

Abstract
Introduction
Mucopolysaccharidosis I (MPS I) is a lysosomal storage disorder characterized by deficient α-L-iduronidase activity leading to accumulation of poorly degraded dermatan and heparan sulfate glycosaminoglycans (GAGs). MPS I is associated with significant cervical spine disease, including vertebral dysplasia, odontoid hypoplasia, and accelerated disc degeneration, leading to spinal cord compression and kypho-scoliosis. The objective of this study was to establish the nature and rate of progression of cervical vertebral bone disease in MPS I using a canine model.
Methods
C2 vertebrae were obtained post-mortem from normal and MPS I dogs at 3, 6 and 12 months-of-age. Morphometric parameters and mineral density for the vertebral trabecular bone and odontoid process were determined using micro-computed tomography. Vertebrae were then processed for paraffin histology, and cartilage area in both the vertebral epiphyses and odontoid process were quantified.
Results
Vertebral bodies of MPS I dogs had lower trabecular bone volume/total volume (BV/TV), trabecular thickness (Tb.Th), trabecular number (Tb.N) and bone mineral density (BMD) than normals at all ages. For MPS I dogs, BV/TV, Tb.Th and BMD plateaued after 6 months-of-age. The odontoid process appeared morphologically abnormal for MPS I dogs at 6 and 12 months-of-age, although BV/TV and TMD were not significantly different from normals. MPS I dogs had significantly more cartilage in the vertebral epiphyses at both 3 and 6 months-of-age. At 12 months-of-age, epiphyseal growth plates in normal dogs were absent, but in MPS I dogs they persisted.
Conclusions
In this study we report reduced trabecular bone content and mineralization, and delayed cartilage to bone conversion in MPS I dogs from 3 months-of-age, which may increase vertebral fracture risk and contribute to progressive deformity. The abnormalities of the odontoid process we describe likely contribute to increased incidence of atlanto-axial subluxation observed clinically. Therapeutic strategies that enhance bone formation may decrease incidence of spine disease in MPS I patients.
Keywords: Spine, trabecular bone, odontoid process, glycosaminoglycans, development, mucopolysaccharidosis
1. Introduction
The mucopolyaccharidoses (MPS) are a subset of lysosomal storage disorders characterized by deficiencies in enzymes that degrade glycosaminoglycans (GAGs) [1]. While disease severity varies depending on the specific type of MPS, manifestations can occur in the skeleton, liver, cardiorespiratory system, eyes, ears, central nervous system, and other sites. Mucopolysaccharidosis I (MPS I), also known as Hurler Syndrome (Hurler-Sheie and Scheie Syndromes are attenuated forms), is characterized by deficient α-L-iduronidase (IDUA) activity, leading to accumulation of poorly degraded dermatan and heparan sulfate glycosaminoglycans [2].
Along with other morbidities, MPS I is associated with significant spine disease [3]. Radiologically, manifestations include vertebral dysplasia and subluxation, and accelerated disc degeneration, leading to spinal cord compression, and kyphotic and scoliotic deformities [3–5]. In the cervical spine, hypoplasia of the odontoid process has been reported and is associated with atlanto-axial subluxation and spinal cord compression [3, 6–9]. Surgical correction of spine disease is required in ~10% of patients with the severe form of MPS I (Hurler syndrome) at a median age of 4 years, ~15% of patients with intermediate-severity MPS I at a median age of 13 years, and ~15% of attenuated-severity MPS I at a median age of 21 years [10]. The lower reported incidence in patients with the most severe form of MPS I likely reflects their earlier mortality.
Current, clinically available treatment options for MPS I include hematopoietic stem cell transplantation (HSCT), and enzyme replacement therapy (ERT). However, these treatments have proven relatively ineffective at correcting spine disease [4, 5, 11–13]. While abnormalities of the bones of the spine in MPS I patients have been noted radiologically, these descriptions are often vague, and the nature and progression of the pathological changes is poorly understood. Improved understanding of spine disease progression in MPS I and the underlying pathophysiology will lead to more rapid diagnosis and identification of new therapeutic targets.
Established animal models of MPS I include knockout mouse models and naturally occurring canine and feline models [14]. MPS I dogs have a mutation in intron 1 of the IDUA gene [15]. MPS I dogs show radiographic and gross pathological evidence of spine disease, including abnormalities of the vertebral bones [16]. These abnormalities are particularly prevalent in the cervical spine. Spinal cord compression is also a common characteristic [17]. Therefore, MPS I dogs present an excellent, clinically relevant model for investigating the pathogenesis and treatment of spine disease in MPS I.
In our previous work, we demonstrated that the vertebral bodies of MPS VII (beta-glucuronidase deficiency) dogs have large cartilaginous lesions in the ventral and dorsal regions of the epiphyses at 6 months-of-age [18]. We hypothesized that these lesions represent delayed conversion of cartilage to bone during postnatal development. While the musculoskeletal manifestations of MPS VII are more severe than those in MPS I, likely due to the additional accumulation of chondroitin sulfate, it remained unknown whether these cartilaginous vertebral lesions are also present in MPS I. In human MPS I patients, lesions in the form of abnormal soft tissue have been noted at the tip of the odontoid process, which may suggest abnormal or delayed bone formation [3].
