

Abstract
The systematic use of antivitamin K anticoagulants (AVK) as rodenticides caused the selection of rats resistant to AVKs. The resistance is mainly associated to genetic polymorphisms in the Vkorc1 gene encoding the VKORC1 enzyme responsible for the reduction of vitamin K 2,3-epoxide to vitamin K. Five major mutations, which are responsible for AVK resistance, have been described. Possible explanations for the biological cost of these mutations have been suggested. This biological cost might be linked to an increase in the vitamin K requirements. To analyze the possible involvement of VKORC1 in this biological cost, rVKORC1 and its major mutants were expressed in Pichia pastoris as membrane-bound proteins and their catalytic properties were determined for vitamin K and 3-OH-vitamin K production. In this report, we showed that mutations at Leu-120 and Tyr-139 dramatically affect the vitamin K epoxide reductase activity. Moreover, this study allowed the detection of an additional production of 3-hydroxyvitamin K for all the mutants in position 139. This result suggests the involvement of Tyr-139 residue in the second half-step of the catalytic mechanism corresponding to the dehydration of vitamin K epoxide. As a consequence, the biological cost observed in Y139C and Y139S resistant rat strains is at least partially explained by the catalytic properties of the mutated VKORC1 involving a loss of vitamin K from the vitamin K cycle through the formation of 3-hydroxyvitamin K and a very low catalytic efficiency of the VKOR activity.
Keywords: VKORC1, Mutation, Vitamin K, Hydroxyvitamin K, Catalytic mechanism
Abbreviations: AVK, antivitamin K anticoagulant; VKORC1, vitamin K epoxide reductase; vit K, vitamin K; vit K>O, vitamin K2,3-epoxide; OH-vit K, hydroxyvitamin K; vit KH2, vitamin K hydroquinone; VKDP, vitamin K-dependent protein.
Highlights
▸ L120Q and Y139C/F/S mutations of rVKORC1 dramatically affect VKOR activity. ▸ Y139C/F/S mutations lead to the production of 3-hydroxyvitamin K. ▸ Tyr-139 residue is involved in the dehydration of vitamin K epoxide. ▸ VKORC1 mutations in position 139 lead to increase in vitamin K requirements. ▸ Rat carriers for Y139C/F/S are models to study consequences of vitamin K deficiency.
1. Introduction
The systematic use of antivitamin K anticoagulants (AVK) as rodenticides since 50 years caused the selection of Rattus norvegicus rats resistant to AVKs [1,2]. This resistance is found worldwide and is supported by two major mechanisms in rats 1/ an accelerated detoxification system involving cytochrome P-450 [3,4] and 2/ the inefficiency of AVKs on VKORC1, their molecular target. This last mechanism has been observed in numerous countries but appears to be major in Western Europe, even if cohabitation of these two mechanisms had been demonstrated in Denmark [5].
The Vkorc1 gene encodes the vitamin K epoxide reductase (VKORC1) [6,7]. This enzyme is responsible for reducing vitamin K-2,3-epoxide (vit K>O) to the enzymatically activated form, the vitamin K hydroquinone (KH2). This reaction is the limiting step of the recycling of vitamin K [8]. KH2 is a cosubstrate for carboxylation of the vitamin K-dependent proteins (VKDP) including four clotting factors (II, VII, IX, X). Post-translational modification of glutamate to gamma carboxyl glutamate is required for the activity of VKDP. Inhibition of VKORC1 by AVK results in partially under-carboxylated vitamin K-dependent blood clotting factors and thus in severe bleeding problems.
In Western Europe, five major mutations in Vkorc1 (i.e., L120Q, L128Q, Y139C, Y139F, Y139S) which were shown to be responsible for the resistance to AVKs [9–12] (Table 1) have been observed in large geographical areas [1,2]. L128Q mutation was found in Scotland, L120Q in Hampshire and Berkshire, Y139S in Wales, Y139F in France and Belgium and Y139C in Denmark and Germany. The spreading of the resistance in wild rodent populations depends on the benefit due to the mutations in the presence of the selection pressure (i.e., a rodenticide treatment with AVKs) and on the potential biological cost associated to the mutation in the absence of the selection pressure. Such a biological cost associated to Vkorc1 mutations could provoke a decrease in the allelic frequency of resistance alleles in the absence of AVK treatment. Yet the in vivo studies suggested a negative effect of Vkorc1 mutations on the vitamin K recycling leading to an increase in the vit K requirements. The homozygous Welsh resistant rats had a 20-fold increase in vit K requirements compared to susceptible rats [13] while the increased vit K requirements of the homozygous Scottish rats has been reported intermediate compared with that in homozygous Welsh rats [14,15]. For the Danish/German resistant rats, various studies reported also an increase in the vit K requirements. Nevertheless, depending on the studies, this increase requirement was moderate to severe [14–16]. Moreover, various fitness trade-offs associated with AVKs resistance were described in rats. In some resistant rats, retarded growth [16,17], modification of the reproductive capacities [18,19] vascular calcification [20] were observed associated to the resistance to AVKs.
Table 1.
