

Abstract
Transcript leaders (TLs) can have profound effects on mRNA translation and stability. To map TL boundaries genome-wide, we developed TL-sequencing (TL-seq), a technique combining enzymatic capture of m7G-capped mRNA 5′ ends with high-throughput sequencing. TL-seq identified mRNA start sites for the majority of yeast genes and revealed many examples of intragenic TL heterogeneity. Surprisingly, TL-seq identified transcription initiation sites within 6% of protein-coding regions, and these sites were concentrated near the 5′ ends of ORFs. Furthermore, ribosome density analysis showed these truncated mRNAs are translated. Translation-associated TL-seq (TATL-seq), which combines TL-seq with polysome fractionation, enabled annotation of TLs, and simultaneously assayed their function in translation. Using TATL-seq to address relationships between TL features and translation of the downstream ORF, we observed that upstream AUGs (uAUGs), and no other upstream codons, were associated with poor translation and nonsense-mediated mRNA decay (NMD). We also identified hundreds of genes with very short TLs, and demonstrated that short TLs were associated with poor translation initiation at the annotated start codon and increased initiation at downstream AUGs. This frequently resulted in out-of-frame translation and subsequent termination at premature termination codons, culminating in NMD of the transcript. Unlike previous approaches, our technique enabled observation of alternative TL variants for hundreds of genes and revealed significant differences in translation in genes with distinct TL isoforms. TL-seq and TATL-seq are useful tools for annotation and functional characterization of TLs, and can be applied to any eukaryotic system to investigate TL-mediated regulation of gene expression.
Regulation of gene expression controls cellular fate and fitness. Post-transcriptional regulation of messenger RNAs (mRNAs) can have large effects on gene expression (via protein output) by modulating mRNA translation, stability, and localization. For example, gene-specific translational efficiencies vary over 100-fold genome-wide (Ingolia et al. 2009), and mRNA half-lives range from a few minutes to many hours. Despite the increasing evidence for pervasive post-transcriptional regulation of gene expression in eukaryotes, most genome-wide studies to date have focused on transcriptional aspects of gene expression. Quantitative genome-scale assays for post-transcriptional control mechanisms are beginning to transform our understanding of the regulation of gene expression but are not yet able to capture several important aspects.
Post-transcriptional regulation of gene expression is largely governed by features of the noncoding portions of mRNA, both downstream from [3′ UTR and poly(A) tail] and upstream of (transcript leader [TL] or 5′ UTR) the open reading frame (ORF). Because some TLs contain upstream ORFs (uORFs) that are translated, it is more accurate to refer to “5′ UTRs” as TLs. TLs are particularly important for translation initiation. During translation initiation in eukaryotes, a cap-binding complex (eIF4F) binds to the TL via the 5′ methyl-7-guanosine (m7G) cap and facilitates recruitment of a small ribosomal subunit and its associated eukaryotic initiation factors (eIFs) to form a pre-initiation complex (PIC). The PIC scans in a net 5′-to-3′ direction until it locates a start codon (AUG), triggering complex rearrangements that eventually result in formation of an elongating 80S ribosome (for review, see Jackson et al. 2010). Scanning PICs can be captured by upstream AUGs (uAUGs), leading to decreased initiation from the main protein-coding ORF. A few uAUGs have well-characterized translational regulatory functions, including those found in the TLs of the stress-responsive transcription factors GCN4 and ATF4 (for review, see Hinnebusch 2005). Other TLs allow specific genes to be efficiently translated under conditions of widespread translational inhibition (Gilbert et al. 2007). Although most examples of TL-mediated translational control come from small-scale studies, recent developments in genome-wide technologies have revealed widespread post-transcriptional regulation by TLs (Calvo et al. 2009; Thoreen et al. 2012).
Understanding the full range of TLs' impact on post-transcriptional regulation of gene expression will require accurate, genome-wide annotations. Previous efforts to define TLs in yeast include full-length cDNA sequencing (Miura et al. 2006), 5′ serial analysis of gene expression (5′SAGE) (Zhang and Dietrich 2005), and computational identification of transcript boundaries from measurements using tiling microarrays (Xu et al. 2009) or RNA-seq (Nagalakshmi et al. 2008). Importantly, the latter two approaches limited each gene to only one TL, whereas the first two approaches observed widespread TL heterogeneity. More than 99% of genes analyzed by Miura et al. (2006) and 95% of genes in Zhang and Dietrich (2005) had more than one TL. Such heterogeneity is consistent with studies of individual genes (Hahn et al. 1985), indicating that one TL per gene is an oversimplification in many cases.
Here we introduce a method to study TLs on a genomic scale, TL-seq, and demonstrate its utility in Saccharomyces cerevisiae, identifying one or more TLs for the majority of genes. Surprisingly, we observed hundreds of genes with very short TLs and showed that this feature leads to initiation at downstream AUGs, often culminating in nonsense-mediated mRNA decay (NMD). Of TLs identified by TL-seq, <15% contained at least one uAUG, significantly fewer than expected by chance. When TLs contain uAUGs, they tend to be conserved, reduce translation, and target the transcript for NMD. In addition, we determined the extent of intragenic TL heterogeneity and identified many new examples in yeast, including ORF-internal transcription start sites (TSSs) that may produce alternative protein variants. Finally, using translation-associated transcript leader sequencing (TATL-seq), we identified hundreds of cases where one gene encodes multiple TL isoforms, and showed that the majority of these variants are associated with distinct translational activities in vivo.
Results
Defining TLs
To facilitate identification of TLs on a genomic scale, we developed TL-seq, an adaptation of 5′ RACE for deep-sequencing platforms. TL-seq takes advantage of the unique m7G-protected 5′-5′ triphosphate linkage in the mRNA cap to biochemically distinguish and physically separate mRNA 5′ ends from other RNA species (Fig. 1A). Fragmented and size-selected RNA is first treated with a phosphatase, reducing the majority of 5′ RNA ends to hydroxyl groups, leaving m7G-capped 5′ ends intact. Subsequent treatment with a pyrophosphatase cleaves two of the three phosphates from the cap, yielding a 5′ monophosphorylated RNA. This RNA species is a substrate for RNA ligase, enabling selective ligation to the 5′ monophosphate (formerly capped) RNA fragments and not the 5′ hydroxyl fragments. 5′ adaptor ligation causes an increase in RNA fragment length that can be resolved by PAGE, enabling purification (Fig. 1B). Finally, these ligated RNA fragments are converted to DNA and deep-sequenced using standard techniques.
Figure 1.
