

Abstract
A water-soluble selenoxide (DHSox) having a five-membered ring structure enables rapid and selective conversion of cysteinyl SH groups in a polypeptide chain into SS bonds in a wide pH and temperature range. It was previously demonstrated that the second-order rate constants for the SS formation with DHSox would be proportional to the number of the free SH groups present in the substrate if there is no steric congestion around the SH groups. In the present study, kinetics of the SS formation with DHSox was extensively studied at pH 4–10 and 25 °C by using reduced ribonuclease A, recombinant hirudin variant (CX-397), insulin A- and B-chains, and relaxin A-chain, which have two to eight cysteine residues, as polythiol substrates. The obtained rate constants showed stochastic SS formation behaviors under most conditions. However, the rate constants for CX-397 at pH 8.0 and 10.0 were not proportional to the number of the free SH groups, suggesting that the SS intermediate ensembles possess densely packed structures under weakly basic conditions. The high two-electron redox potential of DHSox (375 mV at 25 °C) compared to l-cystine supported the high ability of DHSox for SS formation in a polypeptide chain. Interestingly, the rate constants of the SS formation jumped up at a pH around the pKa value of the cysteinyl SH groups. The SS formation velocity was slightly decreased by addition of a denaturant due probably to the interaction between the denaturant and the peptide. The stochastic behaviors as well as the absolute values of the second-order rate constants in comparison to dithiothreitol (DTTred) are useful to probe the chemical reactivity and conformation, hence the folding, of polypeptide chains.
Keywords: Disulfide, Oxidative folding, Redox potential, trans-3, 4-Dihydroxyselenolane oxide
Abbreviations: 1S, 2S, 3S, and 4S, ensembles of SS intermediates with one, two, three, and four SS bonds, respectively; 1S°, 2S°, and 3S°, ensembles of SS intermediates of CX-397 with one, two, and three kinetically formed SS bonds, respectively; AEMTS, 2-aminoethyl methanethiosulfonate; CD, circular dichroism; CX-397, recombinant hirudin variant CX-397; DHSox, trans-3,4-dihydroxyselenolane oxide; DHSred, reduced DHSox; DTTox, oxidized dithiothreitol; DTTred, dl-dithiothreitol; ESI, electron spray ionization; Gdn-HCl, guanidine hydrochloride; GSSG, oxidized glutathione; HPLC, high-performance liquid chromatography; HV-1, recombinant hirudin variant-1; HV-3, recombinant hirudin variant-3; Ins-A, insulin A-chain; Ins-B, insulin B-chain; N, native protein; NHE, normal hydrogen electrode; pI, isoelectric point; R, reduced polypeptide; R°, reduced CX-397 at acidic conditions; Rlx-A, relaxin A-chain; RNase A, ribonuclease A; S−, thiolate; SeSe, diselenide; SH, thiol; SS, disulfide; TFA, trifluoroacetic acid; Tris, tris(hydroxymethyl)aminomethane.
Graphical abstract
Highlights
▸ DHSox was applied as a SS-forming reagent for five polythiol peptides and DTTred. ▸ The SS formation velocities depend on the kind of substrate and solvent conditions. ▸ The SS-formation rate constants are proportional to the number of free SH groups. ▸ Folded structures and SH pKa modify the stochastic nature and absolute values of the rate constants. ▸ DHSox is a useful probe of chemical reactivity and intermediate structures in oxidative protein folding.
1. Introduction
A small globular protein, which has several disulfide (SS) linkages in the native state (N), spontaneously folds into the functional structure from the denatured and reduced form (R) through SS formation (i.e., oxidation) and thiol (SH)/SS exchange (i.e., SS reshuffling and/or SS rearrangement) reactions [1,2]. By chemically trapping the transient SS intermediates, we can elucidate the folding processes in principle. In such oxidative folding study in vitro, disulfide reagents, such as trans-4,5-dihydroxy-1,2-dithiane (DTTox) and oxidized glutathione (GSSG), are generally used as a SS-forming reagent, which gradually produces native SS bonds in a polypeptide chain via inter- and intramolecular SH/SS exchange reactions under neutral to weakly basic conditions (ca. pH 7–9) [3–9]. However, under these typical folding conditions, it is not easy to observe the key folding intermediates on the oxidative folding pathways because SS formation (i.e., intermolecular SH/SS exchange) and SS rearrangement (i.e., intramolecular SH/SS exchange) reactions proceed cooperatively.
In contrast, we recently demonstrated that the SS formation process can be temporarily separated from the SS rearrangement process by using a water-soluble cyclic selenoxide, i.e., trans-3,4-dihydroxyselenolane oxide (DHSox), a highly strong and selective oxidant to the cysteinyl SH groups of polypeptide chains [10]. When DHSox was applied to the oxidative folding of bovine pancreatic ribonuclease A (RNase A), which has four SS linkages (C26–C84, C40–C95, C65–C72, C58–C110) in the native state, rapid (within 1 min), quantitative (or stoichiometric), and irreversible intramolecular SS formation progressed at pH 8.0 and 5–45 °C [11]. After completion of the SS formation, the SS rearrangement reaction took place in the partially oxidized SS intermediates. In addition, the second-order rate constants for the SS formation reaction were found to be proportional to the number of the free SH groups present in the SS intermediates at 25 °C [10,12], suggesting that the early folding intermediates of RNase A do not have rigid structures. Similar stochastic SS formation with DHSox was also observed at pH 7.0 in the oxidative folding of a recombinant hirudin variant (CX-397) having three SS linkages (C6–C14, C16–C28, C22–C39) in the native state [13].
The reaction mechanism of SS formation in a polypeptide chain using DHSox [13] is illustrated in Fig. 1. The reaction proceeds in two steps: the first step [step 1], which corresponds to the attack of the cysteinyl SH group to the selenoxide to form a transient unstable intermediate having a Se–S linkage, and the second step (step 2), which corresponds to the intramolecular rearrangement of the thioselenurane intermediate to produce a SS linkage with elimination of reduced DHS (DHSred) and a water molecule. The stochastic behavior of the SS formation suggested that the rate-determining step is step 1 [13]. Thus, as far as a polypeptide chain is in a random-coil state, the reaction velocity for the SS formation would simply depend on the probability of the molecular contact between DHSox and the SH groups in a polypeptide chain. Formation of a thioselenurane intermediate has recently been characterized by May and coworkers [14] in the SS formation reaction of GSH with phenylaminoalkyl selenoxide.
Fig. 1.
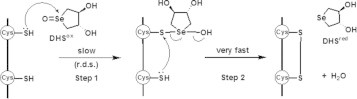
The reaction mechanism of SS bond formation in a polypeptide chain using DHSox as an oxidant.
In the present study, to obtain further insights on the phenomenon of the stochastic SS formation using DHSox, kinetics of the SS formation reaction was extensively studied at pH 4–10 and 25 °C for five model polypeptides: RNase A, CX-397, insulin A-chain (Ins-A), relaxin A-chain (Rlx-A), and insulin B-chain (Ins-B), which have eight, six, four, four, and two cysteine (Cys) residues, respectively. Primary sequences of the model peptides are shown in Fig. 2. RNase A [15], one of benchmark proteins in folding study, is a typical monomeric and basic protein (ca. pI = 9.3) consisting of 124 amino acid residues. CX-397 [16], which has a hybrid amino acid sequence of hirudin variant-1 (HV-1) and -3 (HV-3) with three SS linkages, is a small acidic protein (ca. pI = 4.1) of 66 amino acid residues. Ins-A (theoretical pI = 4.0) and Ins-B (theoretical pI = 6.9), the peptides constituting bovine pancreatic insulin [17], are 21- and 30-amino acid peptides containing four (C6, C7, C11, C20) and two (C7, C19) Cys residues, respectively. Relaxin (human gene 2 relaxin) [18] is a small peptide hormone that is composed of two peptides, Rlx-A and -B chains. This peptide hormone is a member of the insulin superfamily and plays a key role in remodeling connective tissue during parturition. Rlx-A (theoretical pI = 9.3) consists of 24 amino acid residues with four (C10, C11, C15, C24) Cys residues. Reduced RNase A, CX-397, Ins-A, Rlx-A, and Ins-B with no SS bond were allowed to react with controlled amounts of DHSox to form a maximum of four, three, two, two, and one SS bonds, respectively. From the kinetic analysis of the SS formation, it was revealed that the SS formation does not always follow the same kinetic behaviors, indicating that the velocity of SS formation is influenced by several factors, such as the structure formed in a polypeptide chain, the pKa of the cysteinyl SH groups, and a denaturant added in the reaction solution. Based on the data obtained, advantageous features of DHSox as a probe for the structure and reactivity of the folding intermediates of SS-containing proteins are discussed.
Fig. 2.

Primary amino acid sequences of RNase A, CX-397, Ins-A, Rlx-A, and Ins-B.
2. Materials and methods
2.1. Materials
Bovine pancreatic RNase A and insulin were purchased from Sigma Aldrich, Japan and used without purification. A recombinant hirudin variant (CX-397) [16] was provided from Pharmaceuticals and Biotechnology Laboratory, Japan Energy Co., Ltd. and was used without further purification. Relaxin A-chain (Rlx-A) was obtained by solid-phase peptide synthesis by application of the usual Fmoc protocol. DHSox [19] and AEMTS [20] were synthesized according to the literature methods. All other reagents were commercially available and used without further purification.
2.2. Preparation of reduced polypeptides
Reduced RNase A and CX-397 were prepared by following the procedure previously described [12,13].