Ki values for recombinant mutated rVKORC1 enzymes toward warfarin (first generation AVK) and difenacoum (second generation AVK), previously reported by Hodroge et al. [12]. These AVKs inhibited VKOR activity in a non-competitive manner
| Protein | Warfarin (μM) | Difenacoum (μM) |
|---|---|---|
| rVKORC1 | 0.5 | 0.03 |
| L120Q | >100 | 0.89 |
| L128Q | 4.0 | 0.07 |
| Y139C | >100 | 0.16 |
| Y139F | >100 | 0.10 |
| Y139S | >100 | 0.09 |
The objective of this work was to study the involvement of the different mutations of Vkorc1 in the biological cost specifically described in various strains of wild rats resistant to AVKs. To analyze the causes of an increase in the vit K requirements, we analyzed in this study the catalytic properties of the wild type and mutated VKORC1 and linked these properties to such fitness outcomes observed in the different strains of resistant rats.
2. Materials and methods
2.1. Chemicals
Vit K1 was purchased from Sigma–Aldrich (Saint Quentin Fallavier, France). Vit K1 (Phylloquinone) was converted to vit K1>O according to [32]. 2-OH-vit K1 and 3-OH-vit K1 were synthesized from vit K1>O according to [22]. Methanol HPLC grade, and acetic acid (analysis grade) were obtained from Merck (Germany).
2.2. Heterologous expression of recombinant rVKORC1 proteins
Recombinant rVKORC1 and its five major mutants were expressed in Pichia pastoris and yeast microsomes preparations were performed as previously described [12]. Expression level quantification of VKORC1 proteins were determined by Western blotting, as described previously [21]. Microsomal proteins were separated on 12% SDS–polyacrylamide gel electrophoresis, transferred onto Immobilon-P membranes and probed with anti-c-myc antibodies (Invitrogen, Cergy Pontoise, France). The resulting immunocomplexes were visualized using alkaline phosphatase-conjugated anti-mouse immunoglobulins as secondary antibodies and a BCIP/NBT solution. Quantification of the stained bands was performed by densitometry using the Scion Image software. The relative intensity (RI) of the signal was correlated to the quantity of microsomal proteins. The relation was linear from 0 to 50 μg for microsomal proteins containing WT-VKORC1 or mutated VKORC1. To evaluate the expression level of VKORC1 proteins, the expression of wild type VKORC1 (WT-VKORC1) was designated as the basal expression. For the quantification of all the mutants, the same unique pool of yeast microsomes containing WT-VKORC1 was used. Therefore, its expression factor was by definition 1. The expression level of the mutated VKORC1 was evaluated by comparison to the expression of WT-VKORC1. For this purpose, various amounts (from 0 to 50 μg) of microsomal proteins containing WT-VKORC1 and various amounts (depending on the expression level) of microsomal proteins containing one of the mutated VKORC1 were analyzed on the same western blot. Two linear relations (RI = a × quantity of microsomes loaded) were obtained, the first one for microsomes containing WT-VKORC1 (characterized by a specific slope aWT), the second one for microsomes containing the mutated VKORC1 (characterized by a slope a mut). Ratio amut/aWT allowed us to determine the expression factor characterizing the expression level of the mutated VKORC1 in the microsomal fraction compared to the expression level of the WT-VKORC1.
2.3. Vitamin K epoxide reductase activity assays
Microsomal VKOR activity was assayed as previously described. Briefly, standard reactions were performed in 200 mM Hepes buffer (pH 7.4) containing 150 mM KCl, 1 mM dithiothreitol, 0.75–2 g L−1 of yeast microsomal proteins containing recombinant VKORC1 or 1 g L−1 of rat liver microsomal proteins. The reaction was started by the addition of vit K1>O solution in 1% Triton X-100. After incubation at 37 °C for 30 min, the reaction was stopped by adding of 4 mL of iced 1:1 isopropanol/hexane solution. After centrifugation at 5000g for 10 min, the hexane layer was removed and dried under nitrogen. The dry residue was dissolved in 0.2 mL of isopropanol.
2.4. Chromatography
The Agilent 1200 HPLC was equipped with a SunFire reversed-phase C8 column (4.6 mm × 150 mm, 5 μm, Waters, Milford, MA, USA). The mobile phase was a mixture of 1‰ acetic acid in water (A) and 1‰ acetic acid in methanol (B) with gradients programmed as follows: initial 7% A maintained for 1 min, decreased to 2.5% A in 7 min. The flow rate was 1 mL/min. The column temperature was 45 °C. The injection volume was 20 μL. The temperature of the autosampler tray was set to 5 °C and the samples were protected from the daylight. The effluent from the LC column was diverted to waste for the first 1.5 min after the injection to avoid contamination with non-volatile salts and background interferences.
2.5. Ion trap mass spectrometry
The LC/MS/MS experiment was performed on a 1100 Series LC/MSD ion Trap VL with an atmospheric pressure chemical ionisation (APCI) interface and a LCMS Chemstation software from Agilent Technologies (Palo Alto, CA, USA). The ionization mode was positive. The interface and MSD were as follows: nebulizer pressure, 60 psi (N2); dry gas, 5 L/min (N2); dry gas temperature, 350 °C; spray capillary voltage, 4000 V; corona needle, 4000 nA; collision-induced dissociation, 1 V. Collision gas in the trap was helium with a pressure of 6 × 10−6 mbar.
