

Abstract
A series of 4-aryl-2-benzoyl-imidazoles were designed and synthesized based on our previously reported 2-aryl-4-benzoyl-imidazole (ABI) derivatives. The new structures reversed the aryl group and the benzoyl group of previous ABI structures and were named as reverse ABI (RABI) analogs. RABIs were evaluated for biological activity against 8 cancer cell lines including multidrug-resistant cancer cell lines. In vitro assays indicated that several RABI compounds had excellent antiproliferative properties with IC50 values in the low nanomolar range. The average IC50 of the most active compound 12a is 14 nM. In addition, the mechanism of action of these new analogs was investigated by cell cycle analysis, tubulin polymerization assay, competitive mass spectrometry binding assay and molecular docking studies. These studies confirmed that these new RABI analogs maintain their mechanisms of action by disrupting tubulin polymerization, similar to their parental ABI analogs.
Introduction
Cancer remains one of the leading causes of mortality worldwide.1–2 While current therapies are effective in treating early stage cancers, the efficacy against advanced cancers, especially multidrug-resistant cancers is limited. Thus, developing novel anticancer agents that can effectively overcome multidrug resistance will provide significant improvement of quality of life in cancer patients.
We previously reported the discovery of ABI analogs targeting the colchicine binding site in tubulin as potent antiproliferative agents.3–8 Compared with existing tubulin-targeting agents such as paclitaxel, colchicine, or vinblastine, ABI compounds have comparable in vitro and in vivo potency but can effectively circumvent several clinically relevant multidrug resistant mechanisms, including drug resistance mediated by P-glycoprotein (Pgp), multidrug resistance-associated proteins (MRPs), and breast cancer resistant proteins (BCRP).5–6 ABI compounds have also shown excellent oral bioavailability5, a potential advantage over existing tubulin inhibitors which can only be administrated by intravenous injection. To further optimize the potency of ABI analogues and to gain further insight in their structure–activity relationships (SARs), we designed and synthesized several new series of ABI analogs (summarized in Figure 1) by introducing three major changes to the parental ABI scaffold as described below.
Figure 1.
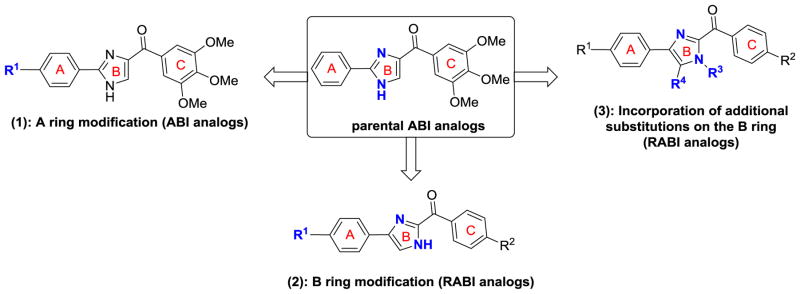
Design protocol for synthesis of RABI analogs
First, we varied the substitutions at the para-position on the A-ring of ABI analogs. This was accomplished by using previously established synthetic strategies. 3–4, 7 Second, we reversed the two major substitutions on the B-ring to produce the 4-aryl-2-benzoyl-imidazoles (reverse ABI, or RABI) compounds. We developed a one-pot synthetic strategy to synthesize RABI analogs in good yields based on the literature for synthesizing similar scaffold.9 Finally, we systematically incorporated additional substitutions in the B-ring of the RABI analogs to determine molecular shape/conformational requirements for their anticancer potency. Biological testing of those RABI compounds revealed their excellent antiproliferative activity against several cancer cell lines including multidrug-resistant cancer cell lines. Mechanism of action of RABIs was investigated using cell cycle analysis, tubulin polymerization assay, competitive mass spectrometry binding assay and molecular modeling. These studies showed that their antitumor activity was achieved through the antimitotic effect by the inhibition of tubulin polymerization, similar to their parental ABI analogs.
Chemistry
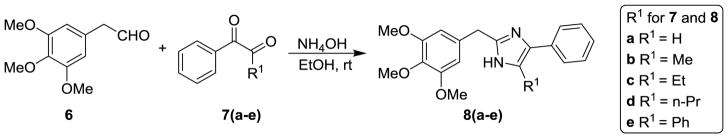
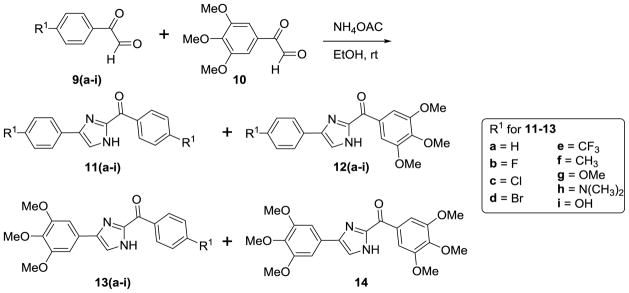
The general synthesis of the A ring modified analogs (5a-c) of ABI compounds is outlined in Scheme 1 using the same protocol as the method reported previously.3–4, 7 The general synthesis of the substituted imidazoles (8a-e) follows Scheme 2. A series of diketones (7a-e) 10 in ethanol reacted with 3,4,5-trimethoxy benzeneacetaldehyde 6 and ammonium hydroxide to generate a series of substituted imidazoles. 11 RABI compounds (11-14) were synthesized utilizing a one-pot, one-step reaction, which is outlined in Scheme 3.9 The arylglyoxal 12 reacts with 3, 4, 5-trimethoxyphenyl glyoxal in the presence of ammonium acetate in ethanol to give four products with similar yields around 20% in one pot. The ratio of compounds 12a-i to 13a-i is approximately 1:1. Two dimensional 1H-13C heteronuclear multiple bond correction spectroscopy (HMBC) NMR experiments were used to distinguish the structures between 12a-i and 13a-i (Figure S1, supplementary data). Strategies to incorporate additional substitutions on the B-ring of the RABI compounds are shown in Scheme 4. In Scheme 4, there are three conditions to introduce substitution to the N1-position. In condition a, compound 12a react with methyl iodide, ethyl bromide, and benzyl bromide in the presence of sodium hydride in anhydrous THF to generate compounds 15a-c.7 In condition b, compound 12a reacts with n-propyl iodide, i-propyl iodide and cyclopentyl bromide in the presence of potassium carbonate in acetonitrile to generate compounds 15d-f.13–14 In condition c, copper iodide, cesium carbonate and a ligand are used to introduce a pyridine ring or a thiophene ring to N1-position of compound 12a to make compound 15g-h.15 A similar method to that in Scheme 3 was employed to synthesize a series of 5-substituted RABI compounds (17a-c) as shown in Scheme 5.
Scheme 1.
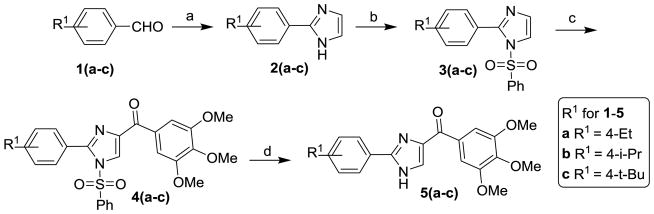
Reagents and conditions: (a) oxalaldehyde, NH4OH, EtOH, 0 °C-rt; (b) NaH, PhSO2Cl, THF, 0 °C-rt; (c) t-BuLi, substituted benzoyl chloride, THF, −78 °C; (d) Bu4NF, THF, rt.
Scheme 2.
Scheme 3.
Scheme 4.
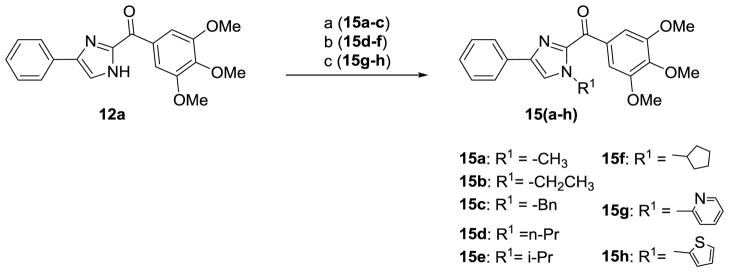
Reagents and conditions: (a) NaH, CH3l for 15a, CH3CH2Br for 15b and BnBr for 15c; (b) K2CO3, ACN, CH3CH2CH2l for 15d, (CH3)2CHI for 15e and cyclopentyl bromide for 15f; (c) Cul, Cs2CO3, DMF, Ligand, 2-bromo-pyridine for 15g, 2-bromothiophene for 15h.
Scheme 5.
Biological Results and Discussion
All the compounds were evaluated for their cytotoxicity in human melanoma cell lines and prostate cancer cell lines. Colchicine, vinblastine and docetaxel, the well-known antimitotic agents were included in the assays, serving as positive controls and as basis for comparison. The results are summarized in Tables 1–4.
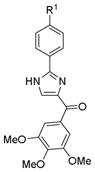
Table 1.
In vitro Growth Inhibitory Effects of ABI compounds with A ring substitution
| Structure | ID | R1 | IC50 ± SEM (nM)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| LNCaP | PC3 | PPC1 | DU145 | A375 | MDA-MB-435 | MDA-MB-435/LCC6MDR1 | Average IC50 | |||
|
5aa3 | H | 152 ± 35 | 288 ± 30 | 133 ± 6 | 196 ± 29 | 160 ± 20 | ND | ND | 186 |
| 5da3 | Me | 12 ± 1 | 9 ± 0.4 | 15 ± 1 | 11 ± 0.1 | 9 ± 2 | 5 ± 1 | 11 ± 2 | 10 | |
| 5a | Et | 9 ± 1 | 13 ± 1 | 15 ± 1 | 25 ± 2 | 14 ± 5 | 25 ± 4 | 27 ± 3 | 18 | |
| 5b | i-Pr | 171 ± 34 | 136 ± 20 | 173 ± 0.3 | 482 ± 40 | ND | 312 ± 4 | 250 ± 4 | 254 | |
| 5c | t-Bu | 423 ± 71 | 436 ± 115 | 294 ± 2 | 1698 ± 400 | ND | 3691 ± 60 | 3074 ± 50 | 1603 | |
ND: Not Determined
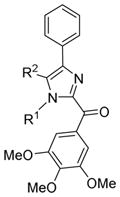
Table 4.
