

Abstract
Most mammalian genes produce multiple mRNA isoforms derived from alternative pre-mRNA splicing, with each alternative exon controlled by a complex network of regulatory factors. The identification of these regulators can be laborious and is usually carried out one factor at a time. We have developed a broadly applicable high-throughput screening method that simultaneously identifies multiple positive and negative regulators of a particular exon. Two minigene reporters were constructed: One produces green fluorescent protein (GFP) from the mRNA including an exon, and red fluorescent protein (RFP) from the mRNA lacking the exon; the other switches these fluorescent products of exon inclusion and exclusion. Combining results from these two reporters eliminates many false positives and greatly enriches for true splicing regulators. After extensive optimization of this method, we performed a gain-of-function screen of 15,779 cDNA clones and identified 40 genes affecting exon 18 of Discs large homolog 4 (Dlg4; also known as post-synaptic density protein 95 [Psd-95]). We confirmed that 28 of the 34 recoverable clones alter reporter splicing in RT-PCR assays. Remarkably, 18 of the identified genes encode splicing factors or RNA binding proteins, including PTBP1, a previously identified regulator of this exon. Loss-of-function experiments examining endogenous Dlg4 transcripts validated the effects of five of eight genes tested in independent cell lines, and two genes were further confirmed to regulate Dlg4 splicing in primary neurons. These results identify multiple new regulators of Dlg4 splicing, and validate an approach to isolating splicing regulators for almost any cassette exon from libraries of cDNAs, shRNAs, or small molecules.
Pre-mRNA splicing patterns are highly regulated to produce functionally distinct gene products during development or in response to extracellular stimuli (Black 2003; Chen and Manley 2009). Most splicing alterations are determined by interactions between cis-regulatory elements in the pre-mRNA and trans-acting protein factors that can affect spliceosome assembly and splice site choices. Well-characterized splicing regulators include the SR proteins and members of the hnRNP group of proteins (Chen and Manley 2009; Cooper et al. 2009; Nilsen and Graveley 2010). However, there are many hundreds of RNA binding proteins encoded in the human genome, and only a fraction of them have been examined for activity in splicing.
An alternative exon is usually controlled by multiple proteins, and the intronic sequences surrounding the exon can contain a variety of conserved regulatory elements. Genome-wide profiling methods can identify many targets of individual splicing regulators (Hartmann and Valcarcel 2009; Darnell 2010; Witten and Ule 2011). However, systemically identifying the multiple positive and negative regulators of a particular alternative exon is a challenge. A variety of screening strategies have yielded only one or two new factors targeting the exon and thus were relatively insensitive for identifying larger sets of splicing regulators (Kar et al. 2006; Wu et al. 2006; Oberdoerffer et al. 2008; Topp et al. 2008). The development of cataloged libraries and high-throughput robotic systems has enabled screening at greater depth and the identification of multiple regulators from a single screen (Warzecha et al. 2009; Moore et al. 2010).
The specificity of single-output splicing reporters is confounded by molecules that alter the overall expression but not the splicing of the reporter transcript. In addition, the isoform measured by a single-output reporter must be produced at a low basal level relative to its alternative, allowing a small change in exon use to yield a relatively large fold effect in reporter output. Consequently, these reporters usually screen for either activators or repressors but not both. A pair of single-output reporters, each encoding a different fluorescent protein, can overcome some of these limitations (Kuroyanagi et al. 2010). However, using two minigenes imposes additional experimental complexities, such as integration of the two reporter genes into different genomic loci with different copy numbers.
A dual-output reporter, where both spliced isoforms are assayed, allows screening for changes in isoform ratio and can thus detect both increases and decreases in exon inclusion. Screening by these ratios is more sensitive for identifying changes in exons whose basal splicing is at intermediate levels, and can also reduce false positives that alter overall reporter expression. An elegant dual-fluorescence splicing reporter was applied in forward genetic screens of Caenorhabditis elegans (Kuroyanagi et al. 2006). The design of this reporter required insertion of complete open reading frames (ORFs) for green fluorescent protein (GFP) and red fluorescent protein (RFP) into each of mutually exclusive exons, which may not work well for mammalian cassette exons. Dual-output reporters produce false positives derived from differences in the translation or stability of the two protein products (Stoilov et al. 2008). These false positives are not eliminated by experimental replications and can make up the majority of hits from a screen, making the subsequent validation of true positives labor intensive.
We have developed a broadly applicable cell-based high-throughput screening (HTS) method to simultaneously identify multiple activators and repressors of an alternative exon. We minimized systematic variation associated with fluorescent screening to allow more accurate detection of moderate splicing changes. Our method greatly reduces false positives while maintaining a high sensitivity in detecting regulators. Using this system, we carried out a genome-wide gain-of-function screen for factors regulating exon 18 of Dlg4, a key regulator of synaptic assembly. We previously showed that this exon is controlled by the polypyrimidine tract binding proteins PTBP1 and PTBP2, which control Dlg4 expression during neuronal development (Zheng et al. 2012). However, there are likely to be cofactors required with the PTB proteins to control Dlg4 splicing in immature neurons, and the widespread expression of Dlg4 mRNA in non-neuronal cells makes it possible that different factors control exon 18 in different cellular contexts. We have now identified multiple new regulatory proteins affecting exon 18 splicing.
Results
Construction of dual-fluorescence minigene reporters
We previously constructed a dual-fluorescence reporter (pflareA) (Stoilov et al. 2008). The pflareA minigene contains GFP and RFP reading frames and an alternative exon of interest (Fig. 1A). The GFP start codon is split between two constitutive exons that flank the alternative exon such that the GFP ORF is initiated for translation only when the alternative exon is skipped. In this case, the RFP ORF is silent as translation does not reinitiate downstream. When the alternative exon is included, the GFP ORF loses its start codon, allowing ribosomes to initiate at the downstream RFP ORF. This arrangement allows the GFP and RFP levels to report the exclusion and inclusion of the alternative exon, respectively. In screening for modulators of splicing, a change in the ratio of the two fluorescence proteins indicates a change in exon inclusion.