2.6. Kinetics
Kinetic parameters (Km and Vmax) were obtained from at least two separate experiments after the addition of increasing amounts of vit K>O (0.003–0.2 mM) to the standard reaction. The estimation of the kinetic parameters was achieved by the incubation of at least nine different concentrations of vit K>O. Incubations were performed in duplicate. Data were fitted by nonlinear regression using the Michaelis–Menten model using the curve fitter R-fit program [33].
3. Results
3.1. Expression of recombinant mutant rVKORC1 proteins
To assess the functional properties of the wild type rVKORC1 and its five major spontaneous mutants (L120Q-, L128Q-, Y139C-, Y139F- and Y139S-rVKORC1) detected in wild rats resistant to AVKs in Western Europe, these proteins were overexpressed as c-myc-fused proteins in P. pastoris. All the mutated proteins were expressed in the microsomal fraction with an expected molecular mass of approximately 20 kDa. Depending on the mutation, the levels of expression of the recombinant protein in the microsomal fraction were different. Thus, we monitored the expression level of each mutated protein by western blot analysis, as described previously [21] and the level of expression was comprised between 1 and 10 (Fig. 1).
Fig. 1.
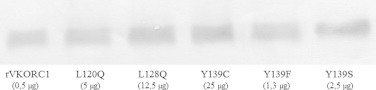
Semi-quantitative analysis of wild type or mutated VKORC1 proteins abundance in yeast microsomes after normalization of the expression of recombinant proteins in the microsomal fractions by western blot. Various amounts of microsomal proteins were loaded in order to obtain the same detected signal.
3.2. Mass spectral properties of vit K1, vit K1>O, 2-OH-vit K1 and 3-OH-vit K1
To investigate the formation of OH-vit K by the recombinant rVKORC1 proteins, we established an LC–MS/MS-based method allowing the detection of 2-OH-vit K1, 3-OH-vit K1, vit K1>O and vit K1. Therefore, the chromatographic conditions previously described by Hodroge et al. [12,21] allowing the detection of vit K1 and vit K1>O only were slightly modified. Under the new chromatographic conditions, the protonated 2-OH-vit K1 (m/z 469) and 3-OH-vit K1 (m/z 469) molecules obtained by chemical synthesis as described by [22] were detected, respectively, at 3.7 and 5.2 min (data not shown). The MS2 spectra of the 2-OH-vit K1 or 3-OH-vit K1 469 molecular ions were identical and showed mainly one fragment ion at m/z 451 (−18) attributed to the loss of H2O (Fig. 2). For all the analyses performed in this study, identification criteria for 2-OH-vit K1 were the retention time at 3.7 min and the product ion 469>451 (m/z(+) = 451); identification criteria for 3-OH-vit K1 were the retention time at 5.2 min and the product ion 469>451 (m/z(+) = 451) (Fig. 3). Under the new chromatographic conditions, the product ion from vit K1>O 467>307 (m/z) and the product ion from vit K1 451>187 (m/z) identified previously [12,21] were detected, respectively, at 7.1 and 8.2 min. For vit K1, 2-OH-vit K1 and 3-OH-vit K1, linearity and accuracy were tested from 25 to 2000 ng/mL (n = 20). The response was linear throughout the concentration range tested with a coefficient of correlation (r2) above 0.99. Accuracy was between 80% and 120% of the theoretical concentrations.
Fig. 2.
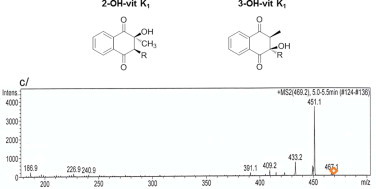
Chemical structure of 2- OH-vit K1 and 3-OH-vit K1 and MS2 spectrum of 2- and 3-OH-vit K1 (c). MS2 data were obtained from the 469,2 m/z ion as the precursor for collision-induced dissociation.
Fig. 3.
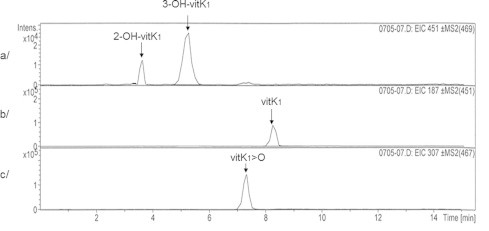
Chromatograms of 2- and 3-OH-vit K1 (a), vit K1 (b) and vit K1>O (c) chemical standards.
3.3. Production of vit K by recombinant mutant rVKORC1 proteins
Recombinant wild type rVKORC1 and its five major mutants were all able to reduce vit K1>O to vit K. The reaction rates of recombinant wild type or mutated rVKORC1 towards vit K1>O followed the Michaelis–Menten model allowing the determination of kinetic parameters (Table 2). Vmax values indicated in this table are Vmax calculated after normalization using the expression factor determined previously by immunoblot analysis. The Km values of recombinant L120Q-, L128Q-, Y139F- and Y139S-rVKORC1 were similar to that of the recombinant WT-rVKORC1. Only the Km of Y139C-rVKORC1 was significantly higher (8-fold) to that of WT-rVKORC1, whereas its Vmax value was unmodified. On the contrary, the Vmax value of recombinant L120Q-, Y139F- and Y139S-rVKORC1 were 5, 2.5 and 5.5-fold lower than that of WT- rVLORC1, respectively.
Table 2.