In vitro Growth Inhibitory Effects of RABI compounds with B ring substitution
| Structure | ID | R1 | R2 | IC50 ± SEM(nM)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| LNCaP | PC3 | PPC1 | DU145 | A375 | WM164 | MDA-MB-435 | MDA-MB-435/LCC6M DR1 | ||||
|
15a | Me | H | 10 ± 1 | 16 ± 1 | 13 ± 0.2 | 26 ± 3 | 9 ± 2 | 33 ± 5 | 16 ± 2 | 18 ± 2 |
| 15b | Et | H | 29 ± 20 | 25 ± 4 | 30 ± 8 | 66 ± 2 | 28 ± 3 | 16 ± 3 | 26 ± 3 | 33 ± 4 | |
| 15c | Bn | H | 66 ± 6 | 72 ± 5 | 77 ± 2 | 160 ± 41 | 104 ± 17 | 34 ± 7 | 94 ± 18 | 156 ± 18 | |
| 15d | n-Pr | H | 49 ± 10 | 26 ± 6 | 10 ± 4 | 72 ± 13 | 42 ± 3 | 14 ± 3 | 37 ± 2 | 44 ± 4 | |
| 15e | i-Pr | H | 62 ± 8 | 53 ± 9 | 15 ± 3 | 114 ± 20 | 38 ± 4 | 23 ± 5 | 18 ± 3 | 44 ± 4 | |
| 15f |
|
H | 51 ± 6 | 56 ± 1 | 63 ± 4 | 167 ± 16 | 135 ± 13 | 56 ± 13 | 134 ± 15 | 161 ± 19 | |
| 15g |
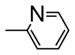
|
H | 20 ± 5 | 11 ± 3 | 8 ± 2 | 37 ± 6 | 14 ± 3 | 6 ± 1 | 11 ± 3 | 15 ± 5 | |
| 15h |
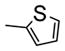
|
H | ND | ND | ND | ND | 20 ± 2 | 8 ± 2 | 25 ± 3 | 27 ± 3 | |
| 17a | H | Me | 938 ± 65 | 1617 ± 144 | 860 ± 5 | 2001 ± 163 | 1302 ± 106 | 1897 ± 116 | 1634 ± 102 | 1586 ± 104 | |
| 17b | H | Et | 2029 ± 880 | 3654 ± 192 | 2078 ± 90 | 5079 ± 635 | 2151 ± 48 | 5514 ± 35 | 8795 ± 23 | 5114 ± 27 | |
| 17c | H | n-Pr | 3094 ± 330 | 12360 ± 7566 | 11410 ± 5918 | 16350 ± 6724 | 36977 ± 73 | 19595 ± 91 | 30540 ± 103 | 14270 ± 362 | |
ND: Not Determined
Effect of Substitutions on the A Ring of ABI Analogs
As shown in Table 1, Compound 5a (average IC50 = 18 nM) maintained potent activity in most cancer cells compared with compound 5da3, 6(average IC50 = 10nM) which has a methyl substitution on A ring para position. Introducing a larger isopropyl group (5b, average IC50 = 254 nM) into this position on the A ring caused a 20-fold decrease of potency compared with compound 5da. Further increasing the size of this substitution using a tert butyl group to this position resulted in a 100-fold loss of activity compared with 5da. The activity trend in terms of para substitution in this A ring is Me > Et > H > i-Pr > t-Bu, clearly suggesting a relatively small binding pocket to the receptor around the A-ring, with a methyl group being the optimal size. Since A ring modification did not generate a more potent compound than 5da, we decided to design and synthesize analogs by modifying the B ring.
The Ketone Linker Remains Critical for RABI Analogs
We previous reported that a ketone linker is essential for the activity of the parental ABI and its related analogs,16 to test whether this is still a critical requirement, we synthesized five RABI analogs containing a methylene linker instead of a carbonyl linker (Scheme 2). The biological activity of these five compounds was shown in Table 2. All of them were basically inactive, consistent with results reported previously which indicated the essential role of the carbonyl linker.
Table 2.
In vitroGrowth Inhibitory Effects of RABI compounds with methylene linker
| Structure | ID | R1 | IC50 ± SEM (μM)
|
||
|---|---|---|---|---|---|
| A375 | MDA-MB-435 | MDA-MB-435/LCC6MDR1 | |||
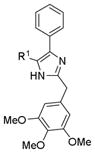
|
8a | H | 10.2 ± 0.4 | ND | ND |
| 8b | Me | > 50 | ND | ND | |
| 8c | Et | ND | > 50 | > 50 | |
| 8d | n-Pr | ND | 10.9 ± 0.1 | 15.9 ± 0.2 | |
| 8e | Ph | ND | > 50 | > 50 | |
ND: Not Determined
Effect of Substitutions on the A Ring of RABIs
After confirming the essential role of the carbonyl linker in the RABI analogs, we converted the methylene linker to carbonyl linker using a slightly modified approach as shown in Scheme 3 and produced RABI analogs 11-13. All RABI compounds were evaluated for their antiproliferative activity and the results were shown in Table 3. Compound 12a was most potent with IC50 values ranging from 6 to 24 nM. Introducing an electron withdrawing group with increasing sizes to the para position on the A ring (4-fluro, 4-chloro, 4-bromo and 4-trifluromethyl) generally decreased activity (compare 12b-e with 12a). Among the three halogen substituted RABI compounds, the trend of activity was F<Cl<Br. Introduction of an electron donating group such as methyl to the compound maintained the activity (12f, IC50: 10–27 nM), while the introduction of methoxy and dimethylamino group caused loss of activity to some extent (12g, IC50: 30–210 nM; 12h, IC50: 96–263 nM). Compound 12i with phenol group as A ring was the least potent one among compounds 12a-12i. These results are consistent with the structure-activity relationships identified in the parental ABI analogs (Table 1 and earlier studies3, 7).
Table 3.
In vitro Growth Inhibitory Effects of RABI compounds without B ring substitution
| Structure | ID | R1 | R2 | IC50 ± SEM (nM)
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| LNCaP | PC3 | PPC1 | DU145 | A375 | WM164 | MDA-MB-435 | MDA-MB-435/LCC6MD R1 | ||||
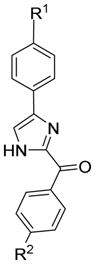
|
11a | H | H | > 50000 | > 50000 | > 50000 | > 50000 | > 50000 | > 50000 | > 50000 | > 50000 |
| 11b | F | F | > 50000 | > 50000 | > 50000 | > 50000 | > 50000 | > 50000 | > 50000 | > 50000 | |
| 11c | Cl | Cl | > 50000 | > 50000 | > 50000 | > 50000 | 35674 ± 665 | > 50000 | > 50000 | > 50000 | |
| 11d | Br | Br | 16930 ± 6183 | 18940 ± 1068 | 13210 ± 706 | 25490 ± 5144 | 24960 ± 35 | 26320 ± 211 | 14355 ± 178 | 17814 ± 155 | |
| 11e | CF3 | CF3 | > 50000 | > 50000 | > 50000 | > 50000 | > 50000 | > 50000 | > 50000 | 26460 ± 533 | |
| 11f | Me | Me | 3762 ± 1720 | 5159 ± 386 | 2405 ± 308 | 6541 ± 460 | 2535 ± 30 | 3693 ± 18 | 3020 ± 23 | 3271 ± 33 | |
| 11g | OMe | OMe | 6419 ± 4365 | 23370 ± 1471 | 38150 ± 2325 | 9839 ± 503 | > 50000 | > 50000 | > 50000 | > 50000 | |
| 11h | N(CH3)2 | N(CH3)2 | ND | ND | ND | ND | > 50000 | > 50000 | > 50000 | > 50000 | |
| 11i | OH | OH | ND | ND | ND | ND | 38769 ± 97 | 48218 ± 113 | 47986 ± 104 | 21421 ± 93 | |
| 12a | H | 3,4,5-(OMe)3 | 6 ± 1 | 14 ± 1 | 13 ± 0.3 | 22 ± 2 | 9 ± 6 | 14 ± 3 | 24 ± 3 | 11 ± 2 | |
| 12b | F | 3,4,5-(OMe)3 | 114 ± 7 | 196 ± 6 | 134 ± 0.1 | 353 ± 10 | 197 ± 15 | 298 ± 13 | 320 ± 14 | 263 ± 10 | |
| 12c | Cl | 3,4,5-(OMe)3 | 22 ± 6 | 64 ± 7 | 51 ± 1 | 125 ± 12 | 51 ± 13 | 63 ± 11 | 106 ± 15 | 75 ± 11 | |
| 12d | Br | 3,4,5-(OMe)3 | 15 ± 5 | 33 ± 2 | 30 ± 1 | 66 ± 2 | 29 ± 7 | 31 ± 4 | 58 ± 14 | 43 ± 11 | |
| 12e | CF3 | 3,4,5-(OMe)3 | 47 ± 35 | 93 ± 3 | 74 ± 2 | 210 ± 18 | 123 ± 18 | 143 ± 8 | 120 ± 15 | 175 ± 9 | |
| 12f | CH3 | 3,4,5-(OMe)3 | 13 ± 1 | 19 ± 1 | 18 ± 0.3 | 30 ± 4 | 13 ± 2 | 14 ± 3 | 27 ± 11 | 21 ± 5 | |
| 12g | OMe | 3,4,5-(OMe)3 | 30 ± 14 | 61 ± 4 | 54 ± 1 | 210 ± 147 | 33 ± 8 | 41 ± 12 | 55 ± 9 | 59 ± 18 | |
| 12h | N(CH3)2 | 3,4,5-(OMe)3 | 96 ± 6 | 118 ± 17 | 120 ± 12 | 263 ± 16 | 141 ± 23 | 129 ± 19 | 200 ± 20 | 162 ± 12 | |
| 12i | OH | 3,4,5-(OMe)3 | 219 ± 101 | 155 ± 23 | 122 ± 6 | 518 ± 10 | 487 ± 35 | 549 ± 24 | 669 ± 31 | 455 ± 29 | |
| 13a | 3,4,5-(OMe)3 | H | 195 ± 91 | 632 ± 42 | 408 ± 11 | 1301 ± 264 | 1023 ± 23 | 1273 ± 30 | 4606 ± 78 | 5770 ± 63 | |
| 13d | 3,4,5-(OMe)3 | Br | 131 ± 175 | 371 ± 247 | 106 ± 3 | 429 ± 0.2 | 136 ± 12 | 177 ± 16 | 186 ± 11 | 161 ± 11 | |
| 13g | 3,4,5-(OMe)3 | OMe | 708 ± 334 | 10390 ± 6646 | 5685 ± 325 | >50000 | 35414 ± 106 | 36007 ± 98 | 10956 ± 96 | 11428 ± 87 | |
| 13h | 3,4,5-(OMe)3 | N(CH3)2 | ND | ND | ND | ND | 47878 ± 563 | 46131 ± 98 | 29175 ± 88 | 40618 ± 112 | |
| 13i | 3,4,5-(OMe)3 | OH | ND | ND | ND | ND | > 50000 | > 50000 | > 50000 | > 50000 | |
| 14 | 3,4,5-(OMe)3 | 3,4,5-(OMe)3 | > 50000 | > 50000 | > 50000 | > 50000 | > 50000 | > 50000 | > 50000 | > 50000 | |
ND: Not Determined
Effect of Substitutions on the C Ring of RABIs
Several RABI compounds (11a-11i) which did not have the 3, 4, 5-trimethoxy moiety on C ring showed poor activity as shown in Table 3. The RABI compounds with unsubstituted phenyl ring as A ring and C ring caused complete loss of activity (11a, IC50>50 μM). Compounds with a single halogen substituent in the para position of A and C rings lost activity completely (IC50 > 5 μM for 11b-d). When methyl, methoxy, trifluoromethyl, dimethylamino, or hydroxyl was introduced to the para position of the A and C rings, the activity was also lost. All these results suggested the essential role of 3, 4, 5-trimethoxy substituents on the C ring. Interestingly, when 3,4,5-trimethoxy group was put on the A ring, the activity of two compounds 13a and 13d returned to some extent (13a, IC50: 195–5770 nM; 13d, IC50: 131–429 nM) even though there is no 3,4,5-trimethoxy substituents on C ring, perhaps suggesting that the orientation of the compound changed when binding to tubulin. One results worth noting is that when 3,4,5-trimethoxy group was introduced to both A ring and C ring, the activity was lost completely. This is consistent with results in previous sections that a bulky A-ring containing a 3,4,5-trimethoxy moiety cannot be tolerated at the receptor.
