

Abstract
Purpose
Reactive oxygen species (ROS) are thought to contribute to the pathogenesis of necrotizing enterocolitis (NEC). Mitochondria as a major source of intracellular ROS and apoptotic signaling during oxidative stress in NEC have not been investigated. We sought to determine: (1) the effects of oxidative stress on intestinal mitochondrial apoptotic signaling, and (2) the role of growth factors in this process.
Methods
We used Swiss Webster mice pups, and rat intestinal epithelial (RIE)-1, mitochondrial DNA-depleted RIE-1 cell line (RIE-1-ρ°) and human fetal intestinal epithelial cells (FHs74 Int) for our studies.
Results
H2O2 induced apoptosis and ROS production. ROS-mediated activation of apoptotic signaling was significantly attenuated with mitochondrial silencing in RIE-1-ρ° cells. Growth factors, especially IGF-1, attenuated this response to H2O2 in intestinal epithelial cells.
Conclusions
Our findings suggest that mitochondria are a major source of intestinal apoptotic signaling during oxidative stress and modulating mitochondrial apoptotic responses may help ameliorate the effects of NEC.
Keywords: Intestinal epithelial cells, Mitochondrial apoptotic signaling, Necrotizing enterocolitis, Oxidative stress, Reactive oxygen species, Growth factors
INTRODUCTION
Increased production of free radicals and impaired antioxidant protection has been demonstrated in inflammatory bowel diseases, indicating a central role for reactive oxygen species (ROS) released by intestinal epithelial cells and neutrophils in their pathogenesis [1,2]. ROS have also been implicated in the pathogenesis of necrotizing enterocolitis (NEC) [3], a devastating disease affecting low birth-weight premature infants and frequently resulting in sepsis and death. Increased ROS have been shown to disrupt mucosal barrier function of intestinal epithelial cells characterized by increased permeability, impaired wound healing and imbalanced antioxidant response [4–10].
The mechanisms behind ROS-mediated cell death are complex, and involve activation or dysfunction of cellular components, receptors, signal transduction pathways and DNA damage. There are multiple pathways leading up to a cell's demise, which frequently involve oxidative stress induced by either prolonged exposure to or high concentrations of ROS. There are two major endogenous ROS sources—mitochondria and cytosolic NADPH family of oxidases. The vast majority of intracellular ROS production is derived from the mitochondrial electron transport chain. Oxidative stress, characterized by a sharp rise in ROS, coupled with mitochondrial dysfunction and nuclear DNA damage, can contribute to a labile cell redox state, leading to apoptosis whenever a certain threshold for ROS accumulation is reached within a cell.
Mitochondrial dysfunction in intestinal epithelial cells has not been clearly elucidated. Recently, our laboratory examined the role of oxidative stress on mitochondrial homeostasis in intestinal epithelial cells during NEC, and we have found that cytokine (e.g., TNF-α)-mediated ROS generation in intestinal epithelial cells is predominantly of mitochondrial origin, and that TNF-α also induces significant mitochondrial functional derangements, including activation of various mitochondrial apoptotic molecules [11]. Our next aim was to determine the effects of H2O2-induced oxidative stress on mitochondrial apoptotic signaling.
We have previously shown that insulin-like growth factor (IGF)-1 pretreatment significantly attenuated oxidative stress-induced intestinal epithelial cell apoptosis, and we also observed stabilization of mitochondrial membrane potential, suggesting that growth factor protective effect may in part affect mitochondrial apoptotic contribution in stressed cells [12]. Hence, we sought to determine whether two growth factors predominant in breast milk, IGF-1 and epidermal growth factor (EGF), would have an attenuating effect on oxidative stress-induced activation of mitochondrial apoptotic signaling in human fetal and rat intestinal epithelial cells.