The objective of this study was to undertake a comprehensive investigation into the progression of cervical spine vertebral bone disease in MPS I dogs during postnatal development, focusing on both the trabecular bone and the odontoid process.
2. Materials and Methods
2.1. Animals
The dogs used in this study were raised at the School of Veterinary Medicine at the University of Pennsylvania, under NIH and USDA guidelines for the care and use of animals in research. MPS I dogs were identified at birth by DNA mutation analysis. Most normal controls were littermates of MPS I dogs or had at least one parent in common. A total of 29 dogs were studied. Euthanasia was performed at each of 3 ages: 3 months (n=5 normals, n=4 MPS I), 6 months (n=5 normals and n=4 MPS I), or 12 months (n=6 normals and n=5 MPS I) using 80 mg/kg of sodium pentobarbital in accordance with the American Veterinary Medical Association guidelines. The cervical spine was dissected out immediately following euthanasia and the C2 vertebra was isolated and frozen at −20°C.
2.2 Micro-Computed Tomography (MicroCT)
Excised C2 vertebrae were thawed, cleaned of surrounding tissue and scanned using high-resolution microCT (VivaCT40; Scanco Medical AG, Brüttisellen, Switzerland). Sequential axial images through the vertebral body were obtained using an isotropic voxel size of 19 μm, an integration time of 380 ms, peak tube voltage of 70 kV, current of 0.114 mA, and an acquisition of 1000 projections per 180°. A three-dimensional Gaussian filter of 1.2 with a limited, finite filter support of 2 was used for noise suppression, and mineralized tissue was segmented from air or soft tissue using a threshold of 220. Image acquisition commenced from the spongiosa adjacent to the caudal epiphyseal growth plate, or the caudal vertebral endplate (where growth plates had closed), and extended cranially for 100 slices.
Standard 3D morphometric analyses were performed using Scanco software to calculate bone volume/total volume (BV/TV), trabecular thickness (Tb.Th), trabecular spacing (Tb.Sp) and trabecular number (Tb.N). Apparent mineral density (bone mineral density, BMD) and material mineral density (tissue mineral density, TMD) were also determined and calibrated against hydroxyapatite (HA) standards (0–784 mg HA/cm3).
For each vertebra, the entire odontoid process was imaged at 19 μm isotropic resolution using the above settings. As there were no distinct trabecular or cortical zones evident within these structures at any age examined, volumes of interest were defined as the entire structure extending from the vertebral body interface, and analyzed to determine BV/TV, BMD and TMD. Volumes of interest excluded large cartilaginous regions noted to be present near the tip of the odontoid in the 3-month-old samples.
Significant effects of disease and age for measured parameters were established via 2-way ANOVAs (GraphPad Prism 5.02; GraphPad Software Inc, La Jolla, USA). Where a significant effect of either factor was detected, post-hoc pairwise tests were performed. Differences were considered significant for p<0.05.
2.3 Histology
Following microCT imaging, C2 vertebral bodies were fixed in 4% paraformaldehyde for one week, then decalcified using formic acid/EDTA (Formical 2000; Decal Chemical Corporation, Tallman, USA). A 3 mm-thick mid-sagittal slab was isolated, and divided into 2 regions encompassing either the odontoid process or the vertebral body, which were then processed for paraffin-embedded histology. Sections 10 μm thick were double-stained with Alcian blue and picrosirius red to demonstrate GAG and collagen, respectively, then imaged and analyzed under bright field light microscopy (Eclipse 90i; Nikon, Tokyo, Japan). The area of cartilage in the caudal epiphysis, inclusive of the growth plate, as a percentage of the total epiphysis area was quantified for 3 and 6 month-old samples (ImageJ; NIH, Bethesda, USA). Significant effects of disease and age were established as described in 2.2. The percent cartilage in the odontoid was calculated only at 3 months-of-age (at 6 months both MPS I and normals were fully calcified), with differences between normal and MPS I samples established using an unpaired t-test (p<0.05).
3. Results
3.1 Trabecular Bone
Observed histologically and via 3D microCT reconstructions, C2 vertebral bodies appeared to have less trabecular bone volume than normals at each of three ages examined (Figure 1). These observations were supported by the results of quantitative morphometric analyses (Figure 2). BV/TV was lower for MPS I dogs than normals at 3, 6 and 12 months-of-age (55%, 60% and 49% of normal respectively, all p<0.05, Figure 2A). Similarly, Tb.Th was lower for MPS I dogs than normals at 3, 6 and 12 months-of-age (84% (not significant), 93% (not significant) and 76% (p<0.05) of normal respectively, Figure 2B). Tb.Sp was greater for MPS I dogs than normals at 3, 6 and 12 months-of-age (1.2, 1.4 and 1.5-fold normal respectively, all p<0.05, Figure 2C). Tb.N was lower for MPS I dogs than normals at 3, 6 and 12 months-of-age (81%, 70% and 64% of normal respectively, all p<0.05, Figure 2D). Bone mineral density was lower for MPS I dogs than normals at 3, 6 and 12 months-of-age (71%, 78% and 61% of normal respectively, all p<0.05, Figure 2E). Tissue mineral density was not significantly different between normal and MPS I dogs at any age (Figure 2F). For normal dogs, BV/TV, Tb.Th and BMD increased significantly from 3 to 6, and from 6 to 12 months-of-age; however, for MPS I dogs these properties initially increased significantly from 3 to 6 months-of-age then plateaued. Tissue mineral density increased significantly for both normal and MPS I dogs from 6 to 12 months-of-age, but not from 3 to 6 months-of-age. Trabecular spacing remained unchanged with age for normal dogs. For MPS I dogs, Tb.Sp showed an increasing trend with age but this was not significant. Tb.N was significantly greater at 12 months-of age compared to 3 months-of-age for normal dogs, but remained static across all ages for MPS I dogs.
