared with three controls (RW10780, RW11206, and RW11175). The strain overexpressing nhr-28 shows 15%–30% extension of lifespan relative to the various controls (all P < 10−5 by log-rank test). (D) Strain OP203 (containing a fosmid with GFP-tagged nhr-76) showed a 7%–15% increase in mean lifespan relative to two controls (RW10780 and RW11206) (P < 0.01 against either). Lifespan data shown are from strains that were backcrossed twice to wild-type; each lifespan experiment was performed twice before backcrossing and gave similar results (Supplemental Table 5).
Modulation of the activity of three of the nine (ELT-3, UNC-62, and SKN-1) has previously been shown to increase lifespan. ELT-3 is involved in age-regulated changes in the hypodermis, and increased expression of ELT-3 (via knockdown of repressors ELT-5 or ELT-6) extends lifespan (Budovskaya et al. 2008). HOX cofactor UNC-62 is involved in age-regulated changes in the intestine, and unc-62 knockdown in adults increases lifespan (Curran and Ruvkun 2007; Van Nostrand et al. 2013). SKN-1 is involved in mediating the oxidative stress response, and overexpression of an activated form of SKN-1 increases lifespan (Tullet et al. 2008; Przybysz et al. 2009).
To validate the role of the remaining six candidate aging regulators (nhr-28, pqm-1, fos-1, C01B12.2, nhr-77, and nhr-76), we asked whether increasing or decreasing their activity can increase lifespan. We first performed RNAi to determine if reduction in activity increased lifespan. We confirmed that knockdown of unc-62 in adults can significantly extend lifespan as reported previously (Curran and Ruvkun 2007), but did not observe reproducible extension of lifespan for any of the novel candidates (Supplemental Table 4).
To assay the lifespan phenotype of overexpression, we obtained a transgenic strain from the modENCODE Consortium that carries a low-copy randomly integrated fosmid that contains the desired transcription factor (with a C-terminal GFP tag), additional flanking genes as described below, and rescue marker unc-119. Strains containing fosmids with pqm-1, fos-1, C01B12.2, or nhr-77, had wild-type lifespan (Supplemental Table 5), whereas strains containing either the nhr-28 or the nhr-76 fosmid showed significant extension of lifespan (15%–30% and 6%–15%, respectively; each P < 0.01 by log-rank test in three independent experiments) (Fig. 6C,D).
The first new candidate aging regulator, nhr-28, encodes a nuclear hormone receptor expressed in the pharynx, hypodermis, and intestine (Reece-Hoyes et al. 2007). NHR-28 ChIP-seq experiments performed on L4 larvae identified binding sites associated with 297 target genes that change expression with age (1.9-fold enriched, P = 5.6 × 10−36). The fosmid containing GFP-tagged NHR-28 also contains ace-1 (encoding a class A acetylcholinesterase) and sur-7 (encoding a cation diffusion facilitator protein). The read density of sequences across the nhr-28 gene region indicates that there are approximately 15 integrated copies of the fosmid in the modENCODE strain.
The second new candidate aging regulator is nhr-76, which encodes a nuclear hormone receptor expressed in body wall muscles, the intestine, excretory gland cell, pharynx, seam cells, and vulval muscles (Miyabayashi et al. 1999). The fosmid containing nhr-76 also contains the majority of the K11H12.9 transcript (which encodes a protein kinase of unknown function) and is present at three or four copies in the modENCODE strain. By use of the naïve Bayes–derived method that selects the top 500 NHR-76 peaks, the NHR-76 ChIP-seq data set was the second-most significantly enriched for age-regulated genes out of all of the modENCODE data sets (2.4-fold enriched, P = 1.3 × 10−15). However, by use of the first method that analyzed only the 50 significant low-complexity targets from the NHR-76 ChIP-seq experiment, only 12 were found to show altered expression with age (3.3-fold enriched, P = 0.00017). Thus, NHR-76 is an example where analysis using the naïve Bayes–derived method is more sensitive in revealing candidate regulators.
A resource for identifying regulators of changes in C. elegans expression profiling experiments
There are currently over 1000 C. elegans expression profiling experiments listed in the GEO database showing transcriptional changes at various developmental stages, at different growth conditions, and in different mutant backgrounds. For each of these, the ChIP-seq screening approach described above could be used to identify candidate causal regulators responsible for the observed expression changes. We have created a website through which users can perform a candidate regulator screen for any expression profiling experiment of interest (http://celegansrwas.stanford.edu).
Discussion
In C. elegans, the modENCODE Consortium has provided a compendium of 98 ChIP-seq data sets for 57 transcription factors. In this work, we provide further evidence that integrating information across these ChIP-seq data sets can be highly informative for driving biological insights. By analyzing these data sets, we found that the number of transcription factors bound to a single DNA region (termed its complexity score) can vary widely, from one to 54. Low-complexity binding sites are enriched for characterize factor-responsive expression, can be used to uncover mechanisms leading to regulation of distinct genes by a transcription factor between developmental stages and in different tissues, and can be screened to find novel transcriptional regulators of genes identified in expression profiling experiments. Each of these three analyses is enabled by the availability of a large number of ChIP-seq data sets.