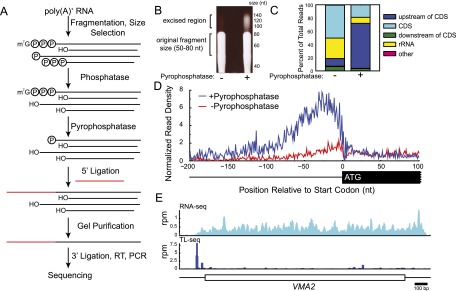
TL-seq preferentially recovers capped 5′ ends. (A) Schematic of TL-sequencing (TL-seq). (B) Fragmented RNA (50–80 nt) was treated or mock-treated with pyrophosphatase. Subsequently, both reactions were treated with RNA ligase and a 45-nt adaptor. The gel is overexposed to visualize the shift (bracket). Size markers for a DNA ladder are indicated to the right of the gel. (C) Distribution of where 5′ ends of reads map with and without inclusion of pyrophosphatase. (D) Genes were aligned by their annotated translation start codon and the distribution of reads calculated with or without pyrophosphatase. (E) Comparison of RNA-seq and TL-seq profiles for VMA2. Ordinate is in reads per million reads (rpm); scale bar, lower right.
As anticipated, TL-seq functioned in a strongly pyrophosphatase-dependent manner. Omission of pyrophosphatase from the enzymatic steps substantially decreased the yield of ligated material (Fig. 1B). Pyrophosphatase treatment resulted in an enrichment of reads upstream of annotated yeast ORFs compared with the untreated sample (Fig. 1C). Metagene analysis revealed a pyrophosphatase-dependent density of reads directly upstream of the start codon of ORFs. The maximum of this read density is 25–30 nucleotides (nt) upstream of annotated ORF start codons (Fig. 1D), on par with previous estimates of TL lengths in yeast (Miura et al. 2006; Nagalakshmi et al. 2008).
While 68% of reads mapped upstream of ORFs, others mapped to presumably uncapped transcripts (e.g., rRNA, 9%), indicating the presence of non-TSS-generated background reads in the library. To increase the signal-to-noise ratio in our TL data, we implemented a peak-calling algorithm that, for each gene, defines an expected background distribution and then identifies regions with significantly higher read density than expected. This method of computationally filtering background reads is analogous to methods commonly used to identify sites of significant enrichment in ChIP-seq data (Johnson et al. 2007). The identified regions of high read density (peaks) were then analyzed.
Peaks upstream of ORFs exhibited known TSS characteristics. There is a stereotypical nucleosome distribution about TSSs in yeast (Yuan et al. 2005). Comparing TSSs predicted by TL-seq with genome-scale nucleosome density maps showed the expected features of this characteristic distribution, including periodic placement of nucleosomes both upstream of and downstream from the TSS as well as a 5′-nucleosome-free region (5′ NFR) (Supplemental Fig. S1A). In addition, a peak of Reb1-binding sites ∼100 nt upstream of TL-seq peaks was apparent and consistent with reports of a fixed-distance relationship between binding of this transcription factor and TSSs in yeast (Koerber et al. 2009; Rhee and Pugh 2011; data not shown). For 2619 ORFs, TL-seq identified a single peak upstream of the annotated AUG. The TL annotations for these monopeak genes were highly reproducible in both length and abundance between biological replicates (Spearman's ρ ∼ 0.94, ρ ∼ 0.98, respectively) (Supplemental Fig. S1B) and in good agreement with TL lengths determined by high-density tiling array analysis (Spearman's ρ ∼ 0.6) (Supplemental Fig. S1C; Xu et al. 2009). Unlike tiling array methods, which cannot reliably distinguish alternative TSSs, TL-seq readily identified genes with at least two distinct TL peaks upstream of their ORF, which included 6.3% of genes. This number should be treated as a lower bound as it requires that intragenic TL variants be spaced >50 nt apart, a limitation imposed by the peak-calling algorithm. By simply examining read abundance, >99% of genes had reads from more than one position upstream of the coding sequence (CDS), most separated by only a few nucleotides. Collectively, these data show that TL-seq and an associated peak-calling algorithm are useful tools for genome-scale identification of TSSs in eukaryotic cells.
Transcription initiation within ORFs
Unexpectedly, 41% of TL-seq peaks mapped within ORF boundaries. While such internal peaks were generally of lower abundance and significance (P-value from peak-calling), they persisted as a substantial fraction of peaks even at more stringent significance cutoffs (Supplemental Fig. S2A). In bulk, these internal peaks showed a nucleosome signature with the same characteristics as canonical TSSs, including a periodicity both upstream and downstream and a 5′ NFR, although this signal was decreased compared with 5′ TL peaks (Supplemental Fig. S2B). Using peak-specific nucleosome signature scores as an orthogonal metric of TSSs, we estimate that 6% of genes exhibit internal transcription (Supplemental Fig. S2C; Supplemental Extended Experimental Procedures), and this number is in good agreement with previous observations of the fraction of internal TSSs (6%) estimated from a large-scale cDNA sequencing approach (Miura et al. 2006). Furthermore, there was significant overlap between internal-TSS-containing genes (P = 2.9 × 10−22, Fisher's exact test) as well as internal TSS positions (Supplemental Table S3) identified by the two studies. This overlap is notably high, given that the previous study pooled cDNAs sequenced from yeast in rich media and undergoing meiosis. Thus, our results support the existence of a substantial number of internal peaks within ORFs.
The internal peaks were recovered from a protocol that enriches for RNA species containing both poly(A) tails and m7G caps, the hallmarks of translatable eukaryotic mRNAs. To assay for translation initiation on these 5′ truncated transcripts, we examined ribosome distributions on internal TL peak-containing ORFs by ribosome footprint profiling (Ribo-seq). Ribo-seq uses deep sequencing of ribosome-protected mRNA fragments to reveal the precise positions of mRNA-associated ribosomes (Ingolia et al. 2009). Ribo-seq of glucose-starved yeast shows a high density of ribosomes at the initiation codon of all genes (Fig. 2A), but not at internal AUGs (Fig. 2A, inset), thus providing a tool for the identification of the sites of initiating ribosomes (PP Vaidyanathan, B Zinshteyn, MK Thompson, WV Gilbert, in prep.). Consistent with the internal TL-seq peaks existing as the 5′ ends of translated mRNAs, ribosome footprint density was elevated at the first downstream AUG, regardless of whether it is in frame or out of frame to the annotated full-length ORF (Fig. 2B,C). For those internal transcripts where the predicted first encountered AUG is out-of-frame, the density of ribosomes at this AUG was much higher than the first in-frame AUG (Fig. 2C, inset), as expected from the 5′ to 3′ scanning model of translation initiation.
Figure 2.
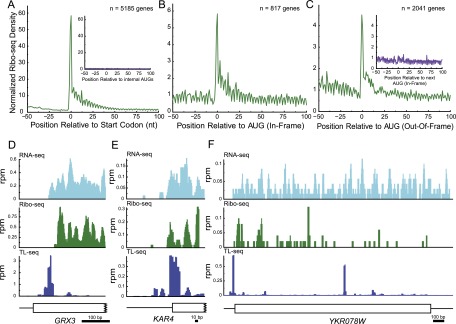
Three types of internal transcription start sites (TSSs) identified by TL-seq. (A) Ribosome footprint density aligned relative to annotated start codons for Ribo-seq from glucose-starved yeast. Ribosomes accumulate at initiation AUGs but not internal AUGs (inset). (B) Ribosome footprint density for internal TL genes whose first AUG is in-frame with the annotated start codon. (C) Ribosome footprint density for internal TL genes whose first AUG is out-of-frame with the annotated start codon. (Inset) The first in-frame AUG for these same peaks. (D) Misannotated N termini. RNA-seq, Ribo-seq, and TL-seq support a TSS starting internal to the annotated AUG. (E) N-terminal peak. TL-seq called a TSS just inside of the annotated ORF. RNA-seq and Ribo-seq support such an internal TSS. (F) TL-seq identified a second internal TSS.
