

Abstract
BACKGROUND
Significant data supports the health benefits of selenium although supplementation trials have yielded mixed results. GPx-1, whose levels are responsive to selenium availability, is implicated in cancer etiology by human genetic data. Selenium’s ability to alter the phosphorylation of the H2AX, a histone protein that functions in the reduction of DNA damage by recruiting repair proteins to the damage site, following exposure to ionizing radiation and bleomycin was investigated.
METHODS
Human cell lines that were either exposed to selenium or were transfected with a GPx-1 expression construct were exposed to ionizing radiation or bleomycin. Phosphorylation of histone H2AX was quantified by flow cytometry and survival by the MTT assay. Phosphorylation of the Chk1 and Chk2 checkpoint proteins was quantified by western blotting.
RESULTS
In colon-derived cells, selenium increases GPx-1 and attenuated H2AX phosphorylation following genotoxic exposures while the viability of these cells was unaffected. MCF-7 cells and transfectants that express high GPx-1 levels were exposed to ionizing radiation and bleomycin, and H2AX phosphorylation and cell viability were assessed. GPx-1 increased H2AX phosphorylation and viability following the induction of DNA damage while enhancing the levels of activated Chk1 and Chk2.
CONCLUSIONS
Exposure of mammalian cells to selenium can alter the DNA damage response and do so by mechanisms that are dependent and independent of its effect on GPx-1.
GENERAL SIGNIFICANCE
Selenium and GPx-1 may stimulate the repair of genotoxic DNA damage and this may account for some of the benefits attributed to selenium intake and elevated GPx-1 activity.
1. Introduction
Selenium has long been a focus of efforts to develop effective strategies to reduce cancer incidence and mortality. This is due to a large body of evidence indicating that selenium provided to animals in low, non-toxic doses can suppress the development of tumors, and do so in most organs examined. Moreover, the protection offered by selenium supplementation occurs in response to a wide variety of DNA damaging exposures, including chemicals and radiation [1]. The benefits of selenium to humans are also supported by epidemiological studies revealing an inverse association between dietary selenium intake and cancer incidence [2]. Less clear are the benefits of selenium supplementation, as early promising results indicating that selenium may prevent prostate cancer [3, 4] were not supported by the large prostate cancer prevention trial, SELECT [5].
While the mechanisms of cancer prevention with selenium remain unknown and are probably due to several distinct mechanisms, it is likely that the repair of DNA damage is one means by which protection is provided. DNA lesions in the form of double strand and single strand breaks, as well as aberrations in chromosome structure and number disrupt genetic integrity and are needed to be repaired efficiently before a cell enters cell division. Other defects result in replication errors that can lead to potentially carcinogenic mutations. Once DNA damage is detected, signaling pathways are initiated to stimulate processes that coordinately remove and repair the damaged site.
Seo et al. reported in 2002 that selenium could stimulate the formation of DNA repair complexes and enhanced DNA repair following UV exposure in human fibroblasts [6]. Subsequently, selenium has been shown to protect cells in vitro against genotoxicity induced by exposure to phthalates [7], bleomycin [8], doxorubicin [9] and inhibit mutagenesis in bone marrow cells following exposure of mice to carboplatin [10]. Using lower doses (30 nM), selenium has also been shown to protect against X-ray [11] and UV-induced mutagenesis [12]. The same dose of selenium also reduced the levels of UV-induced DNA damage in human MCF-7 cells as measured by the reduction in the formation of micronuclei following irradiation [12]. Recently, de Rosa et al showed that human prostate-derived LNCaP cells were protected against UV induced genotoxicity when pre-treated with selenium and that selenium stimulated the excision of oxidative DNA lesions [13].
Many of the biological properties of selenium are due to its role as a constituent of selenium-containing proteins where selenium in the form of selenocysteine often resides at the enzyme’s active site [14]. In humans, there are 25 selenoproteins [15], many of whose activities have been implicated in several human diseases, including cancer [2, 16]. Among these, the first and best characterized is the cytosolic glutathione peroxidase (GPx-1) which uses reducing equivalents from glutathione to detoxify peroxides and whose activity can also impact cellular signaling pathways [17]. Genetic variations in GPx-1 have been shown to be associated with several diseases, including cancer [18, 19] and it’s ectopic expression has been shown to protect DNA from UV-induced DNA damage [12]. These data, as well as the responsiveness of GPx-1 translation to selenium levels, support the possibility that some of the benefits attributed to selenium are due to that elements impact on GPx-1 activity and function.
In order to gain insights into the protective functions of selenium and GPx-1, we examined the effects of selenium levels and GPx-1 activity on human cell lines exposed to either ionizing radiation (IR) or bleomycin, an antibiotic which specifically causes DNA strand breaks [20]. Among the endpoints examined was phosphorylation of histone H2AX, which is post-translationally modified by phosphorylation at the sites of DNA damage, where it is believed to serve as a scaffold for the recruitment of DNA repair enzymes [21, 22]. Phosphorylated H2AX plays a critical role in assembling the repair proteins at site of double strand breaks (DSBs) [23–25] and an impaired H2AX protein can significantly attenuate the repair process. H2AX activation also stimulates cell cycle check point proteins to stall the cell division and prevents chromatin breaks or chromosomal aberrations [21, 26]. The kinetics of loss of foci of phospho-H2AX after the induction of DSB can be indicative of an efficient DNA repair process [27] although there are some limitations to this approach of determining cellular DNA repair capacity [28]. Therefore, the phosphorylation of this protein has become a popular assay for the measure of DNA damage and a tool to investigate its repair.