Effect of Substitutions on the B Ring of RABIs
Modifications on the B ring in two different sites were investigated: the N1- and 5-position of the imidazole ring. Introducing methyl, ethyl, or propyl at the 5-position of the imidazole ring resulted in inactive compounds (17a-c) as shown in Table 4. A trend in activity for the substituted alkyl groups: Me>Et>Pr, was also observed, which suggests big bulky groups at this position are detrimental to activity. In contrast, when different substitution groups were introduced to the N1-position of the imidazole ring, the activity was maintained with only minimal loss compared with the parent compound, 12a. First some alkyl groups were tried on the N1-position including methyl, ethyl, n-propyl, i-propyl and cyclopentyl groups. Introduction of methyl did not affect the activity compared with 12a (15a, IC50: 9–26 nM), while the activity began to lose as the size of the alkyl groups increased, suggesting that a bulky alkyl group at this position was unfavorable. Benzyl group, which was also a relatively big group at the N1-position of the imidazole ring, decreased the activity (15c, IC50: 34–160 nM). Surprisingly, when the substituents were changed from alkyl groups to heterocyclic rings, the ‘effect of big size’ disappeared. The introduction of a pyridine ring to the N1-position generated a very potent compound with IC50 ranging from 6 to 37 nM. The introduction of a thiophene ring also produced a potent compound with IC50 ranging from 8 to 20 nM. The excellent activity of 15g and 15h promises the future optimization at this position using other ring systems.
Effect of RABI Compounds against Multidrug-Resistant Melanoma Cells
Pgp-mediated drug efflux represents a major mechanism for cancer cells to prevent the buildup of effective intracellular drug concentrations. We have previously shown that the parent ABI analogs can effectively overcome a variety of clinically relevant multidrug resistant mechanisms including Pgp-mediated drug resistance.5–6 To determine whether the new RABI analogs maintain this ability, we compared the activity of the RABI compounds against multidrug-resistant melanoma cells (MDA-MB-435/LCCMDR1) and their parental nonresistant cancer cells (MDA-MB-435). This pair of cell lines have been well validated and widely used to assess abilities of drugs overcoming Pgp-mediated MDR.17–20 Compound 12a, 12d and 12e together with colchicine, vinblastine and paclitaxel were tested on both the MDR melanoma cells and their parental melanoma cell lines (Table 5). Compound 12a, 12d and 12e showed much better resistance index (0.5 for 12a, 0.7 for 12d, 0.8 for 12e) than colchicine (58.5), vinblastine (27.5) and paclitaxel (69.3). Although colchicine, vinblastine, and paclitaxel showed excellent activity in nonresistant melanoma cell lines, they were much less potent in resistant melanoma cell lines. In contrast, compounds 12a, 12d and 12e showed comparable or even better activity against Pgp-overexpressed melanoma cells than their parental, nonresistant melanoma cells.
Table 5.
In vitro Growth Inhibitory Effects of RABI Compounds Comparison to Other Anticancer Drugs on Multidrug-Resistant Melanoma Cells and Parent Cell Line
| ID | IC50 ± SEM (nM)
|
||
|---|---|---|---|
| MDA-MB-435 | MDA-MB-435/LCC6MDR1 | Resistance Index | |
| 12a | 24 ± 3 | 11 ± 2 | 0.5 |
| 12d | 58 ± 14 | 43 ± 11 | 0.7 |
| 12e | 27 ± 11 | 21 ± 5 | 0.8 |
| colchicine | 11 ± 2 | 643 ± 9 | 58.5 |
| vinblastine | 0.4 ± 0.1 | 11 ± 1 | 27.5 |
| paclitaxel | 4 ± 1 | 277 ± 41 | 69.3 |
Mechanism of Action Studies on RABIs
Since the parental ABI analogs kills cancer cells by inhibiting mitotic process, we hypothesized that RABIs maintain their mechanism of action. To test this hypothesis, we first performed the cell cycle analysis after the treatment of RABIs on PC3 cells. Cell cycle distribution was determined by propidium iodide (PI) staining. Treated cells were fixed with 70% ice-cold ethanol and the fixed cells were stained with PI in the presence of RNase A. Cell cycle distribution was analyzed by fluorescence-activated cell sorting (FACS) analysis. Compound 5a, 12a, 12d, 12f, 15a, and 15b were treated on PC3 cells for 24 h. Four different concentrations 1, 10, 50, and 100 nM of each compound were chosen to examine the dose effect. Results indicated that RABIs arrest cells in the G2/M phase (Figure. 2A). In the vehicle treated group, about 18% of PC3 cells were distributed in the G2/M phase. RABIs increased the proportion of cells in G2/M phase up to 70% approximately in a concentration-dependent manner (Figure. 2B). The potency of the different concentrations in arresting cells in the G2/M phase positively correlated with in vitro cell growth inhibitory activity.
Figure 2.

Effect of Compounds 5a, 12a, 12d, 12f, 15a and 15b on Cell Cycle
Based on their effect on cell cycle distribution, we next investigated the effect on tubulin polymerization of RABI analogs. To determine the effect of drug on tubulin polymerization, a fluorescence-enhanced tubulin polymerization assay kit was used. The control drug, vinblastine inhibited tubulin polymerization and destabilized microtubule, while paclitaxel promoted microtubule stability (Figure S2, supporting information). RABI compounds, 12a and 15a inhibited tubulin polymerization as the tubulin destabilizer, vinblastine in a concentration-dependent manner (Figure S2, supporting information) and 15a showed more potent inhibitory effect than 12a. In the competitive mass spectrometry binding assay, the amount of unbound colchicine in the presence or absence of any competitor would explain whether there is the competition of compounds and colchicine to bind in tubulin. 12a and 15g competed effectively with colchicine for tubulin binding (Figure S3, supporting information) with potency similar to podophyllotoxin. Vinblastine, the negative control, did not inhibit colchicine binding to tubulin. Collectively, these data confirmed that new RABI analogs maintain their anti-mitotic mechanisms of action, most likely binding to the colchicine binding site in tubulin, as is the case for the parental ABI analogs. 6–7
Molecular Modeling Studies
One surprising results from the analysis of the structure-activity relationships for the RABI analogs is that incorporating a 5- or 6-membered heterocyclic ring substitution at the N1-position in the B-ring produced highly active compounds, while both similar-sized alkyl substitutions or a larger benzyl substitution resulted in significantly reduced activity (Table 4). To better understand how the N1-substituted RABI analogues interact with tubulin, the potential binding modes for two of the most potent compounds, 12a and 15g, were investigated at colchicine binding site in tubulin dimer using Schrodinger 2011 molecular modeling suite (Schrodinger, Inc., New York, NY). Both compounds were docked into two different tubulin crystal structure (PDB ID code: 1SA0 or 3HKD), representing two potential binding geometries for colchicine site ligands. Interestingly, non-substituted RABI analog 12a demonstrated best glide docking score of −9.2 in 3HKD compared with a score of −6.4 in 1SA0, while substituted RABI analog 15g showed best glide score of −9.0 in 1SA0 and could not fit into the binding pocket of 3HKD.
The overview of the binding site of 12a and TN-16 (native ligand of 3HKD) is shown in Figure 3A. This binding pocket is located on the interface between the α- and β-subunits of the tubulin dimer and extended slightly out of the β-subunit.21–22 Figure 3B illustrated the close view of the potential binding pose. Generally, 12a (green stick) overlapped well with TN-16 (blue stick). The A ring of 12a went deep into the pocket and overlapped very well with the first phenyl ring of TN-16. There are very little space between the A-ring and its surrounding amino acids in the β-subunit (Figure 3B), and this explains why little tolerance is allowed for larger substitution in the A-ring for both ABI and RABI compounds. The imidazole ring overlapped well with the pyrrolidinedione ring of TN-16. A potential hydrogen bond is formed between the imidazole NH of 12a and VAL238 in β-H7 (Figure 3B), similar to the one formed between the native ligand TN-16 and VAL238 in β-H7. This hydrogen bond stabilized the interaction of 12a with the binding pocket. The 3,4,5-trimethoxybenzoyl group (C ring) of 12a extends toward the α/β-tubulin interface, similar to the mode of the active parental ABI analogs.4, 7
Figure 3.

(A) The overview of the binding modes of 12a and the native ligand TN-16 in tubulin crystal structure 3HKD. (B) The close view of the potential binding pose of 12a and TN-16 in 3HKD. (C) The overview of the binding modes of 15g and the native ligand colchicine in tubulin crystal structure 1SA0.(D) The close view of the potential binding pose of 15g and colchicine in 1SA0.
