

The hormone erythropoietin (EPO) promotes red blood cell production. The liver is the major source of EPO during embryogenesis. After birth, however, the kidney assumes this role, and hepatic EPO production is largely silenced. This switch has profound medical implications. For example, 20 million Americans have renal disease, of whom 10 to 20% are anemic. Treatment with pharmacological doses of recombinant EPO is costly, requires parenteral administration, and has been linked to cardiovascular side effects and increased mortality (1).
EPO transcription is regulated by HIF (hypoxia-inducible factor). The HIFα subunit is normally prolyl hydroxylated by a PHD (prolyl hydroxylase domain) protein, leading to its polyubiquitination by the von Hippel–Lindau protein (pVHL) and subsequent degradation. Under hypoxic conditions, however, PHD activity decreases and HIFα accumulates. HIFα, bound to HIFβ, transcriptionally activates genes such as EPO. The canonical HIF family members, HIF1α and HIF1β, and most HIF target genes are ubiquitously expressed. In contrast, expression of EPO and HIF2α, which regulates EPO in both the liver and kidney, is more restricted.
PHD2 is the primary HIFα prolyl hydroxylase and is required for embryonic development (2). Silencing PHD2 in adult mice causes increased renal, but not hepatic, EPO production and polycythemia (overproduction of red blood cells) (3, 4). PHD1 and PHD3 are nonessential genes that fine-tune the HIF response (5).
The PHDs belong to a superfamily of ~70 2-oxoglutarate–dependent dioxygenases (2). These enzymes can be inhibited with druglike molecules, some of which induce hepatic EPO synthesis in anephric mice (6). Conceivably this reflects their ability to inactivate the PHDs. However, inactivation of other 2-oxoglutarate–dependent di-oxygenases, including enzymes that affect chromatin, might also be involved.
To determine whether PHD loss is sufficient to reactivate hepatic EPO production, we created mice with livers lacking PHD1, PHD2, PHD3, or combinations thereof by using null (Phd1 and Phd3) and floxxed (Phd2) alleles (5). Phd2 or, as a control, Vhl, was inactivated by using Cre recombinase driven by a hepatocyte-specific transgene (7). Hepatic inactivation of PHD1, PHD2, or PHD3 alone did not increase EPO or hematocrit values (Fig. 1, A and B, and fig. S1). Consistent with previous data (3), hematocrits were modestly increased in mice lacking PHD1 and PHD3 (Fig. 1B and fig. S1). Loss of all three PHDs, however, dramatically increased EPO and hematocrit values (Fig. 1, A and B, and fig. S1). Indeed, these EPO concentrations vastly exceeded those achieved after renal PHD2 inactivation (4, 5, 7).
Fig. 1.
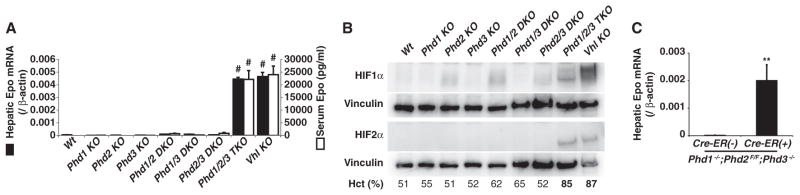
Hepatic EPO synthesis. (A) Real-time reverse transcription polymerase chain reaction (RT-PCR) analysis of hepatic Epo mRNA (solid bars) and serum Epo concentrations (open bars) in 4-week-old male mice with indicated genotypes. n = 3 mice per group. Pound symbols indicate P < 0.0001 comparing indicated genotype versus wild type (wt). DKO, double knockout; TKO, triple knockout. Note that Phd2 KO and Vhl KO are liver-specific. Error bars indicate 1 SEM. (B) Immunoblots of livers from 4-week-old mice as in (A). Mean hematocrits (Hct) are also shown. (C) Real-time RT-PCR analysis of hepatic Epo mRNA from 4-week-old mice with indicated genotypes 1 week after tamoxifen treatment. n = 3. **P < 0.01. Error bars indicate 1 SEM.
HIFα concentrations, particularly HIF2α concentrations, were conspicuously elevated in livers lacking pVHL or all three PHDs, suggesting that exceeding a threshold amount of HIF2α is sufficient to drive hepatic Epo expression (Fig. 1, A and B). The development of polycythemia in mice expressing a stabilized version of HIF2α in the liver supports this idea (8). Although hepatic Vhl inactivation causes benign tumors (7), we have not observed tumors in PHD-defective mice.
The albumin promoter that we used to express Cre becomes active during late embryogenesis, before completion of the switch from the liver to the kidney as the main EPO source, raising the possibility that PHD inactivation can prevent, but not reverse, silencing of the hepatic Epo locus. To address this, we inactivated Phd2 in 4- to 8-week-old Phd1−/−; Phd2 flox/flox;Phd3−/− mice by using a ubiquitously expressed Cre–estrogen receptor fusion protein, which was activated by tamoxifen (Fig. 1C) or by tail vein injection of an adenovirus encoding green fluorescent protein (GFP)–Cre (fig. S1). Both interventions reactivated hepatic EPO synthesis (Fig. 1C and fig. S1). These effects were specific because they were not seen in Phd2+/+ mice or in mice given an adenovirus encoding GFP alone (fig. S1).
These findings, together with earlier work, suggest that PHD2 inactivation is sufficient to induce near maximal renal EPO production whereas inactivation of all three PHDs is needed to reactivate hepatic EPO production. Thus, drugs that inhibit all three PHDs will probably be required to treat anemia linked to chronic renal failure, whereas drugs that inhibit PHD2 alone might suffice in nephric patients.
Certain features of HIF agonists suggest that they may be safer than pharmacological doses of EPO. For example, HIF activates a suite of genes dedicated to erythropoiesis and might therefore promote red blood cell production at lower circulating EPO concentrations. Moreover, HIF activates genes, such as inducible nitric oxide synthase, that might lower blood pressure and cardiovascular risk. Whether any of these theoretical benefits are real remains to be determined in clinical trials.
Supplementary Material
Footnotes
W.G.K. owns equity in and is a paid consultant for Fibrogen, Incorporated, which is developing PHD inhibitors. W.G.K. is a coinventor on a patent related to clinical use of PHD inhibitors that was licensed to Fibrogen, Incorporated.
References and Notes
- 1.Bohlius J, et al. Lancet. 2009;373:1532. doi: 10.1016/S0140-6736(09)60502-X. [DOI] [PubMed] [Google Scholar]
- 2.Kaelin WG, Jr, Ratcliffe PJ. Mol Cell. 2008;30:393. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 3.Takeda K, et al. Blood. 2008;111:3229. doi: 10.1182/blood-2007-09-114561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Minamishima YA, et al. Blood. 2008;111:3236. doi: 10.1182/blood-2007-10-117812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minamishima YA, et al. Mol Cell Biol. 2009;29:5729. doi: 10.1128/MCB.00331-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Safran M, et al. Proc Natl Acad Sci USA. 2006;103:105. [Google Scholar]
- 7.Haase VH, Glickman JN, Socolovsky M, Jaenisch R. Proc Natl Acad Sci USA. 2001;98:1583. doi: 10.1073/pnas.98.4.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim WY, et al. EMBO J. 2006;25:4650. doi: 10.1038/sj.emboj.7601300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.