2. Materials and Methods
2.1. Cell Culture
The human colon cancer cell line HCT116 was cultured in McCoy’s 5A medium (Mediatech) supplemented with 5% FBS. The non-transformed colon epithelial cell line NCM460 was cultured in Ham’s F12 nutrient medium (Gibco) supplemented with 20% FBS, 18.5ug/ml insulin and 0.4ug/ml hydrocortisone. The human breast carcinoma-derived cell line MCF-7 and its derivative MCF-7GPx-1 were cultured in Modified Eagle’s medium (Gibco) supplemented with 10% FBS (Gemini Bio-Products). The selenium concentration of the serum used was determined to be 152 nM by graphite furnace atomic absorption spectrometry conducted at the Texas A&M Veterinary Diagnostic Laboratory at College Station, Texas. The baseline, unsupplemented media therefore contained 7.6 nM, 15.2 nM or 30.4 nM selenium when cells are incubated in 5%, 10%, or 20% FBS, respectively. MCF-7GPx-1 was previously generated by transfection with a GPx-1 expression construct containing the NH2-terminal 5 alanine repeat variation and the codon 198 polymorphism resulting in a proline at that position [29]. Transfectants were maintained in culture medium containing 200 μg/ml G418. The cells were either irradiated using a Cs-137 irradiator (J.L. Sheperd and Associates irradiator Model 143–68) or exposed to bleomycin as indicated in the text.
2.2. Enzyme assays
GPx activity was determined by a coupled spectrophotometric assay with hydrogen peroxide substrate, glutathione reductase and reduced β-nicotinamide adenine dinucleotide phosphate (NADPH). In the text, we refer to the cellular activity determined by this assay as “GPx” activity as it is comprised of the enzymatic activity of GPx-1 as well as other members of the GPx family of proteins that may be expressed in the particular cells being analyzed. The change in NADPH levels due to oxidation was measured at 340 nm and GPx activity was expressed as nmoles of NADPH oxidized per minute per milligram protein. For all experiments, cells were plated in triplicate, and where indicated, treated with 30 nM selenium in the form of sodium selenite for 3 days prior to exposures. Protein lysates were prepared by sonication in 0.1M sodium phosphate, pH 7.0 and quantified using the Bradford assay (Bio-Rad, Hercules, CA). MCF-7GPx-1 cells were maintained in G418 which was removed from the culture media 2 days before plating the cells to reduce any impact of that antibiotic on selenoprotein synthesis [30].
2.3. Phospho-H2AX flow cytometry
Measurement of H2AX phosphorylation was performed by P-H2AX-FITC flow cytometry [31]. Experimental cells were trypsinized at the indicated time points and washed to remove residual media. The cells were resuspended in PBS with 1.6% paraformaldehyde at 37°C for 10 minutes and washed in 0.5% bovine serum albumin (BSA)-PBS, resuspended in 90% methanol and maintained at 4°C for 30 minutes and stored at −20°C. On the day of the staining, the cells were washed with 0.5% BSA-PBS and incubated with 2 μg/ml of anti- phospho-H2AX Ser 139 antibodies (Millipore, Billerica, MA) conjugated with the FITC fluorochrome for 1 hr in the dark at 4°C. The cells were then washed to remove excess antibody and FITC was measured either on a Becton Dickinson LSR or a CyAn ADP analyzer (Beckman Coulter Inc., CA) in the UIC Flow cytometry facility.
2.4. MTT assay
The MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was used to determine cell survival and proliferation. Cells were plated in triplicate in 25cm2 flasks, irradiated or treated with bleomycin as indicated and re-plated into 96 well plates at 2000 cells/well. For the bleomycin treatments, harvested cells were plated into 96 well plates in media with or without 10 μg/ml bleomycin. Cell viability assays were performed at 5 days post-irradiation or 2 days post bleomycin treatment by incubation in 20 μl of MTT (5mg/ml) for 3 hours at 37° C. The culture medium was removed and 150 μl of DMSO was added to each well and incubated at room temperature for another 5 minutes. Optical density was measured using a spectrophotometer at 560 nm and a reference at 650 nm. The same procedure was simultaneously performed on media alone to determine background absorbance.
2.5. Western blot analysis
Cell lysates were prepared from treated cells following irradiation or bleomycin treatment by incubating the cell pellet in cell lysis buffer supplemented with protease inhibitor cocktail (Cell Signaling Technology, MA) on ice for 30 minutes. Protein extracts were electrophoresed and transferred to PVDF membrane (Millipore). The membrane was probed with antibodies for GPx-1 (AbCam, MA), phospho-Chk1 (Ser 296) and phospho-Chk2 (Thr-68) from Cell Signaling Technology, MA. The resulting protein bands were detected using the chemiluminescent detection kit (GE Healthcare, NJ) and equal protein loading on gels was confirmed by blotting the same filters with anti- β-actin antibody (Abcam, MA)
2.6 Statistics
All assays were performed on triplicate cell cultures which were plated and treated independently. Experiments were repeated at least three times. Error bars denote standard deviations and significance was calculated using the Student’s T-test.