Reduced Ins-A and Ins-B were obtained as follows. To a solution of insulin (3–4 mg) dissolved in 500 μL of a 100 mM Tris–HCl/1 mM EDTA buffer solution at pH 8.5 containing 4 M Gdn-SCN as a denaturant was added an excess amount of DTTred (7–8 mg). The reaction mixture was incubated at 28 °C for 60 min. The resulting solution containing reduced chains of insulin was desalted to 0.1 M aqueous ammonia purged with nitrogen by passing through the column packed with Sephadex G15 resin. The desalted peptide solution (7 mL) was acidified to pH 3 with 100 μL of acetic acid and lyophilized. The resulting white solid materials were dissolved in 2.5 mL of 0.1% aqueous trifluoroacetic acid (TFA) solution containing 20% acetonitrile. Insoluble precipitates were settled out by centrifugation (14,000 rpm) for 10 min. Reduced Ins-A and Ins-B were separated by HPLC equipped with a Tosoh TSKgel ODS-100V 4.6 × 150 mm reverse phase column using a 77:23 (v/v) mixture of 0.1% TFA in water (eluent A) and 0.1% TFA in acetonitrile (eluent B) at a flow rate of 1.0 mL/min. A solvent gradient (a ratio of eluent B linearly increased from 23% to 35% in 0–15 min, from 35% to 37% in 15–20 min, from 37% to 40% in 20–23 min, and from 40% to 100% in 23–24 min) was applied. The separated peptide fractions detected by UV absorption at 280 nm were collected and lyophilized. The resulting white powder materials of Ins-A and Ins-B (purity > 95% estimated by HPLC analysis) were stored at −30 °C.
Reduced Rlx-A was prepared as follows. The Rlx-A obtained by solid-pahse peptide synthesis was dissolved in 500 μL of a 100 mM Tris–HCl/1 mM EDTA buffer solution at pH 8.5 containing 8 M urea as a denaturant and was treated with DTTred (7–8 mg) at 28 °C for 60 min. After insoluble precipitates were settled out by centrifugation (14,000 rpm) for 10 min, the resulting solution containing a reduced Rlx-A was desalted to 0.1 M acetic acid through the column packed with Sephadex G15 resin. The desalted peptide solution (7–8 mL) was then lyophilized. The obtained white solid materials were dissolved in 2.5 mL of 0.1% aqueous TFA containing 20% acetonitrile. Insoluble precipitates were settled out by centrifugation (14,000 rpm) for 10 min. Rlx-A was isolated by HPLC equipped with a Tosoh TSKgel ODS-100V 4.6 × 150 mm reverse phase column using a 80:20 (v/v) mixture of 0.1% TFA in water (eluent A) and 0.1% TFA in acetonitrile (eluent B) at a flow rate of 0.7 mL/min. A solvent gradient (a ratio of eluent B linearly increased from 20% to 36% in 0–15 min, from 36% to 39% in 15–20 min, from 39% to 40% in 20–23 min, and from 40% to 100% in 23–24 min) was applied. The fractionated reduced Rlx-A detected by UV absorption at 275 nm was collected and lyophilized. The obtained white powder material of Rlx-A (purity > 90% estimated by HPLC analysis) was stored at −30 °C.
To prepare solutions of reduced Ins-A, Ins-B, and Rlx-A at pH 4.0–10.0, 0.5–1.0 mg of each chain in the reduce form (R) was dissolved in 0.5–1.0 mL of a variable pH buffer solution containing 6 M urea. The stock solution was diluted threefold with the same pH buffer solution without urea. The concentration of R was determined by UV absorbance at 275 nm using the molar extinction coefficients at 275 nm (ɛ275) determined by Ellman's assay [21]; 3100, 3040 (2960 at pH 10.0), and 1650 M−1 cm−1 for reduced Ins-A, Ins-B, and Rlx-A, respectively. The R solution was immediately used in the short-term oxidation experiments described below.
2.3. Short-term oxidation experiment
A quench-flow instrument (a Unisoku quench-mixer equipped with a mixer controller unit) [10] was set up by loading the R solution, a DHSox solution in the corresponding buffer solutions at pH 4.0–10.0 in the presence or absence of a denaturant, an aqueous solution of AEMTS (a SH-blocking reagent) (7–8 mg/mL), and a drive solution (the same buffer solution) in vessels A, B, C, and D, respectively. 200 mM acetate buffer solution at pH 4.0, 100 mM Tris–HCl buffer solution at pH 7.0 and 8.0, and 25 mM NaHCO3 buffer solution at pH 10.0 were employed. The vessels were maintained at 25.0 ± 0.1 °C by a thermostated circulating water-bath system. Oxidation reaction of R was initiated by rapidly mixing 50 μL each of R and DHSox solutions. After a certain period of time, the oxidation reaction was quenched by the reaction with 109 μL of the AEMTS solution. The reaction time was varied from 100 ms to 60 s. Completion of the AEMTS blocking was confirmed at pH 4.0–10.0 based on the kinetic analysis as reported previously [12]. The sample solutions obtained for RNase A and CX-397 were acidified at pH 3–4 by addition of 8–15 μL of 2 M acetic acid, while the sample solutions obtained for Ins-A Ins-B, and Rlx-A were diluted with 830 μL of 0.1% aqueous TFA. These sample solutions were stored at −30 °C until the samples were analyzed by HPLC (vide infra). The dead mixing or quenching time was less than 300 ms [10].
2.4. Oxidation of DTTred with DHSox
To determine the second-order rate constant for the oxidation of DTTred with DHSox, the UV absorbance change was detected by using a single-mixing stopped-flow instrument (BioLogic SFM-20) equipped with a deuterium lamp as a light source. The flow cell has a path length of 10 mm. The measurement was carried out at 25 °C and pH 4.0–10.0 by changing the relative concentration of DTTred. The absorption change due to generation of DTTox was recorded at 310 nm by using a monochromator. The reaction was initiated by mixing 70 μL of DHSox (0.2 mM) and 70 μL of DTTred (0.5–8.0 mM), and monitored for 1.0–20 s. The obtained data were fitted to a single exponential curve, and the obtained reaction velocities were plotted against the concentration of DTTred to determine the second-order rate constant.
2.5. HPLC analysis
Sample solutions of RNase A and CX-397 from the short-term experiments were analyzed by HPLC using a cation-exchange column according to the literature [12,13]. On the other hand, sample solutions of Ins-A, Ins-B, and Rlx-A were directly injected to HPLC equipped with a 1 mL sample solution loop and a Tosoh TSKgel ODS-100V 4.6 × 150 reverse phase column. A different solvent gradient was applied for each peptide chain. For Ins-A, eluent A was 0.1% TFA in water, eluent B was 0.1% TFA in acetonitrile, and a ratio of eluent B was linearly increased from 22% to 28% in 0–15 min, from 28% to 40% in 15–20 min, and kept constant in 20–23 min at a flow rate of 1.0 mL/min. For Ins-B, the same eluents were employed, and a ratio of eluent B linearly increased from 20% to 36% in 0–15 min, from 36% to 39% in 15–20 min, and from 39% to 40% in 20–23 min at a flow rate of 1.0 mL/min. For Rlx-A, eluent A was 0.1% TFA in water, eluent B was 0.1% TFA in 60% acetonitrile, and a ratio of eluent B was linearly increased from 33.3% to 38.3% in 0–15 min, from 38.3% to 41% in 15–20 min, and kept constant in 20–23 min at a flow rate of 0.7 mL/min. The fractionated folding intermediates were detected by UV absorption at 280 (Ins-A and -B) or 275 (Rlx-A) nm. The recorded signals were integrated and analyzed.
2.6. Characterization of disulfide intermediates
The disulfide intermediates fractionated by HPLC were collected, desalted to 0.1 M acetic acid, and lyophilized. The molecular mass of each separated intermediate was measured on a Jeol JMS-T100LP mass spectrometer operated in the ESI(+) mode. Since modification of the folding intermediates with AEMTS caused an increment of the molecular mass by 76 Da per a free SH group, the number of the SS bonds for each fractionated intermediate could be determined based on the observed mass number.
2.7. Enzymatic digestion of 3S intermediates of CX-397
To investigate difference in SS-bond components of the intermediate ensembles generated at pH 4.0 and 8.0, the enzymatic digestion of 3S intermediates ensemble of CX-397 was carried out by following the literature [22]. The desalted and lyophilized 3S intermediates (0.3 mg/mL), which were obtained by the reaction of R with five equivalents of DHSox, were digested at 25 °C with thermolysin (30 μg/mL) for 16 h in 50 mM N-ethylmorpholine/acetate buffer at pH 6.4. The concentrations of 3S intermediates and thermolysin were 0.3 mg/mL and 30 μg/mL, respectively in the reaction solution. The sample solution was acidified to pH 2–3 with 15% aqueous TFA, and 10 μL of the resulting solution containing the digested peptide fragments was directly injected to HPLC equipped with a 20 μL sample solution loop and a Tosoh TSKgel ODS-100V 4.6 × 150 reverse phase column using 0.1% formic acid in water (eluent A) at a flow rate of 0.8 mL/min. The concentration gradient (a ratio of eluent B (0.1% formic acid in acetonitrile) linearly increased from 0% to 10% in 0–30 min, from 10% to 40% in 30–55 min, and from 40% to 50% in 55–58 min) was applied.