Figure 1. Histology and microCT reconstructions of C2 vertebral bone.

Mid-sagittal, histological sections, double-stained with Alcian blue (glycosaminoglycans) and picrosirius red (collagen), from normal and MPS I dogs aged 3, 6 and 12 months (A, B, E, F, I and G), with 3D microCT reconstructions of trabecular bone for the same samples (C, D, G, H, K and L). Lower trabecular bone volume is apparent for MPS I samples at all ages, but is most striking at 12 months-of-age. All histological images are oriented with the ventral (anterior) side at the bottom; ep = caudal vertebral end plate. Scale bars = 2 mm (histology) or 1 mm (microCT). Dashed lines indicate approximate location of odontoid process attachment.
Figure 2. MicroCT analysis of C2 vertebral trabecular bone.
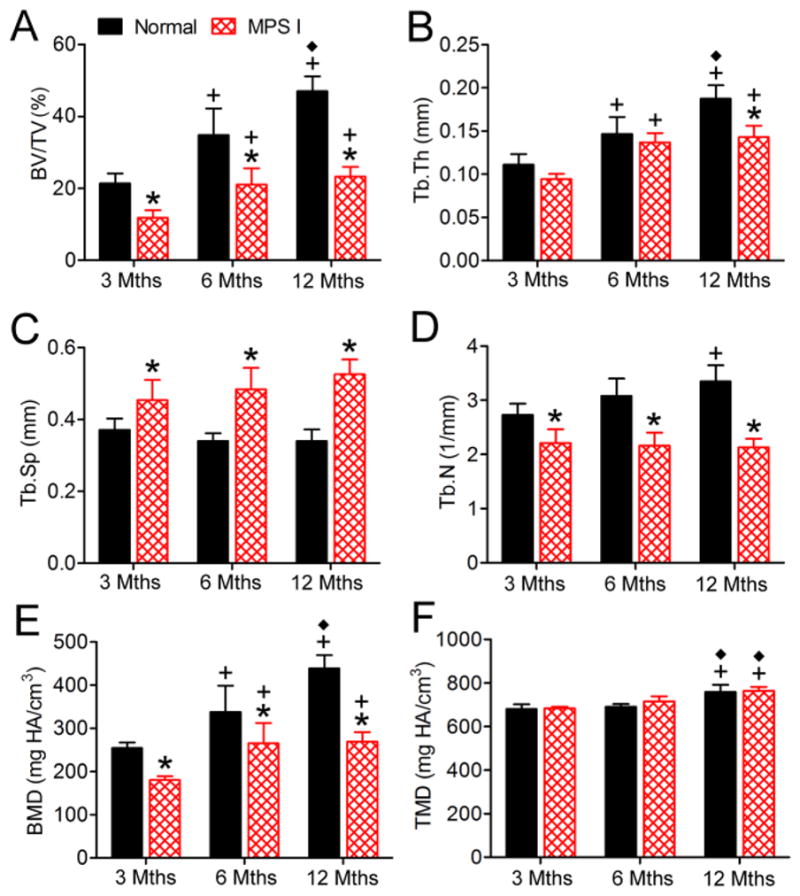
A. Bone volume/total volume (BV/TV). B. Trabecular thickness (Tb.Th). C. Trabecular spacing (Tb.Sp). D. Trabecular number (Tb.N). E. Bone mineral density (apparent density, BMD). F Tissue. mineral density (material density, TMD). *p<0.05 vs normal; +p<0.05 vs 3 months; and ◆p<0.05 vs 6 months.
3.2 Odontoid Process
Observed histologically and via 3D microCT reconstructions, the odontoid process appeared morphologically abnormal in MPS I dogs compared to normals, particularly at 6 and 12 months-of-age (Figure 3). These abnormalities included reduced overall size, narrowness in the lateral aspect, irregular surface morphology and reduced cartilage at the ventral articulating surface (arrows, Figure 3E, F, I and J). However, quantitative analyses (Figure 4) demonstrated no significant differences in BV/TV, BMD or TMD between MPS I dogs and normals at any of the 3 ages examined. With age, BV/TV increased significantly from 3 to 6 months-of-age, but not from 6 to 12 months-of-age for both normal and MPS I dogs. Bone mineral density increased significantly from 3 to 6, and from 6 to 12 months-of-age for normal dogs, and from 3 to 6 but not from 6 to 12 months-of-age for MPS I dogs. Tissue mineral density increased significantly from 3 to 6, and from 6 to 12 months-of-age in normal dogs, and from 6 to 12 months-of-age in MPS I dogs.