3. Results
3.1. Selenium protects human colon-derived cell lines against ionizing radiation
In order to investigate the effects of selenium supplementation on DNA damage and the cell survival of human colon cell lines, the tumor derived HCT116 and non-transformed NCM460 colon epithelial cells were exposed to an additional 30 nM selenium in the form of sodium selenite for 3 days. This dose and duration was chosen because it represents a low level of supplementation that is similar to that already present in the standard culture media, it results in the approximate maximal stimulation of the GPx-1 protein and has been shown to result in the protection of other cell types [12, 13, 29, 32]. Cells were treated with additional selenium, exposed to 4 Gy of ionizing radiation (IR) and the phosphorylation of histone H2AX was quantified by flow cytometry.
At one hour post-irradiation, H2AX phosphorylation (pH2AX) was seen to approximately double for both the HCT116 (Figure 1A) and NCM460 (Figure 1B) cells. As also seen in that figure, pre-incubation with selenium reduced the amount of pH2AX observed following IR for both cell types, data that is consistent with a protective effect for selenium. To assess the presumed protection provided by selenium supplementation, cell proliferation and survival were evaluated. The standard media of the HCT116 and NCM460 cells was supplemented with 30nM selenium for 3 days and the cells were exposed to 1, 3 and 5Gy of IR, maintained in culture for 5 days at which time cell viability was determined by MTT assay. Selenium supplementation resulted in increased HCT116 cell numbers at 5 days post irradiation and for all tested exposures following irradiation (Figure 1C), but in contrast, had no protective effect on the post-irradiation cell numbers for the NCM460 cells (Figure 1D).
Figure 1. Selenium supplementation affects pH2AX levels and cell viability in HCT116 and NCM460 cells exposed to radiation.

HCT116 (A) and NCM460 (B) cells were pre-treated with or without additional 30nM sodium selenite for 3 days and exposed to 4Gy of ionizing radiation and after 1hr, pH2AX levels were measured by flow cytometry. HCT 116 (C) and NCM460 (D) cells were either untreated or treated with 30nM NaSe and exposed to 0, 1, 3, 5 Gy ionizing radiation, replated into 96 well plates and MTT assays were performed on day 1 and 5. *P<0.05, Ψ P<0.005 and § P<0.0005 from No IR control. Error bars indicate the S.D.
3.2. Selenium supplementation attenuated the bleomycin-induced phosphorylation of H2AX
Bleomycin exposure was used to evaluate the generality of the results obtained with IR. This drug is a commonly used chemotherapeutic that binds DNA and generates reactive oxygen molecules that cause single and double strand breaks [20]. Both HCT116 and NCM460 cells were treated with 30 nM selenium for 3 days, exposed to 10 μg/ml of bleomycin for 1 hour after which the cells were fixed and stained with pH2AX antibody for flow cytometry analysis (Figure 2A and 2B). As observed when these cells were irradiated, pre-incubation with additional selenium resulted in a decrease in bleomycin-induced pH2AX phosphorylation. For both cell lines examined, selenium had no effect on bleomycin toxicity when doses ranging from 0.5–100 μg/ml were examined (Figure 2, C and D).
Figure 2. Selenium pre-treatment reduces bleomycin induced pH2AX fluorescence in HCT116 and NCM460 cells but does not affect cell viability.

HCT116 (A) and NCM460 (B) were either untreated or treated with 30nM selenium for 3 days and exposed to 10μg/ml bleomycin for 1hr; cells were fixed and stained for pH2AX. The graphs represent the mean fluorescent intensity of the cells stained for pH2AX as determined by flow cytometry. HCT116 (C) and NCM460 (D) cells were pretreated with or without a supplemental 30 nM selenium for 3 days before treating with 10μg/ml bleomycin. MTT assays were performed at 48 hrs post-bleomycin treatment. § P<0.0005 from Control, ¥ P<0.05 form bleomycin. Error bars indicate the S.D.
3.3. GPx-1 stimulates H2AX phosphorylation
Work by us and others have implicated the stimulation of the GPx-1 selenoprotein as part of the mechanism by which selenium reduces DNA damage following exposure to genotoxic stimuli. The incubation of both HCT116 and NCM460 with additional selenium resulted in a 5–8 fold increase in GPx enzyme activity in those cells (Figure 3A) and a considerable contribution to that increase in enzyme activity was likely due to an increase in GPx-1, as determined by western blotting with GPx-1 specific antibodies (Figure 3C). NCM460 and HCT116 represent different stages of carcinogenesis, are cultured in different media and were derived from different individuals; these differences introduce variables that make biological comparisons difficult to interpret. To further investigate the role of GPx-1 in protecting cells from DNA damage under controlled conditions, we made use of the previous observation that human MCF-7 breast carcinoma cells produce negligible GPx-1 protein or activity, presenting the opportunity to compare these cells to those that express ectopic GPx-1 due to transfection [29, 32]. As seen in Figure 3C, analysis of protein extracts derived from MCF-7 cells does not reveal a GPx-1 band in MCF-7 cells, it is easily detected in GPx-1 transfected MCF-7 cells (MCF-7GPx-1) and that GPx-1 expression could be stimulated in these cells by incubation in media supplemented with selenium (Figure 3C). Previously, we demonstrated that GPx-1 expression in these cells conferred resistance to DNA damage as determined by the appearance of micronuclei in UV-irradiated cells [12].