2.8. Potentiometric titration
The two-electron reduction potential (E°) for DHSox was determined by the potentiometric titration method described in the literature [23,24] with sodium dithionite (Na2S2O4) as a reducing titrant. Measurement was performed in a two-electrode cell system with a single compartment containing a platinum spiral wire indicator electrode and an Ag/AgCl electrode connected to a millivolt meter (Toa HM-70V). 10 mM of sodium dithionite was titrated into the cell containing 35 mL of 1 mM DHSox in 100 mM sodium acetate buffer solution at pH 5.5, with a 30 min equilibration period between each additional aliquot. The obtained equivalent point was corrected to the redox potential of DHSox with respect to the normal hydrogen electrode (NHE).
2.9. Measurement of CD and fluorescence spectra
To characterize structures of the SS intermediates, the fluorescence spectrum was measured on a Jasco FP-6200 spectrofluorometer by using a quartz cell (light path length = 10 mm) under the conditions of 275 nm for the excitation wavelength, 280–400 nm for the emission wavelength range, 60 nm/min for the scan speed, 1 nm for the band width, 1 s for the response, and 5 nm for the slit widths of both the excitation and emission. The circular dichroism (CD) spectrum was recorded on a Jasco J-820 spectropolarimeter by using a quartz cell (light path length = 1 mm) under the conditions of 245–195 nm for the wavelength range, 50 nm/min for the scan speed, 1 nm for the band width, and 1 s for the response. Sample solutions for the spectral analyses were prepared by using a 10-fold diluted buffer solution with respect to that used for the short-term oxidation experiments.
3. Results
3.1. Short-term oxidation of reduced polythiol peptide chains with DHSox
Short-term oxidation experiments of RNase A at pH 4.0–10.0 and CX-397 at pH 7.0 using DHSox as an oxidant have already been described in the literatures [12,13]. The kinetic analysis revealed that the SS formation reaction took place through a stochastic mechanism, indicating that the SS-formation velocity is proportional to the number of the SH groups present along the chain. In this study, to obtain further evidences for the stochastic SS formation, short-term oxidation experiments using DHSox were carried out at pH 4.0–10.0 for additional polythiol peptide models. We chose peptides with a rather small number of Cys residues in order to perform the kinetic analysis of the SS formation reaction as accurate as possible.
Reduced RNase A (RRNase) and CX-397 (RCX-397), with eight and six cysteinyl SH groups, respectively, were obtained by reduction of the native proteins (N) with DTTred in the presence of 4 M guanidine thiocyanate as a denaturant. Similarly, reduced Ins-A (RIns-A) and Ins-B (RIns-B), with four and two cysteinyl SH groups, respectively, were obtained from native bovine pancreatic insulin. On the other hand, reduced Rlx-A (RRlx-A) having four SH groups was synthesized by standard solid-phase peptide synthesis and purified by HPLC after the reductive treatment.
The SS formation (oxidation) reaction of R was initiated by addition of stoichiometric amounts of DHSox (0.5–4 equiv. with respect to R) at 25 °C and pH 4.0–10.0 by using a quench-flow instrument [10]. The reaction was carried out in the presence of urea (2 M) for Ins-A, Ins-B, and Rlx-A in order to prevent aggregation. It should be noted that the experiments could not be performed for Ins-A at pH 4.0 even in the presence of 6 M urea due to aggregation. A reaction time of the oxidation was controlled to be 100 ms–60 s. The generated SS intermediates with one, two, three, or four SS bonds were chemically trapped with AEMTS [20], which can rapidly and irreversibly modify each free SH group into SSCH2CH2NH3+ form. This modification brought an increase in the molecular mass by 76 Da as well as one positive charge per one SH group. Therefore, the number of the blocked SH groups (hence the number of SS bonds) can be easily identified by mass spectrometry as well as HPLC. A cation-exchange column was used in the HPLC analysis for the SS intermediates of RNase A and CX-397, while a reverse-phase column was used for the SS intermediates of Ins-A, Ins-B, and Rlx-A.
HPLC chromatograms of the sample solutions obtained for CX-397, Ins-A, Rlx-A, and Ins-B when the R's were reacted with 2, 1, 1, and 0.5 equiv. of DHSox, respectively, are shown in Fig. 3. R, 1S (with one SS bond), 2S (with two SS bonds), and 3S (with three SS bonds) intermediates were clearly separated for CX-397 depending on the number of the SS bonds by the cation-exchange column (Fig. 3A). A marginal amount (2%) of N, resulting from occasional generation of the three native SS linkages, was observed, suggesting that SS formation reaction almost statistically progressed. A small peak (symbol x) of an impurity, which was originated from R, was also observed. However, since its population was very low, the peak was ignored in the kinetic analysis below. On the other hand, the SS intermediates (R, 1S, and 2S) of Ins-A, Rlx-A, and Ins-B were separated roughly depending on the number of the blocked SH groups by using the reverse-phase column (Fig. 3B–D). Although the peaks of the impurities (<9%) originating from their sources were observed, their populations did not change during the reaction with DHSox. Therefore, these peaks were ignored in the kinetic analysis. It should be noted that the 1S, 2S, and 3S intermediates are not homogeneous but heterogeneous ensembles having one, two, and three SS bonds, respectively. For example, the SS intermediates of Ins-A and Rlx-A having four Cys residues would theoretically contain six and three SS isomers for 1S and 2S, respectively. This can be actually presumed from the HPLC chromatograms. On the other hand, 1S of Ins-B having just two cysteine residues was a sole product. The each SS intermediate fractionated by HPLC was isolated, and the number of SS bond was assigned by ESI(+)-MS spectrometry.
Fig. 3.
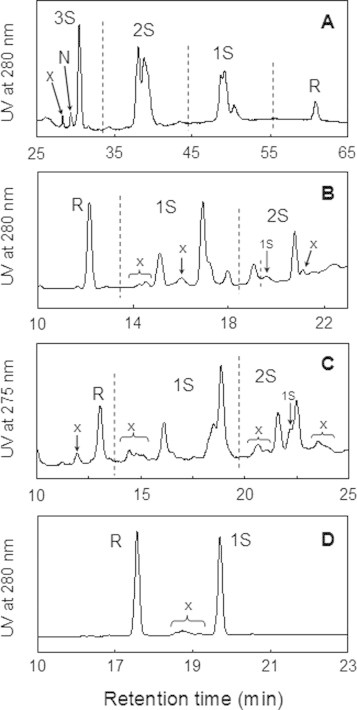
HPLC chromatograms obtained from the short-term oxidation experiments of (A) CX-397, (B) Ins-A, (C) Rlx-A, and (D) Ins-B using DHSox as an oxidant at 25 °C. Reaction conditions: (A) pH = 8.0 (100 mM Tris–HCl buffer), reaction time = 20 s, [RCX-397]0 = [DHSox]0/2 = 41.6 μM; (B) pH = 8.0 (100 mM Tris–HCl buffer with 2 M urea), reaction time = 40 s, [RIns-A]0 = [DHSox]0 = 23.3 μM; (C) pH = 7.0 (100 mM Tris–HCl buffer with 2 M urea), reaction time = 10 s, [RRlx-A]0 = [DHSox]0 = 30.7 μM; and (D) pH = 8.0 (100 mM Tris–HCl buffer with 2 M urea), reaction time = 30 s, [RIns-B]0 = 2[DHSox]0 = 31.0 μM. See the text for details of the HPLC analysis conditions. The peaks with symbol x were impurities derived from the peptide sources and remained unchanged during the SS formation reactions.
Relative populations of the SS intermediate ensembles of CX-397, Ins-A, Rlx-A, and Ins-B as a function of the reaction time are shown in Fig. 4. It is clearly seen that the SS formation reaction with DHSox progresses rapidly, irreversibly, and quantitatively for all model peptides. The comparable results were obtained in the short-term oxidation experiments using different equivalents of DHSox or under different pH conditions (see Supporting information). Similar behaviors could also be observed for RNase A [10,12]. Reaction schemes of the SS formation for RNase A, CX-397, Ins-A, Rlx-A, and Ins-B are given in Eqs. (1)–(5).
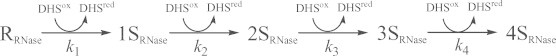 |
(1) |
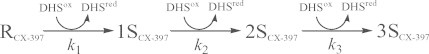 |
(2) |
| (3) |
| (4) |
| (5) |
Fig. 4.
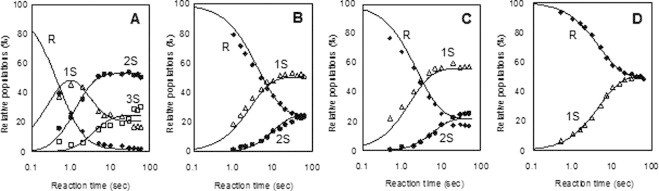
Relative populations of SS intermediates of (A) CX-397, (B) Ins-A, (C) Rlx-A, and (D) Ins-B at 25 °C as a function of the reaction time. Reaction conditions: (A) pH = 8.0 (100 mM Tris–HCl buffer), [RCX-397]0 = [DHSox]0/2 = 53.1 μM; (B) pH = 8.0 (100 mM Tris–HCl buffer with 2 M urea), [RIns-A]0 = [DHSox]0 = 23.3 μM; (C) pH = 7.0 (100 mM Tris–HCl buffer with 2 M urea), [RRlx-A]0 = [DHSox]0 = 30.7 μM; and (D) pH = 8.0 (100 mM Tris–HCl buffer with 2 M urea), [RIns-B]0 = [DHSox]0 = 31.0 μM. Curves were drawn by simulation using the second-order rate constants obtained by fitting the experimental data to reaction schemes of Eqs. (2)–(5).