MATERIALS & METHODS
Cell culture
Rat intestinal epithelial-1 (RIE-1) cells (a gift from Dr. Kenneth D. Brown; Cambridge Research Station, Cambridge, U.K.) were maintained in Dulbecco's modified Eagle medium supplemented with 5% fetal bovine serum (FBS). Cells were maintained at 37°C under an atmosphere containing 5% CO2. Tissue culture media and reagents were obtained from Invitrogen (Carlsbad, CA). Human fetal intestinal epithelial cell line (FHs74 Int) was purchased from American Type Culture Collection (Manassas, VA), maintained in Hybri-Care Medium (Modified Dulbecco's medium) supplemented with 30 ng/ml EGF and 10% FBS. Tissue culture media and reagents were obtained from Mediatech, Inc (Herndon, VA). Hydrogen peroxide (H2O2), mouse monoclonal anti-β-actin antibody and other reagents were purchased from Sigma (St. Louis, MO).
In vivo NEC model
Timed pregnant Swiss-Webster mice were purchased (Charles River Labs, Pontage, MI), pup littermates were obtained from litters at birth and randomized to control or NEC groups. Control animals were maternally reared. All mice in NEC group were hand-fed with KMR liquid milk replacer formula (0.3 cc/g/day; q 3 h). To induce NEC, pups were stressed twice daily with hypoxia by placing them in a plexi glass chamber, breathing 5% oxygen for 10 min. At 96 h, mice in NEC group developed abdominal distention, hematochezia, respiratory distress and lethargy. All surviving mice were sacrificed and distal ileal sections were harvested for assays. Segments of ileum were fixed in formalin and stored in 70% ethanol for paraffin embedding. The remaining ileum was snap frozen in liquid nitrogen for protein analysis. Histologic sections were assessed and scored for severity of NEC by a pathologist (H.K.H.) in a blinded fashion.
Mitochondrial DNA-depleted rat intestinal epithelial cells (RIE-1-ρ°)
To determine whether mitochondria are the predominant source of intracellular ROS, cell death and apoptotic signaling during oxidative stress in intestinal epithelial cells, we established RIE-1-ρ° from parental RIE-1 cells. Silencing of mitochondrial function was achieved by maintaining RIE-1 cells in isolation Dulbecco's modified Eagle medium supplemented with 5% FBS, ethidium bromide (EtBr; 0.1 μg/mL), uridine (50 μg/mL), pyruvate (100 μg/mL) and glucose (4.5 g/L). To confirm successful mtDNA depletion in RIE-1-ρ° cell population, cells were evaluated with FACS cell sorting method for EtBr binding after 14–16 passages and cell lysates were analyzed for decreased mitochondrial cytochrome oxidase subunit 1 protein level using Western blot analysis (data not shown).
Western blot analysis
RIE-1, mitochondrial DNA-damaged RIE-1-ρ° and FHs74 Int cells were treated with H2O2 (500 μM, 30 min) alone or in combination with IGF-1 (100 nM, 30 min) or EGF (500 ng/mL, 30 min). Cell lysates were clarified with centrifugation (13200 rpm, 20 min at 4°C) and stored at −80°C. Protein concentrations were determined using the method described by Bradford [13]. Equal amounts of total protein (20–30 μg) were loaded onto NUPAGE 4–12% Bis-Tris Gel and transferred to PVDF membranes, incubated in a blocking solution for 1 h (Tris-buffered saline containing 5% nonfat dried milk and 0.1 % Tween 20), incubated with primary antibody overnight at 4°C, and then incubated with horseradish peroxidase-conjugated secondary antibody. Anti-β-actin antibody, total Akt and ERK were used for protein loading control. APAF-1, apoptosis-inducing factor (AIF), cytochrome C, p-Akt, p-ERK and ATP synthase β-subunit were used to probe membranes. The immune complexes were visualized by ECLPlus (Amersham Biosciences, Piscataway, NJ). Quantitative densitometric analyses of all Western blot bands were performed using ImageJ (Image Processing and Analysis in Java software, National Institutes of Health, Bethesda, MD).