Figure 3. Histology and microCT reconstructionsf theo odontoid process.

Mid-sagittal, histological sections, double-stained with Alcian blue (glycosaminoglycans) and picrosirius red (collagen), from normal and MPS I dogs aged 3, 6 and 12 months (A, B, E, F, I and G), with microCT reconstructions of the same samples (C, D, G, H, K and L). MPS I samples appear smaller and narrower, and had irregular surface morphology at 6 and 12 months-of-age. Decreased cartilage on the ventral articulating surface (arrows) was also evident at these ages for MPS I samples. At 3 months-of-age, delayed calcification was apparent towards the tip of the MPS I samples. For all images, ventral (anterior) is towards the bottom and caudal (posterior) is towards the right. Dashed lines indicate approximate location of vertebral body attachment. All scale bars = 2 mm.
Figure 4. MicroCT analysis of the odontoid process.
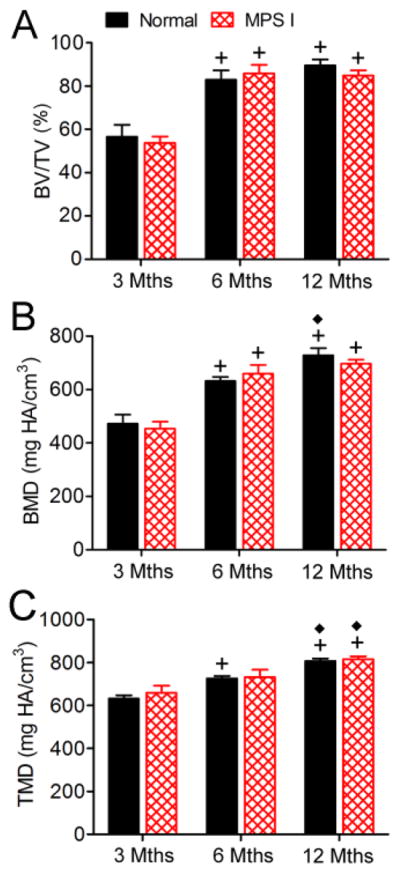
A. Bone volume/total volume (BVTV). B. Bone mineral density (apparent density, BMD). C. Tissue mineral density (material density, TMD). *p<0.05 vs normal; +p<0.05 vs 3 months; and ◆p<0.05 vs 6 months.
3.3 Delayed Cartilage to Bone Conversion
Observed histologically, MPS I dogs had increased cartilaginous regions in the epiphyses, particularly in the ventral regions, at 3 and 6 months-of-age compared to normals (asterisks, Figures 5A–D). Quantitative analyses revealed greater cartilage area as a percentage of the total epiphysis area in MPS I dogs compared to normals at 3 and 6 months-of-age (1.3 and 1.7-fold normal respectively, both p<0.05, Figure 6A). At 12 months-of age, epiphyseal growth plates in normal dogs were absent; however, the growth plates persisted in all five MPS I dogs at this age (representative Figures 5E and 5F). In the odontoid process, at 3 months-of-age, there was slightly greater cartilage area for MPS I dogs compared to normals (1.2-fold normal, not significant, Figure 6B).
Figure 5. Cartilage to bone conversion in the C2 vertebral epiphysis.

Mid-sagittal histological sections double-stained with Alcian blue (glycosaminoglycans) and picrosirius red (collagen) illustrating increased cartilage in the ventral epiphyses (*) at 3 months-of-age (A and B) and at 6 months-of-age (C and D). At 12 months-of age, the epiphyseal growth plate in normal animals was absent (E); however, in MPS I animals, this growth plate persisted (F). Scale bars = 500 μm (A–D) and 1 mm (E and F); gp = growth plate and ep = caudal vertebral endplate. All images are oriented with the ventral side at the bottom.
Figure 6. Quantification of cartilage area in the C2 vertebral epiphysis and odontoid process.

A. Cartilage area (inclusive of the growth plate) as a percentage of the total epiphysis area was determined from mid-sagittal Alcian blue/picrosirius red-stained histological sections, for 3 and 6 month-old samples. B. Cartilage area in the odontoid process as a percentage of total tissue area was determined from mid-sagittal Alcian blue/picrosirius red-stained histological sections, for 3 month-old samples. *p<0.05 vs normal; +p<0.05 vs 3 months.
4. Discussion
Spine disease is prevalent among mucopolysaccharidosis I patients [19]. However, quantitative studies of vertebral bone pathology in MPS I animal models are rare. To our knowledge this is the first study to examine bone pathology in a large animal model of MPS using microCT. Herati et al. demonstrated that MPS I dogs have a higher incidence of vertebral abnormalities compared to normals observed on plain radiographs [16]. These abnormalities included pedicle widening, beaking at the vertebral rim and facet joint fusion. C2 vertebral body lengths for MPS I dogs were 91% of normal (not significant) [16]. MPS I mice have thickened vertebral arches and spinal stenosis [20]. In the current study we extend these important findings to provide the first quantitative evidence of significantly reduced trabecular bone volume and mineral density in the cervical spines of MPS I dogs from relatively early in postnatal skeletal development. At 3 months-of-age, MPS I dogs had significantly lower bone volume and mineral density. In normal dogs these properties increased progressively with age, but in MPS I dogs they plateaued after 6 months-of-age, and at 12 months-of-age were ~50% and 60% of normal respectively. These results complement previous observations: osteopenia has been noted in the spines and long bones of skeletally mature MPS I dogs on plain radiographs [11, 21]. The reduced vertebral bone volume and mineralization may place MPS I patients at increased risk of vertebral fracture.