In addition, we redefined 296 highly occupied target (HOT) regions that are bound by 38 or more factors (>65%). These HOT regions were associated with various types of housekeeping genes (including ribosomal proteins, sl-2 splice leader transcripts, and snoRNAs), in agreement with the previous observation of HOT regions as associated with essential genes that are broadly and highly expressed (Gerstein et al. 2010).
Low-complexity targets correlate better with factor-responsive expression
The subset of transcription factor binding targets that are factor-responsive (i.e., activated or repressed by the factor in gain-of-function or loss-of-function experiments) can be used to infer functions and tissue-specificities of the transcription factor itself, as they represent targets for which expression will respond to altered regulator activity. However, the fraction of genes that are bound by a transcription factor and that are also responsive to changes in expression of the factor can range from 4% to >50% (Spitz and Furlong 2012).
Previous work has shown that prediction of factor-responsive targets from an individual ChIP-seq experiment can be improved by incorporating additional information, such as co-correlated expression across hundreds of different microarray studies (Lai et al. 2010; Cheng et al. 2011; Marbach et al. 2012). Here, we show that by integrating 98 ChIP-seq data sets for 57 transcription factors, we can identify low-complexity binding sites that significantly improve the fraction of factor-responsive targets. In one instance (SKN-1 targets in L1 larvae), enrichment for activation by SKN-1 was only observed for the low-complexity sites. These results illustrate insights using the aggregated results from the entire ChIP-seq compendium that are not possible using ChIP-seq data for just one transcription factor. Further, if the properties of transcription factor binding in humans are similar to C. elegans, our results suggest that incorporating binding site complexity for each bound region will improve analysis of transcription factor function in humans.
In this work we used a cutoff of eight or less transcription factors bound, based on analysis of HLH-1, to define low-complexity targets. Importantly, this criterion effectively segregated SKN-1 targets, indicating that the definition of low complexity targets works for other transcription factors as well. However, depending on the desired application, alternative complexity cutoffs could be chosen that shift the balance toward either greater sensitivity or specificity in identifying factor-responsive targets.
Mechanisms for tissue-specific regulation of downstream targets by UNC-62
Analyses of ChIP-seq experiments across multiple cell-types can reveal binding sites unique to individual cell-types, which can be further studied to reveal insight into causal mechanisms. For example, analysis of MYC binding in two human cell-types revealed that GATA1 and TAL1 motifs correlated with MYC binding in K562 cells, whereas motifs for TEAD1 and the AP-1 complex correlated with binding specific to HeLaS3 cells (Shao et al. 2012). We observed that in C. elegans, UNC-62 showed dramatically different tissue specificities among targets when assayed in different developmental stages. unc-62 is alternatively spliced to produce two transcripts that include either exon 7a or 7b. By using ChIP-seq data, we observe that UNC-62 generally binds to one set of targets in the L2 or L3 larval stages and to a distinct set in adults. Our results suggest a model for tissue specificity of UNC-62 binding in which UNC-62(7b) binds along with known cofactor HOX gene LIN-39 at one set of genes during early larval development, and UNC-62(7a) binds to a different set of genes in the intestine at an altered consensus motif without LIN-39 co-occupancy in adults.
Although we focused on UNC-62 as a proof-of-concept, the transcription factors SKN-1, PHA-4, and FOS-1 also showed enrichment for different tissues among their targets in different developmental stages. In addition, as many of the 57 transcription factors were only profiled in one stage, it is likely that more transcription factors will be found to bind to different targets at different points during development. Alternative isoforms that affect DNA binding domains and combinatorial binding with cofactors are common mechanisms by which transcription factors regulate distinct sets of targets in different contexts (Taneri et al. 2004; Spitz and Furlong 2012). Thus, the type of analysis described here could provide insights into how other regulators achieve regulation of distinct sets of genes at different times and in different cell-types.
Screening ChIP-seq data sets for candidate transcription factors responsible for expression differences observed in expression profiling experiments
DNA microarray and high-throughput RNA sequencing technologies are commonly used to generate a list of genes that change expression in a mutant or altered growth condition. For such experiments, one often would like to identify the critical upstream regulators driving these changes in expression. One can gain insight into potential upstream regulators by searching for high levels of overlap between genes with altered expression and DNA regions bound by each transcription factor from a compendium of ChIP-seq experiments (Lachmann et al. 2010; Zambelli et al. 2012). These candidate upstream regulators can be further studied to confirm their role in regulating the genes that comprise the expression profile of interest. We validated this approach for HLH-1 and SKN-1 gene regulatory circuits, observing that this approach was successful for SKN-1 targets in L1 larvae only when low-complexity binding sites were used.