Cell death detection ELISA
RIE-1, RIE-1-ρ°and FHs74 Int cells were plated onto 24-well plates for 24 h prior to H2O2 treatment. Cells were treated with H2O2 (500 μM) for 30 min. DNA fragmentation was evaluated using a Cell Death Detection ELISAPlus kit (Roche Molecular Biochemicals, Indianapolis, IN) according to manufacturer's instructions.
DCFH flow cytometry for ROS generation analysis
Dichlorofluorescein (DCF) fluorescence was used to determine intracellular ROS generation based on the oxidation of 2',7'-dichloro-dihydro-fluorescein (DCFH-DA) to a fluorescent 2',7'-DCF. RIE-1 and RIE-1-ρ° (1×106) cells were treated with H2O2. Cells were trypsinized, washed and incubated with 10 μM DCFH-DA for 15 min at 37°C. To quantify the intensity of DCF fluorescence, the samples were analyzed on a Becton-Dickenson FACScan flow cytometer (488 nm Argon-ion laser, 525 ± 10 nm band pass emission filter).
RESULTS
ROS-induced activation of mitochondrial apoptotic signaling
H2O2 treatment resulted in increase in mitochondrial APAF-1, AIF and cytochrome C levels in RIE-1 cells. In contrast, a dramatic decrease in these mitochondrial apoptotic markers was noted in mitochondrial DNA-depleted RIE-1-ρ° cells after H2O2 treatment (Fig. 1A). We have previously shown increased baseline expression levels of APAF-1 and cytochrome C predominantly in mitochondrial isolates of RIE-1 cells, with overall diminished baseline levels of these markers in mitochondrial DNA-depleted RIE-1-ρ° cells [11].
Figure 1. Intestinal cell mitochondrial apoptotic activation during NEC.

(A) H2O2 treatment resulted in increase in mitochondrial APAF-1, AIF and cytochrome C levels in RIE-1 cells (1×104). In contrast, dramatic decreases were noted in mitochondrial DNA-depleted RIE-1-ρ-° cells. (B) Extracted total proteins from ileal segments were analyzed by Western blotting for the expression of AIF, APAF-1 and cytochrome C. NEC induced significant increase in intestinal mitochondrial apoptotic signaling (*p<0.05 vs. control). Equal β-actin levels and densitometry indicate even loading.
Next, we wanted to determine the effects of experimental NEC on intestinal mitochondrial apoptotic pathway activation. NEC resulted in significant upregulation of intestinal mitochondrial apoptotic signaling with increased expression of AIF, APAF-1 and cytochrome C levels, confirmed by densitometric analysis (Fig. 1B).
Mitochondrial silencing significantly reduces RIE-1 cell death and ROS production
We had previously demonstrated that H2O2 treatment leads to significant cell death and intracellular ROS production in RIE-1 cells. Next, we wanted to determine whether mitochondrial silencing has any effect on H2O2-induced intestinal cell apoptosis and intracellular rise in ROS. Mitochondrial DNA-depleted RIE-1-ρ° cells demonstrated attenuated responses to H2O2-induced intestinal cell apoptosis and overall decreased baseline cell apoptosis, suggesting mitochondria as the predominant source of apoptotic signaling in RIE-1 cells (Fig. 2A). Similarly, intracellular rise in ROS generation during oxidative stress was significantly diminished in mitochondria-silenced cells, indicating that damaged mitochondria contribute to majority of the intracellular ROS in RIE-1 cells (Fig. 2B).
Figure 2. Mitochondria contribution to apoptosis and ROS generation during oxidative stress.