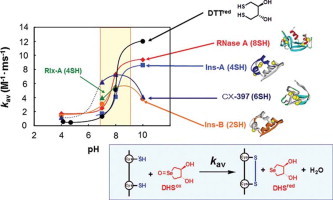
The second-order rate constants, k1 for R → 1S, k2 for 1S → 2S, k3 for 2S → 3S, and k4 for 3S → 4S, were subsequently determined by fitting the experimental data. The obtained rate constants (k1–k4) are listed in Table 1. Good agreement between the simulation curves and the plots of experimental data shown in Fig. 4 clearly indicates accuracy of the postulated reaction schemes. The obtained rate constants k1–k4, k1–k3, and k1–k2 for RNase A, CX-397, and Ins-A and Rlx-A, respectively, which can form the maximum of two to four SS bonds, were found to be basically proportional to the number of the free SH groups present in the reactive polypeptide intermediates except for CX-397 at pH 8.0 and 10.0. For example, the ratios of k1 vs. k2 are roughly 2:1 for Ins-A and Relx-A. In Fig. 5, the averaged rate constants kav, which correspond to the second-order rate constants for formation of one SS bond with DHSox, are plotted against the solution pH. The rate constants monotonously increase with increasing the pH, except for the cases of CX-397 and Ins-B at 10.0. Fig. 5 further shows that kav of RNase A, CX-397, Ins-A, and Ins-B obviously jumps up in a narrow pH range of 7–9.
Table 1.
The second-order rate constants for oxidation (SS formation) of polypeptide chains (RNase A, CX-397, Ins-A, Rlx-A, and Ins-B)a and DTTredb with DHSox as an oxidant at 25 °C.
| pH | Denaturant | k1 (mM−1 s−1) | k2 (mM−1 s−1) | k3 (mM−1 s−1) | k4 (mM−1 s−1) | kav (mM−1 s−1)c | |
|---|---|---|---|---|---|---|---|
| 4.0 | None | 7.1 ± 0.4 | 4.9 ± 0.3 | 3.0 ± 0.2 | 1.7 ± 0.1 | 1.7 ± 0.1 | |
| RNase Ad | 7.0 | None | 10.7 ± 0.5 | 7.3 ± 0.4 | 4.7 ± 0.3 | 2.6 ± 0.2 | 2.5 ± 0.2 |
| 8.0 | None | 30.6 ± 0.9 | 20.1 ± 0.7 | 14.9 ± 0.8 | 7.4 ± 0.4 | 7.3 ± 0.4 | |
| 10.0 | None | 42.6 ± 2.3 | 29.6 ± 2.3 | 17.9 ± 1.1 | 8.2 ± 0.5 | 9.4 ± 1.1 | |
| 4.0 | None | 4.3 ± 0.2 | 2.6 ± 0.1 | 1.2 ± 0.1 | 1.4 ± 0.1 | ||
| 2 M Gdn-HCl | 2.3 ± 0.2 | 1.4 ± 0.1 | 0.63 ± 0.05 | 0.7 ± 0.06 | |||
| 7.0 | Nonee | 9.8 ± 0.5 | 6.6 ± 0.3 | 2.8 ± 0.3 | 3.1 ± 0.2 | ||
| 2 M urea | 3.1 ± 0.2 | 1.7 ± 0.1 | 1.1 ± 0.1 | 1.0 ± 0.1 | |||
| CX-397 | 8.0 | None | 18.6 ± 1.0 | 10.0 ± 0.6 | 3.6 ± 0.6 | 5.4 ± 0.4f | |
| 2 M urea | 11.5 ± 0.9 | 6.2 ± 0.5 | 2.2 ± 0.1 | 3.0 ± 0.2f | |||
| 2 M Gdn-HCl | 9.8 ± 0.6 | 5.0 ± 0.3 | 2.7 ± 0.2 | 2.8 ± 0.2 | |||
| 10.0 | None | 12.4 ± 0.7 | 7.2 ± 0.5 | 3.2 ± 0.5 | 3.8 ± 0.3f | ||
| 7.0 | 2 M urea | 3.4 ± 0.3 | 1.7 ± 0.2 | 1.7 ± 0.2 | |||
| Ins-A | 8.0 | 2 M urea | 8.2 ± 0.3 | 4.0 ± 0.3 | 4.1 ± 0.2 | ||
| 10.0 | 2 M urea | 17.8 ± 0.3 | 8.2 ± 0.3 | 8.6 ± 0.2 | |||
| Rlx-A | 7.0 | 2 M urea | 8.6 ± 0.3 | 3.6 ± 0.1 | 4.0 ± 0.1 | ||
| 8.0 | 2 M urea | 11.3 ± 0.8 | 5.6 ± 0.4 | 5.6 ± 0.4 | |||
| 4.0 | 2 M urea | 1.2 ± 0.1 | 1.2 ± 0.1 | ||||
| 7.0 | 2 M urea | 6.2 ± 0.2 | 6.2 ± 0.2 | ||||
| Ins-B | 8.0 | 2 M urea | 7.3 ± 0.5 | 7.3 ± 0.5 | |||
| 10.0 | 0.2 M urea | 4.7 ± 0.4 | 4.7 ± 0.4 | ||||
| 2 M urea | 4.1 ± 0.2 | 4.1 ± 0.2 | |||||
| 4 M urea | 2.6 ± 0.1 | 2.6 ± 0.1 | |||||
| 4.1 | None | 0.55 ± 0.15 | 0.55 ± 0.15 | ||||
| 4.7 | None | 0.45 ± 0.01 | 0.45 ± 0.01 | ||||
| DTTred | 7.0 | None | 1.3 ± 0.1 | 1.3 ± 0.1 | |||
| 8.0 | None | 5.1 ± 0.2 | 5.1 ± 0.2 | ||||
| 10.0 | None | 12.0 ± 1.0 | 12.0 ± 1.0 |
The values of k1–4 were determined by fitting the data from the short-term oxidation experiments to the reaction schemes of Eqs. (1)–(5).
The values of k1 were determined by the stopped-flow experiments.
The averaged second-order rate constants for SS formation of the dithiols with DHSox at 25 °C obtained by using the equations, (k1/4 + k2/3 + k3/2+ k4)/4 for RNase A, (k1/3 + k2/2 + k3)/3 for CX-397, (k1/2 + k2)/2 for Ins-A and Rlx-A, and k1 for Ins-B and DTTred.
Data quoted from Ref. [13].
The oxidation did not follow stochastic reaction.
Fig. 5.
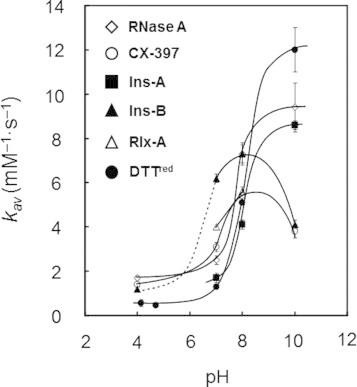
The pH dependence of the kav values obtained at 25 °C by the short-term oxidation experiments using DHSox as an oxidant. The curves were manually drawn for convenience.
3.2. Oxidation kinetics of DTTred
To compare the second-order SS-formation rate constants of polythiol peptide chains with that of a low molecular-weight dithiol compound, we carried out short-term oxidation of DTTred using DHSox as an oxidant (Eq. (6)) [19].
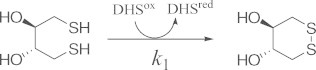 |
(6) |
When 0.125–4.0 mM DTTred was reacted with 0.1 mM DHSox at 25 °C and pH 7.0, the UV absorbance at 310 nm due to formation of DTTox became constant within 10 s, suggesting that the SS formation (oxidation) reaction completed in a short time. The rate constants (k1) for the SS formation of DTTred (Table 1) were determined by plotting the observed pseudo first-order rate constants (kobs) against the concentration of DTTred (Fig. 6). Similar experiments were carried out at pH 4.1, 4.7, 8.0, and 10.0. The obtained second-order rate constants are also plotted in Fig. 5, showing a similar jump of the rate constant at around pH 8 to that observed for the polythiol peptide chains.
Fig. 6.
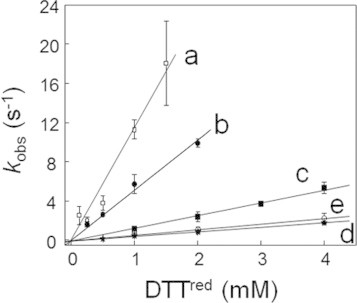
Linear plots showing the dependence of observed pseudo first-order rate constants (kobs) for the oxidation of DTTred (0.125–4.0 mM) with DHSox (0.1 mM). (a) pH 10.0, (b) pH 8.0, (c) pH 7.0, (d) pH 4.7, and (e) pH 4.1.
3.3. A redox potential of DHSox
From the above kinetic analyses of the SS formation, it was obvious that DHSox enables rapid, quantitative, and irreversible SS formation for various polythiol substrates. To further confirm the high oxidizing ability of DHSox, the two-electron redox potential of DHSox was measured by potentiometric titration using sodium dithionite (Na2S2O4) as a reducing titrant at 25 °C and pH 5.5 in an aqueous medium. The obtained titration curve is shown in Fig. 7. The value of the redox potential obtained for DHSox, (375 ± 15 mV vs. NHE) was consistent with previously reported values of several selenoxide compounds (380–490 mV) [23,24]. Compared with the redox potentials of l-cystine (−238 mV) and DTTox (−327 mV) at pH 7.0 [25], the redox potential of DHSox was prominently higher, supporting that DHSox has high potential of SS formation for polythiol substrates.