Figure 3. Levels of GPx-1 protein and enzyme activity in cells with or without selenium supplementation.

HCT116 and NCM460 cells were treated with 30nM selenium for 3 days and protein extracts were assayed for GPx enzyme activity (3A) and GPx-1 protein levels by western blotting with anti-GPx-1 antibodies (3C, a–c). The levels of GPx activity in MCF-7 and MCF-7 cells transfected with a GPx-1 expression construct with and without pre-incubation with an additional 30 nM selenium are presented in 3B and GPx-1 protein levels in the same extracts are determined by western blotting in 3C. Error bars indicate the S.D.
As seen in Figure 4A, exposure of MCF-7 to IR significantly increased pH2AX by approximately 2 fold, one hour post exposure. In contrast to the data obtained in colon-derived cells, selenium supplementation did not reduce post-IR H2AX phosphorylation in GPx-1 expressing or GPx-1 null MCF-7 cells. What was apparent by comparing the pH2AX levels in MCF-7 vs. MCF-7GPx-1 was that even though the baseline levels of pH2AX were similar in the two cell lines, pH2AX levels were significantly higher in those cells expressing high levels of GPx-1 as compared to IR exposed MCF-7 cells. Performing these same experiments inducing DNA damage with bleomycin yielded very similar results: increased H2AX phosphorylation with ectopic GPx-1 expression and no effect of selenium supplementation on pH2AX levels following treatment with bleomycin (Figure 4B).
Figure 4. GPx-1 expression increases the levels of H2AX phosphorylation following exposure to ionizing radiation.

pH2AX levels were measured by flow cytometry in MCF-7 and MCF-7GPx-1 cells with or without selenium treatment and after 1 hr after exposure to 4Gy of ionizing radiation (A and B). *P<0.001 from control; § P<0.0001 from no IR control. Error bars indicate the S.D.
The lack of an effect seen in MCF-7 cells following selenium supplementation may be due to the absence of expression of GPx-1 in those cells. Furthermore, the levels of that enzyme’s activity seen in unsupplemented MCF-7GPx-1 cells, which is likely to be much higher than that observed in the colon-derived cells even after selenium (Figure 3), are perhaps sufficient to achieve the maximal pH2AX induction such that the additional stimulation in GPx-1 levels achieved with selenium supplementation is inconsequential.
3.4. Selenium/GPx-1 enhances MCF-7 viability following DNA damage
In order to assess the effects of selenium and GPx-1 on cell viability following exposure to IR, MCF-7 and MCF-7GPx-1 cells were pre-treated with an additional 30 nM selenium for 3 days prior to radiation exposures of 3 Gy and 5 Gy and viability was assessed by MTT assays performed at 5 days post-IR exposure. MCF-7 cells were more sensitive to IR than MCF-7GPx-1 cells expressing elevated GPx-1 activity (Figure 5) and selenium supplementation did not offer any protection for MCF-7 cells against IR when viability was determined at 5 days post-IR. MCF-7GPx-1 cells were clearly more resistant to IR than MCF-7 cells as observed with both doses of IR when assayed at 5 days (Fig 5B) post-IR. When pre-treated with selenium, MCF-7 cells exhibited increased viability compared to untreated cells at the 5 Gy dose (Figure 5A).
Figure 5. The effect of selenium and GPx-1 expression on viability of the MCF-7 cells exposed to radiation.
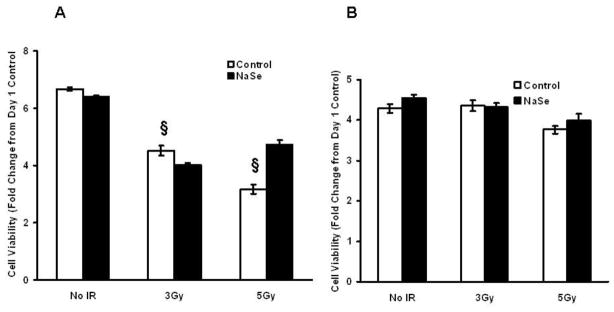
MCF-7 (A) and MCF-7GPx-1 (B) cells were either untreated or treated with 30 nM selenium and exposed to 3 or 5 Gy of ionizing radiations. The cells were then replated into 96 well plates and MTT assays were performed on day 5. § P<0.0001 from no IR control. Error bars indicate the S.D.