Unlike 12a which does not possess a large N1-substitution, the much “fatter” RABI 15g cannot fit into the cylinder shaped binding pocket in 3HKD, but dock reasonably well into the shallower pocket in 1SA0. The potential binding mode of 15g was shown in Figure 3C and 3D. Figure 3C showed the general view of the binding site of 15g and the native ligand colchicine in 1SA0. Figure 3D illustrated the closed view of the potential binding pose. Interestingly, in this binding mode, part of 15g (green stick) overlapped well with the native ligand colchicine (blue stick), while the original A ring of 15g extended out of the colchicine binding pocket into the α/β-tubulin interface. The pyridine ring substitute on the N1-position occupied the site where the 7-membered ring in colchicine binds, while the 3,4,5-trimethoxybenzoyl group (C ring) of 15g overlapped very well with the 3,4,5-trimethoxyphenyl ring in colchicine. A hydrogen bond between the oxygen of 4-OMe in 15g and SH group of β-CYS241 stabilized the interaction. A similar hydrogen bond was also observed between the oxygen of one methoxy group in colchicine and SH of β-CYS241. This binding mode is well consistent with the structure-activity relationships observed for substituted RABI compounds: a smaller, alkyl substitution could not fill this region of the binding pocket and lacks the planar geometry required for binding, while too large a benzyl substitution could not fit into the pocket and also lacks the needed geometry. A 5- or 6-membered heterocyclic ring has the desirable shape and size to fit into the pocket well. These data also suggest that while the original “A ring” may not be critical for binding when a suitable N1-substitution is present, an optimized substitution replacing this original A ring moiety may take advantage of the added interactions between the ligand and receptor and provide even better ligand than 15g. Work towards this direction is current ongoing and will be reported in the future.
Conclusions
In summary, novel RABI analogs were designed and synthesized based on rational structural modification of previous ABI analogs. Structure-activity relationships (Figure 4) were investigated by introducing different substituents into the A, B and C rings. Several RABIs showed excellent antiproliferative activity which was comparable to existing tubulin-targeting agents such as paclitaxel, colchicine, or vinblastine but could overcome Pgp-mediated multiple drug resistance effectively. Among them compound 12a was the most potent one with IC50 in the low nanomolar range, while compound 15g provided very interesting insights in future optimization of these analogs. Mechanism of action studies confirmed that RABI analogs maintain their ability to inhibit tubulin polymerization at colchicine binding site and arresting cells in G2/M phase. These results strongly suggest that novel RABI analogs can be further developed as a promising antitumor agent for the more efficacious treatment of advanced cancers.
Figure 4.
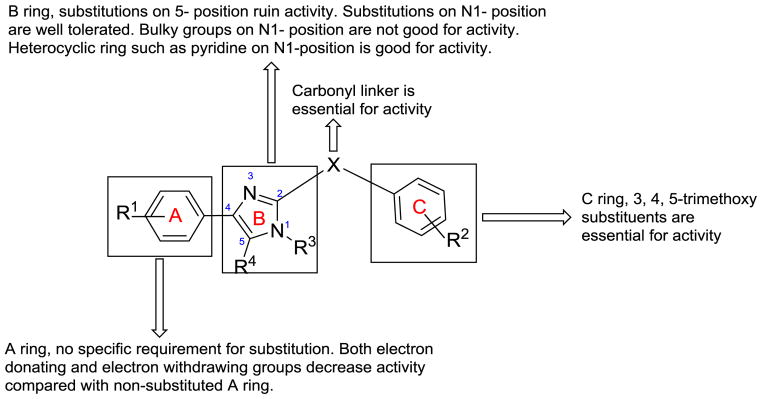
SAR of RABI analogues
Experimental Section
General. All reagents were purchased from Sigma-Aldrich Chemical Co., Fisher Scientific (Pittsburgh, PA), Alfa Aesar (Ward Hill, MA), and AK Scientific (Mountain View, CA) and were used without further purification. Routine thin layer chromatography (TLC) was performed on aluminum-backed Uniplates (Analtech, Newark, DE). NMR spectra were obtained on a Varian Inova-500 spectrometer (Agilent Technologies, Santa Clara, CA) or a Bruker Ascend 400 (Billerica, MA) spectrometer. Chemical shifts are reported as parts per million (ppm) relative to TMS in CDCl3. High Resolution Mass spectra were collected on a Waters Xevo G2-S Tof instrument. The purity of the final compounds was tested via Agilent series HPLC system (Agilent 1100 Series, Agilent 1100 Chemstation, Agilent Technology Co, Ltd.) installed with a photodiode array detector. Four RP-HPLC methods were conducted using a Phenomenex 5 μm C-18 column (250 mm × 4.6 mm) at ambient temperature and a flow rate of 1.0 mL/min. HPLC1: solvent A (water) and solvent B (methanol), 0–30 min 90% B. HPLC2: solvent A (water) and solvent B (methanol), 0–30 min 85% HPLC3: solvent A (water) and solvent B (methanol), 0–30 min 80% B. HPLC4: solvent A (water) and solvent B (methanol), 0–15 min 50–100% B (linear gradient), 15–25 min 100% B, 25–28 min 100–50% B, 28–33 min 50% B. UV detection at 254 nm. Purities of the compounds were established by careful integration of areas for all peaks detected and are reported for each compound in the following section.
General Procedure for the Preparation of 2-Aryl-1H-imidazole (2a-c)
To a solution of appropriate benzaldehyde 1 (100 mmol) in ethanol (350 mL) at 0 °C was added a solution of 40% oxalaldehyde in water (12.8 mL, 110 mmol) and a solution of 29% ammonium hydroxide in water (1000 mmol, 140 mL). After stirring for 2–3 days at room temperature, the reaction mixture was concentrated and the residue was subjected to flash column chromatography with dichloromethane as eluent to yield the titled compound as a yellow powder. Yield: 20–40%.
General Procedure for the Preparation of 2-Aryl-1-(phenylsulfonyl)-1H-imidazole (3a-c)
To a solution of 2-aryl-1Himidazole2 (20 mmol) in anhydrous THF (200 mL) at 0°C was added sodium hydride (60% dispersion in mineral oil, 1.2 g, 30 mmol) and stirred for 30 min. Benzenesulfonyl chloride (2.82 mL, 22 mmol) was added, and the reaction mixture was stirred overnight. After dilution by 100 mL of saturated NaHCO3 solution (aqueous), the reaction mixture was extracted by ethyl acetate (500 mL). The organic layer was dried over magnesium sulfate and concentrated. The residue was purified by flash column chromatography (hexane: ethyl acetate 2:1) to give a pale solid. Yield: 40–50%.
General Procedure for the Preparation of Aryl (2-Aryl-1-(phenylsulfonyl)-1Himidazol-4-yl) Methanone (4a-c)
To a solution of 2-aryl-1-(phenylsulfonyl)-1H-imidazole (6.0 mmol) 3 in anhydrous THF (30 mL) at −78 °C was added 1.7 M tert-butyl lithium in pentane (5.3 mL, 9.0 mmol) and stirred for 10 min. Appropriate substituted benzoyl chloride (7.2 mmol) was added at −78 °C and stirred for overnight. The reaction mixture was diluted with 100 mL of saturated NaHCO3 solution (aqueous) and extracted by ethyl acetate (200 mL). The organic layer was dried over magnesium sulfate and concentrated. The residue was purified by flash column chromatography (hexane: ethyl acetate 4:1) to give a white solid. (Note. Due to the limited amount of starting material or the difficulty of separation, the following products formed in this step were used without further purification as a mixture for the next step. Yield: 15%–40%.
General Procedure for the Preparation of Aryl (2-Aryl-1Himidazol-4-yl) Methanone (5a-c)
To a solution of aryl (2-aryl-1-(phenylsulfonyl)-1H-imidazol-4-yl) methanone (2.0 mmol) 4 in THF (20.0 mL) was added 1.0 M tetrabutyl ammoniumfluoride (4.0 mmol) and stirred overnight. The reaction mixture was diluted by 50 mL of saturated NaHCO3 solution (aqueous) and extracted by ethyl acetate (100 mL). The organic layer was dried over magnesium sulfate and concentrated. The residue was purified by flash column chromatography (hexane: ethyl acetate 3:1) or recrystallized from water and methanol to give a white solid. Yield: 80–95%.
(2-(4-ethylphenyl)-1H-imidazol-4-yl)(3,4,5-trimethoxyphenyl) Methanone (5a)
1H NMR (500 MHz, CDCl3) δ 10.40 (br, 1H), 7.92 (d, J = 8.0 Hz, 2H), 7.85 (s, 1H), 7.36 (d, J = 8.0 Hz, 2H), 7.27 (s, 2H), 3.99 (s, 3H), 3.97 (s, 6H), 2.75 (q, d = 7.5 Hz, 2H), 1.313(t, d=7.5 Hz, 3H); Exact Mass for C21H22N2O4: 366.158; HRMS: [M+H]+: 367.1764; HPLC1: tR 4.97 min, purity 97.8%.
(2-(4-isopropylphenyl)-1H-imidazol-4-yl)(3,4,5-trimethoxyphenyl) Methanone (5b)
1H NMR (500 MHz, CDCl3) δ 10.38 (br, 1H), 7.82 (d, J = 8.0 Hz, 2H), 7.75 (s, 1H), 7.38 (d, J = 8.0 Hz, 2H), 7.15 (s, 2 H), 3.88 (s, 3H), 3.86 (s, 6H), 2.90 (m, 1H), 1.331(d, d = 6.5 Hz, 6H); Exact Mass for C22H24N2O4: 380.1700; HRMS: [M+H]+: 381.1818; HPLC1: tR 5.26 min, purity 97.5%.
(2-(4-tertbutylphenyl)-1H-imidazol-4-yl)(3,4,5-trimethoxyphenyl) Methanone (5c)
1H NMR (500 MHz, CDCl3) δ 10.42 (br, 1 H), 7.94 (d, J = 8.0 Hz, 2H), 7.84 (s, 1H), 7.54 (d, J = 8.0 Hz, 2H), 7.25 (s, 2H), 3.98 (s, 3H), 3.96 (s, 6H), 1.385(s, 9H); Exact Mass for C23H26N2O4: 394.1900; HRMS: [M+H]+: 395.2054; HPLC2: tR 7.75 min, purity >99%.
General procedure for the synthesis of 5-(alkyl or aryl)-4-phenyl-2-(3,4,5-trimethoxybenzyl)-1H-imidazole and 4-(alkyl or aryl)-5-phenyl-2-(3,4,5-trimethoxybenzyl)-1H-imidazole (8a-e)
To a solution of the aldehyde 6 (5 mmol) in ethanol (20 mL) at 0°C was added the phenyl alkyl dione 7 (5.5 mmol) and a solution of 29% ammonium hydroxide in water (50 mmol, 7 mL). After stirring for 2–3 days at room temperature, the reaction mixture was concentrated and the residue was subjected to flash column chromatography with dichloromethane as eluent to yield the titled compound as a yellow powder. Yield: 20–30%.