Figure 1.

A high-throughput screening strategy for regulators of alternative splicing. Two dual-fluorescence minigenes, pflareA (A) and pflareG (B), are constructed to report splicing of an alternative exon with opposite outputs. In the pre-mRNA schematics, exons in boxes are connected by horizontal lines representing introns. After splicing, the exon-included and exon-skipped mRNA isoforms contain different open reading frames (yellow highlight). Exon 18 is spliced at similar levels in the pflareA-exon 18 and the pflareG-exon 18 mRNAs (A,B). However, the pflareA-exon 18 reporter expresses low RFP and high GFP, whereas the pflareG-exon 18 reporter expresses high RFP and low GFP, upon transient expression in N2a cells. (C) The framework for the cell-based high-throughput screens to identify splicing regulatory factors.
We found that additional nonsplicing mechanisms could alter the GFP and RFP levels in a pflareA screen (Stoilov et al. 2008). For example, changes in the translational efficiency of the two reading frames, or changes in the stability of the two proteins, could change the GFP/RFP ratio without altering splicing of the reporter. To increase the specificity of identifying splicing regulators, we developed a counter screen with a complementary reporter. In the new reporter, pflareG, the start codon of the GFP ORF is within the alternative exon, so GFP reports exon inclusion (Fig. 1B). Conversely, RFP reports exon exclusion in pflareG. In a pflareG screen, splicing activators and repressors increase and decrease the GFP/RFP ratio, respectively. In parallel screens, a splicing regulator will alter the GFP/RFP ratios reported by pflareA and pflareG in opposite directions. Except the sequences surrounding the start codon of GFP, pflareA and pflareG have the same tandem GFP and RFP ORFs. Changes in translation or protein stability that shift the GFP/RFP ratio of one reporter are unlikely to change the ratio in the opposite direction using the other reporter. Thus, screening with both reporters and identifying the overlapping hits should filter out false positives and enrich for true splicing regulators (Fig. 1C).
In previous work, we showed that the splicing of Dlg4 exon 18 is controlled by the PTB proteins during neuronal development (Zheng et al. 2012). The sequence surrounding exon 18 is highly conserved in regions that extend beyond the putative PTB binding sites and presumably serves to recruit other regulatory factors. Experiments in PTB knockout mice also indicate that additional factors are controlling this exon (data not shown). To identify some of these factors, we set up a high-throughput screen for modulators of exon 18 splicing.
We constructed pflareA and pflareG reporters for Dlg4 exon 18. We inserted exon 18 and its flanking intronic sequences into pflareG to make the pflareG-exon18 reporter. The native exon 18 has three AUG start codons in frame with the GFP ORF of pflareG, allowing GFP to report exon inclusion. In mouse N2a neuroblastoma cells expressing this reporter, 73% of the pflareG-exon18 mRNA skips exon 18. Consequently, the N2a cells produced bright RFP and dim GFP (Fig. 1B). The basal level of exon 18 inclusion in the minigene mRNA (27%) is lower than in the endogenous Dlg4 transcript (66%), possibly due to promoter differences, to uncharacterized regulatory elements that were not included in the minigene, or to nonsense mediated mRNA decay of the exon18-skipped isoform (Zheng et al. 2012). The same Dlg4 sequence was inserted into the pflareA reporter to make pflareA-exon 18. For this construct, the three ATG codons of exon 18 were mutated to TTG, causing the exon 18–plus transcripts to express RFP (Fig. 1A). Skipping the exon in pflareA-exon18 generates the AUG codon for GFP. Exon 18 was included at equal levels in transcripts from pflareA-exon 18 and pflareG-exon 18, indicating that the mutations did not strongly affect exon 18 splicing (Fig. 1A,B). As expected, when pflareA-exon18 was transfected into N2a cells, it produced strong GFP expression and weak RFP expression (Fig. 1A). Similar pflare reporter gene pairs can be constructed for almost any exon.
Selection of stable cell clones expressing pflare-exon 18
We next derived stable cell lines expressing each reporter and tested their ability to be efficiently reverse transfected (Fig. 1C). We used N2a cells, which express many neuronal markers, possibly including neuronal cofactors for exon 18 splicing. As shown in Supplemental Figure 1, we identified the most effective transfection reagent, optimized the cell density, and determined the times of maximum expression from the transfected plasmid.
Cell clones stably expressing each reporter varied in their GFP and RFP expression (Fig. 2A). This variation may result from different levels of trans-acting factors in the clonal lines. To test this, we assayed the splicing of the endogenous Dlg4 exon 18 in each cell line (Supplemental Fig. 2). The “percent-spliced-in” (PSI) value of exon 18 was quite similar between cell clones and showed no correlation with the GFP/RFP ratio (Pearson correlation coefficient −0.05). This suggests that the main source of GFP/RFP variability is not clonal variation of trans-acting factors, but instead may be due to different numbers or sites of genomic integration of the plasmid, leading to changes in transcription and processing.
Figure 2.

Selection of stable cell lines. (A) Basal GFP and RFP expression vary between different pflareG-exon 18 stable cell clones. (B–E) Changes in the GFP/RFP ratios of B11 (blue), C9 (purple), E11 (green), and F7 (black) cells after reverse transfection of a PTBP1 expressing vector (PTBP1), mock transfection without DNA (Mock), transfection of a empty control vector, or no transfection (no TFX). (F) Percent-spliced-in values of exon 18 in the pflareG-exon 18 mRNA in B11, C9, E11, and F7 cells after different transfection.