Surprisingly, a different response to supplemental selenium and GPx-1 was obtained when these same cells were challenged with bleomycin. MCF-7 and MCF-7GPx-1 cells were pre-treated with selenium for 3 days, exposed to 0–100 μg/ml of bleomycin for 48 hours and viability was assessed by the MTT assay. MCF-7 cells were more sensitive to bleomycin, with cell growth reduced by 50–60% of the untreated cells at 10, 50 and 100 μg/ml concentrations of bleomycin (Figure 6A). In contrast, MCF-7GPx-1 cells (Fig 6B) were much less affected by bleomycin treatment where only 10–20 % less cell growth was observed at higher doses (Figure 6B). Selenium treatment protected MCF-7 cells at all doses above 1 μg/ml of bleomycin, while the benefit of supplemental selenium was less apparent for the GPx-1 expressing cells (Figure 6A and 6B). Figure 6C is a composite of the data demonstrating the increase in viability following exposure to bleomycin when cells were exposed to added selenium or expressed GPx-1.
Figure 6. Selenium and GPx-1 protect MCF-7 and MCF-7GPx-1 cells against bleomycin induced cytotoxicity.

MCF7 (A) and MCF-7GPx-1 (B) cells were either treated or untreated with selenium for 3 days, exposed to 10μg/ml bleomycin for 1 hr and the MTT assay was performed 48 hrs after bleomycin addition. Figure C is a composite of data from A and B demonstrating the enhanced protection of selenium and GPx-1 against bleomycin toxicity. Error bars indicate the S.D. and * indicates P< 0.05 for the difference between treated and control cultures.
The results presented above indicated that GPx-1 enhanced the phosphorylation of H2AX following genotoxic challenge, yet was protective against the cytotoxic/cytostatic consequences of those challenges. Unlike the bleomycin studies where cells are continuously incubated in the drug, the single exposure to IR permitted the evaluation of whether H2AX phosphorylation levels returned to baseline after continued incubation, pH2AX levels were assessed at 4 and 8 hours post IR. As seen in Figure 7, 4 hours after IR, pH2AX levels were only slightly higher than those seen prior to exposure for both cell types, even though the pH2AX was approximately twice as much in GPx-1 over-expressing cells at the one hour time point.
Figure 7. Selenium and GPx-1 enhance H2AX phosphorylation following ionizing radiation.
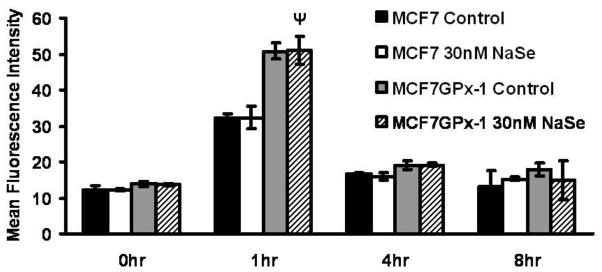
MCF-7 or MCF-7GPx-1 cells were irradiated with 4 Gy of ionizing radiations and H2AX phosphorylation was determined by flow cytometry at the indicated time points. Ψ P<0.005 from MCF7 1hr control. Error bars indicate the S.D.
3.5. GPx-1 enhances the phosphorylation of the Chk1 and Chk2
As an initial screen for possible downstream targets of GPx-1 and/or selenium that can promote increased survival and reduced mutagenesis, we examined the levels of phosphorylated proteins involved in the response to DNA damage. Chk1 and Chk2 are proteins that are activated following DNA damage; Chk1 is activated by ATR (ataxia-telangiectasia and Rad3-related) primarily in response to the detection of single strand breaks bulky lesions, while Chk2 is activated by ATM (ataxia-telangiectasia mutated) following the detection of double strand breaks in DNA [33]. Baseline levels of phosphorylated Chk1 was detected in significantly higher levels in GPx-1 expressing cells (Figure 8). Chk1 levels increased following either selenium supplementation or IR exposure in MCF-7 cells, but not in MCF-7GPx-1 cells (Figure 8). In contrast, Chk2 levels were only detected following IR in MCF-7GPx-1 cells and not at all in MCF-7 cells. Thus, GPx-1 was required for the increased phosphorylation of Chk2 seen in response to IR in MCF-7GPx-1 cells (Figure 8).
Figure 8. GPx-1 enhances the levels of Chk1 and Chk2.

MCF-7 and MCF-7GPx-1 cells were either untreated or treated with 30nM selenium and exposed to 4 Gy of ionizing radiations. Cell lysates were prepared 1 hr post-radiation and analyzed for levels of phosphorylated Chk1 and Chk2 by western blotting with the respective antibodies.
4. Discussion
The studies presented in this manuscript were initiated in order to gain a better understanding of the role of selenium and the GPx-1 selenoprotein in the response to DNA damage. We initially began by examining the effects of selenium supplementation on the response of two colon-derived human cell lines, one transformed and the other immortalized but not transformed, and two different sources of DNA damage, IR and bleomycin. As shown in Figures 1 and 2, there was a similar effect of selenium on H2AX phosphorylation following exposure to IR and bleomycin, that being reduced levels of pH2AX after the exposures. Previously, it was shown that treatment of HL-60 cells with selenium, also in the form of sodium selenite, increased H2AX phosphorylation [34]. The dose of selenium used in that study was 500 nM, over 15 times the 30 nM used in our studies and one that is likely to cause rather than prevent oxidative damage. The 30 nM dose used herein was sufficient to increase GPx enzyme activity (Figure 3) and GPx-1 protein levels (Figure 8), even when combined with the selenium provided by the 10% serum used in the culture media and is still less than is typically present in serum. Thus, the data presented using cultured cells is therefore most comparable to studies comparing individuals with different baseline selenium levels or providing selenium-deficient individuals with supplemental selenium, circumstances that have previously revealed cancer prevention efficacy [35].