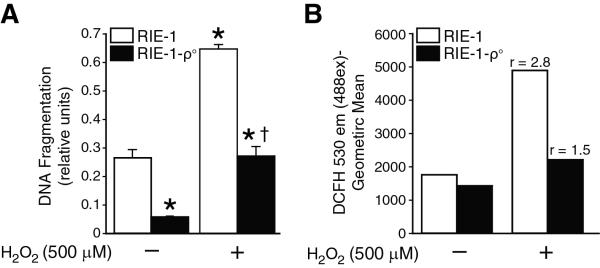
(A) RIE-1 and mitochondria silenced cells (RIE-1-ρ-°) were treated with H2O2 (500 μM) as before. Apoptosis was measured by DNA fragmentation ELISA. Data represent triplicate determinations (mean ±SEM; *p<0.05 vs. control, †p<0.05 vs. H2O2-treated RIE-1 cells). The RIE-1-ρ-° cells demonstrated decreased cell death after treatment with H2O2. (B) The ratio of stimulated v. baseline ROS levels increased (r>1) in H2O2-treated RIE-1 cells with significantly attenuated ROS production in RIE-1-ρ-° cells.
Effects of IGF-1 and EGF on ROS-mediated cell death and mitochondrial apoptotic signaling
We have previously shown that oxidative stress induces activation of anti-apoptotic PI3-K/Akt signaling pathway, causing mitochondria-mediated intestinal epithelial cell apoptosis via oxidant-induced mitochondrial membrane potential drop. Moreover, we demonstrated enhanced survival of intestinal epithelial cells with IGF-1 pretreatment, and PI3-K-dependent restoration of mitochondrial membrane potential. Hence, we now wanted to determine whether growth factor pretreatment would affect intestinal epithelial cell survival via mitochondria-dependent manner. We first examined growth factor-dependent pathway activation in RIE-1 cells stimulated with IGF-1- and EGF, with enhanced activation of PI3-K/Akt survival pathway observed only with IGF-1 pretreatment of cells (Fig. 3A). This effect was not observed in EGF-pretreated cells. With IGF-1 pretreatment alone and in combination with EGF, we observed significant attenuation of DNA fragmentation and RIE-1 cell death, whereas, EGF pretreatment alone did not demonstrate any protective effect during oxidative stress in RIE-1 cells (Fig. 3B).
Figure 3. Effects of IGF-1 and EGF on cell death and mitochondrial apoptotic signaling in H2O2-treated RIE-1 cells.

(A) IGF-1 pretreatment resulted in enhanced anti-apoptotic PI3-K/Akt pathway activation in RIE-1 cells by Western blotting. (B) Pretreatment with IGF-1 showed attenuation of RIE-1 cell apoptosis during oxidative stress, as shown by cell death ELISA. Data represent triplicate determinations (mean ±SEM; *p<0.05 vs. control, †p<0.05 vs. H2O2-treated RIE-1 cells). (C) IGF-1 and EGF pretreatment of RIE-1 cells during oxidative stress demonstrated reduced expression levels only in AIF and cytochrome C proapoptotic mitochondrial molecules by Western blotting. Equal β-actin levels and densitometry indicate even loading.
Next, we examined the expression of mitochondrial apoptotic markers in growth factor-pretreated RIE-1 cells during oxidative stress. Western blot analysis showed that IGF-1 and EGF significantly attenuated cytochrome C and AIF expression levels in RIE-1 cells during H2O2-induced oxidative stress without altering APAF-1 and ATP synthase b expression levels confirmed by densitometric analysis (Fig. 3C). These findings suggest that growth factors, IGF-1 and EGF, may not directly affect expression levels of mitochondrial apoptotic markers, but might exert their anti-apoptotic effects via different, mitochondria-independent mechanisms.
IGF-1 and EGF-pretreated FHs74Int cells show increased survival during oxidative stress
Lastly, we wanted to compare and validate our findings in fetal human intestinal epithelial cells. As shown in Figure 4A, we found significant cell death with H2O2-induced oxidative stress in FHs74 Int cells. Furthermore, we also found significantly decreased apoptosis with growth factor pretreatment; this protective effect was both IGF-1- and EGF-dependent (Fig. 4B). Interestingly, there was no significant difference between IGF-1 and EGF groups. These results were in contrast to our RIE-1 data, where the protective anti-apoptotic effect observed during oxidative stress was primarily IGF-dependent only.