To assess their ability to function in the HTS, we tested cell lines for their response to the expression of PTBP1, a known repressor of exon 18. The results for several pflareG-exon 18 cell lines (B11, C9, E11, and F7) are shown in Figure 2. The GFP/RFP ratio was measured in each cell line after transfection with a PTBP1 expressing plasmid or after mock transfection (application of Lipofectamine 2000 without DNA). PTBP1 represses exon 18 splicing, and as expected, B11, E11, and F7 cells exhibited reduced GFP/RFP after PTBP1 expression, but to different degrees (Fig. 2B). In contrast, the C9 cells exhibited an increased GFP/RFP ratio with PTBP1 expression. Mock transfection had minimal effect on the GFP/RFP ratio, indicating the response of the C9 cells was not due to application of Lipofectamine 2000 (Fig. 2C). However, transfection of a control empty vector also led to increased GFP/RFP ratio in C9 and E11 cells (Fig. 2D). This effect of plasmid transfection on the GFP/RFP ratio could mask the exon repression by PTBP1. Indeed, when the PTBP1 transfection was normalized to the control empty vector transfection, C9 and E11 along with B11 and F7 all showed clear patterns of exon 18 repression by PTBP1 (Fig. 2E). These changes in exon 18 splicing were confirmed by RT-PCR (Fig. 2F). By this assay, the splicing of exon 18 did not change between the mock or vector control transfections for either C9 or E11 cells (Fig. 2E). Thus for these cells, changes in GFP/RFP were not always due to changes in splicing. These experiments indicate that transfection with a control vector is a more appropriate benchmark than mock transfection when assessing GFP/RFP changes in the screen. The response of F7 cells to PTBP1 transfection was smaller than B11 cells (Fig. 2E,F), perhaps due to lower transfection efficiency. Thus, we used the B11 cells for the HTS. After PTBP1 transfection, the GFP/RFP ratio of B11 cells remained unchanged in the first 20 h, decreased substantially in the next 24 h, and then stabilized (Fig. 2E). This paralleled the expected kinetics of gene expression from transfected plasmids (see Supplemental Fig. 1B). Splicing of exon 18 in the stable clones was somewhat less responsive to PTBP1 transfection than in the original N2a cells. This is in part due to the stable cell clones exhibiting lower transfection efficiency than the parental N2a line. Similar optimization was done to identify a suitable pflareA-exon 18 line (data not shown).
Screening the Mammalian Gene Collection Library
The Mammalian Gene Collection (MGC) full-length cDNA library consisted of 15,779 individual human and mouse cDNA clones with some duplicates. The library was deposited onto 44 384-well plates in a one-well–one-cDNA format. On each plate, we used four wells for the human PTBP1 cDNA plasmid as positive controls, and four wells containing no vector as mock transfection negative controls. Four wells of empty control vector yielded a baseline of GFP/RFP used to measure fluorescent ratio changes in experimental wells of the same plate. The pflareG-exon 18 and the pflareA-exon 18 stable cell line were each deposited on one set of library plates for reverse transfection. Plates containing media alone were used for background subtraction. To improve the signal-to-noise ratio of the method, we did extensive optimization of the data acquisition and fluorescence background subtraction, as described in the online supporting information.
We imaged both GFP and RFP in each well at 16, 22, 40, 46, and 64 h post-transfection. At each time point, the GFP/RFP ratio of each well was calculated relative to the empty control vector transfection. This M value, indicative of the splicing change, was plotted against the product of the GFP and RFP intensities, or A value, which assesses the overall expression of the reporter (Rine Dudoit 2002). The data for the pflareG-exon 18 screen are shown in Figure 3. Over time, the group of mock transfections remained clustered within a narrow range of reporter expression and with their M value around zero. In contrast, the control wells receiving the PTBP1 cDNA deviated in M value from both the mock group and the mean of all the wells. This divergence was observed as early as 22 h and continued until 64 h post-transfection (Fig. 3). Importantly, the PTBP1 wells exhibited decreased GFP/RFP ratios in the pflareG-exon 18 reporter as expected for repression of exon 18. Most of the library cDNA clones distributed around zero for the change in GFP/RFP ratio, similar to the mock transfection group. The distribution of the overall fluorescence intensities shifted to higher A values with time, reflecting proliferation of screened cells. There were also wells exhibiting A values deviating from the main distribution, indicating that some clones were altering cell viability or the overall expression of the reporter. Most importantly, some cDNA clones caused the GFP/RFP ratio to diverge from the negative controls, indicating that they might be altering splicing of the reporter.
Figure 3.

Scatter plots of the M value versus the A value for the pflareG-exon 18 reporter at 16, 22, 40, 46, and 64 h after transfection of the MGC cDNA library (black), the spiked-in PTBP1 cDNA (red), or mock transfection (blue).
We calculated a local false-discovery rate (FDR) for individual genes by comparing their M values to the population distribution of M values, and defined significant changes in the GFP/RFP ratio. Setting the local FDR at 0.05, the pflareG-exon 18 screen defined 466 clones with increased GFP/RFP ratio (putative activator clones) and 196 clones with decreased GFP/RFP ratio (putative repressor clones). Among the repressor clones, 155 were the PTBP1 positive controls, and 41 clones were from the MGC cDNA library. Given the total of 176 PTBP1 controls in the screen, the sensitivity for the pflareG-exon 18 screen was ∼88% (155/176) with a local FDR of 0.05. From the pflareA-exon 18 screen, we identified 53 activator clones and 231 repressor clones. Ninety-one repressor clones were from the MGC library. The other 140 repressor clones were the PTBP1 positive controls, making the sensitivity of the pflareA-exon 18 screen ∼80% (140/176).
To filter out false positives, we selected clones identified as positive hits in both screens. These included 28 clones from the MGC library that scored as activators in both screens and 21 clones that scored as repressors in both screens (Fig. 4A,B). Some genes had multiple hits, as the MGC library contains both human and mouse homologs of some genes. Accounting for repetitive hits, our screen identified 23 unique activator genes and 17 unique repressor genes (Table 1), including the PTBP1 clones from the MGC library. Considering the spiked-in PTBP1 controls, 124 scored as positive in both screens. Thus the sensitivity of the combined screens was 70% (124/176), roughly equal to the product of the sensitivities of the individual screens.