However, we note that several factors can limit the success of this approach. First, many transcription factors have not yet been profiled in ChIP-seq experiments by modENCODE. Second, some have different sets of targets in different tissues or at different times of development. For these factors, the conditions used for the ChIP-seq experiment need to be matched to the conditions used for the expression profiling experiment. Finally, in some cases changes in the activity of an initial factor will initiate a cascade of changes in downstream regulators. In this case, expression profiling of worms mutant for the first factor will reveal the entire set of genes involved in the transcriptional cascade, including not only the primary targets of the first factor but also genes that are indirectly regulated. This cascade will hinder one's ability to identify the initial manipulation.
We observed a significant overlap between SKN-1 targets in the L1 stage and SKN-1–responsive genes in adults under oxidative stress, which could be larger if data sets matched for the same stage were used. However, it is infeasible to obtain ChIP-seq data for all factors in a large number of stages or conditions. Thus, our results with SKN-1 shows that upstream regulators can be identified even when the ChIP-seq and expression profiling experiments are performed using different conditions.
Genomics screen for aging regulators
Next, we applied this method to the identification of new candidate regulators of aging in C. elegans. A variety of techniques have been utilized to identify specific regulators that are responsible for causing changes in expression in old age. For instance, a motif-driven approach was used to identify modules whose presence correlated with genes that changed expression with age (Adler et al. 2007). This led to the identification of the binding site for the transcription factor complex NF-KB as enriched in nine of 10 tissues queried, suggesting that NF-KB is a candidate master regulator of aging in multiple tissues. NF-KB binding activity increases with age in various tissues, and chemical inhibition of NF-KB activity led to a rejuvenation of aging phenotypes in the epidermis, suggesting that NF-KB is an important regulator of gene expression as well as detrimental phenotypes in old age (Adler et al. 2007).
In C. elegans, Budovskaya et al. (2008) found that GATA sequence motifs were enriched upstream of age-regulated genes, and identified GATA transcription factor ELT-3 as one of the factors responsible for expression changes between young and old worms. Expression of elt-3 decreases with age, but worms that retain high levels of expression of elt-3 in old age (due to knockdown of upstream repressors elt-5 or elt-6) have increased lifespan, suggesting that ELT-3 may also be an important regulator of aging.
In this study, we compared genes altered during aging with genes bound by transcription factors in ChIP-seq data sets generated by the modENCODE Consortium, identifying nine transcription factors with enriched binding to age-regulated genes. Two (ELT-3 and UNC-62) have previously been shown both to be directly responsible for changes in expression of downstream targets in old age as well as to modulate lifespan (Curran and Ruvkun 2007; Budovskaya et al. 2008; Van Nostrand et al. 2013). A third (SKN-1) is linked to aging through its role as an oxidative stress protective factor with decreased activity with age, and overexpression of constitutively active SKN-1 increases lifespan (Tullet et al. 2008; Przybysz et al. 2009). In addition to these three, we found that strains containing fosmids for two transcription factors (nuclear hormone receptors NHR-28 and NHR-76) had significantly extended lifespan. Although they did not affect lifespan in simple knockdown or overexpression experiments, two of the remaining transcription factors (PQM-1 and FOS-1) may play a role in aging through their involvement in pathogen response, as pathogenicity significantly limits lifespan in C. elegans (Garigan et al. 2002; Shapira et al. 2006; Kao et al. 2011; Sanchez-Blanco and Kim 2011). Recent work has also implicated FOS-1 in modulation of lifespan by dietary restriction (Uno et al. 2013).
In summary, experimental evidence for as many as seven of the nine candidate regulators supports a role in the worm aging process. Further work will be required to determine whether the candidate transcription factors identified in this ChIP-seq screen are directly responsible for changes in expression of their downstream targets seen in the normal aging process. Although in this work we explore the model system of aging, the ChIP-seq screening approach described here could be used to help untangle complex regulatory networks responsible for expression changes in other expression profiles by identifying a small number of transcription factor candidates for further experimental study.
Methods
ChIP-seq data sets and analysis
Ninety-eight ChIP-seq data sets for 57 transcription factors were obtained from the modENCODE Consortium as of 4/30/12 (http://submit.modencode.org/submit/public/list or http://data.modencode.org/). Binding data were mapped to WS220 coordinates using scripts available from WormBase (ftp://ftp.sanger.ac.uk/pub2/wormbase/software/Remap-between-versions/) (Harris et al. 2010). Binding sites were then associated with annotated WS220 transcripts if the position of maximum read density within the binding site was (1) located within the minimum of 5 kb upstream of the annotated transcription start site or the distance to the nearest annotated protein coding gene or (2) contained within the gene body (up to the annotated transcription stop site).