5-phenyl-2-(3,4,5-trimethoxybenzyl)-1H-imidazole and 4-phenyl-2-(3,4,5-trimethoxybenzyl)-1H-imidazole (8a)
1H NMR (500 MHz, Chloroform-d) δ 7.91 – 7.62 (m, 2H), 7.43 – 7.34 (m, 2H), 7.27 – 7.21 (m, 2H), 6.51 (s, 2H), 4.12 (s, 2H), 3.86 (s, 3H), 3.84 (s, 6H); Exact Mass for C19H20N2O3: 324.1500; HRMS: [M+H]+: 325.1684.
5-methyl-4-phenyl-2-(3,4,5-trimethoxybenzyl)-1H-imidazole and 4-methyl-5-phenyl-2-(3,4,5-trimethoxybenzyl)-1H-imidazole (8b)
1H NMR (500 MHz, Chloroform-d) δ 7.55 (d, J = 7.6 Hz, 2H), 7.43 – 7.37 (m, 2H), 7.25 (d, J = 6.7 Hz, 1H), 6.49 (s, 2H), 4.03 (s, 2H), 3.84 (d, J = 1.3 Hz, 3H), 3.81 (d, J = 1.2 Hz, 6H), 2.42 (s, 3H); Exact Mass for C20H22N2O3: 338.1600; HRMS: [M+H]+: 339.1799.
5-ethyl-4-phenyl-2-(3,4,5-trimethoxybenzyl)-1H-imidazole and 4-ethyl-5-phenyl-2-(3,4,5-trimethoxybenzyl)-1H-imidazole (8c)
1H NMR (500 MHz, Chloroform-d) δ 8.47 (s, 0.42H), 8.39 (s, 0.58H), 7.86 – 7.62 (m, 2H), 7.41 (m, 2H), 7.35 – 7.31 (m, 1H), 6.54 (m, 2H), 4.10 (m, 2H), 3.90 – 3.83 (m, 9H), 2.79 (m, 2H), 1.38 – 1.17 (m, 3H); Exact Mass for C21H24N2O3: 352.1800; HRMS: [M+H]+: 353.1912.
4-phenyl-5-propyl-2-(3,4,5-trimethoxybenzyl)-1H-imidazole and 5-phenyl-4-propyl-2-(3,4,5-trimethoxybenzyl)-1H-imidazole (8d)
1H NMR (400 MHz, Chloroform-d) δ 8.63 (s, 0.48H), 8.50 (s, 0.52H), 7.70 – 7.64 (m, 1H), 7.41 (m, 3H), 7.37 – 7.32 (m, 1H), 6.52 (d, J = 1.3 Hz, 2H), 4.08 (d, J = 5.1 Hz, 2H), 3.87 (m, 3H), 3.86 – 3.84 (m, 6H), 2.73 (m, 2H), 1.88 – 1.75 (m, 1H), 1.64 (m, 1H), 0.98 (m, 3H); Exact Mass for C22H26N2O3: 366.1900; HRMS: [M+H]+: 367.2011.
4, 5-diphenyl-2-(3,4,5-trimethoxybenzyl)-1H-imidazole (8e)
1H NMR (400 MHz, Chloroform-d) δ 7.44 – 7.36 (m, 4H), 7.28 – 7.20 (m, 6H), 6.50 (s, 2H), 4.08 (s, 2H), 3.78 (m, 9H); Exact Mass for C25H24N2O3: 400.1800; HRMS: [M+H]+: 401.1977.
General procedure for the preparation of (4 or 5)-aryl-2-aryloyl-(1H)-Imidazole derivatives (11-14)
To ammonium acetate (10 mmol) in ethanol (5 ml) and water (0.3 ml) was added arylglyoxal hydrate 9(1 mmol) in ethanol (5 ml) and 3, 4, 5-trimethoxyphenyl glyoxal hydrate 10 (1 mmol) in ethanol (10 ml). The mixture was stirred at room temperature for 30–45 min. The reaction was stopped after the consumption of the starting material monitored by TLC. The mixture was then extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated to get the crude product. The crude was purified by flash chromatography (dichloromethane:methanol 50:1).
Phenyl-(4-phenyl-1H-imidazol-2-yl)methanone and phenyl-(5-phenyl-1H-imidazol-2-yl)methanone (11a)
1H NMR (500 MHz, DMSO-d6): d 13.80 (s, 0.25H), 13.63 (s, 1H), 8.60 (d, J = 7.76 Hz, 2H), 8.47 (d, J = 7.7 Hz, 0.5H,), 8.08 (s, 1H,), 7.97 (d, J = 7.95 Hz, 0.5H), 7.94 (d, J = 7.64 Hz, 2H), 7.79 (s, 0.25H), 7.69 (t, J = 7.1 Hz, 1H), 7.66 (t, J = 7.6 Hz, 0.25H), 7.60 (t, J = 7.6 Hz, 2H), 7.57 (t, J = 8.1 Hz, 0.5H), 7.47 (t, J = 7.55 Hz, 0.5H), 7.42 (t, J = 7.7 Hz, 2H), 7.37 (t, J = 7.1 Hz, 0.25H), 7.28 (t, J = 7.3 Hz, 1H); Exact Mass for C16H12N2O: 248.095; HRMS: [M+H]+: 249.1058; HPLC4: tR 10.83 min, purity 97.6%.
(4-fluorophenyl)(4-(4-fluorophenyl)-1H-imidazol-2-yl)methanone and (4-fluorophenyl)(5-(4-fluorophenyl)-1H-imidazol-2-yl)methanone (11b)
1H NMR (400 MHz, Chloroform-d) δ 10.68 (s, 1H), 10.52 (s, 1H), 8.93 – 8.82 (dd, J = 5.89, 8.64 Hz, 2H), 8.72 (dd, J = 5.60, 8.70 Hz, 0.39H), 7.89 (dd, J = 5.39, 8.72 Hz, 2H), 7.63 (dd, J = 5.05, 8.25 Hz, 0.46H), 7.59 (d, J = 2.2 Hz, 0.29 H), 7.55 ( d, J = 2.8 Hz, 1H) 7.25–7.20 (m, 2H), 7.20 – 7.13 (m, 2H); Exact Mass for C16H10F2N2O: 284.0761; HRMS: [M+H]+: 285.0830; HPLC4: tR 13.97 min, purity 98.0%.
(4-chlorophenyl)(4-(4-chlorophenyl)-1H-imidazol-2-yl)methanone and (4-chlorophenyl)(5-(4-chlorophenyl)-1H-imidazol-2-yl)methanone(11c)
1H NMR (400 MHz, Chloroform-d) δ 10.70 (s, 0.65 H), 10.55 (s, 1H), 8.78 (d, J = 8.65 Hz, 2H), 8.63 (s, 1H), 7.87 (s, 2H), 7.66 – 7.52 (m, 5H), 7.50 – 7.39 (m, 2H); Exact Mass for C16H10Cl2N2O: 316.017; HRMS: [M+H]+: 317.0292; HPLC4: tR 16.30 min, purity 98.8%.
4-bromophenyl-(4-(4-bromophenyl)-1H-imidazol-2-yl)ketone and 4-bromophenyl-(5-(4-bromophenyl)-1H-imidazol-2-yl)methanoe(11d)
1H NMR (500 MHz, DMSO-d6) δ 13.91(s, 0.16H),13.73 (s, 1H), 8.51 ( d, J = 8.4 Hz, 2H), 8.42 (d, J = 8.3 Hz, 0.32H), 8.16 (s, 1H), 7.93 (d, J = 8.15 Hz, 0.32H), 7.89 (d, J = 8.35 Hz, 2H),7.83 (d, J = 8.45 Hz, 2H), 7.80 (d, J = 8.4 Hz, 0.32H), 7.79 (s, 0.16H), 7.67(d, J = 8.05 Hz, 0.32H), 7.62 (d, J = 8.35 Hz, 2H); Exact Mass for C16H10Br2N2O: 403.916; HRMS: [M+H]+: 404.9241; HPLC4: tR 16.53 min, purity 95.5%.
(4-(trifluoromethyl) phenyl)(4-(4-(trifluoromethyl)phenyl)-1H-imidazol-2-yl)methanone and (4-(trifluoromethyl)phenyl)(5-(4-(trifluoromethyl)phenyl)-1H-imidazol-2-yl)methanone (11e)
1H NMR (400 MHz, Chloroform-d) δ 10.83 (s, 0.38H), 10.60 (s, 1H), 8.78 (d, J = 8.19 Hz, 2H), 8.64 (d, J = 8.13 Hz, 0.48H), 7.93 (d, J = 8.34 Hz, 2H), 7.76 (d, J = 8.56 Hz, 2H), 7.66 – 7.59 (m, 3H); Exact Mass for C18H10F6N2O: 384.0697; HRMS: [M+H]+: 385.0790; HPLC1: tR 7.76 min, purity 98.3%.
p-tolyl(4-p-tolyl-1H-imidazol-2-yl)methanone and p-tolyl(5-p-tolyl-1H-imidazol-2-yl)methanone(11f)
1H NMR (400 MHz, Chloroform-d) δ 10.96 (s, 1H), 10.73 (s, 1H), 8.71 (d, J = 8.26 Hz, 2H), 8.54 (d, J = 8.23 Hz, 2H), 7.82 (d, J = 8.11 Hz, 2H), 7.67 – 7.49 (m, 4H), 7.37 (t, J = 7.62, 7.62 Hz, 4H), 7.30 (s, 1H), 7.28 (s, 2H), 7.26 (s, 1H), 2.49 (d, J = 4.52 Hz, 6H), 2.42 (d, J = 4.95 Hz, 6H); Exact Mass for C18H16N2O: 276.1263; HRMS: [M+H]+: 277.1385; HPLC3: tR 10.27 min, purity 98.7%.
(4-methoxyphenyl)(4-(4-methoxyphenyl)-1H-imidazol-2-yl)methanone and (4-methoxyphenyl)(5-(4-methoxyphenyl)-1H-imidazol-2-yl)methanone (11g)
1H NMR (400 MHz, Chloroform-d) δ 10.50 (s, 1H), 10.38 (s, 1H), 8.77 (d, J = 8.90 Hz, 2H), 8.60 (d, J = 8.89 Hz, 2H), 7.77 (s, 1H), 7.75 (s, 1H), 7.48 (d, J = 8.75 Hz, 1H), 7.45 – 7.36 (m, 2H), 7.03 – 6.87 (m, 5H), 3.86 (s, 2H), 3.85 (s, 2H), 3.84 (s, 2H), 3.80 (s, 2H), 3.79 (s, 2H); Exact Mass for C18H10F6N2O: 384.0697; HRMS: [M+H]+: 385.0790; HPLC2: tR 6.05 min, purity 97.2%.