Although selenium was able to reduce the levels of pH2AX following genotoxic exposures, only in the case of HCT116 cells was there a significant improvement in cell viability, and this was apparent only following exposure to IR and not bleomycin (Figures 1 and 2). In humans, selenium status was shown to be inversely associated with IR-associated DNA damage, but only for women who were carriers of a defective BRCA1 gene that predisposes to cancer and is involved in DNA repair [36]. The complexities that are encountered when comparing humans or cells derived from different individuals prompted us to study selenium and GPx-1 status using the MCF-7 cell line, which permitted the examination of selenium and GPx-1 effects in identical genetic backgrounds.
Pre-treatment of MCF-7 and MCF-7GPx-1 cells with selenium followed by either IR or bleomycin indicated that selenium supplementation did not significantly alter H2AX phosphorylation, either at baseline or following genotoxic exposure. Surprisingly, and contrary to expectations, pH2AX levels after exposure to either IR or bleomycin were approximately twice as high in GPx-1 expressing cells as compared to MCF-7 cells with negligible GPx activity. Given the anti-oxidant function of GPx-1 [17] and our previous work demonstrating enhanced protection against UV-induced micronuclei formation [12], we investigated this phenomenon further by assessing cell viability of these same cells following IR and bleomycin exposure. As seen in Figures 5 and 6, GPx-1 was protective as assessed by cell viability determined with the MTT assay.
If one takes the phosphorylation of H2AX as an indicator of DNA damage, the elevated levels of pH2AX and increased survival observed in cells expressing GPx-1 following genotoxic exposure would seem contradictory. Although there are inherent limitations in measuring H2AX phosphorylation as a measure of DNA damage and repair due to the selection of appropriate time points and cell cycle effects [37, 38], one possible explanation for these data is that the increased H2AX phosphorylation observed in GPx-1 expressing cells is a reflection of enhanced signaling leading to H2AX phosphorylation which would result in accelerated recruitment of the DNA repair machinery and subsequently less damage and better survival. Some support of this notion can be derived from observations of the effects of enhanced selenium levels and GPx-1 expression on the levels of the Chk1 and Chk2 proteins, proteins involved in the cellular response to DNA damage [39]. Minimally, Chk2 is activated by phosphorylation of ATM in response to double strand breaks while Chk1 is activated by ATR in response to single stranded DNA [40]. As seen in Figure 8, levels of phosphorylated Chk1 are higher in GPx-1 expressing cells and those levels did not change post irradiation, while the induction of phospho-Chk2 following IR was significantly more robust in GPx-1 expressing cells. These results are consistent with the type of DNA damage that principally occurs following IR, double strand breaks, and that IR exposure induced Chk2 phosphorylation but not that of Chk1. Moreover, the increased phosphorylation of Chk2 following IR observed with selenium supplementation of MCF-7 cells indicates that there is a GPx-1 independent effect of selenium, although it should be noted that the enhancement of Chk2 phosphorylation by selenium supplementation was considerably more dramatic in GPx-1 expressing cells (Figure 8). Collectively, the results presented indicated that selenium increased H2AX, Chk1 and Chk2 phosphorylation, as well as increased cell viability following exposure to DNA damage. This, and selenium’s ability to stimulate GPx-1 protein levels, may indicate that an increase in the cellular response to DNA damage is one plausible mechanism by which selenium protects against the toxic and carcinogenic effects of diverse DNA damaging agents [41–43] and this subject has recently been reviewed [44]. However, it should also be considered that selenium may induce these changes by selenoprotein-independent processes as well as the stimulation of the levels and activities of other selenoproteins. Many of the selenoproteins are involved in redox processes and redox reactions have been shown be important modifiers of the DNA repair response [45], with one particular selenoprotein, thioredoxin reductase, being one such candidate [46].
An intriguing possibility raised by the data presented in this manuscript is whether GPx-1 may in fact be able to promote tumor growth under certain circumstances. This might occur by a number of different mechanisms, including the protection of tumor cells from oxidative stress, the stimulation of growth promoting pathways by altering growth-related signaling pathways and perhaps protecting tumor cell DNA from damage. The potentially beneficial and harmful effects of GPx-1 activity have recently been reviewed [17] and is supported by our data indicating a significant association between total GPx activity in the prostate and the Gleason score, and indicator of the aggressiveness of the cancer of that organ [47]. These issues indicate the need for further investigation to improve our understanding of the benefits of selenium and role of GPx-1 in human disease.
Highlights.