Catalytic properties of wild type and mutated rVKORC1. To determine the VKOR activity and the production of OH-vit K, standard reactions were performed in 200 mM Hepes buffer (pH 7.4) containing 150 mM KCl and 0.75–2 g L−1 of microsomal proteins containing membrane wild type or mutant VKORC1. Kinetic parameters (Km, Vmax and Vmax/Km) of the vit K or 3-OH-vit K production were evaluated by incubation of increasing concentrations of vit K>O (from 0 to 200 μM). The Vmax values determined at saturating concentration of vit >O substrate were evaluated after normalization of the VKORC1 expression. Each data point represents the mean ± 2SD of three individual determinations. *p < 0.02 compared to VKORC1.
| Recombinant proteins | Production of vit K |
Production of 3-OH-vit K |
||||||
|---|---|---|---|---|---|---|---|---|
| Km (μM) | Vmax (pmol/min/mg) | Vmax/Km (nL/min/mg) | Vmax/Km (%/WT) | Km (μM) | Vmax (pmol/min/mg) | Vmax/Km (nL/min/mg) | ||
| WT | 7.2 ± 2.5 | 11.6 ± 0.6 | 1611 | 100 | – | <0.08 | – | >145 |
| L120Q | 25.0 ± 4.0 | 0.9 ± 0.01* | 36 | 2.2 | – | <0.08 | – | >10 |
| L128Q | 12.1 ± 1.0 | 9.6 ± 1.0 | 793 | 49 | – | 0.13 ± 0.02 | 10.7 | 73 |
| Y139C | 60.0 ± 6.0* | 5.1 ± 0.3 | 85 | 5.3 | 57.6 ± 3.0 | 3.90 ± 0.80* | 65.0 | 1.3 |
| Y139F | 17.8 ± 4.5 | 1.9 ± 0.2* | 107 | 6.6 | 23.1 ± 4.7 | 0.63 ± 0.09* | 35.4 | 3.0 |
| Y139S | 13.1 ± 1.3 | 0.8 ± 0.1* | 61 | 3.8 | 19.5 ± 2.5 | 0.98 ± 0.09* | 74.8 | 0.8 |
3.4. Production of OH-vit K by recombinant mutant rVKORC1 proteins
Because the formation of OH-vit K from vit K>O in the presence of rat liver microsomes was previously described, we analyzed the involvement of the wild type and mutant rVKORC1 in this production. We beforehand verified that our standard incubation performed in the absence of substrate (i.e., vit K1>O) or in the presence of microsomal proteins of non-recombinant yeasts or when incubation time was 0 min did not result in a production of 2-OH-vit K1 or 3-OH-vit K1. To investigate the VKORC1-dependent production of OH-vit K, the formation of these products after incubation of 200 μM of vit K1>O was evaluated in the presence of recombinant wild type or mutant rVKORC1. When incubation was performed in the presence of WT-rVKORC1 or L120Q- or L128Q-rVKORC1, virtually no 2-OH-vit K1 or 3-OH-vit K1 was detected but vit K1 was found as described previously (Fig. 4a). When incubation of vit K1>O was performed in the presence of Y139C-, Y139F-, or Y139S-rVKORC1, two different reaction products were detected, corresponding, respectively, to 3-OH-vit K1 and vit K1 (Fig. 4b). Formation of 2-OH-vit K1 was not detected for rVKORC1 mutated in position 139. The 3-OH-vit K1 production in the presence of Y139C-, Y139F-, or Y139S-rVKORC1 was linear according to the incubation time and to the concentration of proteins (data not shown). The reaction rates of recombinant rVKORC1 mutated in position 139 for the production of 3-OH-vit K1 followed the Michaelis–Menten model allowing the determination of kinetic parameters (Table 2). The Km values of recombinant Y139C-, Y139F- and Y139S-rVKORC1 evaluated for the production of 3-OH-vit K1 were identical to those determined for the production of vit K1. The Vmax values of recombinant Y139C-, Y139F- and Y139S-rVKORC1 were, respectively 3.9, 0.6 and 1.0 pmol/min/mg (i.e., corresponding to ∼80%, 30% and 120% of the production of vit K1 in the same conditions) (Table 2). The production of 3-OH-vit K1 catalyzed by rVKORC1 mutated in position 139 was inhibited in a non-competitive manner by difenacoum, an antivitamin K molecule, with a Ki similar to that obtained for the production of vit K1 (Table 1) [12].
Fig. 4.
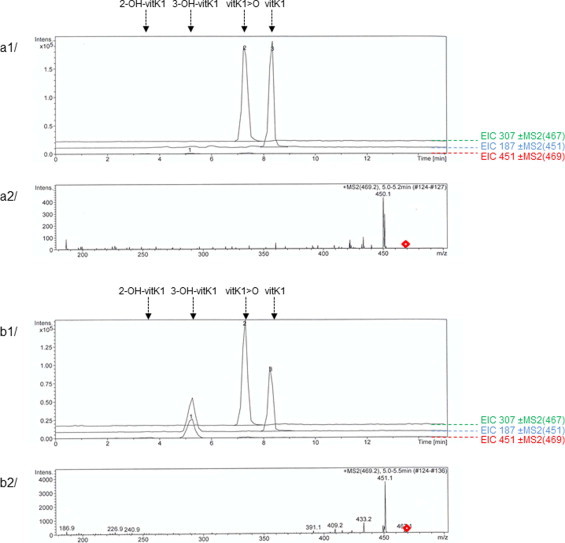
Chromatograms obtained after analysis of the incubation of vit K1>O with wild type rVKORC1 (a1) or Y139C-rVKORC1 (b1) and their respective MS2 spectrums (a2 and b2) obtained from the 469,2 m/z ion as the precursor for collision-induced dissociation. Microsomal proteins of P. pastoris yeast cells expressing the recombinant enzyme were incubated in the presence of 200 μM of vit K>O as described under Materials and Methods. Retention times for 2-OH-vit K1, 3-OH-vit K1, vit K1>O and vit K1 were, respectively, 3.5, 5.2, 7.1 and 8.2 min.