(4-(dimethylamino)phenyl)(4-(4-(dimethylamino)phenyl)-1H-imidazol-2-yl)methanone and (4-(dimethylamino)phenyl)(5-(4-(dimethylamino)phenyl)-1H-imidazol-2-yl)methanone(11h)
1H NMR (500 MHz, DMSO-d6) δ 13.17 (s, 0.35H), 13.12 (s, 1H), 8.64 (d, J = 8.95 Hz, 2H), 8.50 (d, J = 8.95 Hz, 1H), 7.80 – 7.69 (m, 4H), 7.50 (s, 1H), 6.82 (d, J = 8.99 Hz, 2H), 6.76 (t, J = 7.70, 7.70 Hz, 5H), 3.07 (s, 6H), 3.05 (s, 4H), 2.95 (s, 4H), 2.93 (s, 6H) ); Exact Mass for C18H10F6N2O: 384.0697; HRMS: [M+H]+: 385.0790; HPLC1: tR 5.43 min, purity 95.3%.
(4-hydroxyphenyl)(4-(4-hydroxyphenyl)-1H-imidazol-2-yl)methanone and (4-hydroxyphenyl)(5-(4-hydroxyphenyl)-1H-imidazol-2-yl)methanone(11i)
1H NMR (500 MHz, DMSO-d6) δ 13.37 (s, 0.37H), 13.29 (s, 1H), 10.39 (s, 1H), 9.46 (s, 1H), 8.60 (d, J = 7.88 Hz, 3H), 8.47 (s, 1H), 7.79 (s, 2H), 7.73 (d, J = 7.48 Hz, 4H), 7.55 (s, 1H), 6.92 (d, J = 8.02 Hz, 4H), 6.81 (d, J = 8.03 Hz, 4H); Exact Mass for C16H12N2O3: 280.0800; HRMS: [M+H]+: 281.0967; HPLC2: tR 3.54 min, purity 99.8%.
(4-phenyl-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone and (5-phenyl-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone (12a)
1H NMR (400 MHz, Chloroform-d) δ 10.63 (s, 0.48H), 10.47 (s, 1H), 8.19 (s, 2H), 7.98 (s, 1H), 7.82 (t, J = 1.67, 1.67 Hz, 1H), 7.81 (t, J = 1.11, 1.11 Hz, 1H), 7.60 – 7.53 (m, 1H), 7.51 (d, J = 1.97 Hz, 1H), 7.46 – 7.30 (m, 3H), 7.29 – 7.22 (m, 1H), 3.95 (s, 5H), 3.91 (s, 3H), 3.91 (s, 3H), 3.89 (s, 1H); Exact Mass for C19H18N2O4: 338.1267; HRMS: [M+H]+: 339.1423; HPLC2: tR 6.40 min, purity 95.0%.
(4-(4-fluorophenyl)-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone and (5-(4-fluorophenyl)-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone (12b)
1H NMR (400 MHz, Chloroform-d) δ 10.60 (s, 0.30H), 10.46 (s, 1H), 8.15 (s, 2H), 7.97 (s, 1H), 7.77 (dd, J = 5.35, 8.90 Hz, 2H), 7.58 – 7.50 (dd, J = 5.10, 8.20 Hz, 0.47 H), 7.48 (s, 0.46 H), 7.46 (s, 1H), 7.45 (s, 1H), 7.10 – 7.02 (m, 2H), 3.94 (s, 6H), 3.92 (s, 3H), 3.91 (s, 2H), 3.89 (s, 1H); Exact Mass for C19H17FN2O4: 356.1172; HRMS: [M+H]+: 357.1245; HPLC2: tR 6.90 min, purity 99.3%.
(4-(4-chlorophenyl)-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone and (5-(4-chlorophenyl)-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone (12c)
1H NMR (400 MHz, Chloroform-d) δ 10.57 (s, 0.33H), 10.45 (s, 1H), 8.15 (s, 2H), 7.97 (s, 0.49H), 7.81 – 7.68 (m, 2H), 7.51 – 7.47 (m, 1H), 7.36 – 7.30 (m, 2H), 3.94 (s, 6H), 3.91 (s, 3H), 3.89 (s, 0.75H); Exact Mass for C19H17ClN2O4: 372.0877; HRMS: [M+H]+: 373.0992; HPLC4: tR 15.76 min, purity 95.6%.
(4-(4-bromophenyl)-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone and (5-(4-bromophenyl)-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone(12d)
1H NMR (400 MHz, Chloroform-d) δ 10.77 (s, 0.36H), 10.59 (s, 1H), 8.24 (s, 2H), 8.06 (s, 1H), 7.78 (d, J = 1.86 Hz, 1H), 7.76 (d, J = 1.98 Hz, 1H), 7.69 – 7.47 (m, 4H), 4.03 (s, 6H), 4.01 (s, 3H), 4.00 (s, 2H), 3.99 (s, 1H); Exact Mass for C19H17BrN2O4: 416.0372; HRMS: [M+H]+: 417.0496; HPLC1: tR 6.37 min, purity 96.7%.
(4-(4-(trifluoromethyl)phenyl)-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone and (5-(4-(trifluoromethyl)phenyl)-1H-imidazol-2-yl)(3,4,5trimethoxyphenyl)methanone (12e)
1H NMR (400 MHz, Chloroform-d) δ 10.90 (s, 0.16H), 10.67 (s, 1H), 8.26 (s, 2H), 8.08 (s, 0.36H), 8.01 (d, J = 7.30 Hz, 2H), 7.80–7.88 (m, 0.79H), 7.76 – 7.62 (m, 3H), 4.08 – 3.95 (m, 11H); Exact Mass for C20H17F3N2O4: 406.114; HRMS: [M+H]+: 407.1319; HPLC2: tR 9.60 min, purity 95.1%.
(4-p-tolyl-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone and (5-p-tolyl-1Himidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone (12f)
1H NMR (400 MHz, Chloroform-d) δ 10.57 (s, 0.77H), 10.44 (s, 1H), 8.18 (s, 2H), 7.96 (s, 1H), 7.71 (d, J = 1.87 Hz, 1H), 7.69 (d, J = 1.88 Hz, 1H), 7.47 (d, J = 2.44 Hz, 2H), 7.22 (s, 1H), 7.16 (s, 1H), 3.94 (s, 6H), 3.92 (s, 3H), 3.90 (s, 3H), 3.89 (s, 2H); Exact Mass for C20H20N2O4: 352.1423; HRMS: [M+H]+: 353.1527; HPLC1: tR 5.63 min, purity 95.9%.
(4-(4-methoxyphenyl)-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone and (5-(4-methoxyphenyl)-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone (12g)
1H NMR (400 MHz, Chloroform-d) δ 10.60 (s, 1H), 10.50 (s, 1H), 8.27 (s, 2H), 8.05 (s, 1H), 7.84 (s, 1H), 7.82 (s, 1H), 7.59 (d, J = 8.87 Hz, 1H), 7.54 (d, J = 1.90 Hz, 1H), 7.51 (d, J = 2.31 Hz, 1H), 7.05 – 6.97 (m, 4H), 4.04 (s, 5H), 4.01 (s, 3H), 4.00 (s, 3H), 3.98 (s, 2H), 3.89 (s, 2H), 3.88 (s, 3H); Exact Mass for C20H20N2O5: 368.1372; HRMS: [M+H]+: 369.1572; HPLC4: tR 13.78 min, purity 96.5%.
(4-(4-(dimethylamino)phenyl)-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone and (5-(4-(dimethylamino)phenyl)-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone (12h)
1H NMR (500 MHz, Chloroform-d) δ 10.67 (s, 1H), 10.54 (s, 0.49H), 8.28 (s, 1H), 8.03 (s, 2H), 7.78 (s, 1H), 7.76 (s, 1H), 7.53 (d, J = 8.79 Hz, 2H), 7.50 (d, J = 1.56 Hz, 1H), 6.78 (m, 4H), 4.03 (s, 4H), 3.99 (s, 8H), 3.97 (s, 3H), 3.04 (s, 6H), 3.02 (s, 4H); Exact Mass for C21H23N3O4: 381.1689; HRMS: [M+H]+: 382.1842; HPLC1: tR 5.53 min, purity 96.0%.
(4-(4-hydroxyphenyl)-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone and (5-(4-hydroxyphenyl)-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone)(12i)
1H NMR (500 MHz, Chloroform-d) δ 11.19 (s, 1H), 10.75 (s, 1H), 8.23 (s, 2H), 7.98 (s, 2H), 7.77 (s, 1H), 7.75 (s, 1H), 7.54 (d, J = 5.13 Hz, 3H), 7.49 (s, 1H), 6.90 (t, J = 9.24, 9.24 Hz, 4H), 4.01 (s, 6H), 3.99 (s, 3H), 3.97 (s, 5H), 3.96 (s, 3H); Exact Mass for C19H18N2O5: 354.1216; HRMS: [M+H]+: 355.1378; HPLC2: tR 4.46 min, purity 98.1%.
phenyl(4-(3,4,5-trimethoxyphenyl)-1H-imidazol-2-yl)methanone and phenyl(5-(3,4,5-trimethoxyphenyl)-1H-imidazol-2-yl)methanone(13a)
1H NMR (400 MHz, Chloroform-d) δ 10.67 (s, 1H), 10.51 (s, 1H), 8.68 – 8.63 (m, 2H), 8.53 – 8.49 (m, 1H), 7.62 – 7.53 (m, 2H), 7.53 – 7.44 (m, 5H), 7.04 (s, 2H), 6.75 (s, 1H), 3.89 (s, 6H), 3.89 (s, 3H), 3.83 (s, 2H), 3.82 (s, 3H);Exact Mass for C19H18N2O4: 338.1267; HRMS: [M+H]+: 339.1348; HPLC2: tR 5.23 min, purity 97.4%.
(4-bromophenyl)(4-(3,4,5-trimethoxyphenyl)-1H-imidazol-2-yl)methanone and (4-bromophenyl)(5-(3,4,5-trimethoxyphenyl)-1H-imidazol-2-yl)methanone(13d)
1H NMR (400 MHz, Chloroform-d) δ 10.66 (s, 1H), 10.57 (s, 1H), 8.72 – 8.62 (m, 2H), 8.56 – 8.50 (m, 1H), 7.80 – 7.75 (m, 1H), 7.75 – 7.67 (m, 4H), 7.57 (q, J = 2.04 Hz, 2H), 7.12 (s, 2H), 6.82 (s, 1H), 3.99 (s, 6H), 3.98 (s, 4H), 3.93 (s, 2H), 3.92 (s, 3H); Exact Mass for C19H17BrN2O4: 416.0372; HRMS: [M+H]+: 417.0454; HPLC2: tR 7.28 min, purity 97.9%.