Closer examination of the putative internal transcripts on a gene-by-gene basis identified four broad categories of internal TL peaks. The first category was that of 5′ misannotation (Fig. 2D). In these cases, the annotated translation initiation codon is likely incorrect, as RNA-seq and Ribo-seq data, as well as decreased amino acid conservation, corroborate the TL-seq annotation of a predominant TSS 3′ to the annotated TSS. A second category was that of extreme N-terminal peaks in which the major peak identified by TL-seq mapped within the first 100 bases of the ORF. This class includes genes that are known to produce internal transcripts generating N-terminal protein variants, such as KAR4 (Fig. 2E; Gammie et al. 1999), FUM1 (Wu and Tzagoloff 1987), and HTS1 (Chiu et al. 1992). The majority (≥85%) of internal TSSs were N-terminal (≤100 nt into an ORF) (Supplemental Fig. S2D). A third potentially interesting class of internal peaks mapped to loci that appear to generate much shorter distinct second transcripts (Fig. 2F). Northern analysis for three genes in this category revealed multiple transcripts of distinct sizes (Supplemental Fig. S3). A fourth class of ORF-internal TL-seq peaks could be attributed to RNA-ligase–dependent capture bias (Supplemental Fig. S4) and were subsequently filtered (see Supplemental Methods).
Genes with short TLs exhibit inefficient start codon recognition
TL-seq revealed an unexpected class of genes with very short TLs (≤12 nt), which are interesting from a translation initiation perspective. A ribosomal small subunit correctly positioned at an initiation codon protects at least 12 nt 5′ to the AUG-occupied ribosomal P-site (Legon 1976). In addition, the capped 5′ ends of most translating mRNAs are thought to be bound by eIFs that facilitate ribosome recruitment. Given the physical constraints of the factors involved, it seemed likely that short TL genes would initiate translation inefficiently at the cap-proximal AUG. Consistent with this prediction, artificial 5′ truncation of the PGK1 TL to ≤21 nt has been shown to reduce the efficiency of translation from the first start codon in yeast (van den Heuvel et al. 1989). We reasoned that the frame of the second downstream AUG would determine the susceptibility of a short TL mRNA to NMD (Fig. 3A). If a ribosome missed the cap-proximal AUG and initiation occurred at a downstream AUG in-frame with the annotated ORF, termination would occur at the annotated stop codon. However, if the first recognized AUG was out-of-frame, 98% of the time ribosomes would terminate at a premature termination codon (PTC), which leads to degradation by the NMD pathway in yeast (Leeds et al. 1991).
Figure 3.

Short TL genes are enriched for nonsense-mediated mRNA decay (NMD) targets. (A) Model predicting why short TL genes with a second out-of-frame AUG are NMD targets (not to scale). Failure to identify the cap-proximal AUG in short TLs results in scanning and recognition of a second, downstream AUG. If the second AUG is out-of-frame, it results in premature termination and NMD. For simplicity, the eIF4F complex is drawn on the cap during scanning, though some or all of its subunits may remain associated with the small ribosomal subunit (Jackson et al. 2010; Aitken and Lorsch 2012). (B) Fold change in steady-state mRNA levels for short TL genes in upf1Δ cells. Genes with short TLs exhibit a significant shift toward increased RNA levels only when the next AUG encountered is out-of-frame. The number of genes in each group is indicated in parentheses. (C) Ribosome density analysis of genes with a second AUG out-of-frame, using Ribo-seq from glucose-starved yeast. The dotted line shows all genes; solid lines show short TL genes with second AUG out-of-frame. Fold up indicates genes whose mRNAs are increased in the upf1Δ microarray data.
According to the above logic, short TL genes should be targets of the NMD pathway only when the second AUG within the ORF is out-of-frame. To test this prediction, we analyzed published microarray data for changes in transcript levels in yeast deleted for each of three factors required for NMD (Upf1, Upf2, or Upf3) (He et al. 2003). Genes with short TLs exhibited a significant shift toward increased steady-state mRNA levels in NMD-deficient upf1Δ yeast strains, consistent with our model (P = 0.0009) (Fig. 3B). As predicted, a significant shift was observed only when the second AUG was out-of-frame (Fig. 3B). We confirmed the microarray results by quantitative RT-PCR (qRT-PCR): Eight of nine genes behaved as expected (Supplemental Fig. S5). Increased mRNA levels for short TL genes were observed in all examined NMD-deficient strains (Supplemental Fig. S5; data not shown) and also for short TL genes identified by other TL annotations (P = 10−7) (Xu et al. 2009; data not shown).
Some short TL genes' mRNA levels were more affected by inactivation of NMD than others. According to our model, NMD sensitivity should relate to the extent of in-frame versus out-of-frame initiation. As predicted, genes whose mRNA levels increased in upf1Δ exhibited decreased ribosome density at the first annotated AUG concomitant with increased ribosomal density at the downstream out-of-frame AUG (Fig. 3C). This result is consistent with a direct role for out-of-frame translation leading to NMD of short TL genes. We note that while TL-seq identified a TL ≤12 nt for short TL genes, the Ribo-seq density, as well as RNA-seq density (data not shown), indicate the existence of longer TL isoforms for these genes as well, likely a result of TL heterogeneity. Although we focused our analysis on TLs ≤12 nt, which is the minimum length between the ribosomal P site and mRNA exit site, it is likely that >12 nt are required for simultaneous interaction of eIF4F with the cap and 40S subunits with the mRNA (Legon 1976; Kozak and Shatkin 1977; Lazarowitz and Robertson 1977). Consistent with this view, analysis of TL lengths up to 20 nt yielded similar results (data not shown). Thus, mRNAs with short TLs represent a new class of NMD substrate, revealing a new functional role for NMD.
uAUGs are conserved inhibitory elements for translation
Having established TL-seq as a method for defining TSSs, we examined the relationships between TL features and translation activity genome-wide. To systematically investigate the translational activity of TLs, we developed translation-associated transcript leader sequencing (TATL-seq) (Fig. 4A). TL-seq was performed on each of seven fractions across a polysome gradient, which differentially sediments mRNAs according to the number of ribosomes bound. Because translation initiation is thought to be rate-limiting for translation of most genes, more efficiently translated mRNA isoforms associate with heavier polysomes. The distribution of 3916 TL peaks for 3651 genes was quantified across a polysome gradient, thus determining TL isoform–specific sedimentation, and presumptively, information about the translational activity of individual TL isoforms. As expected, TL abundance was highly correlated between adjacent gradient fractions and less correlated between fractions with large differences in translation activity (e.g., nontranslating mRNAs in fraction 1 and efficiently translated mRNAs in fraction 7) (Fig. 4B,C). TATL-seq thus enables de novo TL annotation while simultaneously testing those TLs' translational activity in a single experiment.