3.5. Production of vit K and OH-vit K by rat liver microsomes
In order to compare the properties of recombinant rVKORC1 proteins with those of the native VKORC1, we evaluated the production of vit K and OH-vit K in the presence of 200 μM of vit K1>O by liver microsomes from susceptible rats, from rats homozygous for Y139F and from rats homozygous for Y139C. Results obtained are presented in Fig. 5. The production of vit K1 from vit K1>O was observed for all rat liver microsomes (Fig. 5). Production of vit K1 for liver microsomes from rat homozygous for Y139C and from rat homozygous for Y139F was, respectively, ∼3- and 6-fold lower. For all the rat liver microsomes used in this study, the production of 2-OH-vit K1 from vit K1>O was never detected. Production of 3-OH-vit K1 was observed only in the presence of liver microsomes from rats homozygous for Y139C or for Y139F (Fig. 5). Production of 3-OH-vit K1 for liver microsomes from rat homozygous for Y139C and from rat homozygous for Y139F corresponded, respectively, to ∼ 50% and 28% of the production of vit K1 in the same conditions.
Fig. 5.
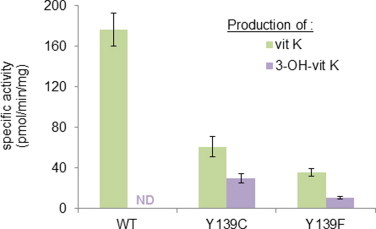
Production of vit K and 3-OH-vit K by liver microsomes from susceptible rats, from rats homozygous for Y139F and from rats homozygous for Y139C incubated in the presence of 200 μM of vit K1>O.
4. Discussion
In 1983, Fasco et al. [22] observed an in vitro warfarin-dependent production of OH-vit K in rat liver microsomes incubated in the presence of vit K>O. This production was only observed with warfarin-resistant rat liver microsomes, not with those from normal rats. They suggested that this warfarin-dependent production is catalyzed by VKOR. Nevertheless, they failed to demonstrate in vivo the involvement of VKOR activity in this production [23] and they were not able to explain the association between this production and the resistance to AVK, especially since the cause of the resistance of the rats used in this study was at that time unknown [22,23]. Therefore we expressed herein the wild type rVKORC1 and its five major spontaneous mutants associated to the resistance to AVK in Rattus norvegicus [1,2] and determined the functional consequences of these mutations on the recycling of vit K. We considered in this study the production of vit K catalyzed by VKORC1, but also the production of the metabolically inactive 2- or 3-OH-vit K.
In this study, we demonstrate the role of VKORC1 in the synthesis of OH-vit K. When standard incubations of vit K>O was performed in the presence of liver microsomes of rat homozygous for Y139F or recombinant VKORC1 enzymes mutated in position 139 (i.e., Y139C-, Y139F- and Y139S-rVKORC1), a second metabolite, in addition to vit K, was detected. The identification criteria of this second metabolite coincided totally with those of the 3-OH-vit K chemical standard. No OH-vit K formation was observed in incubations using liver microsomes from susceptible rats as previously described by Fasco et al. [22], and also in incubations using recombinant wild type-rVKORC1 and recombinant L120Q- and L128Q-rVKORC1. Because wild-type-, L120Q- and L128Q-rVKORC1 do not catalyze the production of OH-vit K from vit K>O, it appears that it is the absence of the Tyr-139 residue which leads to the production of 3-OH-vit K, in addition to the production of vit K.