Fig. 7.
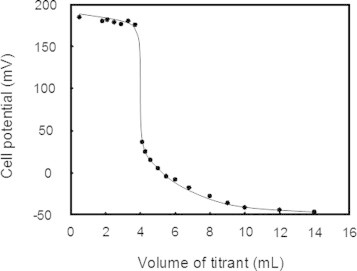
A potentiometric titration curve. The experiment was carried out at 25 °C in 100 mM sodium acetate at pH 5.5 containing 1.0 mM DHSox titrated with 10 mM Na2S2O4 as a titrant. See the experimental section for the details.
4. Discussion
4.1. Comparison of DHSox with previous SS-forming reagents
DHSox, a water-soluble selenoxide with five-membered ring, was applied as a SS-forming reagent for five polythiol peptides and DTTred in this study. In each case, rapid, quantitative, and irreversible SS formation was clearly observed. In the previous oxidative folding study of SS-containing proteins, disulfide reagents, such as DTTox and GSSG, were usually applied [3–9]. In addition to these conventional reagents, new types of the reagents with improved reactivity, such as polymer-bound SS-containing reagents [26], aromatic SS reagents [27], glutathione derivatives [28], and short peptides with a -Cys-Xaa-Xaa-Cys- motif [29], have been developed. Very recently, diselenide (SeSe) reagents, such as oxidized selenoglutathione [25,30], dipyridyl diselenide [31], and selenocystamine [31], have also been applied to oxidative folding of proteins in vitro and in vivo. However, the redox potentials of disulfides (−265 to −236 mV) and diselenides (−407 to −352 mV) are comparable or rather lower than that of l-cystine (−238 mV) [25]. In other examples, sulfoxide reagents with high redox potential, such as dimethyl sulfoxide (160 mV) [32] and polymer-bound methyl sulfoxide [33], are sometimes applied for oxidation of thiol substrates.
In contrast, selenoxides possess much higher redox potentials than the aforementioned reagents. May and coworkers [23] and Singh and coworkers [24] reported the redox potentials of various selenoxide reagents (380–490 mV) and suggested that selenoxides can be useful as a SS-forming reagent to thiols, such as benzenethiol and GSH. The redox potential of DHSox (375 ± 15 mV at 25 °C) measured in this study was found to be comparable, supporting the high oxidizing ability of DHSox to generate SS bonds. Indeed, the second-order rate constants (3.5–4.1 mM−1 s−1 at pH 7.0) reported by May and coworkers [23] for the oxidation of GSH with phenylaminoethyl selenoxide and its derivatives were similar to those determined in this study (kav = 1.3–6.2 mM−1s−1 at pH 7.0) for the oxidation of polythiol peptides with DHSox.
The high redox potential of DHSox is advantageous for temporary separation of the SS formation phase from the SS rearrangement phase in oxidative folding of SS-containing proteins. Indeed, by applying DHSox for the folding of RNase A, we have already succeeded in separation of the two folding phases clearly and characterization of the key folding intermediates much easier than when the conventional reagents with a low redox potential were applied [10–12]. On the other hand, formation of N is usually very slow by using DHSox because it does not catalyze the SS rearrangement. To overcome this limitation of DHSox as an oxidative folding reagent, a thiol reagent, such as cysteine and glutathione (GSH), should be added to the reaction solution after completion of the SS formation.
4.2. Stochastic SS formation
The SS formation reaction of polythiol peptides with DHSox would take place in a stochastic manner if there is no steric congestion [13]. This was confirmed for various polythiol peptides in this study (see Table 1). The rate constants, k1 and k2, for Ins-A were roughly in a ratio of 2:1 in a range of pH 7.0–10.0. Similarly, the k1 and k2 for Rlx-A at pH 7.0 and 8.0 were proportional to the number of the free SH groups existing in the reactive species. The same behaviors can be seen for the cases of RNase A at pH 4–10 and CX-397 at pH 4.0 and 7.0. However, the rate constants k1–k3 of CX-397 obtained at pH 8.0 and 10.0 were not in a ratio of 3:2:1. The reasons for the non-stochastic behaviors are discussed in the following sections.
Provided that the SS formation of peptides with DHSox proceeds stochastically, the averaged second-order rate constants (kav) obtained for any polypeptides should be similar to that obtained for small dithiols under the same conditions. This is indeed the case as the values of k1 obtained for DTTred are roughly comparable to the kav values for the polypeptide models (Fig. 5), supporting the stochastic SS formation mechanism. However, it would also be notable that the value of k1 for DTTred at pH 10.0 is significantly larger than the kav values determined for polythiol peptides. This may reflect the redox potential of DTTox (−327 mV), which is lower than that of l-cystine (−238 mV) [24].
4.3. Effects of pH on the SS formation velocity
Acidity (deprotonation liability) of a SH group is an important factor for the SS formation velocity because the thiolate (S−) form is much sensitive to an oxidant than the neutral thiol (SH) form. The pKa value of a cysteinyl SH group is 8.22 [34], and the ratio of the reactive thiolate species rapidly increases in a narrow pH range around the pKa value. Therefore, the rate constant of the SS formation would increase significantly at the pH. In fact, when the averaged rate constants (kav) were plotted against the pH, jumps of the kav values were clearly observed at around the pKa value of the SH group (Fig. 5). However, the magnitude of the kav jump was too small if the kav value was proportional to the thiolate concentration. This suggests that the SS formation is accelerated by protonation of DHSox [13].
Fig. 5 also shows that the behavior of the kav jump is slightly different between the polythiol peptide substrates. In the case of RNase A and Ins-A, the kav jumps were observed at around pH 8, which is close to the pKa values for a cysteinyl SH group (pKa = 8.22) [34]. The pH range was also close to that of the kav jump observed for DTTred (pKa = 9.15) [35]. On the other hand, the kav jump was observed at a lower pH in the case of Ins-B. Similar behavior would be expected for Rlx-A. This can be explained by the enhanced reactivity of the SH groups due to the basic amino acid residues located in the proximity of the SH groups. For example, the primary sequences of Ins-B and Rlx-A involves His-Lys-Cys-Gly-Ser-His (the sequential number from 5 to 10) and Lys-Cys-Cys-His (the sequential number from 9 to 12), respectively (Fig. 2), which contain Lys and His residues close to Cys. These basic amino acid residues would decrease the pKa values of the Cys residues [36]. Thus, the cysteinyl SH groups of Rlx-A and Ins-B would be expected to have lower pKa values on the average.
According to the stochastic mechanism, almost the same rate constants of SS formation should be obtained by using DHSox regardless of the sort and length of the amino acid sequence. In reality, however, each cysteinyl SH group in a polypeptide would have a slightly different pKa value, which makes the polypeptide as a whole different in the chemical reactivity to form a SS bond. As a result, large deviation of the kav values could be observed in the pH range close to the pKa value of the SH groups. This in turn suggests the possibility that the absolute value of the rate constant for the SS formation of a polypeptide with DHSox provides valuable information about the reactivity of the SH groups irrespective of the length of the polypeptide chain.
Fig. 5 also shows that the kav of CX-397 and Ins-B obviously decrease at the pH higher than 8. This can be explained by the steric factors as discussed below.
4.4. Effects of conformation in a polythiol peptide
The ratios of the rate constants k1–k3 for CX-397 at pH 8.0 and 10.0 were about 5:3:1 and 16:9:4, respectively, showing non-stochastic behaviors. This would indicate that the bimolecular process for the formation of SS intermediates shown in Fig. 1 (step 1), or the subsequent unimolecular process (step 2), is disturbed by local or global structure formation under basic conditions. To examine the adequacy of this assumption, short-term oxidation experiments of CX-397 and the kinetic analysis were carried out at pH 8.0 in the presence of a denaturant (2 M urea). The obtained rate constants k1–k3 based on the reaction scheme of Eq. (2) were 11.5, 6.2, and 2.2 mM−1 s−1, respectively, still showing non-stochastic behaviors (Table 1). However, in the presence of 2 M Gdn-HCl, which is a more efficient denaturant than urea, the rate constants became 9.8, 5.0, and 2.7 mM−1s−1, respectively, which are nearly in a ratio of 3:2:1, suggesting the stochastic SS formation. The result strongly suggested that the SS intermediates of CX-397 under basic conditions have densely packed conformation whereas the intermediates lose the conformation in the presence of a strong denaturant. In the meantime, the absolute values of the rate constants obtained in the presence of the denaturants were obviously smaller than those obtained in the absence. The effects of a denaturant on the SS formation velocity are discussed later. Scheraga and coworkers previously reported that the SS intermediate ensembles (1S, 2S, and 3S) generated in the oxidative folding processes of hirudin variant (HV-1) have stable structures at pH 8.3 and 12 °C [37]. Chang and coworkers also reported that generation of SS folding intermediates of hirudin at pH 8.3 and 23 °C induces the intramolecular packing with hydrophobic collapse [38]. Our results would support that the structures are not so stable to tolerate against a strong denaturant.