We addressed the issue as to how selenium and selenoproteins may impact DNA damage repair by focusing on H2AX phosphorylation
sing GPx-1 null and cells expressing GPx-1 due to transfection permitted the evaluation of effects of selenium and GPx-1 independently
oth H2AX phosphorylation and cell survival were evaluated
GPx-1 effects on the Chk1 and Chk2 DNA damage repair proteins were observed
Acknowledgments
This work was supported by a grant from the National Institutes of Health [Grant # RO1CA127943] to AMD and Post Doctoral Fellowship from the American Institute for Cancer Research [Grant #10A072] to AJ-M and SB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.El-Bayoumy K. In: Cancer Prevention. De Vita VT, Hellman S, Rosenberg SA, editors. J.B. Lippincott Co; Philadelphia: 1991. pp. 1–15. [Google Scholar]
- 2.Rayman MP. Lancet. 2012;379:1256–68. doi: 10.1016/S0140-6736(11)61452-9. [DOI] [PubMed] [Google Scholar]
- 3.Clark LC, Combs GF, Jr, Turnbull BW, Slate EH, Chalker DK, Chow J, Davis LS, Glover RA, Graham GF, Gross EG, Krongrad A, Lesher JL, Jr, Park HK, Sanders BB, Jr, Smith CL, Taylor JR. JAMA. 1996;276:1957–63. [PubMed] [Google Scholar]
- 4.Duffield-Lillico AJ, Dalkin BL, Reid ME, Turnbull BW, Slate EH, Jacobs ET, Marshall JR, Clark LC. BJU International. 2003;91:608–612. doi: 10.1046/j.1464-410x.2003.04167.x. [DOI] [PubMed] [Google Scholar]
- 5.Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, Parnes HL, Minasian LM, Gaziano JM, Hartline JA, Parsons JK, Bearden JD, 3rd, Crawford ED, Goodman GE, Claudio J, Winquist E, Cook ED, Karp DD, Walther P, Lieber MM, Kristal AR, Darke AK, Arnold KB, Ganz PA, Santella RM, Albanes D, Taylor PR, Probstfield JL, Jagpal TJ, Crowley JJ, Meyskens FL, Jr, Baker LH, Coltman CA., Jr JAMA. 2009;301:39–51. doi: 10.1001/jama.2008.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seo YR, Sweeney C, Smith ML. Oncogene. 2002;21:3663–9. doi: 10.1038/sj.onc.1205468. [DOI] [PubMed] [Google Scholar]
- 7.Erkekoglu P, Rachidi W, De Rosa V, Giray B, Favier A, Hincal F. Free Radic Biol Med. 2010;49:559–66. doi: 10.1016/j.freeradbiomed.2010.04.038. [DOI] [PubMed] [Google Scholar]
- 8.Laffon B, Valdiglesias V, Pasaro E, Mendez J. Biol Trace Elem Res. 2010;133:12–9. doi: 10.1007/s12011-009-8407-9. [DOI] [PubMed] [Google Scholar]
- 9.Santos RA, Takahashi CS. Food Chem Toxicol. 2008;46:671–7. doi: 10.1016/j.fct.2007.09.090. [DOI] [PubMed] [Google Scholar]
- 10.Kumar MS, Pollok KE, Smith ML. Anticancer Res. 2010;30:291–3. [PubMed] [Google Scholar]
- 11.Diamond AM, Dale P, Murray JL, Grdina DJ. Mutation Research. 1996;356:147–154. doi: 10.1016/0027-5107(96)00016-4. [DOI] [PubMed] [Google Scholar]
- 12.Baliga MS, Wang H, Zhuo P, Schwartz JL, Diamond AM. Biol Trace Elem Res. 2007;115:227–42. doi: 10.1007/BF02685998. [DOI] [PubMed] [Google Scholar]
- 13.de Rosa V, Erkekoglu P, Forestier A, Favier A, Hincal F, Diamond AM, Douki T, Rachidi W. Free Radic Res. 2012;46:105–16. doi: 10.3109/10715762.2011.647009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gladyshev VN, Hatfield DL. Curr Protoc Protein Sci. 2001;Chapter 3(Unit 38) doi: 10.1002/0471140864.ps0308s20. [DOI] [PubMed] [Google Scholar]
- 15.Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN. Science. 2003;300:1439–43. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 16.Davis CD, Tsuji PA, Milner JA. Annu Rev Nutr. 2012;32:73–95. doi: 10.1146/annurev-nutr-071811-150740. [DOI] [PubMed] [Google Scholar]
- 17.Lubos E, Loscalzo J, Handy DE. Antioxid Redox Signal. 2011;15:1957–97. doi: 10.1089/ars.2010.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rayman MP. Biochim Biophys Acta. 2009;1790:1533–40. doi: 10.1016/j.bbagen.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 19.Zhuo P, Diamond AM. Biochim Biophys Acta. 2009;115:227–42. doi: 10.1016/j.bbagen.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burger RM, Peisach J, Horwitz SB. J Biol Chem. 1981;256:11636–44. [PubMed] [Google Scholar]
- 21.Lowndes NF, Toh GW. Curr Biol. 2005;15:R99–R102. doi: 10.1016/j.cub.2005.01.029. [DOI] [PubMed] [Google Scholar]
- 22.Thiriet C, Hayes JJ. Mol Cell. 2005;18:617–22. doi: 10.1016/j.molcel.2005.05.008. [DOI] [PubMed] [Google Scholar]