Figure 4.
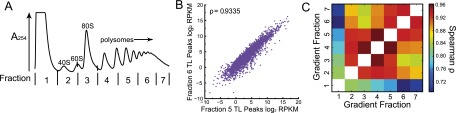
Translation-associated TL-seq (TATL-seq) quantifies translation activity of TLs in vivo. (A) TATL-seq was performed on each of seven fractions across a polysome gradient. (B) Peaks were called on the computationally pooled TATL-seq fractions, and then RPKMs were computed for each fraction individually. The Spearman correlation between two gradient fractions is shown. (C) Heatmap of Spearman's ρ for peak abundance in different TATL-seq fractions.
uAUGs are thought to negatively affect the efficiency of translation initiation at the downstream ORF, but the generality of this effect in yeast has been a subject of debate, partially because previous analyses were restricted to a handful of uAUGs (Cvijović et al. 2007; Lawless et al. 2009). To assess the generality of uAUG-mediated translational repression in rapidly dividing yeast, we examined the polysomal distribution of 773 TL species that contained one or more uAUGs compared with 3143 TL species that lacked uAUGs. uAUG-containing mRNAs showed a substantial and significant increase in sedimentation outside of polysomal fractions (Mann Whitney P = 8.7 × 10−34 for fraction 1, P = 1.1 × 10−14 for fraction 2) (Fig. 5A). In addition to providing a sink for scanning ribosomes, uAUGs that are followed by short coding regions (uORFs) lead to translation termination near the 5′ end of the transcript, which has been shown to elicit mRNA degradation via the NMD pathway (Oliveira and McCarthy 1995). Consistent with uORF-stimulated NMD being a general mechanism, uAUG-containing mRNAs' steady-state levels were significantly increased in NMD-deficient yeast (mean fold change 1.72, P < 10−16) (Fig. 5B).
Figure 5.

uAUGs are an underrepresented and conserved sequence element associated with decreased translation. (A) TATL-seq sedimentation pattern for uAUG-containing and all TLs. Relative abundance (ordinate) is the abundance of a given TL in a fraction divided by its abundance across the entire gradient. (B) Fold change in mRNA steady-state levels for single TL genes, either with uAUG-containing TLs or with all TLs. Numbers in parentheses indicate number of genes. (C) The fraction of uAUG-containing TLs was calculated based on observed TL lengths for single TL genes (red arrow) and 10,000 randomizations of gene-specific TL length (histogram). P-value based on Z-score of the observed value compared to histogram. (D) Conservation of each of 64 possible uNNN trinucleotides in the TL region was calculated using a genome-wide alignment of S. paradoxus, S. mikatae, S. bayanus, and S. cerevisiae and single TL genes. The ordinate is the number of conserved instances over the total occurrences of that trinucleotide. Near-uAUG trinucleotides are highlighted in blue.
An estimated ∼15% of genes with a single TL identified by TL-seq contained one or more uAUGs, which is consistent with previous estimates of uAUG prevalence in yeast (Lawless et al. 2009). This frequency was much lower than the expected value predicted from randomizing gene-specific TL lengths (∼23%, P = 10−69) (Fig. 5C). The observed frequency of uAUGs was also significantly lower than expected by chance given the dinucleotide composition of TLs (P < 10−310) (data not shown). Thus uAUGs are underrepresented in yeast TLs. Nevertheless, some yeast uAUGs are known to have biological functions as translational regulators of gene expression (e.g., GCN4 [Mueller and Hinnebusch 1986] and CPA1 [Wang et al. 1999]). Consistent with potential regulatory importance, uAUG is the most conserved of all possible uNNN codons in the TL region (Fig. 5D). Taken together, these data show that uAUGs are uncommon in yeast TLs, but when uAUGs are present, they tend to be both conserved and functional.
Although translation initiation can occur at near-AUG codons under some circumstances (Chang 2004; Tang 2004; Ingolia et al. 2009), our data do not support a widespread functional role for non-AUG initiation. Upstream-near-AUGs (near-uAUGs) are not highly conserved, unlike uAUGs (Fig. 5D). Furthermore, near-uAUGs were not associated with higher sedimentation in a polysome gradient, nor was there a significant shift in translation efficiency (TE) by Ribo-seq (Supplemental Fig. S6A,B). Similar results were obtained using gene-specific TE values from cells subjected to amino acid starvation (data not shown), a condition in which near-uAUG initiation was proposed to be a frequent event (Ingolia et al. 2009). Finally, genes with near-uAUG codons in their TLs were not shifted toward increased steady-state mRNA levels in NMD-deficient yeast (Supplemental Fig. S6C). Restricting these analyses to near-uAUG codons in favorable initiation contexts did not affect the results. Thus near-uAUG codons do not have an identifiable genome-scale functional role akin to that observed for uAUGs.
Intragenic TL heterogeneity
The TL peak-calling algorithm reduces a distribution of reads to a single peak, thus simplifying the data for downstream analyses. However, this simplification ignored potentially interesting differences in the distribution of reads within TL peaks. In fact, the distribution of reads within TL peaks was variable, with some peaks being more heterogeneous than others. Unlike microarray-based techniques for identifying TSSs, TL-seq allowed us to directly observe and study such variability. To quantify gene-specific TL heterogeneity, we utilized shape index (SI), a metric that incorporates fractional read abundance at every position in a defined region to provide a measure of the heterogeneity therein (Hoskins et al. 2011). The maximum SI score is zero, which indicates that all reads in the region were generated from a single position. Deviations from this distribution result in a decrease in SI score. Using this metric to quantify intragenic TL heterogeneity, the genome-wide distribution of SI scores (Fig. 6A, top) was centered about a modestly heterogeneous average (e.g., SUI3) (Fig. 6B), with a substantial fraction of genes being much more or less heterogeneous (e.g., BSC5 and MSS116, respectively) (Fig. 6B).
Figure 6.
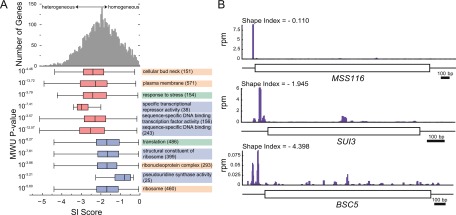
There is intragenic TL heterogeneity. (A) SI scores of GO categories with 10 or more genes were compared to all genes (top histogram). GO categories with a significantly different SI score distribution (Bonferroni-corrected Mann Whitney U P < 0.001) are shown, with number of genes in parentheses. Box and whisker plots indicate quartiles and range; red/blue shading indicates decreased/increased SI. Shading of GO categories indicates process (green), component (orange), or function (blue). (B) Three examples of genes with different shape index (SI) scores.