Figure 4. ROS induces cell death and mitochondrial apoptotic signaling in FHs74 Int cells.
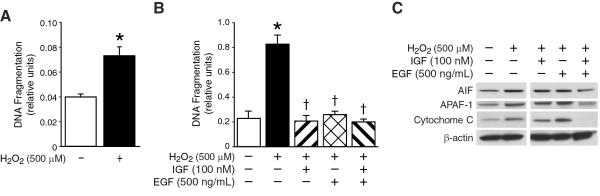
(A) FHs74 Int cells were treated with H2O2 (500 μM) as before. Apoptosis was measured by DNA fragmentation ELISA. Data represent triplicate determinations (mean ±SEM; *p<0.05 vs. control). H2O2 treatment resulted in significant cellular apoptosis. (B) FHs74 Int cells were serum starved, pretreated with IGF-1, EGF or in combination and then treated with H2O2 as before. Apoptosis was measured by DNA fragmentation ELISA. Data represent triplicate determinations (mean ±SEM; *p<0.05 vs. control, †p<0.05 vs. H2O2-treated FHs74 Int cells). IGF-1- and EGF-pretreated FHs74 Int cells showed increased survival during oxidative stress. (C) FHs74 Int cells were serum starved, pretreated with IGF-1, EGF or in combination and then treated with H2O2 as before. H2O2 treatment resulted in significant increases in AIF, APAF-1 and cytochrome C expression by Western blotting. There was significant attenuation of this effect when FHs74 Int cells were pretreated with both IGF-1 and EGF, suggesting synergistic effect on modulating mitochondrial apoptotic pathway activation. Equal β-actin levels indicate even loading.
To determine whether growth factor pretreatment of FHs74 Int cells during H2O2-induced oxidative stress induced any attenuating effect on activation of mitochondrial apoptotic pathways, we analyzed cell lysates for the expression levels of mitochondrial apoptotic markers. Growth factor pretreatment, either IGF-1 or EGF alone, in FHs74 Int cells did not significantly alter the expression of mitochondrial apoptotic markers; however, in combination, IGF-1 and EGF pretreated fetal human intestinal epithelial cells showed significant attenuation of AIF, APAF-1 and cytochrome C mitochondrial apoptotic markers (Fig. 4C). These findings suggest that growth factors can alter mitochondrial apoptotic signaling and play anti-apoptotic role in FHs74 Int cells during oxidative stress.
DISCUSSION
NEC is a challenging disease process with low survival rate that affects predominantly low-birth weight premature infants. Despite many clinical advances made over the years, premature infants are still at high risk of developing NEC occurring at random despite significant implementation of preventive measures such as protocol-based incremental enteral feeding regimen. Mitochondrial insults, including oxidative damage, can lead to significant ROS accumulation and further mitochondrial DNA damage, thus propagating death signaling [14]. Mitochondria are also the arsenal of pro-apoptotic signaling. Mitochondrion and its components are both the targets and mediators of oxidative injury [15]. The role of mitochondria in apoptotic signaling encompasses interpretation and amplification of cellular stress signals coming from various cellular compartments, which associate with mitochondrial plasma membrane by modifying its permeability.
Activation of apoptotic signaling can take shape by several mechanisms that include cytochrome C release and activation of caspase signaling cascade. Non-caspase mechanism of mitochondrial apoptotic signaling involves mitochondrial pro-apoptotic molecules such as AIF, endonuclease G and Smac/DIABLO. In particular, AIF and endonuclease G release from mitochondrion into cytosol and translocation to nucleus induces direct nuclear DNA fragmentation, whereas Smac/DIABLO release into cytosol, coupled with increased ROS generation, further amplifies activated caspase signaling cascade. Such variety of mitochondria-regulated apoptotic signaling allows for programmed cell death execution at the time of cellular stress [16–18]. It becomes apparent that most cell signals that induce death commonly converge on mitochondria to initiate apoptotic pathway. Hence, mitochondria are early mediators of apoptosis and a major source of intracellular ROS.