In our previous work we showed that MPS VII dogs have radiolucent lesions in the vertebral epiphyses [18, 22, 23]. These lesions were shown to be first evident at 1 month-of-age [22], and to persist throughout the animals’ lifetimes even in the presence of retroviral gene therapy [23], which successfully increased survival times and attenuated the severity of other skeletal disease. At 6 months-of-age, these lesions were found to significantly compromise the mechanical stability of the intervertebral joint, strongly implicating them in the progression of spinal deformity [18]. We hypothesized that these lesions represented a delay, and ultimately a failure, to convert cartilage to bone during the normal course of endochondral ossification. In the current study we present evidence of a similar delay in cartilage to bone conversion in MPS I dogs. Specifically, we found a significantly increased cartilage area in the vertebral epiphyses of MPS I dogs relative to normals at 3 and 6 months-of-age. Increased cartilage was most evident in the ventral regions, which was also the location of the most severe lesions in MPS VII dogs. Of note, the peripheral regions of the epiphysis are the last to calcify during development, suggesting that impairment of bone formation rate may occur concomitantly with progressive GAG accumulation as animals age. These findings complement previous work that described increased growth-plate associated cartilage in the vertebra and long bones of MPS I mice [24], although in that study the region analyzed was the sub-epiphyseal region. An additional finding in the current study was the abnormal persistence of the growth plate at 12 months-of-age, also suggestive of delayed cartilage-bone conversion. While the severity of these “lesions” is certainly less than that found in MPS VII, their presence in MPS I may point to common underlying pathophysiology for spine disease in both of these disorders.
While abnormal accumulation of GAGs, both within cells and in the extracellular matrix, undoubtedly is the instigator, the precise mechanisms by which those GAGs disrupt the normal physiological processes that regulate bone development and homeostasis are largely unknown. It has been suggested that dermatan and heparan sulfate accumulation in MPS I may affect bone quality by modulating cathepsin K activity [24]. Cathepsin K is an enzyme secreted by osteoclasts during bone resorption, and its collagenolytic activity is dependent on specific types of GAGs. Collagenolytic activity of cathepsin K is enhanced by chondroitin and keratan sulfates, whereas heparan and dermatan sulfates inhibit it [25]. Since MPS I results in accumulation of dermatan and heparan sulfates that inhibit cathepsin K, this may be one possible explanation for the slower removal of epiphyseal cartilage in MPS I vertebrae. Were this same to hold true for the trabecular bone, then perhaps higher bone volume would be expected in MPS I due to less efficient bone resorption. However, we found that the opposite was true, with significantly less trabecular bone in MPS I vertebrae at all ages examined. Also, significantly lower Tb.N in MPS I dogs at all ages may indicate a deficit in the formation of primary spongiosa, which would likely be independent of osteoclast function. Future studies will comprehensively examine the contribution of osteoclast and cathepsin k dysfunction to the observed pathologies.
There is evidence that GAGs accumulating in MPS drive an inflammatory cascade through activation of the TLR4 signaling pathway [26–28]. Upregulation of TLR4 has been demonstrated in articular chondrocytes and synoviocytes from MPS VI rats [26], TLR4/MPSVII double knockout mice had improved cranial and long bone morphology and growth plate organization compared to MPS VII mice [27]. In MPS VII dogs, the annulus fibrosus (AF) of the intervertebral disc was found to exhibit significantly higher mRNA expression of TLR4 [22]. Both the AFs and vertebral epiphyses of MPS VII dogs had significantly elevated levels of destructive proteases, including cathepsins and matrix metalloproteinases, compared to normal dogs [22]. While the presence of a similar inflammatory cascade in MPS I vertebrae has not been evaluated, upregulation of these destructive proteases could contribute to bone loss.
Alterations in the rate of chondrocyte maturation may contribute to bone pathology in MPS I. During endochondral ossification, chondrocytes progress through stages of differentiation, including proliferation and hypertrophy, before ultimately undergoing apoptosis to be replaced by osteoblasts [29]. This process occurs both within the primary and secondary centers of ossification, and in the growth plate, facilitating longitudinal bone growth. Chondrocyte differentiation is regulated by a complex cascade of signaling events, involving members of fibroblast growth factor, bone morphogenetic protein and transforming growth factor-beta families of molecules, morphogens such as indian hedgehog and parathyroid hormone related peptide, and the Wnt/beta-catenin and retinoic acid pathways [30–33]. These and other factors interact to regulate the rate of chondrocyte proliferation and hypertrophy, vascular invasion and osteoblast recruitment. There is evidence that GAGs, particularly heparan sulfate, perform essential roles in both the distribution and activation of many of these signaling molecules [34, 35]. Future studies should assess whether these pathways are disrupted by abnormal accumulation of GAGs in MPS, leading to delayed and reduced bone formation. There is also evidence that chondrocyte maturation may be affected by an increasingly inflammatory microenvironment: recent evidence suggests that the inflammatory cytokines interleukin-1 beta and tumor necrosis factor alpha inhibit chondrocyte differentiation and bone growth [36, 37]. Impaired growth factor signaling during endochondral ossification may also affect fracture healing capacity in MPS I patients; this could be the subject of future investigations.