Figure 4.

The pflare-exon 18 screens of the MGC cDNA expression library. The pflareG-exon 18 screen (blue) and the pflareA-exon 18 screen (yellow) identified 28 common activator clones (A) and 21 common repressor clones (B) from the MGC cDNA library. The two screens also identified 124 overlapping spiked-in PTBP1 clones (B). The identified genes are enriched for genes involved in RNA splicing (C) and RNA binding (D) compared to the whole MGC gene set. (E) RT-PCR validated changes in pflareG-exon18 mRNA splicing after transient transfection of the identified genes. Note that both human and mouse genes were recovered in the screen. Following nomenclatural rules, genes are in italic font with human genes in all capital letters. (F) A plot of the M values in the pflareG-exon 18 screen versus the ΔPSI measured by RT-PCR. Each dot represents one of the 34 tested genes.
Table 1.
Overlapping hits from the pflareA and pflareG screens
Clones identified in the screen are highly enriched for splicing regulators
As a validation of the procedure, mouse Ptbp1 itself was identified as a positive hit from the MGC library, as was its close homolog Ptbp3. Moreover, many of the other identified genes are known to be involved in splicing, to directly bind to RNA, or both (Table 1). Of the 23 identified activator proteins, SRSF3, HNRNPC, and PUF60 are known splicing regulator proteins, and SNRNP70 and SF3B4 are components of the spliceosome. Of 17 putative repressor proteins, PTBP1, PTBP3, CELF4, SF1, MBNL3, TRA2B, HNRNPK, and RBM10 are all known splicing regulators. To examine the functional clustering of the identified genes, we performed gene enrichment analysis using their annotations from multiple databases, including Gene Ontology (GO), KEGG pathways (networks of molecular interactions), and Interpro (protein domains and functional sites). These were compared to the term frequencies in the entire MGC clone library. Strikingly, genes involved in RNA splicing constitute 30% of the identified genes, but only 2% of total MGC genes (Fig. 4C). Similarly, 45% of the identified genes but only 6% of the MGC genes are characterized as RNA binding (Fig. 4D). Supplemental Table 1 lists all the enriched terms with a Bonferroni-adjusted P-value <0.05. Remarkably, all of the enriched terms are relevant to mRNA splicing, the spliceosome, or RNA binding, indicating a very high specificity of the method in identifying splicing regulators. In contrast, a parallel analysis of genes that were identified in only one of the pflare-exon 18 screens revealed no enriched terms related to splicing, the spliceosome or RNA binding. This strongly validates the approach of enriching true positives from two complementary screens.
To confirm the activity of the individual positive clones, we performed RT-PCR experiments to assay the change in splicing of the pflareG-exon 18 reporter upon overexpression of each candidate regulator. Of the 40 genes, six were not recovered from the library, and we tested the remaining 34. We set the criterion that genes must induce at least a 5% change in PSI value from the negative control with a P-value of <0.05. Using this cutoff, 28 of the 34 tested genes were found to alter the splicing of the reporter, yielding a validation rate of 82% (Fig. 4E; Table 1; Supplemental Fig. 3). Considering the large number of true negatives in the library of 15,779 clones, the recovery of only six false-positive genes indicates a very high specificity for the screen.
To quantify the relationship between the true splicing changes measured by RT-PCR and the changes in GFP/RFP (M), we plotted the M values against the change in PSI for the 34 tested genes (Fig. 4F). These two variables were strongly positively correlated with a Pearson correlation coefficient of 0.86 for the pflareG-exon 18 screen, and 0.83 for the pflareA-exon screen. This indicates that the change in GFP/RFP is a sensitive indicator of changes in exon inclusion.
Validation of the identified regulators on endogenous Dlg4 splicing
Having identified genes whose overexpression altered splicing of the reporters, we tested whether loss of function of these regulators altered endogenous Dlg4 splicing in the opposite direction. We selected four activators and four repressors that exhibited substantial basal levels of expression in N1E neuroblastoma cells and knocked down their expression by RNAi. To control for off-target effects, two small interfering RNAs (siRNAs) were tested for each regulator. After siRNA transfection, the level of each target mRNA was measured by RT-qPCR (Fig. 5A). All the siRNAs were found to yield substantial loss of their target mRNAs, although their efficiency varied. We found that knockdown of three of the four putative activator proteins—DAZAP1, PUF60, and SF3B4—reduced exon 18 inclusion in the endogenous Dlg4 RNA, as expected (Fig. 5B). In spite of relatively efficient knockdown, the loss of SNRNP70 had minimal effect on Dlg4 exon 18 splicing. In contrast, exon 18 splicing was enhanced by knockdown of two of the presumptive repressor proteins, PCBP4 and RBM10 (Fig. 5B). Loss of CELF4 or RBM42 did not have a clear effect on exon 18 splicing in N1E cells. One siRNA targeting the Rbm42 transcript reduced exon 18 splicing. This may be due to an off-target effect of the siRNA. Thus, DAZAP1, PUF60, SF3B4, PCBP4, and RBM10 all clearly regulate splicing of the endogenous Dlg4 transcript and not just the reporter. The effects of SNRNP70, CELF4, and RBM42 may be specific for the reporter. However, it is possible that these factors require greater knockdown efficiency to show an effect on endogenous Dlg4 splicing or that they are active in cells other than N1E cells, where their activity may be redundant.
Figure 5.

RNAi-mediated depletion of selected hits in N1E cells validates their effect on the splicing of endogenous Dlg4 exon 18. (A) Steady-state mRNA levels of the tested genes were significantly decreased in N1E cells at 72 h after transfection of the target-specific siRNAs. Two different siRNAs targeting each gene were examined. After the knockdown with each siRNA, endogenous exon 18 splicing was assayed by RT-PCR (B). The PSI of exon 18 was plotted for each siRNA, and a representative gel is shown. The dashed line marks the basal level of exon 18 inclusion. Values represent mean ± SD (N = 3). (*) P < 0.05 (Student’s t-test, in comparison to the control [Ctr]).