- 23.Celeste A, Petersen S, Romanienko PJ, Fernandez-Capetillo O, Chen HT, Sedelnikova OA, Reina-San-Martin B, Coppola V, Meffre E, Difilippantonio MJ, Redon C, Pilch DR, Olaru A, Eckhaus M, Camerini-Otero RD, Tessarollo L, Livak F, Manova K, Bonner WM, Nussenzweig MC, Nussenzweig A. Science. 2002;296:922–7. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, Eckhaus M, Ried T, Bonner WM, Nussenzweig A. Cell. 2003;114:371–83. doi: 10.1016/s0092-8674(03)00567-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bassing CH, Chua KF, Sekiguchi J, Suh H, Whitlow SR, Fleming JC, Monroe BC, Ciccone DN, Yan C, Vlasakova K, Livingston DM, Ferguson DO, Scully R, Alt FW. Proc Natl Acad Sci U S A. 2002;99:8173–8. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, Yan C, Tepsuporn S, Morales JC, Adams MM, Lou Z, Bassing CH, Manis JP, Chen J, Carpenter PB, Alt FW. Mol Cell. 2006;21:201–14. doi: 10.1016/j.molcel.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Lobrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, Barton O, Jeggo PA. Cell Cycle. 2010;9:662–9. doi: 10.4161/cc.9.4.10764. [DOI] [PubMed] [Google Scholar]
- 28.Markova B, Albers C, Breitenbuecher F, Melo JV, Brummendorf TH, Heidel F, Lipka D, Duyster J, Huber C, Fischer T. Oncogene. 2010;29:739–51. doi: 10.1038/onc.2009.374. [DOI] [PubMed] [Google Scholar]
- 29.Hu YJ, Diamond AM. Cancer Res. 2003;63:3347–51. [PubMed] [Google Scholar]
- 30.Handy DE, Lubos E, Yang Y, Galbraith JD, Kelly N, Zhang YY, Leopold JA, Loscalzo J. J Biol Chem. 2009;284:11913–21. doi: 10.1074/jbc.M900392200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kataoka Y, Bindokas VP, Duggan RC, Murley JS, Grdina DJ. J Radiat Res. 2006;47:245–57. doi: 10.1269/jrr.0628. [DOI] [PubMed] [Google Scholar]
- 32.Zhuo P, Goldberg M, Herman L, Lee BS, Wang H, Brown RL, Foster CB, Peters U, Diamond AM. Cancer Res. 2009;69:8183–90. doi: 10.1158/0008-5472.CAN-09-1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reinhardt HC, Yaffe MB. Curr Opin Cell Biol. 2009;21:245–55. doi: 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou N, Xiao H, Li TK, Nur EKA, Liu LF. J Biol Chem. 2003;278:29532–7. doi: 10.1074/jbc.M301877200. [DOI] [PubMed] [Google Scholar]
- 35.Duffield-Lillicoe A, Reid M, Turnbull J, Combs GJ, Slate E, Fischbach L, Marshall J, Clarke L. Cancer Epidemiol Biomarkers Prev. 2002;11:630–639. [PubMed] [Google Scholar]
- 36.Kotsopoulos J, Chen Z, Vallis KA, Poll A, Ghadirian P, Kennedy G, Ainsworth P, Narod SA. Cancer Causes Control. 2010;21:679–87. doi: 10.1007/s10552-009-9495-8. [DOI] [PubMed] [Google Scholar]
- 37.Darzynkiewicz Z, Traganos F, Zhao H, Halicka HD, Skommer J, Wlodkowic D. Methods Cell Biol. 2011;103:115–47. doi: 10.1016/B978-0-12-385493-3.00006-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ivashkevich A, Redon CE, Nakamura AJ, Martin RF, Martin OA. Cancer Lett. 2012;327:123–33. doi: 10.1016/j.canlet.2011.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bartek J, Lukas J. Cancer Cell. 2003;3:421–9. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 40.Smith J, Tho LM, Xu N, Gillespie DA. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 41.Ferguson LR, Karunasinghe N, Zhu S, Wang AH. Mutat Res. 2012;733:100–10. doi: 10.1016/j.mrfmmm.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 42.Valdiglesias V, Pasaro E, Mendez J, Laffon B. Arch Toxicol. 2010;84:337–51. doi: 10.1007/s00204-009-0505-0. [DOI] [PubMed] [Google Scholar]
- 43.Brozmanova J, Manikova D, Vlckova V, Chovanec M. Arch Toxicol. 2010;84:919–38. doi: 10.1007/s00204-010-0595-8. [DOI] [PubMed] [Google Scholar]
- 44.Bera S, Rosa VD, Rachidi W, Diamond AM. Mutagenesis. 2012 [Epub ahead of print] [Google Scholar]
- 45.Luo M, He H, Kelley MR, Georgiadis MM. Antioxid Redox Signal. 2010;12:1247–69. doi: 10.1089/ars.2009.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arner ES, Holmgren A. Semin Cancer Biol. 2006;16:420–6. doi: 10.1016/j.semcancer.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 47.Jerome-Morais A, Wright ME, Liu R, Yang W, Jackson MI, Combs GF, Jr, Diamond AM. Prostate. 2012;72:1006–12. doi: 10.1002/pros.21506. [DOI] [PMC free article] [PubMed] [Google Scholar]