In this study, we have demonstrated that oxidative stress leads to increased intracellular ROS production by mitochondria and activation of mitochondrial apoptotic signaling pathways in both human fetal and rat intestinal epithelial cells. We also observed attenuated apoptotic response with application of growth factors, IGF-1 and EGF, during oxidative stress-induced apoptosis and decreased expression of mitochondrial apoptotic markers. These findings suggest that during experimental and in vitro NEC, mitochondria play a pivotal role in further propagating oxidative stress and cell death by contributing the vast majority of newly produced intracellular ROS, and activating caspase-dependent and independent apoptotic signaling pathways. Furthermore, exogenous application of two major growth factors abundant in breast milk, IGF-1 and EGF, show significant protective effect, and warrant further in-depth investigations. Successful application of growth factors in the clinical setting of NEC may be of value, and further studies focusing on mitochondrial functional stabilization by the abovementioned and other growth factors may be of significant therapeutic value. Modulating intestinal mitochondrial dysfunction and silencing mitochondrial apoptotic signaling may provide therapeutic benefit to the neonates with NEC.
ACKNOWLEDGEMENTS
The authors thank Karen Martin for manuscript preparation. This work was supported by grants R01 DK61470, R01 DK48498, P01 DK35608 and T32 DK07639 from the National Institutes of Health and a grant 8580 from Shriners Hospital for Children.
Footnotes
*Presented at the XXIIIrd International Symposium of Paediatric Surgical Research, Tokyo, Japan
REFERENCES
- 1.Keshavarzian A, Morgan G, Sedghi S, Gordon JH, Doria M. Role of reactive oxygen metabolites in experimental colitis. Gut. 1990;31(7):786–790. doi: 10.1136/gut.31.7.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keshavarzian A, Sedghi S, Kanofsky J, List T, Robinson C, Ibrahim C, Winship D. Excessive production of reactive oxygen metabolites by inflamed colon: analysis by chemiluminescence probe. Gastroenterology. 1992;103(1):177–185. doi: 10.1016/0016-5085(92)91111-g. doi:S0016508592002841 [pii] [DOI] [PubMed] [Google Scholar]
- 3.Clark DA, Fornabaio DM, McNeill H, Mullane KM, Caravella SJ, Miller MJ. Contribution of oxygen-derived free radicals to experimental necrotizing enterocolitis. Am J Pathol. 1988;130(3):537–542. [PMC free article] [PubMed] [Google Scholar]
- 4.Banan A, Choudhary S, Zhang Y, Fields JZ, Keshavarzian A. Oxidant-induced intestinal barrier disruption and its prevention by growth factors in a human colonic cell line: role of the microtubule cytoskeleton. Free Radic Biol Med. 2000;28(5):727–738. doi: 10.1016/s0891-5849(00)00160-x. doi:S0891-5849(00)00160-X [pii] [DOI] [PubMed] [Google Scholar]
- 5.Banan A, Farhadi A, Fields JZ, Mutlu E, Zhang L, Keshavarzian A. Evidence that nuclear factor-kappa B activation is critical in oxidant-induced disruption of the microtubule cytoskeleton and barrier integrity and that its inactivation is essential in epidermal growth factor-mediated protection of the monolayers of intestinal epithelia. J Pharmacol Exp Ther. 2003;306(1):13–28. doi: 10.1124/jpet.103.047415. doi:10.1124/jpet.103.047415 306/1/13 [pii] [DOI] [PubMed] [Google Scholar]