From the previous oxidative folding study of RNase A at various pHs by using DHSox [12], it was revealed that the SS formation processes consist of two subphases; the kinetic and thermodynamic subphases. In the former subphase, relative populations of the SS isomers are kinetically governed by probabilities. In the latter subphase, on the other hand, the kinetically formed SS isomers are redistributed to slightly stable SS intermediate ensembles via fast SS reshuffling. The SS formation of CX-397 would also progress via similar two-phase processes (Fig. 8). Thus, under acidic conditions, the kinetic intermediates (1S°, 2S°, and 3S°) with no rigid structure would form from R° because only stochastic SS formation proceeds. The produced kinetic intermediates are then converted to the thermodynamic intermediates (1S, 2S, and 3S) through SS reshuffling by changing the solvent pH or standing the solution for a long time. Given the reaction scheme of Fig. 8 for CX-397, the SS components of the 3S intermediates should be different under acidic and basic conditions. From the profile of the HPLC chromatograms, however, the difference in the relative populations of the 3SS isomers between the kinetic and thermodynamic intermediates (i.e. 3S° and 3S, respectively) was not obvious. We, therefore, applied enzymatic digestion for 3S° and 3S obtained at pH 4.0 and 8.0, respectively. As a result, it was found that the populations of the digested peptide fragments are significantly different (see Supporting information), suggesting the existence of some structures under weakly basic conditions. To confirm the structural difference between the kinetic and thermodynamic intermediates of CX-397, CD and fluorescence spectra were measured at pH 4.0 in different reaction times.
Fig. 8.
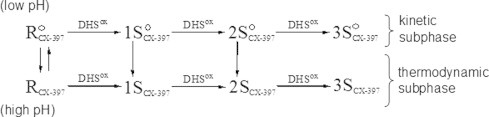
A reaction scheme of the SS formation phase for the oxidative folding of CX-397 using DHSox as an oxidant. and RCX-397 are the unfolded reduced states of CX-397 at acidic and basic pHs, respectively.
The kinetic intermediates (2S° as a major component) were prepared by the oxidation of RCX-397 with two equivalents of DHSox at pH 4.0 for 5 min, and the thermodynamic intermediates (2S as a major component) were obtained by the subsequent incubation at 25 °C for 24 h. During the incubation, the kinetic intermediates would slowly transform to the 2S via SS reshuffling even at pH 4.0 as previously observed in the case of RNase A [12]. The CD spectra of these intermediates did not exhibit any significant differences from that of R, indicating that no secondary structure is present in the both kinetic and thermodynamic intermediates (Fig. 9A). In the fluorescence spectra, on the other hand, the relative intensity at around 310 nm increased obviously during the incubation, suggesting that the structures of the intermediates become compact during the kinetic to thermodynamic transition by SS reshuffling (Fig. 9B). The result supported that the SS formation processes of CX-397 are composed of kinetic and thermodynamic subphases like the case of RNase A [12].
Fig. 9.
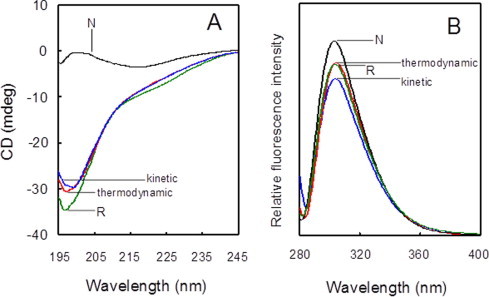
CD and fluorescence spectra of kinetic (blue lines) and thermodynamic (red lines) SS intermediates of CX-397 obtained at pH 4.0 and 25 °C. The reaction times for the oxidation of RCX-397 with DHSox were 5 min and 24 h for the kinetic and thermodynamic intermediates, respectively. For comparison, the respective spectra of R (green lines) and N (black lines) are shown. (A) CD spectra at [R]0 = [DHSox]0/2 = 35.7 μM. (B) Fluorescence spectra at [R]0 = [DHSox]0/2 = 30.0 μM (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
A rate constant k1 of CX-397 at pH 10.0 was smaller than that at pH 8.0. This indicates that R at a high pH already has some structures. The phenomenon that the reduced state (R) has stable conformation under specific conditions has sometime been reported for SS-containing proteins, such as ribonuclease T1 [39].
Fig. 5 further shows that the rate constant (kav) for Ins-B at pH 10.0 is obviously smaller than that at pH 8.0, suggesting that RIns-B also has densely packed structure even in the presence of 2 M urea. Indeed, when the fluorescence spectra of RIns-B in the presence of 0.2, 2, and 4 M urea were measured, intensity of the fluorescence at 310 nm was enhanced (Fig. 10), suggesting more structuring in a lower concentration of urea. The presence of such a structure was previously reported in the 30% trifluoroethanol solution [40].
Fig. 10.
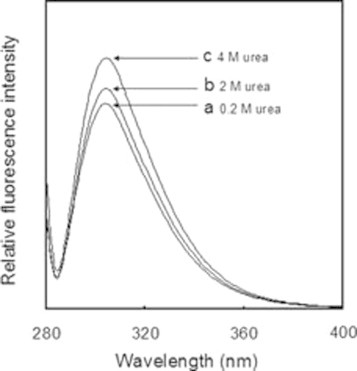
Fluorescence spectra of RIns-B at 10.0 in the presence of (a) 0.2, (b) 2, and (c) 4 M urea. The concentration of RIns-B was 20.5 μM in all conditions.
4.5. Effects of a denaturant
Urea and Gdn-HCl are frequently used as a denaturant as well as an additive preventing aggregation of polypeptides. In this study, 2 M urea was added in the reaction solutions of Ins-A, Rlx-A, and Ins-B, since they are highly liable to aggregate. Therefore, the effects of the denaturant on the SS formation velocity should be clarified. When the values of kav are compared between RNase A and Ins-A, which showed the kav jump at a similar pH range (Fig. 5), the kav values for Ins-A are apparently smaller than those for RNase A. This would indicate that the SS formation velocities with DHSox are decreased by addition of urea. The rate constants k1–k3 of CX-397 obtained in the presence of 2 M urea or Gdn-HCl were smaller than those in the absence of the denaturant. These results suggest that the access of DHSox to the free SH groups in a polypeptide is slightly inhibited probably by the electrostatic interaction between the denaturant and the polypeptide chain.
Similarly, when the concentrations of urea were changed from 0.2 to 4 M, a decrease of the rate constants was observed for Ins-B at pH 10.0, although the robust structure of RIns-B would be significantly destroyed (Fig. 10). To understand the observed SS formation velocity the both acceleration and deceleration effects of a denaturant should be considered. The former effect would be caused by denaturation of the robust structure of the substrate, whereas the latter effect would be brought by the interaction of the denaturant with the substrate. According to the observed decrease in the SS formation velocity for Ins-B in the presence of 4 M urea at pH 10.0, the deceleration effect of a denaturant must be slightly predominant for polythiol peptide chains.
5. Conclusions
In this study, we have applied DHSox to the SS formation reactions of five polythiol peptides (i.e., reduced species of several SS-containing proteins) and a small dithiol DTTred. Detailed kinetic analysis of the SS formation reactions revealed several features of DHSox as a selective SS-forming reagent.
-
(1)
DHSox allows rapid, quantitative, and irreversible SS formation for a wide range of polythiol substrates.
-
(2)
The reaction velocity jumps up at around pH 8, which is near the pKa value of cysteinyl SH groups. However, the pH range of the velocity jump would shift lower when the basic amino acid residues, such as Lys and His, which would modify the pKa of the SH groups, are located in the proximity.
-
(3)
The SS-formation rate constants are simply proportional to the number of the free SH groups present in the reduced or partially oxidized polythiols, confirming that the SS formation normally proceeds stochastically (13). However, the formation of global or local folded structures in the substrates would decelerate the SS formation reaction and modify the stochastic nature.
-
(4)
The velocity of the SS formation slightly decreases in the presence of a denaturant, such as urea and Gdn-HCl, due probably to the interaction between the denaturant and the peptide molecules.
The strong ability of DHSox to generate SS bonds was indeed supported by measurement of the redox potential (375 mV vs. NHE at 25 °C), which is much higher than disulfides, diselenides, and sulfoxides that are utilized in the folding study of SS-reduced proteins. To summarize, DHSox with high oxidation ability is a useful reagent for formation of SS bonds in polythiol peptides. The stochastic behaviors of SS formation with DHSox and the absolute values of the second-order rate constants can be utilized to probe the chemical reactivity and conformation of peptides. Thus, selenoxide reagents would provide valuable information on the folding of versatile SS-containing proteins.
Acknowledgment
This study was supported by JSPS KAKENHI Grant No. 23550198.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Appendix A. Supplementary material
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.fob.2012.12.004.
Appendix A. Supplementary material
Typical HPLC chromatogram obtained by short-term oxidation experiment of Ins-A using 1 equivalent of DHSox for 1 min at 25 °C. The fraction number is corresponding to that in the Table S2. The symbols of x is a unknown impurities.