By using all 98 ChIP-seq data sets, the transcription factor complexity of every nucleotide in the genome was defined as the number of transcription factors that were found to have a significant binding site (Q-value ≤ 10−5) that overlaps that region. The complexity of a binding site was defined as the maximum complexity of any position within the binding site. Overlapping binding sites for the same transcription factor observed in multiple developmental stages were only counted once. For downstream analyses, genes with multiple binding sites of low complexity and intermediate/high complexity were counted for both groups.
Highly occupied target (HOT) regions were defined as contiguous genomic regions bound by at least 65% of the transcription factors considered (38 out of 57). Low-complexity binding sites were defined as those that had no position within the binding site that was enriched in nine or more transcription factors (out of the 57 total). To associate HOT regions with nearby genes, a shorter 1 kb upstream to 500 nt downstream from transcription start site window was used, and the HOT region center was defined as the midpoint of the subregion bound by the maximum number of factors within the HOT region. For enrichment analysis, 19 snRNA SL2 splice leader transcripts (sls-2.#), 76 small and large ribosomal subunit genes (rps-# and rpl-#), and 139 snoRNA transcripts were obtained from WormBase release WS220 (Harris et al. 2010). Gene ontology annotations were obtained from Gene Ontology (The Gene Ontology Consortium 2000). Collagen genes were defined as genes annotated with NCBI KOG3544 (type IV and type XIII collagens).
Statistics and computational tools used
The degree of overlap between a ChIP-seq data set and the various gene lists was calculated using the Matthews correlation coefficient, which provides a summary statistic that includes all four outcomes (true positives, true negatives, false positives, and false negatives) (Baldi et al. 2000). Significance of overlap was determined by Fisher's exact test on the 2 × 2 contingency table (using the R statistics program), approximated with the Yates' χ2 test for data sets with expected and observed overlaps greater than five. P-values for χ2 tests were obtained using the Perl Statistics∷Distributions module. For all analyses, enrichment was determined relative to the set of genes that were both present in the microarray or RNA-seq study and those in the WormBase WS220 release considered for ChIP-seq targets.
To calculate the percentage of overlapping binding sites between ChIP-seq data sets, low-complexity binding sites for a first ChIP-seq data set “A” were compared against all binding sites in a second ChIP-seq data set (“B”). A binding site in the first data set was considered to overlap with a binding site in the second if at least half of the smaller of the two binding sites was contained within the larger binding site. To avoid penalizing for the different number of binding sites identified across experiments, this procedure was repeated to compare low-complexity binding sites in ChIP-seq data set B against all binding sites in ChIP-seq data set A, and the higher of the two percentages was used as the overlap between the two ChIP-seq data sets. To identify sequence motifs in UNC-62 binding sites, the 100-bp window surrounding the position of maximum ChIP-seq read density in each UNC-62 binding site was identified as the core binding region. A total of 200-bp windows on either side of this core region served as the negative control. A de novo motif search was performed using the Peak-Motifs program (Thomas-Chollier et al. 2012), with the setting for 7-mer oligonucleotide length. Motif logos were generated using WebLogo (Crooks et al. 2004).
Gene expression data sets used
For HLH-1, 2128 genes that were significantly induced upon overexpression of HLH-1 in early embryos were obtained (Fukushige et al. 2006; Fox et al. 2007). For SKN-1, four microarrays of oxidative stress (GSM237006-GSM237009), as well as three microarrays of oxidative stress and skn-1 RNAi (GSM237010-GSM237012), were used to identify SKN-1–responsive genes (Park et al. 2009). DNA microarray intensity values were scaled up or down to a target mean intensity value of 500, using only the central 96% of probes (second to 98th percentile). Probes that were not detected in all four control or all three skn-1 RNAi microarrays were discarded, and an unpaired two-sample t-test was then used to identify genes with significantly altered expression upon skn-1 RNAi. Using a cutoff of P-value ≤ 0.01 and requiring a twofold decrease in expression led to the identification of 91 SKN-1–responsive (activated) genes (Park et al. 2009). For analysis of UNC-62–responsive targets, 115 genes with significantly decreased expression and 67 genes with significantly increased expression upon knockdown of unc-62 were obtained from GEO (GSE39574) (Van Nostrand et al. 2013). To perform the screen for candidate regulators of aging, a data set describing genes with significantly altered (both increased and decreased) expression with age was obtained (Budovskaya et al. 2008).
Tissue-enriched gene lists
Data sets of tissue-enriched and tissue-specific genes were obtained from previous publications as follows: tissue-enriched data sets for 25 tissues and tissue sub-types in various developmental stages (Spencer et al. 2011), intestine in L4 larvae (Pauli et al. 2006), muscle in L1 larvae (Roy et al. 2002), neurons and A-class neurons in embryos and L2 larvae (Von Stetina et al. 2007), embryonic touch neurons (Zhang et al. 2002), embryonic motor neurons (Fox et al. 2005), and embryonic AFD and AWD neurons (Colosimo et al. 2004). Annotations of known transcription factor expression profiles were obtained from WormBase (Harris et al. 2010).