Although its transcripts are found in other tissues, the described function of DLG4 protein is in neurons. Thus, we next wanted to confirm the activity of some splicing regulators in primary neuronal culture. This presented some technical challenges, as the transfection efficiency of our standard siRNAs was lower in primary neurons, and the endogenous Dlg4 transcript is subject to nonsense mediated mRNA decay (NMD). To deplete the new regulators from neurons, we used Dharmacon Accell siRNAs, which we have found to work better than standard siRNAs in neurons. Mouse cortical neurons were explanted at embryonic day 15 and plated into 24-well plates. These cultured primary neurons were then transfected at day 0 in vitro (DIV0), and RNA was collected at day 5 in vitro (DIV5). At DIV5, >90% of steady-state Dlg4 mRNA contains exon 18. The exon-skipped isoform is lost to NMD, making it difficult to detect changes in splicing. To better assess changes in the splicing of exon 18, the neuronal cultures were treated with cycloheximide to block NMD and stabilize the exon-skipped isoform. Under these conditions, exon 18 has a PSI value of 73.
We tested Accell siRNAs targeting Dazap1, Celf4, Puf60, and Rbm10 transcripts in mouse neurons. We assayed four different siRNAs for each target, each transfected into a separate well, and isolated RNA to assess loss of the target transcript. None of the Dazap1-targeted siRNAs caused a depletion in the Dazap1 mRNA, and these siRNAs were not studied further. The two most effective siRNAs targeting each of Puf60, Rbm10, and Celf4 were combined, and these siRNA pairs were transfected into neurons and RNA was isolated at DIV5. Assaying the level of the target mRNAs, we found that the combined siRNAs targeting Celf4, Puf60, and Rbm10 effectively depleted these transcripts in primary neurons (Fig. 6A). Next assaying the splicing of Dlg4 exon 18 in the siRNA treated neurons, we found that depletion of Puf60 transcripts decreased exon 18 splicing from 73% to 59% (Fig. 6B), confirming the role of PUF60 protein as a positive regulator of exon 18. Depletion of Rbm10 transcripts stimulated exon 18 inclusion from 73%–86%, confirming its role as a splicing repressor (Fig. 6B). Depletion of Celf4 transcripts did not have a significant effect on exon 18 splicing (data not shown). Thus, it may be a false positive from the screen, although it is possible that it plays a role in Dlg4 splicing in mature neurons, but its function is redundant in the early cultures used here. The results with PUF60 and RBM10 demonstrate that the screen successfully discovered both positive and negative splicing regulators of exon 18.
Figure 6.

PUF60 and RBM10 regulate endogenous exon 18 splicing in cultured primary neurons. (A) Steady-state mRNA levels in primary neurons after transfection of a pair of Accell siRNAs targeting either the Puf60 or Rbm10 transcript. (B) The Puf60 and Rbm10 siRNAs alter endogenous exon 18 splicing in primary neurons at DIV 5. Exon 18 PSI values are plotted and a representative gel is shown. Values represent mean ± SD (N = 3). (*) P < 0.05 (Student’s t-test, in comparison to the control [Ctr]).
Discussion
Skipping of exon 18 in the Dlg4 transcript is required for repression of DLG4 expression outside of neurons, and the controlled induction of this exon during neuronal development is essential to synaptogenesis (Zheng et al. 2012). This exon may also be dynamically regulated in mature neuronal circuits to allow DLG4 expression to change with the demands of synaptic remodeling. These diverse contexts of Dlg4 regulation imply that the regulators of exon 18 extend well beyond the PTB proteins already identified. To begin to identify these factors, we developed a screening strategy for the multiple positive and negative regulators of a cassette exon (Fig. 1C). Our method uses parallel screens with two fluorescent reporters to identify overlapping hits and enrich for true splicing regulators. We applied this strategy to identify trans-acting regulators of Dlg4 exon 18 within the MGC cDNA expression library. We found multiple splicing activators and repressors, in addition to the known regulator PTBP1 and its homolog PTBP3. A cDNA expressing PTBP2, the other PTBP1 paralog known to repress exon 18, was not present in the library (Zheng et al. 2012). This indicates that screens of larger cDNA libraries or screens of short hairpin RNAs (shRNAs) or siRNAs could identify additional regulators of this exon.
Regulators identified in the screen were validated in loss-of-function assays on the endogenous Dlg4 transcript. PUF60 and RBM10 were further confirmed to regulate Dlg4 in primary neurons, where PUF60 stimulates exon 18 splicing, and RBM10 inhibits it. These proteins’ mechanisms of action will be very interesting to examine. PUF60 is a widely expressed splicing factor that can interact with spliceosomal components to promote splicing at weak 3′ splice sites (Hastings et al. 2007; Corsini et al. 2009). In stimulating splicing at the 3′ splice site upstream of exon 18, PUF60 may counter the repressive action of PTBP1, which binds just upstream. The mechanism of the negative action of RBM10 is not clear, but interestingly, this protein was identified as interacting with PTBP2 in a yeast two-hybrid screen (Rual et al. 2005). Thus, this protein may act as a cofactor with the PTB proteins in repressing exon 18. We find that Rbm10 mRNA is expressed in embryonic brain, but is progressively reduced during development (data not shown). This expression profile parallels that of the PTBP1 protein, consistent with the two proteins working together in early neuronal maturation.