Hypoplasia of the odontoid process in MPS I patients has been implicated in the increased incidence of atlanto-axial subluxation, resulting impingement of the spinal cord and neurological deficits [3, 6–9]. While we did not find significant differences in BV/TV or TMD, there were clear abnormalities in the shape and size of the odontoid process in MPS I dogs when compared to normals, particularly at 6 and 12 months-of-age. These abnormalities may be the result of reduced chondrocyte proliferation and aberrant maturation during ossification, and could increase the likelihood of fracture or dislocation, especially when combined with increased ligamentous laxity.
Current clinical treatment strategies for MPS I, which include enzyme replacement therapy and hematopoietic stem cell transplantation, are relatively ineffective at improving the skeletal manifestations of the disorder [4, 5, 11–13]. Experimental treatments, such as neonatal retroviral-mediated gene therapy, have shown some efficacy in reducing vertebral bone disease in MPS I dogs [16]. Intrathecal enzyme replacement therapy from an early age was found to reduce the incidence of spinal cord compression in MPS I dogs [17]. These results and those of the current study highlight the importance of early diagnosis and intervention to prevent the onset and progression of musculoskeletal disease in MPS I. Ongoing work in our laboratory will assess whether enzyme replacement therapy, both in isolation and in combination with anti-inflammatory medications, is effective at preventing or attenuating spine disease in MPS I dogs. In the future, it may be worth considering whether drugs used to treat other metabolic bone diseases such as osteoporosis could also be useful for treating bone disease in MPS I. Indeed, the results of this study would suggest an osteoporosis-like phenotype. However, this would first require a more thorough understanding of the underlying pathophysiology, and is likely further complicated by the skeletal immaturity of prospective patients, and that such treatments may risk disrupting normal developmental processes.
Highlights.
MPS I dogs have lower vertebral trabecular bone volume and mineral density compared to normal dogs from 3 months-of-age.
MPS I dogs exhibit delayed cartilage to bone conversion in the vertebral epiphyses and retain growth plates at 12 months-of-age.
Abnormalities in the size and morphology of the odontoid process are consistent with increased incidence of atlanto-axial subluxation clinically.
Therapeutic strategies that enhance bone formation and mineralization may decrease incidence of spine disease in MPS I patients.
Acknowledgments
This work was funded by a grant from the University of Pennsylvania Center for Orphan Diseases Research and Therapy, and grants from the NIH (P40 OD010939, P30 AR050950 and R01 DK066448). The authors thank Dr Sherry Liu for technical advice on microCT analyses.
Footnotes
Author contributions to this study were as follows: LJS and JAC contributed to conceptual design, performed experiments and drafted the manuscript; MDB, CDA and PO performed experiments and critically revised the manuscript for important intellectual content; MEH contributed to conceptual design, supervised raising of the animals, performed post-mortems and critically revised the manuscript for important intellectual content; EMS, KPP and DME contributed to conceptual design and critically revised the manuscript for important intellectual content. All authors approved the final version of the manuscript prior to submission. MEH owns stock in BioMarin Pharmaceuticals, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Neufeld EF, Muenzer J. The Mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. 8. New York: McGraw-Hill; 2001. pp. 3421–3452. [Google Scholar]
- 2.Scott HS, Bunge S, Gal A, Clarke LA, Morris CP, Hopwood JJ. Molecular genetics of mucopolysaccharidosis type I: diagnostic, clinical, and biological implications. Hum Mutat. 1995;6:288–302. doi: 10.1002/humu.1380060403. [DOI] [PubMed] [Google Scholar]
- 3.Tandon V, Williamson JB, Cowie RA, Wraith JE. Spinal problems in mucopolysaccharidosis I (Hurler syndrome) J Bone Joint Surg Br. 1996;78:938–44. doi: 10.1302/0301-620x78b6.1279. [DOI] [PubMed] [Google Scholar]
- 4.Kachur E, Del Maestro R. Mucopolysaccharidoses and spinal cord compression: case report and review of the literature with implications of bone marrow transplantation. Neurosurgery. 2000;47:223–8. doi: 10.1097/00006123-200007000-00046. discussion 228–9. [DOI] [PubMed] [Google Scholar]