Naïve Bayes responsiveness score
The scoring metric that combines Q-value and complexity was created using a simple two-feature naïve Bayes classification model. For each ChIP-seq binding site, the discrete Q-values were segmented into bins (where a bin from x to y indicates binding sites with x ≤ Q-value < y): 10−2 to 5 × 10−3, 5 × 10−3 to 10−4, 10−4 to 10−5, 10−5 to 10−7.5, 10−7.5 to 10−10, 10−10 to 10−15, 10−15 to 10−25, and 10−25 to 0. Complexity was similarly binned, based upon whether the binding site was bound (in total) by 1, 2, 3–4, 5–6, 7–9, 10–12, 13–15, 16–20, 21–25, 26–31, 32–37, or 38–57 transcription factors.
To initially test the method, naïve Bayes classifiers were independently trained upon each ChIP-seq data set to compare training error for predicting HLH-1–responsive genes across ChIP-seq data sets. After training, for a binding site with Q-value 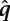 and complexity
and complexity 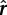 , the classifier yields a probability of that binding site being associated with the HLH-1–responsive (y = 1) or nonresponsive (y = 0) class. The log of the ratio of these probabilities
, the classifier yields a probability of that binding site being associated with the HLH-1–responsive (y = 1) or nonresponsive (y = 0) class. The log of the ratio of these probabilities 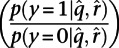 was used as a score for each binding site; the top-scoring 500 binding sites for each factor were used to determine the predictive ability of each ChIP-seq data set on HLH-1–responsive genes.
was used as a score for each binding site; the top-scoring 500 binding sites for each factor were used to determine the predictive ability of each ChIP-seq data set on HLH-1–responsive genes.
For further analyses, the classifier was trained using HLH-1 ChIP-seq targets and HLH-1 factor–responsive genes and then used to score binding sites from the other 97 ChIP-seq data sets. In all downstream analyses, the 500 binding sites with the highest log ratio score were used.
Lifespan analysis
General C. elegans techniques as well as lifespan experiments were performed according to the method previously described (Budovskaya et al. 2008). Unless otherwise noted, lifespan experiments were performed by placing day 1 adult worms (with visible eggs) on NGM plates containing 30 μM 5-fluoro-2′-deoxyuridine (FUDR). Deaths before day 7 of adulthood were censured. Log-rank tests were performed using OASIS (Yang et al. 2011). RNAi clones used were obtained from the Ahringer RNAi library (Kamath et al. 2003) and sequenced to verify proper insertions. RNAi knockdown experiments in adults were performed on NGM plates supplemented with 30 uM FUDR, 100 μg/mL ampicillin, and 2 mM IPTG to induce dsRNA expression.
For overexpression, strains were obtained from modENCODE in which a fosmid containing a transcription factor (with a C-terminal GFP fusion) is overexpressed along with an unc-119 transgene as a selectable marker in an unc-119(ed3) background. As controls, we used four strains (RW11108, RW11206, RW10780, and RW10384) that also contain a biolistically bombarded and integrated unc-119 transgene. These strains lack overexpression of a transcription factor, but instead contain a promoter fusion to a histone-tagged mCherry fluorescent reporter (and showed weak or no expression of mCherry). For the initial screen of all nine transcription factors, the modENCODE strain used for the ChIP-seq experiments was used without backcrossing; strains containing NHR-28 (OP317) or NHR-76 (OP203) fosmids that showed significant extension of lifespan in the initial screen were then backcrossed twice to wild-type (N2) and repeated to confirm extension of lifespan. Strain OP203 had a tendency to move onto the walls of the plate, increasing the number of worms lost in the experiment; for the backcrossed lifespan experiment, a ring of palmitic acid (10 mg/mL) was added around the plate in an attempt to alleviate this concern.
To estimate fosmid insertion copy number, read density in the ChIP-seq input control samples was used in a manner similar to previously described (Sarov et al. 2012). The average read density across sliding 10-kb windows was calculated for the entire genome, and copy number was defined as the ratio between the average read density in windows within the fosmid and the average of all other windows in the genome.
Acknowledgments
We thank the modENCODE Consortium for providing ChIP-seq data. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). We thank Anne Brunet, Arend Sidow, Michael Snyder, and members of the Kim laboratory for helpful discussion, suggestions, and improvements. We thank members of the Kim laboratory and Jeanine Frey for critical reading of the manuscript. E.L.V.N. has been supported by the Stanford Genome Training Program and the Smith Fellowship (Stanford Graduate Fellowships program). Research in the laboratory of S.K.K. is supported by the NHGRI, NIGMS, NIA, and the Glenn Foundation.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.152876.112.