To further explore the mechanism of the production of 3-OH-vit K, we determined the kinetic constants characterizing the production of 3-OH-vit K comparatively to those obtained for the production of vit K. For all the recombinant rVKORC1 mutated in position 139, Km obtained for the production of 3-OH-vit K is similar to that obtained for the parallel production of vit K. Similarity of Km between 3-OH-vit K and vit K productions suggests that the binding site of vit K>O is similar whatever is the reaction product formed by VKORC1. Another argument is the fact that the production of 3-OH-vit K is inhibited by AVK (i.e., difenacoum) in a non-competitive manner similarly to the production of vit K with the same inhibition constant [9,12]. Therefore, it is more likely that the production of 3-OH-vit K is due to a modification of the reaction mechanism than to a non-enzymatic conversion of the free enolate as proposed by Fasco et al. [22]. A theoretical reaction mechanism for converting vit K>O to vit K by VKOR was proposed in 1981 by Silverman [24] based on a chemical model study. This mechanism was revisited later by a quantum chemical study [25]. The theoretical model implied an active site composed of two Cys (i.e., Cys 132 and 135 of the CXXC motif) and can be decomposed into two half-steps (Fig. 6). The first half-step corresponds to the formation of a covalent bond between one of the catalytic Cys132 or 135 and the C2 of vit K>O. The second half-step allows the dehydration of the substrate and the liberation of the reaction product. It implies the intervention of the second Cys and leads to the formation of a disulfide bridge between the two Cys. The production of 3-OH-vit K by VKORC1 is of particular interest because it demonstrates the crucial role of the Tyr-139 amino acid in this reaction mechanism. Tyr-139 residue appears to determine the second half-step of the catalytic mechanism (Fig. 6). Indeed, in the absence of Tyr-139, the second half-step is clearly modified in favour of the formation of 3-OH-vit K. The phenolic alcool of Tyr-139 could be involved in the protonation of the hydroxyl group linked to the C3 of the intermediate produced by the first half step of the catalysis (Fig. 6). In the absence of Tyr-139, another proton, possibly from the medium, should be used in order to produce the vit K but, the formation of 3-OH-vit K, which does not involve such a protonation, is favoured. As a consequence the exchange of proton in which the Tyr-139 is involved is slowed down when Tyr-139 residue is mutated and the production of vit K by the mutated VKORC1 is decreased.
Fig. 6.
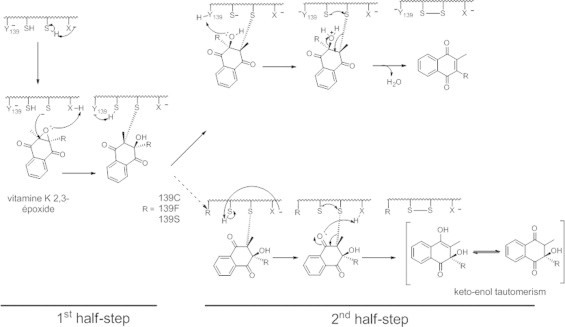
Mechanism for OH-vit K formation by Y139C, Y139F or Y139S.
Because of this additional pathway for vit K>O, the vitamin K status is thus determined by the ability of VKORC1 to reduce vit K>O to vit K, by the abnormal ability of VKORC1 to produce OH-vit K leading to a leak of vit K from its own cycle, and finally by the food input (Fig. 7). It is thus conceivable that a decrease in the VKOR catalytic efficiency (as observed for L120Q, Y139C/F/C) associated or not with an elevated 3-OH-vit K production (as observed for mutations in position 139) might cause an increase in dietary requirements of vit K. Indeed, severe increase in dietary requirements was described in resistant rats from Denmark and Germany (i.e., certainly homozygous for Y139C) [16,19] and from Wales (i.e., certainly homozygous for Y139S) [26]. On the contrary, vit K requirements in resistant rats from Scotland (i.e., homozygous for L128Q) [27] are described to be similar to vit K requirements of susceptible rats. All these observations are totally in agreement with the catalytic results obtained in this study. Furthermore, in rat homozygous for Y139C, retarded growth [16,17] and cardiovascular calcifications [19] were reported. Both are to be considered as the consequence of a deficiency in vit K. Indeed, Matrix Gla protein (MGP), a VKDP which is considered as an inhibitor of the tissue calcification, is effective under its gamma-carboxylated form only [28,29]. When gamma-carboxylation reaction is stopped due to the decrease in the efficiency of the vitamin K cycle (as it is demonstrated for Y139C), MGP is probably undercarboxylated. The presence of undercarboxylated MGP might explain the cardiovascular phenotype observed in Y139C rats [20] and the retarded growth [16,17] associated probably to growth plate closure with excessive mineralization as described for rats treated with warfarin [30]. The absence of such clinical signs in rats homozygous for L120Q argues for a major involvement of the loss of vit K in the vitamin K cycle by the formation of 3-OH-vit K and a minor involvement of the low VKOR activity in the biological cost. Therefore, resistance to AVKs would not be systematically associated with a biological cost. Only the resistance due to a mutation of the Tyr-139 residue might be associated with a biological cost. Such a mutation was described in one human patient requiring a high dose of a vit K antagonist [31]. In this patient, the Tyr-139 residue is replaced by a His residue [31]. This mutation was demonstrated to lead to resistance to AVK without modifying the catalytic efficiency to produce vit K [21]. On the other hand the consequences of this mutation on the production of OH-vit K were not analyzed. It would be interesting to evaluate the OH-vit K formation in patients carriers for this mutation and the clinical consequences associated with such a mutation.
Fig. 7.
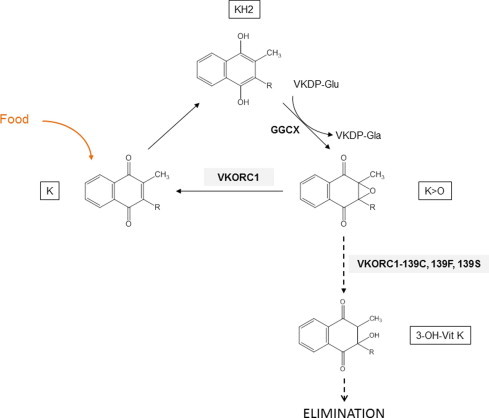
The vitamin K cycle and the specific loss of vit K by the formation of 3-OH-vit K catalyzed by Y139C, Y139F or Y139S.