Putative regulators, such as CELF4 that were successfully depleted by RNAi but did not affect endogenous exon 18, may be false positives that target the reporter RNA but not the native transcript or that have dominant effects from overexpression. However, some may regulate exon 18 in other contexts than those tested. The cells used for validation may have different endogenous factors from the cells where an identified regulator is active. Thus, screening by cDNA overexpression may be more comprehensive than RNAi-based screens, as some true regulators may not be present in the assayed cells to allow the loss of function to be observed. On the other hand, protein overexpression may produce effects not seen under normal physiological conditions. It is thus most informative to examine both gain and loss of function for a putative regulatory factor and to confirm its effects in each cell type of interest. In fact, Dlg4 transcripts are expressed in diverse cell types but are largely lacking exon 18. One goal of our screen was to identify possible regulators for both neuronal and non-neuronal cells.
Interestingly, several core splicing factors, including SNRNP70, SF3B4, and SF1, were also identified in our screen. Previous studies indicate that changes in the concentration of core spliceosomal proteins can affect subsets of alternative exons (Park et al. 2004; Cheng et al. 2007; Hastings et al. 2007; Corioni et al. 2011). Even if the expression of these factors is relatively constant in neuronal development, other mechanisms may control their activity to affect alternative splicing. Several other new regulators, including HNRNPK, SRSF3, TRA2B, and MBNL3, have potential RNA binding motifs within exon 18 or nearby (Stamm et al. 2006; data not shown). Thus, in addition to more detailed characterization of PUF60 and RBM10, it will be interesting to examine the other identified regulators using additional strategies. Understanding the roles of these new regulators of Dlg4 will occupy our attention in the future.
Advantages and disadvantages of the screening strategy
Approaches for identifying splicing modulators are often applicable only to particular exons or capable of detecting only activators or repressors but not both (Kar et al. 2006; Wu et al. 2006; Oberdoerffer et al. 2008; Topp et al. 2008; Chen and Zheng 2009; Warzecha et al. 2009). These methods can have limited sensitivity, requiring large changes in splicing to detect valid hits, and low specificity, where multiple false-positive hits need to be eliminated in subsequent assays (Stoilov et al. 2008). We applied dual-output reporters to increase sensitivity, and used complementary screens to increase specificity. With this approach, a pair of complementary screens filtered 90% of the hits from the individual screens, while decreasing the sensitivity by only 10%–18%. The overlapping hits were largely not those that ranked highly in either individual screen, indicating that the complementary screens provide a sensitive confirmation of true splicing regulators.
We found that for the nonoverlapping hits, the shift in GFP/RFP ratio was largely due to changes in GFP expression. The pflareG exon 18 screen identified more positives than the pflareA exon 18 screen, and the false positives mostly increased the GFP/RFP ratio. This may be due to pflareG exon 18 expressing a lower basal level of GFP than pflareA exon 18, creating a larger dynamic range for false positives that enhance the GFP signal.
Fluorescent protein output allows noninvasive assay of live cells and is relatively easy to apply in a HTS, but it can be less sensitive than chemiluminescence assays (Wehry 1986). To increase the sensitivity of our fluorescent readout and reduce systemic noise, we optimized the data acquisition and normalization for fluorescent detection. One of the most effective adjustments was to derive local background fluorescence signals for individual wells using scaling factors to normalize individual plates to a media-alone control background plate.
Several features of the screen will likely require reoptimization when applying it to new exons. We found substantial variation in reporter expression between stable cell clones (Fig. 2). Multiple clonal lines of each splicing reporter were tested to identify one that gave optimal responses. We found that transfection of an empty vector could alter GFP/RFP ratio of some lines, and we chose lines for screening where this effect was minimal. We also found that the responsiveness of individual cell clones to PTBP1 expression correlated with their transfection efficiency. Thus, in developing screens for exons where no regulators are already known, one could choose cell lines exhibiting the highest transfection efficiency. Finally, we found that the kinetics of reporter expression also needed to be assessed before screening different libraries or in different cells.
Although largely subject to similar regulation, minigene reporters do not always recapitulate the regulation of endogenous exons. The minigene may not contain all of the relevant cis-regulatory elements for the test exon. The minigene sequences flanking the inserted alternative exon may confer responsiveness to trans-acting factors that do not affect the endogenous exon. In testing pflare minigenes containing a different cassette exon, we identified a set of regulators largely different from the exon 18 regulators we found here (E Anderson and D Black, unpubl.). However, both screens identified CELF4 as altering splicing of the reporters. Thus, this protein could be acting through sequences in the reporter backbone. Additional test exons will need to be assessed to determine if there are universal regulators of any exon within a particular reporter backbone. In any case, molecules that affect the splicing of the minigene reporter need confirmation on the endogenous transcripts.
The pflare plasmids can accommodate nearly any cassette exon. We have constructed functional pflareA and pflareG reporters for several other exons (data not shown). However, to generate the correct arrangement of initiation codons, mutagenesis of the test exon may be needed, and this may alter cis-acting splicing regulatory elements. If the alternative exon has an in-frame AUG start codon for the GFP ORF, it must be mutated in the pflareA vector. Conversely, if it does not have an in-frame AUG and Kozak sequence, these must be introduced into pflareG for GFP initiation. The effect of these mutations on splicing needs assessment. However, because the mutations are introduced into only one of the two vectors, a gain of false positives and/or a loss of true positives will manifest in only one of two screens. Since the candidate regulators are selected from the overlap from both screens, the mutagenesis will most likely lead to a loss of true positives rather than the gain of false positives, if it has an effect. Once established, reporter cell lines containing these constructs can be used to screen libraries of siRNAs or shRNAs, and of small molecules, in addition to cDNAs. This approach should allow the identification of a wide range of regulators for an exon of interest.
Methods
Reporter cell lines
Dlg4 exon 18 and its flanking intronic sequences (424 nucleotides upstream of and 443 nucleotides downstream from exon 18) were inserted into the EcoRI and BamHI sites of pflareG or pflareA vectors. Afterward, three ATG codons in exon 18 were mutated to TTG in the pflareA vector using the QuickChange site-directed mutagenesis kit (Stratagene). The resulting pflareG-exon18 and pflareA-exon18 vectors were linearized by DraIII and transfected into N2a cells. Cell colonies stably expressing either reporter were selected by 1 mg/mL G418 and stable expression of GFP and RFP.