References
- Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E, Chang HY 2007. Motif module map reveals enforcement of aging by continual NF-κB activity. Genes Dev 21: 3244–3257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An JH, Blackwell TK 2003. SKN-1 links C. elegans mesendodermal specification to a conserved oxidative stress response. Genes Dev 17: 1882–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldi P, Brunak S, Chauvin Y, Andersen CA, Nielsen H 2000. Assessing the accuracy of prediction algorithms for classification: An overview. Bioinformatics 16: 412–424 [DOI] [PubMed] [Google Scholar]
- Biggin MD 2011. Animal transcription networks as highly connected, quantitative continua. Dev Cell 21: 611–626 [DOI] [PubMed] [Google Scholar]
- Bowerman B, Eaton BA, Priess JR 1992. skn-1, a maternally expressed gene required to specify the fate of ventral blastomeres in the early C. elegans embryo. Cell 68: 1061–1075 [DOI] [PubMed] [Google Scholar]
- Budovskaya YV, Wu K, Southworth LK, Jiang M, Tedesco P, Johnson TE, Kim SK 2008. An elt-3/elt-5/elt-6 GATA transcription circuit guides aging in C. elegans. Cell 134: 291–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Yan KK, Yip KY, Rozowsky J, Alexander R, Shou C, Gerstein M 2011. A statistical framework for modeling gene expression using chromatin features and application to modENCODE datasets. Genome Biol 12: R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Alexander R, Min R, Leng J, Yip KY, Rozowsky J, Yan KK, Dong X, Djebali S, Ruan Y, et al. 2012. Understanding transcriptional regulation by integrative analysis of transcription factor binding data. Genome Res 22: 1658–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosimo ME, Brown A, Mukhopadhyay S, Gabel C, Lanjuin AE, Samuel AD, Sengupta P 2004. Identification of thermosensory and olfactory neuron-specific genes via expression profiling of single neuron types. Curr Biol 14: 2245–2251 [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE 2004. WebLogo: A sequence logo generator. Genome Res 14: 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran SP, Ruvkun G 2007. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet 3: e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox RM, Von Stetina SE, Barlow SJ, Shaffer C, Olszewski KL, Moore JH, Dupuy D, Vidal M, Miller DM III 2005. A gene expression fingerprint of C. elegans embryonic motor neurons. BMC Genomics 6: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox RM, Watson JD, Von Stetina SE, McDermott J, Brodigan TM, Fukushige T, Krause M, Miller DM III 2007. The embryonic muscle transcriptome of Caenorhabditis elegans. Genome Biol 8: R188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushige T, Krause M 2005. The myogenic potency of HLH-1 reveals wide-spread developmental plasticity in early C. elegans embryos. Development 132: 1795–1805 [DOI] [PubMed] [Google Scholar]
- Fukushige T, Brodigan TM, Schriefer LA, Waterston RH, Krause M 2006. Defining the transcriptional redundancy of early bodywall muscle development in C. elegans: Evidence for a unified theory of animal muscle development. Genes Dev 20: 3395–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Foat BC, Bussemaker HJ 2004. Defining transcriptional networks through integrative modeling of mRNA expression and transcription factor binding data. BMC Bioinformatics 5: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber M, Yosef N, Goren A, Raychowdhury R, Thielke A, Guttman M, Robinson J, Minie B, Chevrier N, Itzhaki Z, et al. 2012. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Mol Cell 47: 810–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garigan D, Hsu AL, Fraser AG, Kamath RS, Ahringer J, Kenyon C 2002. Genetic analysis of tissue aging in Caenorhabditis elegans: A role for heat-shock factor and bacterial proliferation. Genetics 161: 1101–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium 2000. Gene ontology: Tool for the unification of biology. Nat Genet 25: 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al. 2010. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 330: 1775–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et al. 2012. Architecture of the human regulatory network derived from ENCODE data. Nature 489: 91–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TW, Antoshechkin I, Bieri T, Blasiar D, Chan J, Chen WJ, De La Cruz N, Davis P, Duesbury M, Fang R, et al. 2010. WormBase: A comprehensive resource for nematode research. Nucleic Acids Res 38: D463–D467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Shi H, Liu J 2009. Two Hox cofactors, the Meis/Hth homolog UNC-62 and the Pbx/Exd homolog CEH-20, function together during C. elegans postembryonic mesodermal development. Dev Biol 334: 535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B 2007. Genome-wide mapping of in vivo protein-DNA interactions. Science 316: 1497–1502 [DOI] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. 2003. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421: 231–237 [DOI] [PubMed] [Google Scholar]