(4-methoxyphenyl)(4-(3,4,5-trimethoxyphenyl)-1H-imidazol-2-yl)methanone and (4-methoxyphenyl)(5-(3,4,5-trimethoxyphenyl)-1H-imidazol-2-yl)methanone(13g)
1H NMR (400 MHz, Chloroform-d) δ 10.80 (s, 1H), 10.66 (s, 1H), 8.87 – 8.79 (m, 2H), 8.73 – 8.67 (m, 1H), 7.56 (d, J = 1.83 Hz, 1H), 7.53 (d, J = 2.32 Hz, 1H), 7.14 (s, 2H), 7.05 (dd, J = 3.59, 8.90 Hz, 3H), 6.83 (s, 1H), 3.99 (s, 6H), 3.96 (s, 3H), 3.95 (s, 3H), 3.94 (s, 2H), 3.92 (s, 2H), 3.91 (s, 3H); Exact Mass for C20H20N2O5: 368.1372; HRMS: [M+H]+: 369.1494; HPLC2: tR 5.94 min, purity 97.4%.
(4-(dimethylamino)phenyl)(4-(3,4,5-trimethoxyphenyl)-1H-imidazol-2-yl)methanone and (4-(dimethylamino)phenyl)(5-(3,4,5-trimethoxyphenyl)-1H-imidazol-2-yl)methanone(13h)
1H NMR (400 MHz, Chloroform-d) δ 11.20 (s, 1H), 10.94 (s, 1H), 8.94 – 8.75 (m, 2H), 8.74 – 8.61 (m, 1H), 7.54 (s, 1H), 7.50 (s, 1H), 7.15 (d, J = 2.41 Hz, 2H), 6.86 (s, 1H), 6.76 (dd, J = 5.30, 9.05 Hz, 3H), 3.99 (s, 7H), 3.93 (s, 4H), 3.91 (s, 6H), 3.14 (s, 6H), 3.13 (s, 4H); Exact Mass for C21H23N3O4: 381.1689; HRMS: [M+H]+: 382.1842; HPLC1: tR 4.82 min, purity 97.8%.
(4-hydroxyphenyl)(4-(3,4,5-trimethoxyphenyl)-1H-imidazol-2-yl)methanone and (4-hydroxyphenyl)(5-(3,4,5-trimethoxyphenyl)-1H-imidazol-2-yl)methanone(13i)
1H NMR (500 MHz, Chloroform-d) δ 11.81 (s, 1H), 11.45 (s, 1H), 8.62 – 8.44 (m, 2H), 8.39 – 8.19 (m, 2H), 7.53 (d, J = 31.95 Hz, 2H), 7.07 (s, 2H), 6.82 (dd, J = 12.16, 30.47 Hz, 6H), 3.90 (s, 5H), 3.88 (s, 8H), 3.84 (s, 6H); Exact Mass for C19H18N2O5: 354.1216; HRMS: [M+H]+: 355.1339; HPLC1: tR 4.00 min, purity 97.4%.
3,4,5-Trimethoxyphenyl-(4-(3,4,5-trimethoxyphenyl)-1H-imidazol-2-yl)methanone and 3,4,5-trimethoxyphenyl-(5-(3,4,5-trimethoxyphenyl)-1H-imidazol-2-yl)methanone(14)
1H NMR (400 MHz, Chloroform-d) δ 10.76 (s, 0.40H), 10.61 (s, 1H), 8.20 (s, 2H), 7.95 (s, 1H), 7.48 (d, J = 1.90 Hz, 0.40H), 7.47 (d, J = 2.34 Hz, 1H), 7.06 (s, 2H), 6.75 (s, 1H), 3.93 (s, 5H), 3.91 (d, J = 0.84 Hz, 4H), 3.89 (s, 1H), 3.87 (s, 2H), 3.86 ( s, 5H) 3.83 (s, 1H), 3.82 (s, 2H); Exact Mass for C22H24N2O7: 428.1584; HRMS: [M+H]+: 429.1677; HPLC3: tR 9.60 min, purity 96.4%.
General procedure for the preparation of (4 or 5)-aryl-2-aryloyl-(1H)-Imidazole derivatives (15a-c)
To a solution of 12a (135 mg, 0.4 mmol)in THF (10 mL) in ice-bath was added sodium hydride (60% dispersion in mineral oil, 28 mg, 0.60 mmol) followed by adding methyl iodide (85 mg, 0.60 mmol) (for 15a) or ethyl iodide (93 mg, 0.60 mmol) (for 15b)or benzyl bromide(102 mg, 0.60 mmol) (for 15c). The resulting reaction mixture was stirred overnight under reflux condition. After dilution by 50 ml of saturated NaHCO3 solution (aqueous), the reaction mixture was extracted by ethyl acetate (100 ml). The organic layer was dried over magnesium sulfate and concentrated. The residue was purified by flash column chromatography.
(1-methyl-4-phenyl-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone (15a)
1H NMR (500 MHz, Chloroform-d) δ 7.97 (d, J = 2.38 Hz, 2H), 7.85 (d, J = 6.01 Hz, 2H), 7.46 – 7.39 (m, 3H), 7.28 (d, J = 2.39 Hz, 1H), 4.16 – 4.10 (m, 3H), 3.99 (d, J = 2.82 Hz, 9H); Exact Mass for C20H20N2O4: 352.1423; HRMS: [M+H]+: 353.1527; HPLC1: tR 6.03 min, purity 96.4%.
(1-ethyl-4-phenyl-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone(15b)
1H NMR (400 MHz, Chloroform-d) δ 7.95 (s, 2H), 7.89 – 7.83 (m, 2H), 7.51 (s, 1H), 7.43 (t, J = 7.61, 7.61 Hz, 2H), 7.36 – 7.31 (m, 1H), 4.56 (q, J = 7.19, 7.19, 7.19 Hz, 2H), 3.99 (s, 6H), 3.98 (s, 3H), 1.30 (t, J = 7.19, 7, 19 Hz, 3H); Exact Mass for C21H22N2O4: 366.158; HRMS: [M+H]+: 367.1725; HPLC1: tR 6.47 min, purity 97.1%.
(1-benzyl-4-phenyl-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone(15c)
1H NMR (500 MHz, Chloroform-d) δ 7.94 (d, J = 1.70 Hz, 2H), 7.84 (d, J = 7.73 Hz, 2H), 7.47 – 7.28 (m, 9H), 5.74 (s, 2H), 3.98 (s, 9H); Exact Mass for C26H24N2O4: 428.1736; HRMS: [M+H]+: 429.1931; HPLC1: tR 6.70 min, purity 95.4%.
General procedure for the preparation of (4 or 5)-aryl-2-aryloyl-(1H)-Imidazole derivatives (15d-f)
To a solution of 12a (135 mg, 0.4 mmol) in ACN (10 mL) was added potassium carbonate (82 mg, 0.60 mmol) followed by n-propyl iodide (82 mg, 0.48 mmol) (for 15d) or i-propyl iodide (82 mg, 0.48 mmol) (for 15e) or cyclopentyl bromide (72 mg, 0.48 mmol) (for 15f). The resulting reaction mixture was stirred overnight under reflux condition. After dilution by 50 ml of saturated NaHCO3 solution (aqueous), the reaction mixture was extracted by ethyl acetate (100 ml). The organic layer was dried over magnesium sulfate and concentrated. The residue was purified by flash column chromatography.
(4-phenyl-1-propyl-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone(15d)
1H NMR (500 MHz, Chloroform-d) δ 7.94 (s, 2H), 7.90 – 7.82 (m, 2H), 7.47 (s, 1H), 7.42 (t, J = 7.67 Hz, 2H), 7.34 – 7.28 (m, 1H), 4.51 – 4.40 (m, 2H), 3.98 (d, J = 1.49 Hz, 9H), 1.96 (h, J = 7.40 Hz, 2H), 1.03 (t, J = 7.42 Hz, 3H); Exact Mass for C22H24N2O4: 380.1700; HRMS: [M+H]+: 381.1897; HPLC1: tR 6.88 min, purity 98.0%.
(1-isopropyl-4-phenyl-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone(15e)
1H NMR (400 MHz, Chloroform-d) δ 7.79 (s, 3H), 7.78 – 7.73 (m, 1H), 7.57 (s, 1H), 7.34 (dd, J = 6.87, 8.44 Hz, 2H), 7.25 – 7.21 (m, 1H), 5.66 – 5.21 (m, 1H), 3.89 (s, 9H), 1.52 (s, 6H); Exact Mass for C22H24N2O4: 380.1700; HRMS: [M+H]+: 381.1937; HPLC1: tR 6.90 min, purity 98.3%.
(1-cyclopentyl-4-phenyl-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone(15f)
1H NMR (500 MHz, Chloroform-d) δ 7.90 – 7.82 (m, 4H), 7.61 (d, J = 1.7 Hz, 1H), 7.42 (td, J = 7.7, 1.7 Hz, 2H), 7.33 – 7.28 (m, 1H), 5.88 – 5.32 (m, 1H), 3.98 (t, J = 1.9 Hz, 9H), 2.64 – 2.20 (m, 2H), 2.00 – 1.77 (m, 6H); Exact Mass for C24H26N2O4: 406.1893; HRMS: [M+H]+: 407.2103; HPLC1: tR 8.07 min, purity 98.3%.
General procedure for the preparation of (4 or 5)-aryl-2-aryloyl-(1H)-Imidazole derivatives (15g-h)
Under inert atmosphere, a Schlenk flask was charged with Cs2CO3 (260 mg, 0.8 mmol), CuI (76 mg, 0.4 mmol), ligand (0.4 mmol), compound 12a (135 mg, 0.4 mmol), 2-pyrimidyl bromide (124 mg, 0.8 mmol) (for 15g) or 2-bromothiophene (130 mg, 0.8 mmol) (for 15h) and DMF (5 mL). The reaction mixture was stirred for 30 min at room temperature, and then heated to 110 °C for 2 days. The reaction mixture was monitored by TLC. After the starting material was completely consumed, the reaction was stopped and the mixture was cooled to room temperature. The reaction mixture was directly passed through a plug of silca gel. After being rinsed with ethyl acetate, the combined filtrate was washed with saturated brine. After the organic layer was dried by sodium sulfate, it was concentrated. The residue was purified by column chromatography on silica gel to provide the desired product.
(4-phenyl-1-(pyridin-2-yl)-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone (15g)
1H NMR (500 MHz, Chloroform-d) δ 8.59 (dt, J = 1.70, 4.33 Hz, 1H), 7.91 (ddt, J = 1.67, 3.99, 7.42 Hz, 3H), 7.86 (d, J = 1.52 Hz, 2H), 7.82 (d, J = 1.57 Hz, 1H), 7.48 – 7.40 (m, 4H), 7.34 (td, J = 1.38, 7.27 Hz, 1H), 3.98 (d, J = 1.62 Hz, 3H), 3.96 (d, J = 1.41 Hz, 6H). Exact Mass for C24H21N3O4: 415.1500; HRMS: [M+H]+: 416.1692; HPLC1: tR 4.85 min purity, 97.9%.