To investigate whether differences in TL peak heterogeneity might be functionally significant, we examined the distribution of SI scores among Gene Ontology (GO) categories. Notably, TLs from genes encoding proteins predicted to have regulatory functions (GO categories “specific transcriptional repressor activity,” “sequence-specific DNA binding,” “response to stress”) were significantly shifted toward greater heterogeneity (Fig. 6B). For these genes, TL heterogeneity might represent an underappreciated mechanism for regulation. In mammals, genes with alternative TL variants are also enriched for regulatory functions (Resch et al. 2009), consistent with what was observed here in yeast. Conversely, genes encoding ribosomal proteins and factors required for ribosome biogenesis and translation (GO categories “ribosome,” “translation,” “pseudouridine synthase activity”) were significantly shifted toward more homogeneous TLs. For such mRNAs, a homogeneous TL population might ensure high levels of expression and/or concerted post-transcriptional regulation of the downstream ORFs.
Intragenic TL heterogeneity can have consequences for translation
Two hundred thirty yeast genes exhibited more than one well-separated peak of TL reads. Such alternative TLs have been shown to confer differences in TE and regulation in a handful of cases, though the generality of this phenomenon is unknown (Rosenstiel et al. 2007; Smith et al. 2009). Many of these 230 genes showed distinct polysome sedimentation patterns. To confirm that these TLs exist as full-length alternative TL mRNAs with the same ORF, we examined RNA-seq density and observed an increase in read density at each position that did not decrease at further downstream positions (data not shown). Of genes with two TL species, the longer TL appeared more poorly translated on average, being 1.6-fold more abundant than the shorter isoform in the nontranslating pool (P = 1.1 × 10−4 Mann Whitney) (Fig. 7A). Many of these apparent differences in translation are likely due to uAUGs: ∼35% of the long TL isoforms contained at least one uAUGs, a greater than twofold enrichment over the genome-wide frequency of uAUGs in TLs. Examples of genes with multiple TL species is shown in Figure 7, B and C, and Supplemental Figure S7. A full list is available in Supplemental Table S4, and a summary of these and other findings is presented in Table 1.
Figure 7.

Intragenic TL heterogeneity leads to different translation behavior in vivo. (A) Average sedimentation pattern for 204 short/long TL pairs. (B) CRZ1 is an example of a gene with multiple TLs showing distinct sedimentation patterns in a polysome gradient. (C) TATL-seq profile of CRZ1 from fractions 1 and 7. (D) TLs are sufficient to confer the translational behavior predicted from TATL-seq (four of six genes). In vivo translation (ordinate) was determined as luciferase activity per unit RNA. Mean and standard deviation of biological triplicates is shown, (*) P < 0.05, Student's t-test.
Table 1.
Summary information from peaks for pooled translation-associated transcript leader (TL)-sequencing (TATL-seq) libraries
To determine whether the alternative TL sequences were sufficient to alter translation activity, translation efficiencies (protein produced per mRNA) were determined for alternative TL variants for six genes using Firefly luciferase (Fluc) reporters. Each TL was inserted upstream of Fluc and downstream from the GAL1 promoter, yielding constructs for inducible expression of mRNAs that differed solely in the TL region. Fluc activity per mRNA varied by almost 25-fold (Fig. 7D), demonstrating that TLs were sufficient to confer large differences in TE in vivo. These results are consistent with in vitro data showing large TL-dependent differences in cap-dependent TE (Rojas-Duran and Gilbert 2012). The largest fold difference between variants of a single gene was seen for CRZ1; the shorter isoform was translated more than 19-fold more efficiently than the longer isoform. In five of six cases, including CRZ1, intragenic TL variants showed significantly different translation efficiencies (Fig. 7D). In four such cases, the TL isoform that was predicted to have higher translation activity by TATL-seq was in fact better translated in the TL construct. For these genes, we conclude that differences in the TL region alone are sufficient to confer the observed differences in TE. The remaining cases may represent instances where other elements (e.g., 3′ UTRs, promoter-dependent mRNP assembly differences) or combinations of elements contribute to the differential translation activities observed by TATL-seq.
Discussion
Mapping of TSSs and TLs is a critical component of functional genome annotation. Here we present techniques for genome-wide, high resolution TSS identification and functional characterization of TLs. Applying the TL-seq and TATL-seq approaches to yeast revealed TLs that alter protein-coding potential, TE, and susceptibility to NMD, thus demonstrating the methods' potential to illuminate TL-mediated post-transcriptional regulation of gene expression. TL-seq has a fast and straightforward library preparation procedure that can be applied to any capped transcriptome, that is, any RNA pool for which 5′ RACE is useful. Because TL-seq and TATL-seq are sequencing-based approaches, they are applicable in any organism with a sequenced genome and do not require prior knowledge of transcribed regions.
The TL-seq approach enabled visualization of TSSs within other annotated transcripts. Such internal TSSs exhibited characteristic nucleosome signatures similar to canonical TSSs, suggesting they are true TSSs and not merely artifacts of the technique. Furthermore, protein products from internal initiation have recently been identified by mass spectrometry, thus corroborating our findings here that such internal transcripts are translated (Fournier et al. 2012). Analysis of transcript 5′ ends also yielded a significant number of ORF-internal reads in Drosophila (16%) (Ni et al. 2010) and human cells (2%–3%) (Kanamori-Katayama et al. 2011). These findings raise the possibility that diverse eukaryotes express internal transcripts, the functions of which are largely uncharacterized.
The majority of internal TSSs fell into the category of N-terminal peaks. The phenomenon of internal TSSs encoding functionally distinct N-terminal protein isoforms was first described for the SUC2 invertase gene (Carlson and Botstein 1982). The N-terminal sequence that is missing from the shorter mRNA isoform encodes a secretory peptide, and thus the upstream TL variant encodes a secreted Suc2p, while the downstream TL encodes cytoplasmic Suc2p. Similar cases have been described for FUM1 (Wu and Tzagoloff 1987), LEU4 (Beltzer et al. 1986), HTS1 (Chiu et al. 1992), TRM1 (Ellis et al. 1987), VAS1 (Chatton et al. 1988), and KAR4 (Gammie et al. 1999). Our data suggest that N-terminal TSSs might be a more common way of creating and regulating protein diversity than previously appreciated. Approximately 1% of TSSs were ≥100 nt internal to the ORF, though it is unclear whether the internal TSS events observed here are mechanistically related to the previously described phenomenon of “cryptic initiation” (Supplemental Text; Kaplan 2003).
TL-seq also identified an unexpected class of genes with extremely short TLs. The physical constraints of translation initiation on short TLs suggest these features pose problems for the initiation machinery, and our results verify this to be the case. Previously it was noted that short TLs of viral and/or artificial mRNAs were associated with decreased initiation at the cap-proximal AUG and increased initiation at downstream AUGs both in vivo and in vitro (Sedman et al. 1990; Kozak 1991; Pestova and Kolupaeva 2002). We demonstrate for the first time that short TLs on natural cellular mRNAs lead to inefficient initiation at the cap-proximal AUG, increased initiation at downstream AUGs in vivo, and targeting of the transcript for decay by NMD. Together, these observations indicate a novel form of TL-mediated post-transcriptional regulation and reveal a new functional role for NMD in yeast.