- Kao CY, Los FC, Huffman DL, Wachi S, Kloft N, Husmann M, Karabrahimi V, Schwartz JL, Bellier A, Ha C, et al. 2011. Global functional analyses of cellular responses to pore-forming toxins. PLoS Pathog 7: e1001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivega I, Dean A 2012. Enhancer and promoter interactions: Long distance calls. Curr Opin Genet Dev 22: 79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz SG, Williams BA, Sternberg PW, Wold BJ 2012. Transcription factor redundancy and tissue-specific regulation: Evidence from functional and physical network connectivity. Genome Res 22: 1907–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvon EZ, Stampfel G, Yanez-Cuna JO, Dickson BJ, Stark A 2012. HOT regions function as patterned developmental enhancers and have a distinct cis-regulatory signature. Genes Dev 26: 908–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma'ayan A 2010. ChEA: Transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics 26: 2438–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F, Chang JS, Wu WS 2010. Identifying a transcription factor's regulatory targets from its binding targets. Gene Regul Syst Bio 4: 125–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei H, Liu J, Fukushige T, Fire A, Krause M 2009. Caudal-like PAL-1 directly activates the bodywall muscle module regulator hlh-1 in C. elegans to initiate the embryonic muscle gene regulatory network. Development 136: 1241–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, et al. 2006. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet 38: 431–440 [DOI] [PubMed] [Google Scholar]
- MacArthur S, Li XY, Li J, Brown JB, Chu HC, Zeng L, Grondona BP, Hechmer A, Simirenko L, Keranen SV, et al. 2009. Developmental roles of 21 Drosophila transcription factors are determined by quantitative differences in binding to an overlapping set of thousands of genomic regions. Genome Biol 10: R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro MF, Meneghini MD, Bowerman B, Broitman-Maduro G, Rothman JH 2001. Restriction of mesendoderm to a single blastomere by the combined action of SKN-1 and a GSK-3β homolog is mediated by MED-1 and -2 in C. elegans. Mol Cell 7: 475–485 [DOI] [PubMed] [Google Scholar]
- Marbach D, Roy S, Ay F, Meyer PE, Candeias R, Kahveci T, Bristow CA, Kellis M 2012. Predictive regulatory models in Drosophila melanogaster by integrative inference of transcriptional networks. Genome Res 22: 1334–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyabayashi T, Palfreyman MT, Sluder AE, Slack F, Sengupta P 1999. Expression and function of members of a divergent nuclear receptor family in Caenorhabditis elegans. Dev Biol 215: 314–331 [DOI] [PubMed] [Google Scholar]
- The modENCODE Consortium. 2010. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330: 1787–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman C, Sun LV, Wang J, de Wit E, Talhout W, Ward LD, Greil F, Lu XJ, White KP, Bussemaker HJ, et al. 2006. Hotspots of transcription factor colocalization in the genome of Drosophila melanogaster. Proc Natl Acad Sci 103: 12027–12032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre N, Brown CD, Ma L, Bristow CA, Miller SW, Wagner U, Kheradpour P, Eaton ML, Loriaux P, Sealfon R, et al. 2011. A cis-regulatory map of the Drosophila genome. Nature 471: 527–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu W, Lu ZJ, Zhong M, Sarov M, Murray JI, Brdlik CM, Janette J, Chen C, Alves P, Preston E, et al. 2011. Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Res 21: 245–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noyes MB, Christensen RG, Wakabayashi A, Stormo GD, Brodsky MH, Wolfe SA 2008. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell 133: 1277–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Tedesco PM, Johnson TE 2009. Oxidative stress and longevity in Caenorhabditis elegans as mediated by SKN-1. Aging Cell 8: 258–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli F, Liu Y, Kim YA, Chen PJ, Kim SK 2006. Chromosomal clustering and GATA transcriptional regulation of intestine-expressed genes in C. elegans. Development 133: 287–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potts MB, Wang DP, Cameron S 2009. Trithorax, Hox, and TALE-class homeodomain proteins ensure cell survival through repression of the BH3-only gene egl-1. Dev Biol 329: 374–385 [DOI] [PubMed] [Google Scholar]
- Przybysz AJ, Choe KP, Roberts LJ, Strange K 2009. Increased age reduces DAF-16 and SKN-1 signaling and the hormetic response of Caenorhabditis elegans to the xenobiotic juglone. Mech Ageing Dev 130: 357–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece-Hoyes JS, Shingles J, Dupuy D, Grove CA, Walhout AJ, Vidal M, Hope IA 2007. Insight into transcription factor gene duplication from Caenorhabditis elegans Promoterome-driven expression patterns. BMC Genomics 8: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, et al. 2000. Genome-wide location and function of DNA binding proteins. Science 290: 2306–2309 [DOI] [PubMed] [Google Scholar]