Library and array construction
The cDNA library was comprised of the “Assay Ready” set of 15779 putatively expressible clones from the MGC collection from Open Biosystems in the pCMVsport6.0 and pCMVsport6.1 vector (Invitrogen). Briefly, cDNAs were arrayed from MGC IMAGE IRAK source plates into 384-well plates using a Genetix Qbot (Molecular Devices) and were subsequently used for plasmid preparation. Plasmid DNA was prepared for screening by using plasmid miniprep consumables (Macherey–Nagel) on a Biomek FX robot (Beckman Coulter), normalized, and spotted into assay plates for screening. Most screening hits were verified by sequencing and subsequent blastn analysis. Further information can be found in Iourgenko et al. (2003).
High-throughput screens and data analysis
MGC cDNA clones (40 ng each) were prespotted in 384-well plates. Ten microliters Opti-MEM per well was first dispensed into the plates using the Multidrop 384 (Thermo LabSystems). Then 0.12 μL Lipofetamine 2000 in 10 μL Opti-MEM was added to each well to mix with DNA. After 25 min, 8000 reporter cells were plated in every well except the A23, A24, B23, and B24 wells. These four wells were dispensed with only culture media and later used to derive a scale factor for normalization. Background plates containing media alone were included in the screens for interplate background subtraction.  is the raw GFP or RFP intensities of a well at row i column j in plate k. The scale factor
is the raw GFP or RFP intensities of a well at row i column j in plate k. The scale factor  for plate k (either a cell plate or a background plate) is calculated based on the A23, A24, B23, and B24 wells as below:
for plate k (either a cell plate or a background plate) is calculated based on the A23, A24, B23, and B24 wells as below:
 |
where  is the average values across all the plates. The normalized GFP and RFP expression values of the stable cells
is the average values across all the plates. The normalized GFP and RFP expression values of the stable cells  after transfection with cDNA x at row i column j in plate c are calculated as below:
after transfection with cDNA x at row i column j in plate c are calculated as below:
where  and
and 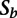 are the scale factors for the cell plates and background plates, respectively,
are the scale factors for the cell plates and background plates, respectively,  is the total number of background plates, and
is the total number of background plates, and  are the raw fluorescence intensities of the background plates. The splicing ratio of the reporter upon expression of cDNA x was then approximated by
are the raw fluorescence intensities of the background plates. The splicing ratio of the reporter upon expression of cDNA x was then approximated by  in the pflareG-exon18 cell and
in the pflareG-exon18 cell and  in the pflareA-exon 18 cell. Meanwhile, the basal splicing level of the reporter at a given time was estimated from the four wells that were transfected with a control empty vector in every plate. Therefore, it was approximated by the mean of
in the pflareA-exon 18 cell. Meanwhile, the basal splicing level of the reporter at a given time was estimated from the four wells that were transfected with a control empty vector in every plate. Therefore, it was approximated by the mean of  in the pflareG-exon 18 cell and the mean of
in the pflareG-exon 18 cell and the mean of 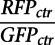 in the pflareA-exon 18 cell for each plate. To examine the action of cDNA x on the splicing of the pflareG-exon 18 reporter, the change in the splicing ratio or the M value
in the pflareA-exon 18 cell for each plate. To examine the action of cDNA x on the splicing of the pflareG-exon 18 reporter, the change in the splicing ratio or the M value  was calculated as
was calculated as
where  was derived from the same plate as cDNA x. Comparing to a control value within the same plate further reduces plate-to-plate variation and the scanner field variation. The M values in the pflareA-exon 18 screen were calculated as
was derived from the same plate as cDNA x. Comparing to a control value within the same plate further reduces plate-to-plate variation and the scanner field variation. The M values in the pflareA-exon 18 screen were calculated as
 indicates a possible increase in splicing by cDNA x, whereas
indicates a possible increase in splicing by cDNA x, whereas  indicates a possible decrease in the splicing. The average fluorescence intensity of cells after transfection, or the A value, was calculated as below:
indicates a possible decrease in the splicing. The average fluorescence intensity of cells after transfection, or the A value, was calculated as below:
We applied local FDR control to determine the cutoff of the M values in calling a positive hit using the R package locfdr.
Validation
Cells were transfected with cDNA plasmids using Lipofectamine 2000 (Invitrogen) and Silencer Select siRNAs using Lipofectamine RNAiMax (Invitrogen). Sequences of the siRNAs are in Supplemental Table 2. Primary cortical neurons were cultured according to the method previously described (Zheng et al. 2010) and transfected at DIV0 with 1 μM Dharmacon Accell siRNAs (Puf60: EQ-051526; Rbm10: EQ-052853; Celf4: EQ-046866; Dazap1: EQ-041766; Thermo Scientific). Total RNA was extracted with TRIzol (Invitrogen). Reverse transcription was performed using Superscript III and followed by quantitative PCR to examine the included and skipped isoforms. The PCR products were resolved on 8% UREA-PAGE gels, imaged on a Typhoon Imager, and quantified by ImageQuant TL. The PCR primers for the pflare reporters are 5′-CGTCGCCGTCCAGCTCGACCAG-3′ and 5′-AAACAGATCTACCATTGGTGCACCTGA-3′. Real-time RT-qPCR was performed according to the method previously described (Zheng and Chen 2009). Term enrichment analysis was performed according to the method previously described (Chen 2010).
Other procedures are available in the Supplemental Methods.
Acknowledgments
We thank C. Gao for help with producing pflareG stable cell clones and W. Hwong for help with picking bacteria clones from the MGC cDNA library. We thank P. Stoilov, E. Anderson, and members of the Black laboratory for helpful discussion. This work was supported in part by grants from the U.S. National Institutes of Health (R01GM097230 to L.C. and R01GM49662 to D.L.B.). D.L.B. is an investigator at the Howard Hughes Medical Institute.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.147546.112.