Why might short TL mRNAs exist, if only to be degraded? Short TL genes present unique post-transcriptional regulatory opportunities for a cell. Their degradation via NMD could be regulated, as conditions such as hypoxia and amino acid starvation have been reported to stabilize NMD substrates in humans (Mendell et al. 2004; Gardner 2008). For reporter mRNAs with short TLs, the efficiency of cap-proximal AUG recognition in vitro can be controlled by the levels of eIF1 (Pestova and Kolupaeva 2002), raising the possibility that the levels or activity of eIF1 in vivo may control the production of full-length protein (and the extent of out-of-frame initiation) for short TL genes. Alternatively, these genes may produce alternate, longer TLs in altered cellular conditions, as has been observed for some yeast genes following nitrogen starvation, pheromone response, and osmotic stress (Law et al. 2005), thus leading to less out-of-frame initiation and less NMD of those mRNAs. The balance between initiation at cap-proximal and downstream AUGs, and the recognition of the latter by NMD, represents an opportunity for concerted regulation of this class of transcripts.
In all systems in which it has been examined, gene-specific TE has been shown to vary substantially (Arava et al. 2003; Hendrickson et al. 2009; Ingolia et al. 2009; Guo et al. 2010; Stadler and Fire 2011; Thoreen et al. 2012). While we do not yet understand all the factors contributing to this wide (greater than 100-fold) variation, here we demonstrate that TLs can have a direct role in explaining some of the observed variation in translation genome-wide.
TATL-seq has great potential to illuminate the mechanisms responsible for translation activity differences and regulation. Since it is a direct measurement of TL isoform–specific translation, it is particularly useful for discerning the relative contributions of multiple isoforms to the overall translation of a gene. Isoform differences are invisible to most conventional approaches (e.g., RNA-seq, polysome microarray, ribosome footprint profiling) and may confound efforts to relate TL properties to ORF-based measurements of translation activity.
Our systematic investigation of intragenic TL variation showed that such variation has significant consequences for translation. These findings support and extend the conclusions from low-throughput studies of yeast and human genes with alternative TLs. We detected TL variants with differing translation even under standard growth conditions in S. cerevisiae, which has relatively low levels of mRNA isoform diversity. In contrast, TL diversity is quite common in mammals; in fact, it was suggested that TSS selection contributes more to mRNA isoform diversity than alternative splicing in some tissues (Pal et al. 2011). Furthermore, in a diverse panel of human tissues, the total number of alternative TLs observed was similar to the numbers of alternative 3′ UTRs and alternatively spliced internal exons (Wang et al. 2008). Intriguingly, the majority of TL variants showed tissue-specific expression patterns. Importantly, because most intragenic TL variants do not change the coding potential of the mRNA, their influences must be felt during the post-transcriptional life of the mRNA, namely, during translation, localization, and/or decay. We anticipate that the TL-seq and TATL-seq methods described here will enable systematic studies of TL regulation and function in eukaryotes.
Methods
Yeast strains and growth conditions
Yeast cultures (Sigma 1278b MAT a ura3 leu2 trp1 his3 and BY4742 Mat α his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) were grown to mid-log (OD600 ∼ 0.5–1.0) phase in YPAD (1% yeast extract, 2% peptone, 0.01% adenine hemisulfate, 2% glucose) at 30°C in flasks with vigorous shaking.
TL sequencing
Polyadenylated mRNA (oligo dT cellulose purified as described) (Sambrook et al. 2001) was fragmented by alkaline hydrolysis. RNA fragments of ∼50–80 nt were gel purified and dephosphorylated with 30 U calf-intestinal phosphatase (CIP; NEB) in 50 μL reactions for 60 min at 37°C followed by phenol:chloroform extraction and isopropanol precipitation. Purified CIP-treated fragments were treated with 25 U tobacco acid pyrophosphatase (TAP; Epicentre) in 50 μL for 2 h at 37°C and then precipitated. Next, a 5′ RNA adaptor was added via ligation in a 20 μL reaction with 20 U of T4 RNA Ligase (NEB) for 1 h at 37°C. Gel purification of a higher-molecular-weight species yielded the ligated RNA, which was then 3′end captured via poly(A) tailing (TL-seq, according to the method previously described in Ingolia et al. [2009]) or ligation with preadenylated adaptor (TL-seq biological replicates and TATL-seq, according to the method previously described in Mayr and Bartel [2009]). cDNA was prepared from ligated RNA (Superscript III, Invitrogen), amplified by 10 or 12 cycles of PCR (Phusion, Finnzymes), and sequenced on an Illumina Genome Analyzer II (TL-seq and TATL-seq) or HiSeq (TL-seq replicates).
The computationally pooled TATL-seq libraries were used for Figures 1E and 2 through 7, as these libraries gave more data for more genes. Analyses gave similar results using TL-seq or pooled TATL-seq data.
Peak-calling algorithm
The peak-calling algorithm was developed with modifications from Johnson et al. (2007). For each gene, an expected background density of reads at a given nucleotide assuming a uniform distribution throughout the feature is
where N is the total number of reads mapping to that feature (including upstream and downstream boundaries), and L is the total length of the feature (including upstream and downstream boundaries). The observed read density, x, at each position was calculated by scanning along the feature using an n-nucleotide window. For analyses here, n = 50 nt; using smaller n yielded a higher fraction of artifactual peaks, while larger n yielded fewer peaks overall. If a window contained more than five times the expected number of reads (i.e., ≥5nλ), a P-value for the enrichment was calculated based on the Poisson distribution:
Consecutive windows of enrichment (P < 0.01) ≥ n nucleotides in length were defined as a peak. The exact nucleotide position of the peak was defined as the mode read in that region.
Monopeak TL-seq genes (used in Figs. 3, 5B–D; Supplemental Figs. S1B, S6B,C) were those genes with exactly one TL-seq peak upstream of the annotated ORF start codon (2619 out of 3434 total ORFs with TL-seq peaks).
uAUG analysis
The expected frequency of uAUGs in TLs was determined by randomizing TL lengths between genes and calculating the fraction of uAUG-containing TLs for 10,000 randomizations. Alternatively, individual TL sequences were shuffled preserving di- or mononucleotide frequency within a given TL sequence using the algorithm described in Altschul and Erickson (1985). P-values were calculated using a z-statistic approximating a normal distribution from the randomizations.
For conservation analysis, TL positions were included if an ungapped genomic alignment existed amongst four yeast species: S. cerevisiae, Saccharomyces paradoxus, Saccharomyces mikatae, and Saccharomyces bayanus. A trinucleotide within a S. cerevisiae TL was deemed “conserved” if the same trinucleotide was present at the same position in the three other yeast species. A trinucleotide was deemed “nonconserved” if one or more yeast species contained a mutation anywhere in that trinucleotide at the aligned position. For each trinucleotide, the frequency of conservation was defined as the number of conserved instances divided by the total number of eligible instances of that trinucleotide (conserved plus nonconserved).