- Roy PJ, Stuart JM, Lund J, Kim SK 2002. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature 418: 975–979 [DOI] [PubMed] [Google Scholar]
- Sanchez-Blanco A, Kim SK 2011. Variable pathogenicity determines individual lifespan in Caenorhabditis elegans. PLoS Genet 7: e1002047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarov M, Murray JI, Schanze K, Pozniakovski A, Niu W, Angermann K, Hasse S, Rupprecht M, Vinis E, Tinney M, et al. 2012. A genome-scale resource for in vivo tag-based protein function exploration in C. elegans. Cell 150: 855–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z, Zhang Y, Yuan GC, Orkin SH, Waxman DJ 2012. MAnorm: A robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol 13: R16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapira M, Hamlin BJ, Rong J, Chen K, Ronen M, Tan MW 2006. A conserved role for a GATA transcription factor in regulating epithelial innate immune responses. Proc Natl Acad Sci 103: 14086–14091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer WC, Zeller G, Watson JD, Henz SR, Watkins KL, McWhirter RD, Petersen S, Sreedharan VT, Widmer C, Jo J, et al. 2011. A spatial and temporal map of C. elegans gene expression. Genome Res 21: 325–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz F, Furlong EE 2012. Transcription factors: From enhancer binding to developmental control. Nat Rev Genet 13: 613–626 [DOI] [PubMed] [Google Scholar]
- Taneri B, Snyder B, Novoradovsky A, Gaasterland T 2004. Alternative splicing of mouse transcription factors affects their DNA-binding domain architecture and is tissue specific. Genome Biol 5: R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Chollier M, Herrmann C, Defrance M, Sand O, Thieffry D, van Helden J 2012. RSAT peak-motifs: Motif analysis in full-size ChIP-seq datasets. Nucleic Acids Res 40: e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, Liu S, Oliveira RP, Baumeister R, Blackwell TK 2008. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell 132: 1025–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno M, Honjoh S, Matsuda M, Hoshikawa H, Kishimoto S, Yamamoto T, Ebisuya M, Yamamoto T, Matsumoto K, Nishida E 2013. A fasting-responsive signaling pathway that extends life span in C. elegans. Cell Rep 3: 79–91 [DOI] [PubMed] [Google Scholar]
- Van Auken K, Weaver D, Robertson B, Sundaram M, Saldi T, Edgar L, Elling U, Lee M, Boese Q, Wood WB 2002. Roles of the Homothorax/Meis/Prep homolog UNC-62 and the Exd/Pbx homologs CEH-20 and CEH-40 in C. elegans embryogenesis. Development 129: 5255–5268 [DOI] [PubMed] [Google Scholar]
- Van Nostrand EL, Sánchez-Blanco A, Wu B, Nguyen A, Kim SK 2013. Roles of the developmental regulator unc-62/Homothorax in limiting longevity in Caenorhabditis elegans. PLoS Genet 9: e1003325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Stetina SE, Watson JD, Fox RM, Olszewski KL, Spencer WC, Roy PJ, Miller DM 3rd 2007. Cell-specific microarray profiling experiments reveal a comprehensive picture of gene expression in the C. elegans nervous system. Genome Biol 8: R135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagmaister JA, Gleason JE, Eisenmann DM 2006. Transcriptional upregulation of the C. elegans Hox gene lin-39 during vulval cell fate specification. Mech Dev 123: 135–150 [DOI] [PubMed] [Google Scholar]
- Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, et al. 2012. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res 22: 1798–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Sym M, Kenyon C 2005. The roles of two C. elegans HOX co-factor orthologs in cell migration and vulva development. Development 132: 1413–1428 [DOI] [PubMed] [Google Scholar]
- Yang JS, Nam HJ, Seo M, Han SK, Choi Y, Nam HG, Lee SJ, Kim S 2011. OASIS: Online application for the survival analysis of lifespan assays performed in aging research. PLoS ONE 6: e23525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip KY, Cheng C, Bhardwaj N, Brown JB, Leng J, Kundaje A, Rozowsky J, Birney E, Bickel P, Snyder M, et al. 2012. Classification of human genomic regions based on experimentally determined binding sites of more than 100 transcription-related factors. Genome Biol 13: R48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambelli F, Prazzoli GM, Pesole G, Pavesi G 2012. Cscan: Finding common regulators of a set of genes by using a collection of genome-wide ChIP-seq datasets. Nucleic Acids Res 40: W510–W515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ma C, Delohery T, Nasipak B, Foat BC, Bounoutas A, Bussemaker HJ, Kim SK, Chalfie M 2002. Identification of genes expressed in C. elegans touch receptor neurons. Nature 418: 331–335 [DOI] [PubMed] [Google Scholar]
- Zhong M, Niu W, Lu ZJ, Sarov M, Murray JI, Janette J, Raha D, Sheaffer KL, Lam HY, Preston E, et al. 2010. Genome-wide identification of binding sites defines distinct functions for Caenorhabditis elegans PHA-4/FOXA in development and environmental response. PLoS Genet 6: e1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]