Typical HPLC chromatogram obtained by short-term oxidation experiment of Rlx-A using 1 equivalent of DHSox for 1 min at 25 °C. The fraction number is corresponding to that in the Table S2. The symbols of x is a unknown impurities.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 47.9 μM at 25 °C and pH 4.0.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/3 = 45.8 μM at 25 °C and pH 4.0.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 43.6 μM at 25 °C and pH 8.0.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/3 = 44.1 μM at 25 °C and pH 8.0.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 52.5 μM at 25 °C and pH 10.0.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 53.6 μM at 25 °C and pH 10.0.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/3 = 44.2 μM at 25 °C and pH 10.0.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/4 = 61.6 μM at 25 °C and pH 4.0 in the presence of 2 M Gdn-HCl.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 41.4 μM at 25 °C and pH 7.0 in the presence of 2 M urea.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/3 = 29.7 μM at 25 °C and pH 8.0 in the presence of 2 M urea.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 32.3 μM at 25 °C and pH 8.0 in the presence of 2 M Gdn-HCl.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/3 = 26.3 μM at 25 °C and pH 8.0 in the presence of 2 M Gdn-HCl.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 23.9 μM at 25 °C and pH 7.0 in the presence of 2 M urea.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 18.7 μM at 25 °C and pH 7.0 in the presence of 2 M urea.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/3 = 18.5 μM at 25 °C and pH 7.0 in the presence of 2 M urea.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 14.5 μM at 25 °C and pH 8.0 in the presence of 2 M urea.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/3 = 14.8 μM at 25 °C and pH 8.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-A as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 10.5 μM at 25 °C and pH 10.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-A as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 16.5 μM at 25 °C and pH 10.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = 2[DHSox]0 = 41.3 μM at 25 °C and pH 4.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 42.1 μM at 25 °C and pH 4.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = 2[DHSox]0 = 21.5 μM at 25 °C and pH 7.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/0.65 = 18.7 μM at 25 °C and pH 7.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 25.7 μM at 25 °C and pH 7.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = 2[DHSox]0 = 25.8 μM at 25 °C and pH 8.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/0.45 = 46.9 μM at 25 °C and pH 8.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = 2[DHSox]0 = 40.0 μM at 25 °C and pH 10.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 40.0 μM at 25 °C and pH 10.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 42.6 μM at 25 °C and pH 10.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = 2[DHSox]0 = 31.6 μM at 25 °C and pH 10.0 in the presence of 0.2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 31.1 μM at 25 °C and pH 10.0 in the presence of 0.2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = 2[DHSox]0 = 37.2 μM at 25 °C and pH 10.0 in the presence of 4 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 31.0 μM at 25 °C and pH 10.0 in the presence of 4 M urea.
UV absorbance changes at 310 nm (panel A) and the plots of the second-order rate constant against the pH obtained at 25 °C by the short-term oxidation of DTTred with DHSox using a stopped-flow instrument. The reaction conditions of panel A were (a) [DTTred]0 = 2 mM, [DHSox]0 = 0.1 mM in 200 mM acetate buffer at pH 4.0, (b) [DTTred]0 = 2 mM, [DHSox]0 = 0.1 mM in 100 mM Tris–HCl buffer at pH 7.0, (c) [DTTred]0 = 2 mM, [DHSox]0 = 0.1 mM in 100 mM Tris–HCl buffer at pH 8.0, and (d) [DTTred]0 = 2 mM, [DHSox]0 = 0.1 mM in 25 mM NaHCO3 buffer at pH 10.0. See the text for details of determination of the second-order rate constants.
HPLC chromatogramss of the peptide fragments of (A) 3S° and (B) 3S digested by thermolysin. For detailed digestion and HPLC conditions, see experimental section in the text.
Observed and expected molecular masses of the folding intermediates and the native state of Ins-A as determined by ESI(+)-TOF-MS spectrometry.
Observed and expected molecular masses of the folding intermediates and the native state of Rlx-A as determined by ESI(+)-TOF-MS spectrometry.
Observed and expected molecular masses of the folding intermediates and the native state of Ins.-B as determined by ESI(+)-TOF-MS spectrometry.
References
- 1.Narayan M., Welker E., Wedemeyer W.J., Scheraga H.A. Oxidative folding of proteins. Acc. Chem. Res. 2000;33:805–812. doi: 10.1021/ar000063m. [DOI] [PubMed] [Google Scholar]
- 2.Welker E., Wedemeyer W.J., Narayan M., Scheraga H.A. Coupling of conformational folding and disulfide-bond reactions in oxidative folding of proteins. Biochemistry. 2001;40:9059–9064. doi: 10.1021/bi010409g. [DOI] [PubMed] [Google Scholar]
- 3.Weissman J.S., Kim P.S. Reexamination of the folding of BPTI: predominance of native intermediates. Science. 1991;253:1386–1393. doi: 10.1126/science.1716783. [DOI] [PubMed] [Google Scholar]
- 4.Rothwarf D.M., Scheraga H.A. Regeneration of bovine pancreatic ribonuclease A. 1. Steady-state distribution. Biochemistry. 1993;32:2671–2679. doi: 10.1021/bi00061a027. [DOI] [PubMed] [Google Scholar]
- 5.Rothwarf D.M., Scheraga H.A. Regeneration of bovine pancreatic ribonuclease A. 3. Dependence on the nature of the redox reagent. Biochemistry. 1993;32:2690–2697. doi: 10.1021/bi00061a029. [DOI] [PubMed] [Google Scholar]
- 6.Ewbank J.J., Creighton T.E. Pathway of disulfide-coupled unfolding and refolding of bovine α-lactalbumi. Biochemistry. 1993;32:3677–3693. doi: 10.1021/bi00065a022. [DOI] [PubMed] [Google Scholar]
- 7.Hevehan D.L., De Bernardez Clark E. Oxidative renaturation of lysozyme at high concentrations. Biotechnol. Bioeng. 1997;54:221–230. doi: 10.1002/(SICI)1097-0290(19970505)54:3<221::AID-BIT3>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 8.van den Berg B., Chung E.W., Robinson C.V., Dobson C.M. Characterization of the dominant oxidative folding intermediate of hen lysozyme. J. Mol. Biol. 1999;290:781–796. doi: 10.1006/jmbi.1999.2915. [DOI] [PubMed] [Google Scholar]
- 9.Kibria F.K., Lees W.J. Balancing conformational and oxidative kinetic traps during the folding of bovine pancreatic trypsin inhibitor (BPTI) with glutathione and glutathione disulfide. J. Am. Chem. Soc. 2008;130:796–797. doi: 10.1021/ja077468n. [DOI] [PubMed] [Google Scholar]
- 10.Iwaoka M., Kumakura F., Yoneda M., Nakahara T., Henmi K., Aonuma H., Nakatani H., Tomoda S. Direct observation of conformational folding coupled with disulphide rearrangement by using a water-soluble selenoxide reagent – a case of oxidative regeneration of ribonuclease A under weakly basic conditions. J. Biochem. 2008;144:121–130. doi: 10.1093/jb/mvn049. [DOI] [PubMed] [Google Scholar]
- 11.Arai K., Kumakura F., Iwaoka M. Kinetic and thermodynamic analysis of the conformational folding process of SS-reduced bovine pancreatic ribonuclease A using a selenoxide reagent with high oxidizing ability. FEBS Open Bio. 2012;2:60–70. doi: 10.1016/j.fob.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arai K., Kumakura F., Iwaoka M. Characterization of kinetic and thermodynamic phases in the prefolding process of bovine pancreatic ribonuclease A coupled with fast SS formation and SS reshuffling. Biochemistry. 2010;49:10535–10542. doi: 10.1021/bi101392w. [DOI] [PubMed] [Google Scholar]
- 13.Arai K., Dedachi K., Iwaoka M. Rapid and quantitative disulfide bond formation for a polypeptide chain using a cyclic selenoxide reagent in an aqueous medium. Chem. Eur. J. 2011;17:481–485. doi: 10.1002/chem.201002742. [DOI] [PubMed] [Google Scholar]
- 14.Cowan E.A., Oldham C.D., May S.W. Identification of a thioselenurane intermediate in the reaction between phenylaminoalkyl selenoxides and glutathione. Arch. Biochem. Biophys. 2011;506:201–207. doi: 10.1016/j.abb.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Raines R.T. Ribonuclease A. Chem. Rev. 1998;98:1045–1065. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- 16.Komatsu Y., Misawa S., Sukesada A., Ohba Y., Hayashi H. CX-397, a novel recombinant hirudin analog having a hybrid sequence of hirudin variants-1 and -3. Biochem. Biophys. Res. Commun. 1993;196:773–779. doi: 10.1006/bbrc.1993.2316. [DOI] [PubMed] [Google Scholar]
- 17.Mayer J.P., Zhang F., DiMarchi R.D. Insulin structure and function. Biopolymers. 2007;88:687–713. doi: 10.1002/bip.20734. [DOI] [PubMed] [Google Scholar]
- 18.Schwabe C., Bullesbach E.E. Relaxin: structures, functions, promises, and nonevolution. FASEB J. 1994;8:1152–1160. doi: 10.1096/fasebj.8.14.7958621. [DOI] [PubMed] [Google Scholar]
- 19.Iwaoka M., Takahashi T., Tomoda S. Syntheses and structural characterization of water-soluble selenium reagents for the redox control of protein disulfide bonds. Heteroatom Chem. 2001;12:293–299. [Google Scholar]
- 20.Bruice T.W., Kenyon G.L. Novel alkyl alkanethiolsulfonate sulfhydryl reagents. Modification of derivatives of l-cysteine. J. Protein Chem. 1982;1:47–58. [Google Scholar]