Shape index
Shape index (as defined by Hoskins et al. [2011]) was used to quantify TL heterogeneity within genes and is defined as
where fi is the frequency of reads from a given nucleotide i. For GO analysis, the SI of all genes within a given GO category (SGD annotations) was compared to the overall distribution of SIs, and GO categories with a P < 0.001 (Mann Whitney U Bonferroni-corrected P-value) were deemed significantly different.
TATL-seq
Polysome gradients were fractionated, and RNA was collected from each of seven fractions. Purified RNA was poly(A) selected and fragmented, as per the TL-seq protocol. To quantify individual TLs across a polysome gradient, peak-calling was performed on the computationally pooled TATL-seq libraries. The abundance of each peak in each fraction was then quantified by calculating the density of reads (RPKM). The relative abundance of a TL in a fraction is equal to its read density in that fraction divided by its read density over all fractions. Specifically, the relative abundance of a TL x in fraction i was defined as
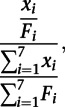 |
where xi is the number of reads mapping to the TL and Fi is the library size of fraction i.
Details of RNA isolation, polysome gradient fractionation, read assignment, peak RPKM determination, Ribo-seq analysis, nucleosome analysis, peak filters, luciferase assays, and more are available in the Supplemental Methods.
Data access
The TL-seq and TATL-seq data, as well as processed TL-lengths, and links to UCSC and SGD Genome Browser-formatted data from these experiments have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under series accession no. GSE39074. The data are also viewable under “Select Tracks” in GBrowse at SGD (http://yeastgenome.org).
Acknowledgments
We thank Stuart Levine and the BioMicro Center for performing the sequencing and for discussion of ChIP-seq peak-calling algorithms; Uttam RajBhandary, Jason Merkin, and Charles Lin for insightful discussions; Chris Burge, Stirling Churchman, and members of the Gilbert Laboratory for critical reading of the manuscript. This work was supported by a National Science Foundation Graduate Research Fellowship award to J.A.A. and a National Institute of General Medical Sciences Grant GM081399 to W.V.G.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.150342.112.
References
- Aitken CE, Lorsch JR 2012. A mechanistic overview of translation initiation in eukaryotes. Nat Struct Mol Biol 19: 568–576 [DOI] [PubMed] [Google Scholar]
- Altschul SF, Erickson BW 1985. Significance of nucleotide sequence alignments: A method for random sequence permutation that preserves dinucleotide and codon usage. Mol Biol Evol 2: 526–538 [DOI] [PubMed] [Google Scholar]
- Arava Y, Wang Y, Storey JD, Liu CL, Brown PO, Herschlag D 2003. Genome-wide analysis of mRNA translation profiles in Saccharomyces cerevisiae. Proc Natl Acad Sci 100: 3889–3894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltzer JP, Chang LF, Hinkkanen AE, Kohlhaw GB 1986. Structure of yeast LEU4. The 5′ flanking region contains features that predict two modes of control and two productive translation starts. J Biol Chem 261: 5160–5167 [PubMed] [Google Scholar]
- Calvo SE, Pagliarini DJ, Mootha VK 2009. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci 106: 7507–7512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson M, Botstein D 1982. Two differentially regulated mRNAs with different 5′ ends encode secreted with intracellular forms of yeast invertase. Cell 28: 145–154 [DOI] [PubMed] [Google Scholar]
- Chang KJ 2004. Translation initiation from a naturally occurring non-AUG codon in Saccharomyces cerevisiae. J Biol Chem 279: 13778–13785 [DOI] [PubMed] [Google Scholar]
- Chatton B, Walter P, Ebel JP, Lacroute F, Fasiolo F 1988. The yeast VAS1 gene encodes both mitochondrial and cytoplasmic valyl-tRNA synthetases. J Biol Chem 263: 52–57 [PubMed] [Google Scholar]
- Chiu MI, Mason TL, Fink GR 1992. HTS1 encodes both the cytoplasmic and mitochondrial histidyl-tRNA synthetase of Saccharomyces cerevisiae: Mutations alter the specificity of compartmentation. Genetics 132: 987–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvijović M, Dalevi D, Bilsland E, Kemp GJL, Sunnerhagen P 2007. Identification of putative regulatory upstream ORFs in the yeast genome using heuristics and evolutionary conservation. BMC Bioinformatics 8: 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis SR, Hopper AK, Martin NC 1987. Amino-terminal extension generated from an upstream AUG codon is not required for mitochondrial import of yeast N2,N2-dimethylguanosine-specific tRNA methyltransferase. Proc Natl Acad Sci 84: 5172–5176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier CT, Cherny JJ, Truncali K, Robbins-Pianka A, Lin MS, Krizanc D, Weir MP 2012. Amino termini of many yeast proteins map to downstream start codons. J Proteome Res 11: 5712–5719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gammie AE, Stewart BG, Scott CF, Rose MD 1999. The two forms of karyogamy transcription factor Kar4p are regulated by differential initiation of transcription, translation, and protein turnover. Mol Cell Biol 19: 817–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner LB 2008. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol Cell Biol 28: 3729–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert WV, Zhou K, Butler TK, Doudna JA 2007. Cap-independent translation is required for starvation-induced differentiation in yeast. Science 317: 1224–1227 [DOI] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466: 835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn S, Hoar ET, Guarente L 1985. Each of three “TATA elements” specifies a subset of the transcription initiation sites at the CYC-1 promoter of Saccharomyces cerevisiae. Proc Natl Acad Sci 82: 8562–8566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Li X, Spatrick P, Casillo R, Dong S, Jacobson A 2003. Genome-wide analysis of mRNAs regulated by the nonsense-mediated and 5′ to 3′ mRNA decay pathways in yeast. Mol Cell 12: 1439–1452 [DOI] [PubMed] [Google Scholar]
- Hendrickson DG, Hogan DJ, McCullough HL, Myers JW, Herschlag D, Ferrell JE, Brown PO 2009. Concordant regulation of translation and mRNA abundance for hundreds of targets of a human microRNA. PLoS Biol 7: e1000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch AG 2005. Translational regulation of GCN4 and the general amino acid control of yeast. Annu Rev Microbiol 59: 407–450 [DOI] [PubMed] [Google Scholar]
- Hoskins RA, Landolin JM, Brown JB, Sandler JE, Takahashi H, Lassmann T, Yu C, Booth BW, Zhang D, Wan KH, et al. 2011. Genome-wide analysis of promoter architecture in Drosophila melanogaster. Genome Res 21: 182–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CUT, Pestova TV 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11: 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B 2007. Genome-wide mapping of in vivo protein-DNA interactions. Science 316: 1497–1502 [DOI] [PubMed] [Google Scholar]