Acknowledgments
This work was supported by Grants from Agence Nationale pour la Recherche (RODENT 2009-CESA-008–03) and by DGER.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Pelz H.J., Rost S., Hünerberg M., Fregin A., Heiberg A.C., Baert K., MacNicoll A.D., Prescott C.V., Walker A.S., Oldenburg J., Müller C.R. The genetic basis of resistance to anticoagulants in rodents. Genetics. 2005;170:1839–1847. doi: 10.1534/genetics.104.040360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rost S., Pelz H.J., Menzel S., MacNicoll A.D., León V., Song K.J., Jäkel T., Oldenburg J., Müller C.R. Novel mutations in the VKORC1 gene of wild rats and mice – a response to 50 years of selection pressure by warfarin? BMC Genet. 2009;6:10–14. doi: 10.1186/1471-2156-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sugano S., Kobayashi T., Tanikawa T., Kawakami Y., Kojima H., Nakamura K., Uchida A., Morishima N., Tamai Y. Suppression of CYP3A2 mRNA expression in the warfarin-resistant roof rat, Rattus Rattus: possible involvement of cytochrome P450 in the warfarin-resistance mechanism. Xenobiotica. 2001;76:5960–5964. doi: 10.1080/00498250110060932. [DOI] [PubMed] [Google Scholar]
- 4.Ishizuka M., Okajima F., Tanikawa T., Min H., Tanaka K.D., Sakamoto K.Q., Fujita S. Elevated warfarin metabolism in warfarin-resistant roof rats (Rattus rattus) in Tokyo. Drug Metab. Dispos. 2007;35:62–66. doi: 10.1124/dmd.106.011775. [DOI] [PubMed] [Google Scholar]
- 5.Heiberg A.C. Anticoagulant resistance: a relevant issue in sewer rat (Rattus norvegicus) control? Pest Manage. Sci. 2009;65:444–449. doi: 10.1002/ps.1709. [DOI] [PubMed] [Google Scholar]
- 6.Rost S., Fregin A., Ivaskevicius V., Conzelmann E., Hörtnagel K., Pelz H.J., Lappegard K., Seifried E., Scharrer I., Tuddenham E.G., Müller C.R., Strom T.M., Oldenburg J. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature. 2004;427:537–541. doi: 10.1038/nature02214. [DOI] [PubMed] [Google Scholar]
- 7.Li T., Chang C.Y., Jin D.Y., Lin P.J., Khvorova A., Stafford D.W. Identification of the gene for vitamin K epoxide reductase. Nature. 2004;427:541–544. doi: 10.1038/nature02254. [DOI] [PubMed] [Google Scholar]
- 8.Oldenburg J., Marinova M., Müller-Reible C., Watzka M. The vitamin K cycle. Vitam. Horm. 2008;78:35–62. doi: 10.1016/S0083-6729(07)00003-9. [DOI] [PubMed] [Google Scholar]
- 9.Lasseur R., Longin-Sauvageon C., Videmann B., Billeret M., Berny P., Benoit E. Warfarin resistance in a French strain of rats. J. Biochem. Mol. Toxicol. 2005;19:379–385. doi: 10.1002/jbt.20104. [DOI] [PubMed] [Google Scholar]
- 10.Lasseur R., Grandemange A., Longin-Sauvageon C., Berny P., Benoit E. Comparison of the inhibition effect of different anticoagulants on vitamin K epoxide reductase activity from warfarin-susceptible and resistant rat. Pest Biochem. Physiol. 2007;88:203–208. [Google Scholar]
- 11.Grandemange A., Kohn M.H., Lasseur R., Longin-Sauvageon C., Berny P., Benoit E. Consequences of the Y139F VKORC1 mutation on resistance to AVKs:in-vivo investigation in the 7th generation congenic Y139F strain. Pharmacogenet. Genomics. 2009;19:742–750. doi: 10.1097/FPC.0b013e32832ee55b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hodroge A., Longin-Sauvageon C., Fourel I., Benoit E., Lattard V. Biochemical characterization of spontaneous mutants of rat VKORC1 involved in the resistance to antivitamin K anticoagulants. Arch. Biochem. Biophys. 2011;515:14–20. doi: 10.1016/j.abb.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 13.Hermodson M.A., Sutties J.W., Link K.P. Warfarin metabolism and vitamin K requirement in the warfarin-resistant rat. Am. J. Physiol. 1969;217:1316–1319. doi: 10.1152/ajplegacy.1969.217.5.1316. [DOI] [PubMed] [Google Scholar]
- 14.Martin A.D. Vitamin K requirement and anticoagulant response in the warfarin-resistant rat. Biochem. Soc. Trans. 1973;1:1206–1208. [Google Scholar]
- 15.Greaves J.H., Ayres P.B. Warfarin resistance and vitamin K requirement in the rat. Lab. Animals. 1973;7:141–148. doi: 10.1258/002367773781008614. [DOI] [PubMed] [Google Scholar]
- 16.Markussen M.D.K., Heiberg A.C., Nielsen R., Leirs H. Vitamin K requirement in Danish anticoagulant-resistant Norway rats (Rattus norvegicus) Pest Manage. Sci. 2003;59:913–920. doi: 10.1002/ps.703. [DOI] [PubMed] [Google Scholar]