- 21.Ellman G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959;82:70–72. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 22.Chang J.-Y., Schindler P., Chatrenet B. The disulfide structures of scrambled hirudins. J. Biol. Chem. 1995;270:11992–11997. doi: 10.1074/jbc.270.20.11992. [DOI] [PubMed] [Google Scholar]
- 23.De S.V., Woznichak M.M., Burns K.L., Grant K.B., May S.W. Selenium redox cycling in the protective effects of organoselenides against oxidant-induced DNA damage. J. Am. Chem. Soc. 2004;126:2409–2413. doi: 10.1021/ja037294j. [DOI] [PubMed] [Google Scholar]
- 24.Kumar S., Singh H.B., Wolmershaeuser G. Protection against peroxynitrite-mediated nitration reaction by intramolecularly coordinated diorganoselenides. Organometallics. 2006;25:382–393. [Google Scholar]
- 25.Beld J., Woycechowsky K.J., Hilvert D. Selenoglutathione: efficient oxidative protein folding by a diselenide. Biochemistry. 2007;46:5382–5390. doi: 10.1021/bi700124p. [DOI] [PubMed] [Google Scholar]
- 26.Annis I., Chen L., Barany G. Novel solid-phase reagents for facile formation of intramolecular disulfide bridges in peptides under mild conditions. J. Am. Chem. Soc. 1998;120:7226–7238. [Google Scholar]
- 27.Gough J.D., Williams Jr R.H., Donofrio A.E., Lees W.J. Folding disulfide-containing proteins faster with an aromatic thiol. J. Am. Chem. Soc. 2002;124:3885–3892. doi: 10.1021/ja016938p. [DOI] [PubMed] [Google Scholar]
- 28.Okumura M., Saiki M., Yamaguchi H., Hidaka Y. Acceleration of disulfide-coupled protein folding using glutathione derivatives. FEBS J. 2011;287:1137–1144. doi: 10.1111/j.1742-4658.2011.08039.x. [DOI] [PubMed] [Google Scholar]
- 29.Cabrele C., Fiori S., Pegoraro S., Moroder L. Redox-active cyclic bis(cysteinyl)peptides as catalysts for in vitro oxidative protein folding. Chem. Biol. 2002;9:731–740. doi: 10.1016/s1074-5521(02)00152-7. [DOI] [PubMed] [Google Scholar]
- 30.Beld J., Woycechowsky K.J., Hilvert D. Diselenides as universal oxidative folding catalysts of diverse proteins. J. Biotechnol. 2010;150:481–489. doi: 10.1016/j.jbiotec.2010.09.956. [DOI] [PubMed] [Google Scholar]
- 31.Beld J., Woycechowsky K.J., Hilvert D. Small-molecule diselenides catalyze oxidative protein folding in vivo. ACS Chem. Biol. 2010;5:177–182. doi: 10.1021/cb9002688. [DOI] [PubMed] [Google Scholar]
- 32.Wood P.M. The redox potential for dimethyl sulfoxide reduction to dimethyl sulfide. Evaluation and biochemical implications. FEBS Lett. 1981;124:11–14. doi: 10.1016/0014-5793(81)80042-7. [DOI] [PubMed] [Google Scholar]
- 33.Verdié P., Ronga L., Cristau M., Amblard M., Cantel S., Enjalbal C., Puget K., Martinez J., Subra G. Oxyfold: a simple and efficient solid-supported reagent for disulfide bond formation. Chem. Asian J. 2011;6:2382–2389. doi: 10.1002/asia.201100202. [DOI] [PubMed] [Google Scholar]
- 34.Tajc S.G., Tolbert B.S., Basavappa R., Miller B.L. Direct determination of thiol pKa by isothermal titration microcalorimetry. J. Am. Chem. Soc. 2004;126:10508–10509. doi: 10.1021/ja047929u. [DOI] [PubMed] [Google Scholar]
- 35.Chau M.-H., Nelson J.W. Direct measurement of the equilibrium between glutathione and dithiothreitol by high performance liquid chromatography. FEBS Lett. 1991;291:296–298. doi: 10.1016/0014-5793(91)81305-r. [DOI] [PubMed] [Google Scholar]
- 36.Bulaj G., Kortemme T., Goldenberg D.P. Ionization-reactivity relationships for cysteine thiols in polypeptides. Biochemistry. 1998;37:8965–8972. doi: 10.1021/bi973101r. [DOI] [PubMed] [Google Scholar]
- 37.Thannhauser T.W., Rothwarf D.M., Scheraga H.A. Kinetic studies of the regeneration of recombinant hirudin variant 1 with oxidized and reduced dithiothreitol. Biochemistry. 1997;36:2154–2165. doi: 10.1021/bi962340w. [DOI] [PubMed] [Google Scholar]
- 38.Chatrenet B., Chang J.-Y. The disulfide folding pathway of hirudin elucidated by stop/go folding experiments. J. Biol. Chem. 1993;268:20988–20996. [PubMed] [Google Scholar]
- 39.Müecke M., Schmid F.X. Folding mechanism of ribonuclease T1 in the absence of the disulfide bonds. Biochemistry. 1994;33:14608–14619. doi: 10.1021/bi00252a029. [DOI] [PubMed] [Google Scholar]
- 40.Dupradeau F.-Y., Richard T., Le Flem G., Oulyadi H., Prigent Y., Monti J.-P. A new B-chain mutant of insulin: comparison with the insulin crystal structure and role of sulfonate groups in the B-chain structure. J. Peptide Res. 2002;60:56–64. doi: 10.1034/j.1399-3011.2002.02990.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Typical HPLC chromatogram obtained by short-term oxidation experiment of Ins-A using 1 equivalent of DHSox for 1 min at 25 °C. The fraction number is corresponding to that in the Table S2. The symbols of x is a unknown impurities.
Typical HPLC chromatogram obtained by short-term oxidation experiment of Rlx-A using 1 equivalent of DHSox for 1 min at 25 °C. The fraction number is corresponding to that in the Table S2. The symbols of x is a unknown impurities.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 47.9 μM at 25 °C and pH 4.0.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/3 = 45.8 μM at 25 °C and pH 4.0.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 43.6 μM at 25 °C and pH 8.0.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/3 = 44.1 μM at 25 °C and pH 8.0.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 52.5 μM at 25 °C and pH 10.0.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 53.6 μM at 25 °C and pH 10.0.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/3 = 44.2 μM at 25 °C and pH 10.0.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/4 = 61.6 μM at 25 °C and pH 4.0 in the presence of 2 M Gdn-HCl.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 41.4 μM at 25 °C and pH 7.0 in the presence of 2 M urea.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/3 = 29.7 μM at 25 °C and pH 8.0 in the presence of 2 M urea.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 32.3 μM at 25 °C and pH 8.0 in the presence of 2 M Gdn-HCl.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/3 = 26.3 μM at 25 °C and pH 8.0 in the presence of 2 M Gdn-HCl.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 23.9 μM at 25 °C and pH 7.0 in the presence of 2 M urea.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 18.7 μM at 25 °C and pH 7.0 in the presence of 2 M urea.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/3 = 18.5 μM at 25 °C and pH 7.0 in the presence of 2 M urea.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 14.5 μM at 25 °C and pH 8.0 in the presence of 2 M urea.
Relative populations of SS intermediates of CX-397 as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/3 = 14.8 μM at 25 °C and pH 8.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-A as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 10.5 μM at 25 °C and pH 10.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-A as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 16.5 μM at 25 °C and pH 10.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = 2[DHSox]0 = 41.3 μM at 25 °C and pH 4.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 42.1 μM at 25 °C and pH 4.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = 2[DHSox]0 = 21.5 μM at 25 °C and pH 7.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/0.65 = 18.7 μM at 25 °C and pH 7.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 25.7 μM at 25 °C and pH 7.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = 2[DHSox]0 = 25.8 μM at 25 °C and pH 8.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/0.45 = 46.9 μM at 25 °C and pH 8.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = 2[DHSox]0 = 40.0 μM at 25 °C and pH 10.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 40.0 μM at 25 °C and pH 10.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 42.6 μM at 25 °C and pH 10.0 in the presence of 2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = 2[DHSox]0 = 31.6 μM at 25 °C and pH 10.0 in the presence of 0.2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0 = 31.1 μM at 25 °C and pH 10.0 in the presence of 0.2 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = 2[DHSox]0 = 37.2 μM at 25 °C and pH 10.0 in the presence of 4 M urea.
Relative populations of SS intermediates of Ins-B as a function of the reaction time. Reaction conditions were [R]0 = [DHSox]0/2 = 31.0 μM at 25 °C and pH 10.0 in the presence of 4 M urea.
UV absorbance changes at 310 nm (panel A) and the plots of the second-order rate constant against the pH obtained at 25 °C by the short-term oxidation of DTTred with DHSox using a stopped-flow instrument. The reaction conditions of panel A were (a) [DTTred]0 = 2 mM, [DHSox]0 = 0.1 mM in 200 mM acetate buffer at pH 4.0, (b) [DTTred]0 = 2 mM, [DHSox]0 = 0.1 mM in 100 mM Tris–HCl buffer at pH 7.0, (c) [DTTred]0 = 2 mM, [DHSox]0 = 0.1 mM in 100 mM Tris–HCl buffer at pH 8.0, and (d) [DTTred]0 = 2 mM, [DHSox]0 = 0.1 mM in 25 mM NaHCO3 buffer at pH 10.0. See the text for details of determination of the second-order rate constants.
HPLC chromatogramss of the peptide fragments of (A) 3S° and (B) 3S digested by thermolysin. For detailed digestion and HPLC conditions, see experimental section in the text.
Observed and expected molecular masses of the folding intermediates and the native state of Ins-A as determined by ESI(+)-TOF-MS spectrometry.
Observed and expected molecular masses of the folding intermediates and the native state of Rlx-A as determined by ESI(+)-TOF-MS spectrometry.
Observed and expected molecular masses of the folding intermediates and the native state of Ins.-B as determined by ESI(+)-TOF-MS spectrometry.