- 5.Weisstein JS, Delgado E, Steinbach LS, Hart K, Packman S. Musculoskeletal manifestations of Hurler syndrome: long-term follow-up after bone marrow transplantation. J Pediatr Orthop. 2004;24:97–101. doi: 10.1097/00004694-200401000-00019. [DOI] [PubMed] [Google Scholar]
- 6.Belani KG, Krivit W, Carpenter BL, Braunlin E, Buckley JJ, Liao JC, Floyd T, Leonard AS, Summers CG, Levine S, et al. Children with mucopolysaccharidosis: perioperative care, morbidity, mortality, and new findings. J Pediatr Surg. 1993;28:403–8. doi: 10.1016/0022-3468(93)90240-l. discussion 408–10. [DOI] [PubMed] [Google Scholar]
- 7.Hite SH, Peters C, Krivit W. Correction of odontoid dysplasia following bone-marrow transplantation and engraftment (in Hurler syndrome MPS 1H) Pediatr Radiol. 2000;30:464–70. doi: 10.1007/s002470000210. [DOI] [PubMed] [Google Scholar]
- 8.Miebach E, Church H, Cooper A, Mercer J, Tylee K, Wynn RF, Wraith JE. The craniocervical junction following successful haematopoietic stem cell transplantation for mucopolysaccharidosis type I H (Hurler syndrome) J Inherit Metab Dis. 2011;34:755–61. doi: 10.1007/s10545-011-9309-5. [DOI] [PubMed] [Google Scholar]
- 9.Thomas SL, Childress MH, Quinton B. Hypoplasia of the odontoid with atlanto-axial subluxation in Hurler’s syndrome. Pediatr Radiol. 1985;15:353–4. doi: 10.1007/BF02386776. [DOI] [PubMed] [Google Scholar]
- 10.Arn P, Wraith JE, Underhill L. Characterization of surgical procedures in patients with mucopolysaccharidosis type I: findings from the MPS I Registry. J Pediatr. 2009;154:859–64. doi: 10.1016/j.jpeds.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 11.Kakkis ED, McEntee MF, Schmidtchen A, Neufeld EF, Ward DA, Gompf RE, Kania S, Bedolla C, Chien SL, Shull RM. Long-term and high-dose trials of enzyme replacement therapy in the canine model of mucopolysaccharidosis I. Biochem Mol Med. 1996;58:156–67. doi: 10.1006/bmme.1996.0044. [DOI] [PubMed] [Google Scholar]
- 12.Sifuentes M, Doroshow R, Hoft R, Mason G, Walot I, Diament M, Okazaki S, Huff K, Cox GF, Swiedler SJ, Kakkis ED. A follow-up study of MPS I patients treated with laronidase enzyme replacement therapy for 6 years. Mol Genet Metab. 2007;90:171–80. doi: 10.1016/j.ymgme.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 13.Thomas JA, Jacobs S, Kierstein J, Van Hove J. Outcome after three years of laronidase enzyme replacement therapy in a patient with Hurler syndrome. J Inherit Metab Dis. 2006;29:762. doi: 10.1007/s10545-006-0457-y. [DOI] [PubMed] [Google Scholar]
- 14.Haskins M, Casal M, Ellinwood NM, Melniczek J, Mazrier H, Giger U. Animal models for mucopolysaccharidoses and their clinical relevance. Acta Paediatr Suppl. 2002;91:88–97. doi: 10.1111/j.1651-2227.2002.tb03117.x. [DOI] [PubMed] [Google Scholar]
- 15.Menon KP, Tieu PT, Neufeld EF. Architecture of the canine IDUA gene and mutation underlying canine mucopolysaccharidosis I. Genomics. 1992;14:763–8. doi: 10.1016/s0888-7543(05)80182-x. [DOI] [PubMed] [Google Scholar]
- 16.Herati RS, Knox VW, O’Donnell P, D’Angelo M, Haskins ME, Ponder KP. Radiographic evaluation of bones and joints in mucopolysaccharidosis I and VII dogs after neonatal gene therapy. Mol Genet Metab. 2008;95:142–51. doi: 10.1016/j.ymgme.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dickson PI, Hanson S, McEntee MF, Vite CH, Vogler CA, Mlikotic A, Chen AH, Ponder KP, Haskins ME, Tippin BL, Le SQ, Passage MB, Guerra C, Dierenfeld A, Jens J, Snella E, Kan SH, Ellinwood NM. Early versus late treatment of spinal cord compression with long-term intrathecal enzyme replacement therapy in canine mucopolysaccharidosis type I. Mol Genet Metab. 2010;101:115–22. doi: 10.1016/j.ymgme.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith LJ, Martin JT, Szczesny SE, Ponder KP, Haskins ME, Elliott DM. Altered lumbar spine structure, biochemistry, and biomechanical properties in a canine model of mucopolysaccharidosis type VII. J Orthop Res. 2010;28:616–22. doi: 10.1002/jor.21030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muenzer J, Wraith JE, Clarke LA, II, CPoMaToM Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123:19–29. doi: 10.1542/peds.2008-0416. [DOI] [PubMed] [Google Scholar]