- Kanamori-Katayama M, Itoh M, Kawaji H, Lassmann T, Katayama S, Kojima M, Bertin N, Kaiho A, Ninomiya N, Daub CO, et al. 2011. Unamplified cap analysis of gene expression on a single-molecule sequencer. Genome Res 21: 1150–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan CD 2003. Transcription elongation factors repress transcription initiation from cryptic sites. Science 301: 1096–1099 [DOI] [PubMed] [Google Scholar]
- Koerber RT, Rhee HS, Jiang C, Pugh BF 2009. Interaction of transcriptional regulators with specific nucleosomes across the Saccharomyces genome. Mol Cell 35: 889–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M 1991. A short leader sequence impairs the fidelity of initiation by eukaryotic ribosomes. Gene Expr 1: 111–115 [PMC free article] [PubMed] [Google Scholar]
- Kozak M, Shatkin AJ 1977. Sequences of two 5′-terminal ribosome-protected fragments from reovirus messenger RNAs. J Mol Biol 112: 75–96 [DOI] [PubMed] [Google Scholar]
- Law GL, Bickel KS, Mackay VL, Morris DR 2005. The undertranslated transcriptome reveals widespread translational silencing by alternative 5′ transcript leaders. Genome Biol 6: R111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawless C, Pearson RD, Selley JN, Smirnova JB, Grant CM, Ashe MP, Pavitt GD, Hubbard SJ 2009. Upstream sequence elements direct post-transcriptional regulation of gene expression under stress conditions in yeast. BMC Genomics 10: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarowitz SG, Robertson HD 1977. Initiator regions from the small size class of reovirus messenger RNA protected by rabbit reticulocyte ribosomes. J Biol Chem 252: 7842–7849 [PubMed] [Google Scholar]
- Leeds P, Peltz SW, Jacobson A, Culbertson MR 1991. The product of the yeast UPF1 gene is required for rapid turnover of mRNAs containing a premature translational termination codon. Genes Dev 5: 2303–2314 [DOI] [PubMed] [Google Scholar]
- Legon S 1976. Characterization of the ribosome-protected regions of 125I-labelled rabbit globin messenger RNA. J Mol Biol 106: 37–53 [DOI] [PubMed] [Google Scholar]
- Mayr C, Bartel DP 2009. Widespread shortening of 3′ UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138: 673–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, Dietz HC 2004. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet 36: 1073–1078 [DOI] [PubMed] [Google Scholar]
- Miura F, Kawaguchi N, Sese J, Toyoda A, Hattori M, Morishita S, Ito T 2006. A large-scale full-length cDNA analysis to explore the budding yeast transcriptome. Proc Natl Acad Sci 103: 17846–17851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller PP, Hinnebusch AG 1986. Multiple upstream AUG codons mediate translational control of GCN4. Cell 45: 201–207 [DOI] [PubMed] [Google Scholar]
- Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M 2008. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni T, Corcoran DL, Rach EA, Song S, Spana EP, Gao Y, Ohler U, Zhu J 2010. A paired-end sequencing strategy to map the complex landscape of transcription initiation. Nat Methods 7: 521–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira CC, McCarthy JE 1995. The relationship between eukaryotic translation and mRNA stability. A short upstream open reading frame strongly inhibits translational initiation and greatly accelerates mRNA degradation in the yeast Saccharomyces cerevisiae. J Biol Chem 270: 8936–8943 [DOI] [PubMed] [Google Scholar]
- Pal S, Gupta R, Kim H, Wickramasinghe P, Baubet V, Showe LC, Dahmane N, Davuluri RV 2011. Alternative transcription exceeds alternative splicing in generating the transcriptome diversity of cerebellar development. Genome Res 21: 1260–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova TV, Kolupaeva VG 2002. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev 16: 2906–2922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resch AM, Ogurtsov AY, Rogozin IB, Shabalina SA, Koonin EV 2009. Evolution of alternative and constitutive regions of mammalian 5′UTRs. BMC Genomics 10: 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HS, Pugh BF 2011. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147: 1408–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Duran MF, Gilbert WV 2012. Alternative transcription start site selection leads to large differences in translation activity in yeast. RNA 18: 2299–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstiel P, Huse K, Franke A, Hampe J, Reichwald K, Platzer C, Roberts RG, Mathew CG, Platzer M, Schreiber S 2007. Functional characterization of two novel 5′ untranslated exons reveals a complex regulation of NOD2 protein expression. BMC Genomics 8: 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW, Irwin N, Janssen K 2001. Molecular cloning, 3rd ed. (ed. Argentine J, et al.), Cold Spring Harbor Laboratory Press, Cold Spring Harbor [Google Scholar]
- Sedman SA, Gelembiuk GW, Mertz JE 1990. Translation initiation at a downstream AUG occurs with increased efficiency when the upstream AUG is located very close to the 5′ cap. J Virol 64: 453–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L, Brannan RA, Hanby AM, Shaaban AM, Verghese ET, Peter MB, Pollock S, Satheesha S, Szynkiewicz M, Speirs V, et al. 2009. Differential regulation of oestrogen receptor β isoforms by 5′ untranslated regions in cancer. J Cell Mol Med 14: 2172–2184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler M, Fire A 2011. Wobble base-pairing slows in vivo translation elongation in metazoans. RNA 17: 2063–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang HL 2004. Translation of a yeast mitochondrial tRNA synthetase initiated at redundant non-AUG codons. J Biol Chem 279: 49656–49663 [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM 2012. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485: 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Heuvel JJ, Bergkamp RJ, Planta RJ, Raué HA 1989. Effect of deletions in the 5′-noncoding region on the translational efficiency of phosphoglycerate kinase mRNA in yeast. Gene 79: 83–95 [DOI] [PubMed] [Google Scholar]
- Wang Z, Gaba A, Sachs MS 1999. A highly conserved mechanism of regulated ribosome stalling mediated by fungal arginine attenuator peptides that appears independent of the charging status of arginyl-tRNAs. J Biol Chem 274: 37565–37574 [DOI] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Tzagoloff A 1987. Mitochondrial and cytoplasmic fumarases in Saccharomyces cerevisiae are encoded by a single nuclear gene FUM1. J Biol Chem 262: 12275–12282 [PubMed] [Google Scholar]
- Xu Z, Wei W, Gagneur J, Perocchi F, Clauder-Münster S, Camblong J, Guffanti E, Stutz F, Huber W, Steinmetz LM 2009. Bidirectional promoters generate pervasive transcription in yeast. Nature 457: 1033–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan G-C, Liu Y-J, Dion MF, Slack MD, Wu LF, Altschuler SJ, Rando OJ 2005. Genome-scale identification of nucleosome positions in S. cerevisiae. Science 309: 626–630 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Dietrich FS 2005. Identification and characterization of upstream open reading frames (uORF) in the 5′ untranslated regions (UTR) of genes in Saccharomyces cerevisiae. Curr Genet 48: 77–87 [DOI] [PubMed] [Google Scholar]