- 17.Smith P., Townsend G., Smith R.H. A cost of resistance in the brown rat? Reduced growth rate in warfarin-resistant lines. Funct. Ecol. 1991;5:441–447. [Google Scholar]
- 18.Heiberg A.C., Leirs H., Siegismund H.R. Reproductive success of bromadiolone-resistant rats in absence of anticoagulant pressure. Pest Manage. Sci. 2006;62:862–871. doi: 10.1002/ps.1249. [DOI] [PubMed] [Google Scholar]
- 19.Jacob J., Endepols S., Pelz H.J., Kampling E., Cooper T.G., Yeung C.H., Redmann K., Schlatt S. Vitamin K requirement and reproduction in bromadiolone-resistant Norway rats. Pest Manage. Sci. 2011;68:378–385. doi: 10.1002/ps.2273. [DOI] [PubMed] [Google Scholar]
- 20.Kohn M., Price R.E., Pelz H.J. A cardiovascular phenotype in warfarin-resistant VKORC1 mutant rats. Artery Res. 2008;2:138–147. doi: 10.1016/j.artres.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hodroge A., Matagrin B., Moreau C., Fourel I., Hammed A., Benoit E., Lattard V. VKORC1 mutations detected in patients resistant to vitamin K antagonists are not all associated with a resistant VKOR activity. J. Thromb. Haemost. 2012;10:2535–2543. doi: 10.1111/jth.12019. [DOI] [PubMed] [Google Scholar]
- 22.Fasco M.J., Preusch P.C., Hildebrandt E., Suttie J.W. Formation of hydroxyvitamin K by vitamin K epoxide reductase of warfarin-resistant rats. J. Biol. Chem. 1983;258:4372–4380. [PubMed] [Google Scholar]
- 23.Preusch P.C., Suttie J.W. Formation of 3-hydroxy-2,3-dihydrovitamin K1 in vivo: relationship to vitamin K epoxide reductase and warfarin resistance. J. Nutr. 1984;114:902–910. doi: 10.1093/jn/114.5.902. [DOI] [PubMed] [Google Scholar]
- 24.Silverman R.B. Chemical model studies for the mechanism of vitamin K epoxide reductase. J. Am. Chem. Soc. 1981;103:5939–5941. [Google Scholar]
- 25.Davis C.H., Deerfield D., 2nd, Wymore T., Stafford D.W., Pedersen L.G. A quantum chemical study of the mechanism of action of vitamin K epoxide reductase (VKOR): transition states. J. Mol. Graph. Model. 2007;26:401–408. doi: 10.1016/j.jmgm.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 26.Greaves J.H., Ayres P.B. Linkages between genes for coat colour and resistance to warfarin in Rattus Norvegicus. Nature. 1969;224:284–285. doi: 10.1038/224284a0. [DOI] [PubMed] [Google Scholar]
- 27.MacNicoll A.D., Dennis N.J., Atterby H., Hudson A., Langton S., Kerins G.S. Fitness effects of different alleles of the warfarin resistance gene-implications for resistance management. In: Pelz H.J., Cowan D.P., Feare C.J., editors. Advances in Vertebrate Pest Management II. Filander Verlag; Furth, Germany: 2001. pp. 171–180. [Google Scholar]
- 28.Sweatt A., Sane D.C., Hutson S.M., Wallin R. Matrix Gla protein (MGP) and bone morphogenetic protein-2 in aortic calcified lesions of aging rats. J. Thromb. Haemost. 2003;1:178–185. doi: 10.1046/j.1538-7836.2003.00023.x. [DOI] [PubMed] [Google Scholar]
- 29.Schurgers L.J., Teunissen K.J., Knapen M.H., Kwaijtaal M., Van Diest R., Appels A., Reutelingsperger C.P., Cleutjens J.P., Vermeer C. Novel conformation-specific antibodies against matrix gamma-carboxyglutamic acid (Gla) protein: undercarboxylated matrix Gla protein as marker for vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2005;25:1629–1633. doi: 10.1161/01.ATV.0000173313.46222.43. [DOI] [PubMed] [Google Scholar]
- 30.Price P.A., Williamson M.K., Haba T., Dell R.B., Jee W.S.S. Excessive mineralization with growth plate closure in rats on chronic warfarin treatment. Proc. Natl. Acad. Sci. USA. 1982;79:7734–7738. doi: 10.1073/pnas.79.24.7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watzka M., Geisen C., Bevans C.G., Sittinger K., Spohn G., Rost S., Seifried E., Müller C.R., Oldenburg J. Thirteen novel VKORC1 mutations associated with oral anticoagulant resistance: insights into improved patient diagnosis and treatment. J. Thromb. Haemost. 2011;9:109–118. doi: 10.1111/j.1538-7836.2010.04095.x. [DOI] [PubMed] [Google Scholar]
- 32.Tishler M., Fieser L., Wendler N. Hydro, oxydo and other derivatives of vitamin K1 related compounds. J. Am. Chem. Soc. 1940;62:2866–2871. [Google Scholar]
- 33.Ihaka R., Gentleman R. A language for data analysis and graphics. J. Comput. Graph. Stat. 1996;5:299–314. [Google Scholar]