- 20.Rowan DJ, Tomatsu S, Grubb JH, Montano AM, Sly WS. Assessment of bone dysplasia by micro-CT and glycosaminoglycan levels in mouse models for mucopolysaccharidosis type I, IIIA, IVA, and VII. J Inherit Metab Dis. 2012 doi: 10.1007/s10545-012-9522-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shull RM, Walker MA. Radiographic findings in a canine model of mucopolysaccharidosis I. Changes associated with bone marrow transplantation. Invest Radiol. 1988;23:124–30. doi: 10.1097/00004424-198802000-00008. [DOI] [PubMed] [Google Scholar]
- 22.Smith LJ, Baldo G, Wu S, Liu Y, Whyte MP, Giugliani R, Elliott DM, Haskins ME, Ponder KP. Pathogenesis of lumbar spine disease in mucopolysaccharidosis VII. Mol Genet Metab. 2012 doi: 10.1016/j.ymgme.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith LJ, Martin JT, O’Donnell P, Wang P, Elliott DM, Haskins ME, Ponder KP. Effect of neonatal gene therapy on lumbar spine disease in mucopolysaccharidosis VII dogs. Mol Genet Metab. 2012 doi: 10.1016/j.ymgme.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson S, Hashamiyan S, Clarke L, Saftig P, Mort J, Dejica VM, Bromme D. Glycosaminoglycan-mediated loss of cathepsin K collagenolytic activity in MPS I contributes to osteoclast and growth plate abnormalities. Am J Pathol. 2009;175:2053–62. doi: 10.2353/ajpath.2009.090211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Z, Yasuda Y, Li W, Bogyo M, Katz N, Gordon RE, Fields GB, Bromme D. Regulation of collagenase activities of human cathepsins by glycosaminoglycans. J Biol Chem. 2004;279:5470–9. doi: 10.1074/jbc.M310349200. [DOI] [PubMed] [Google Scholar]
- 26.Simonaro CM, D’Angelo M, He X, Eliyahu E, Shtraizent N, Haskins ME, Schuchman EH. Mechanism of glycosaminoglycan-mediated bone and joint disease: implications for the mucopolysaccharidoses and other connective tissue diseases. Am J Pathol. 2008;172:112–22. doi: 10.2353/ajpath.2008.070564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simonaro CM, Ge Y, Eliyahu E, He X, Jepsen KJ, Schuchman EH. Involvement of the Toll-like receptor 4 pathway and use of TNF-alpha antagonists for treatment of the mucopolysaccharidoses. Proc Natl Acad Sci U S A. 2010;107:222–7. doi: 10.1073/pnas.0912937107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clarke LA. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology. 2011;50 (Suppl 5):v13–8. doi: 10.1093/rheumatology/ker395. [DOI] [PubMed] [Google Scholar]
- 29.Lefebvre V, Smits P. Transcriptional control of chondrocyte fate and differentiation. Birth Defects Res C Embryo Today. 2005;75:200–12. doi: 10.1002/bdrc.20048. [DOI] [PubMed] [Google Scholar]
- 30.Koyama E, Golden EB, Kirsch T, Adams SL, Chandraratna RA, Michaille JJ, Pacifici M. Retinoid signaling is required for chondrocyte maturation and endochondral bone formation during limb skeletogenesis. Dev Biol. 1999;208:375–91. doi: 10.1006/dbio.1999.9207. [DOI] [PubMed] [Google Scholar]
- 31.Minina E, Kreschel C, Naski MC, Ornitz DM, Vortkamp A. Interaction of FGF, Ihh/Pthlh, and BMP signaling integrates chondrocyte proliferation and hypertrophic differentiation. Dev Cell. 2002;3:439–49. doi: 10.1016/s1534-5807(02)00261-7. [DOI] [PubMed] [Google Scholar]
- 32.Tamamura Y, Otani T, Kanatani N, Koyama E, Kitagaki J, Komori T, Yamada Y, Costantini F, Wakisaka S, Pacifici M, Iwamoto M, Enomoto-Iwamoto M. Developmental regulation of Wnt/beta-catenin signals is required for growth plate assembly, cartilage integrity, and endochondral ossification. J Biol Chem. 2005;280:19185–95. doi: 10.1074/jbc.M414275200. [DOI] [PubMed] [Google Scholar]
- 33.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–6. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 34.Lin X. Functions of heparan sulfate proteoglycans in cell signaling during development. Development. 2004;131:6009–21. doi: 10.1242/dev.01522. [DOI] [PubMed] [Google Scholar]
- 35.Oussoren E, Brands MMMG, Ruijter GJG, der Ploeg ATv, Reuser AJJ. Bone, joint and tooth development in mucopolysaccharidoses: relevance to therapeutic options. Biochim Biophys Acta. 2011;1812:1542–56. doi: 10.1016/j.bbadis.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 36.Simsa-Maziel S, Monsonego-Ornan E. Interleukin-1beta promotes proliferation and inhibits differentiation of chondrocytes through a mechanism involving down-regulation of FGFR-3 and p21. Endocrinology. 2012;153:2296–310. doi: 10.1210/en.2011-1756. [DOI] [PubMed] [Google Scholar]
- 37.MacRae VE, Farquharson C, Ahmed SF. The restricted potential for recovery of growth plate chondrogenesis and longitudinal bone growth following exposure to pro-inflammatory cytokines. J Endocrinol. 2006;189:319–28. doi: 10.1677/joe.1.06609. [DOI] [PubMed] [Google Scholar]