Freely available online through the Genome Research Open Access option.
References
- Black DL 2003. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72: 291–336 [DOI] [PubMed] [Google Scholar]
- Chen L 2010. A global comparison between nuclear and cytosolic transcriptomes reveals differential compartmentalization of alternative transcript isoforms. Nucleic Acids Res 38: 1086–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Manley JL 2009. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nat Rev Mol Cell Biol 10: 741–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zheng S 2009. Studying alternative splicing regulatory networks through partial correlation analysis. Genome Biol 10: R3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D, Cote J, Shaaban S, Bedford MT 2007. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol Cell 25: 71–83 [DOI] [PubMed] [Google Scholar]
- Cooper TA, Wan L, Dreyfuss G 2009. RNA and disease. Cell 136: 777–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corioni M, Antih N, Tanackovic G, Zavolan M, Kramer A 2011. Analysis of in situ pre-mRNA targets of human splicing factor SF1 reveals a function in alternative splicing. Nucleic Acids Res 39: 1868–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsini L, Hothorn M, Stier G, Rybin V, Scheffzek K, Gibson TJ, Sattler M 2009. Dimerization and protein binding specificity of the U2AF homology motif of the splicing factor Puf60. J Biol Chem 284: 630–639 [DOI] [PubMed] [Google Scholar]
- Darnell RB 2010. HITS-CLIP: Panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip Rev RNA 1: 266–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann B, Valcarcel J 2009. Decrypting the genome’s alternative messages. Curr Opin Cell Biol 21: 377–386 [DOI] [PubMed] [Google Scholar]
- Hastings ML, Allemand E, Duelli DM, Myers MP, Krainer AR 2007. Control of pre-mRNA splicing by the general splicing factors PUF60 and U2AF65. PLoS ONE 2: e538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iourgenko V, Zhang W, Mickanin C, Daly I, Jiang C, Hexham JM, Orth AP, Miraglia L, Meltzer J, Garza D, et al. 2003. Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci 100: 12147–12152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar A, Havlioglu N, Tarn WY, Wu JY 2006. RBM4 interacts with an intronic element and stimulates tau exon 10 inclusion. J Biol Chem 281: 24479–24488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroyanagi H, Kobayashi T, Mitani S, Hagiwara M 2006. Transgenic alternative-splicing reporters reveal tissue-specific expression profiles and regulation mechanisms in vivo. Nat Methods 3: 909–915 [DOI] [PubMed] [Google Scholar]
- Kuroyanagi H, Ohno G, Sakane H, Maruoka H, Hagiwara M 2010. Visualization and genetic analysis of alternative splicing regulation in vivo using fluorescence reporters in transgenic Caenorhabditis elegans. Nat Protoc 5: 1495–1517 [DOI] [PubMed] [Google Scholar]
- Moore MJ, Wang Q, Kennedy CJ, Silver PA 2010. An alternative splicing network links cell-cycle control to apoptosis. Cell 142: 625–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR 2010. Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberdoerffer S, Moita LF, Neems D, Freitas RP, Hacohen N, Rao A 2008. Regulation of CD45 alternative splicing by heterogeneous ribonucleoprotein, hnRNPLL. Science 321: 686–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR 2004. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci 101: 15974–15979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rine Dudoit YHY, Callow MJ, Speed TP 2002. Statistical methods for identifying differentially expressed genes in replicated cDNA microarray experiments. Statistica Sinica 12: 111–139 [Google Scholar]
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, et al. 2005. Towards a proteome-scale map of the human protein-protein interaction network. Nature 437: 1173–1178 [DOI] [PubMed] [Google Scholar]
- Stamm S, Riethoven JJ, Le Texier V, Gopalakrishnan C, Kumanduri V, Tang Y, Barbosa-Morais NL, Thanaraj TA 2006. ASD: A bioinformatics resource on alternative splicing. Nucleic Acids Res 34: D46–D55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoilov P, Lin CH, Damoiseaux R, Nikolic J, Black DL 2008. A high-throughput screening strategy identifies cardiotonic steroids as alternative splicing modulators. Proc Natl Acad Sci 105: 11218–11223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topp JD, Jackson J, Melton AA, Lynch KW 2008. A cell-based screen for splicing regulators identifies hnRNP LL as a distinct signal-induced repressor of CD45 variable exon 4. RNA 14: 2038–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warzecha CC, Sato TK, Nabet B, Hogenesch JB, Carstens RP 2009. ESRP1 and ESRP2 are epithelial cell-type-specific regulators of FGFR2 splicing. Mol Cell 33: 591–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehry EL 1986. Molecular fluorescence, phosphorescence, and chemiluminescence spectrometry. Anal Chem 58: 13R–33R [DOI] [PubMed] [Google Scholar]
- Witten JT, Ule J 2011. Understanding splicing regulation through RNA splicing maps. Trends Genet 27: 89–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JY, Kar A, Kuo D, Yu B, Havlioglu N 2006. SRp54 (SFRS11), a regulator for tau exon 10 alternative splicing identified by an expression cloning strategy. Mol Cell Biol 26: 6739–6747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Chen L 2009. A hierarchical Bayesian model for comparing transcriptomes at the individual transcript isoform level. Nucleic Acids Res 37: e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Eacker SM, Hong SJ, Gronostajski RM, Dawson TM, Dawson VL 2010. NMDA-induced neuronal survival is mediated through nuclear factor I-A in mice. J Clin Invest 120: 2446–2456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Gray EE, Chawla G, Porse BT, O’Dell TJ, Black DL 2012. PSD-95 is post-transcriptionally repressed during early neural development by PTBP1 and PTBP2. Nat Neurosci 15: 381–388 [DOI] [PMC free article] [PubMed] [Google Scholar]