- 6.Kruidenier L, Kuiper I, Lamers CB, Verspaget HW. Intestinal oxidative damage in inflammatory bowel disease: semi-quantification, localization, and association with mucosal antioxidants. J Pathol. 2003;201(1):28–36. doi: 10.1002/path.1409. doi:10.1002/path.1409. [DOI] [PubMed] [Google Scholar]
- 7.Kruidenier L, Kuiper I, Van Duijn W, Mieremet-Ooms MA, van Hogezand RA, Lamers CB, Verspaget HW. Imbalanced secondary mucosal antioxidant response in inflammatory bowel disease. J Pathol. 2003;201(1):17–27. doi: 10.1002/path.1408. doi:10.1002/path.1408. [DOI] [PubMed] [Google Scholar]
- 8.Buffinton GD, Doe WF. Depleted mucosal antioxidant defences in inflammatory bowel disease. Free Radic Biol Med. 1995;19(6):911–918. doi: 10.1016/0891-5849(95)94362-h. doi:089158499594362H [pii] [DOI] [PubMed] [Google Scholar]
- 9.D'Odorico A, Bortolan S, Cardin R, D'Inca R, Martines D, Ferronato A, Sturniolo GC. Reduced plasma antioxidant concentrations and increased oxidative DNA damage in inflammatory bowel disease. Scand J Gastroenterol. 2001;36(12):1289–1294. doi: 10.1080/003655201317097146. [DOI] [PubMed] [Google Scholar]
- 10.Koutroubakis IE, Malliaraki N, Dimoulios PD, Karmiris K, Castanas E, Kouroumalis EA. Decreased total and corrected antioxidant capacity in patients with inflammatory bowel disease. Dig Dis Sci. 2004;49(9):1433–1437. doi: 10.1023/b:ddas.0000042242.22898.d9. [DOI] [PubMed] [Google Scholar]
- 11.Baregamian N, Song J, Bailey CE, Papaconstantinou J, Evers BM, Chung DH. Tumor necrosis factor-alpha and apoptosis signal-regulating kinase 1 control reactive oxygen species release, mitochondrial autophagy, and c-Jun N-terminal kinase/p38 phosphorylation during necrotizing enterocolitis. Oxid Med Cell Longev. 2009;2(5):297–306. doi: 10.4161/oxim.2.5.9541. doi:9541 [pii] 10.4161/oxim.2.5.9541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baregamian N, Song J, Jeschke MG, Evers BM, Chung DH. IGF-1 protects intestinal epithelial cells from oxidative stress-induced apoptosis. J Surg Res. 2006;136(1):31–37. doi: 10.1016/j.jss.2006.04.028. doi:S0022-4804(06)00221-6 [pii] 10.1016/j.jss.2006.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical biochemistry. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 14.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005;70(2):200–214. doi: 10.1007/s10541-005-0102-7. doi:BCM70020246 [pii] [DOI] [PubMed] [Google Scholar]
- 15.Fiskum G, Murphy AN, Beal MF. Mitochondria in neurodegeneration: acute ischemia and chronic neurodegenerative diseases. J Cereb Blood Flow Metab. 1999;19(4):351–369. doi: 10.1097/00004647-199904000-00001. doi:10.1097/00004647-199904000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408(6809):239–247. doi: 10.1038/35041687. doi:10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 17.Lemasters JJ. Dying a thousand deaths: redundant pathways from different organelles to apoptosis and necrosis. Gastroenterology. 2005;129(1):351–360. doi: 10.1053/j.gastro.2005.06.006. doi:S0016508505011042 [pii] [DOI] [PubMed] [Google Scholar]
- 18.Lam E, Kato N, Lawton M. Programmed cell death, mitochondria and the plant hypersensitive response. Nature. 2001;411(6839):848–853. doi: 10.1038/35081184. doi:10.1038/35081184. [DOI] [PubMed] [Google Scholar]