(4-phenyl-1-(thiophen-2-yl)-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone (15h)
1H NMR (500 MHz, Chloroform-d) δ 7.87 – 7.76 (m, 4H), 7.52 (d, J = 1.84 Hz, 1H), 7.37 (td, J = 1.83, 7.63, 8.06 Hz, 2H), 7.30 – 7.23 (m, 2H), 7.06 (dd, J = 1.95, 3.59 Hz, 1H), 6.97 (dp, J = 1.70, 5.60 Hz, 1H), 3.89 (d, J = 1.94 Hz, 3H), 3.88 (d, J = 1.95 Hz, 6H); Exact Mass for C23H20N2O4S: 420.1100; HRMS: [M+H]+: 421.1298; HPLC1: tR 6.33 min, purity 98.2%.
General procedure for the preparation of (4 or 5)-alkyl-(5 or 4)-aryl-2-aryloyl-(1H)-Imidazole derivatives (18a-c)
To ammonium acetate (10 mmol) in ethanol (5 ml) and water (0.3 ml) was added phenyl alkyl diones 17(a-c) (1 mmol) in ethanol (5 ml) and 3, 4, 5-trimethoxyphenyl glyoxal hydrate 10 (1 mmol) in ethanol (10 ml). The mixture was stirred at room temperature for 30–45 min. The reaction was stopped after the consumption of the starting material monitored by TLC. The mixture was then extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate and concentrated to get the crude product. The crude was purified by flash chromatography.
(5-methyl-4-phenyl-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone and (4-methyl-5-phenyl-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone (18a)
1H NMR (400 MHz, Chloroform-d) δ 10.43 (s, 0.59H), 10.32 (s, 1H), 8.24 (s, 2H), 8.05 (s, 1H), 7.87 – 7.71 (m, 3H), 7.62 – 7.38 (m, 6H), 4.02 (s, 5H), 4.01 (s, 3H), 3.99 (s, 2H), 3.98 (s, 2H), 3.95 (s, 2H), 2.64 (s, 3H), 2.56 (s, 1H); Exact Mass for C19H17ClN2O4: 372.0877; HRMS: [M+H]+: 373.0992; HPLC2: tR 6.42 min, purity 95.4%.
(5-ethyl-4-phenyl-1H-imidazol-2-yl)(3, 4, 5-trimethoxyphenyl)methanone and (4-ethyl-5-phenyl-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone (18b)
1H NMR (400 MHz, Chloroform-d) δ 10.85 (s, 1H), 8.28 (s, 2H), 8.15 (s, 1H), 7.75 (m, 1H), 7.45–7.30 (m, 5H), 4.00(m, 9H), 3.05 (m, 2H), 1.40 (m, 3H); Exact Mass for C19H17ClN2O4: 372.0877; HRMS: [M+H]+: 373.0992; HPLC1: tR 5.37 min, purity 96.3%.
(4-phenyl-5-propyl-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone and (5-phenyl-4-propyl-1H-imidazol-2-yl)(3,4,5-trimethoxyphenyl)methanone(18c)
1H NMR (400 MHz, Chloroform-d) δ 10.52 (s, 0.52 H), 10.47 (s, 1H), 8.24 (s, 2H), 8.14 (s, 1H), 7.78 (d, J = 7.39 Hz, 2H), 7.60 – 7.43 (m, 5H), 7.36 (t, J = 7.40, 7.40 Hz, 1H), 4.01 (s, 5H), 4.00 (s, 3H), 3.99 (s, 4H), 3.00 – 2.93 (t, J = 7.40, 7.40 Hz, 2H), 2.84 (t, J = 7.50, 7.50 Hz, 1H), 1.94 – 1.87 (m, 1H), 1.87 – 1.78 (m, 2H), 1.07 (t, J = 7.33, 7.33 Hz, 3H), 0.98 – 0.92 (t, J = 7.10, 7.10 Hz, 1H); Exact Mass for C19H17ClN2O4: 372.0877; HRMS: [M+H]+: 373.0992; HPLC: tR 12.46 min, purity 96.1%.
Cell Culture and Cytotoxicity Assay
We examined the antiproliferative activity of the RABI compounds in four human melanoma cell lines (A375 and WM-164, MDA-MB-435 and MDA-MB-435/LCC6MDR1) and four human prostate cancer cell lines (LNCaP, DU 145, PC-3, and PPC-1). All these cell lines were purchased from ATCC (American Type Culture Collection, Manassas, VA). Melanoma cells were cultured in DMEM (Cellgro Mediatech Inc., Herndon, VA), and prostate cancer cells were cultured in RPMI 1640 (Cellgro Mediatech, Inc., Herndon, VA) supplemented with 10% fetal bovine serum (Cellgro Mediatech). Cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2. Then 1000–5000 cells were plated into each well of 96-well plates depending on growth rate and exposed to different concentrations of a test compound for 48 h (fast growing melanoma cells) or 96 h (slow growing prostate cancer cells) in three-five replicates. Cell numbers at the end of the drug treatment were measured by the sulforhodamine B (SRB) assay. Briefly, the cells were fixed with 10% trichloroacetic acid and stained with 0.4% SRB, and the absorbances at 540 nm were measured using a plate reader (DYNEX Technologies, Chantilly VA). Percentages of cell survival versus drug concentrations were plotted, and the IC50 (concentration that inhibited cell growth by 50% of untreated control) values were obtained by nonlinear regression analysis using GraphPad Prism (GraphPad Software, San Diego, CA).
Cell Cycle Analysis
Cell cycle distribution was determined by propidium iodide (PI) staining. Treated cells were washed with PBS and fixed with 70% ice-cold ethanol overnight. Fixed cells were then stained with 20 μg/mL of PI in the presence of RNase A (300 μg/mL) at 37° C. for 30 min. Cell cycle distribution was analyzed by fluorescence-activated cell sorting (FACS) analysis core services at the University of Tennessee Health Science Center, TN.
Molecular Modeling
All molecular modeling studies were performed with Schrodinger Molecular Modeling Suite 2011 (Schrodinger LLC, New York, NY) running on a Dell Linux workstation. We selected tubulin complex with TN16 (PDB code: 3HKD) and tubulin complex with colchicine (PDB code: 1SA0) as our modeling system. RABIs were built and prepared using the Ligprep module, and they were docked into the TN16 site and colchicine site by the Glide module in the Schrodinger Suite. The best docking complexes were subject to restricted molecular dynamics to release any strains by using the Macromodel module with OPLS-2005 force field. The ligand and its surrounding residues within 15 Å were allowed to move freely while the outer atoms are frozen.
Supplementary Material
Acknowledgments
This work was supported by the NIH grant R01CA148706, 1S10RR026377-01, 1S10OD010678-01 and funds from GTx, Inc. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS USED
- ABI
2-aryl-4-benzoyl-imidazoles
- RABI
reverse ABI
- HMBC
heteronuclear multiple-bond correlation spectroscopy
- Pgp
P-glycoprotein
- SAR
structure–activity relationship
- ND
Not Determined
- PI
propidium iodide
- FACS
fluorescence-activated cell sorting
- SRB
sulforhodamine B
Footnotes
Supporting Information Available:
Structure elucidation of 12a; in vitro tubulin polymerization assay; competitive binding at the colchicine site in tubulin for compounds 12a and 15g. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA: a cancer journal for clinicians. 2011;61(2):69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA: a cancer journal for clinicians. 2012;62(1):10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 3.Chen J, Wang Z, Li CM, Lu Y, Vaddady PK, Meibohm B, Dalton JT, Miller DD, Li W. Discovery of novel 2-aryl-4-benzoyl-imidazoles targeting the colchicines binding site in tubulin as potential anticancer agents. Journal of medicinal chemistry. 2010;53(20):7414–7427. doi: 10.1021/jm100884b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen J, Ahn S, Wang J, Lu Y, Dalton JT, Miller DD, Li W. Discovery of novel 2-aryl-4-benzoyl-imidazole (ABI-III) analogues targeting tubulin polymerization as antiproliferative agents. Journal of medicinal chemistry. 2012;55(16):7285–7289. doi: 10.1021/jm300564b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li CM, Lu Y, Chen J, Costello TA, Narayanan R, Dalton MN, Snyder LM, Ahn S, Li W, Miller DD, Dalton JT. Orally bioavailable tubulin antagonists for paclitaxel-refractory cancer. Pharmaceutical research. 2012;29(11):3053–3063. doi: 10.1007/s11095-012-0814-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z, Chen J, Wang J, Ahn S, Li CM, Lu Y, Loveless VS, Dalton JT, Miller DD, Li W. Novel tubulin polymerization inhibitors overcome multidrug resistance and reduce melanoma lung metastasis. Pharmaceutical research. 2012;29(11):3040–3052. doi: 10.1007/s11095-012-0726-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Li CM, Wang J, Ahn S, Wang Z, Lu Y, Dalton JT, Miller DD, Li W. Synthesis and antiproliferative activity of novel 2-aryl-4-benzoyl-imidazole derivatives targeting tubulin polymerization. Bioorganic & medicinal chemistry. 2011;19(16):4782–4795. doi: 10.1016/j.bmc.2011.06.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li CM, Chen J, Lu Y, Narayanan R, Parke DN, Li W, Ahn S, Miller DD, Dalton JT. Pharmacokinetic optimization of 4-substituted methoxybenzoyl-aryl-thiazole and 2-aryl-4-benzoyl-imidazole for improving oral bioavailability. Drug metabolism and disposition: the biological fate of chemicals. 2011;39(10):1833–1839. doi: 10.1124/dmd.110.036616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khalili B, Tondro T, Hashemi MM. Novel one-pot synthesis of (4 or 5)-aryl-2-aryloyl-(1H)-imidazoles in water and tauto-isomerization study using NMR. Tetrahedron. 2009;65(34):6882–6887. [Google Scholar]
- 10.Parkinson EI, Jason Hatfield M, Tsurkan L, Hyatt JL, Edwards CC, Hicks LD, Yan B, Potter PM. Requirements for mammalian carboxylesterase inhibition by substituted ethane-1,2-diones. Bioorganic & medicinal chemistry. 2011;19(15):4635–4643. doi: 10.1016/j.bmc.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pelphrey PM, Popov VM, Joska TM, Beierlein JM, Bolstad ES, Fillingham YA, Wright DL, Anderson AC. Highly efficient ligands for dihydrofolate reductase from Cryptosporidium hominis and Toxoplasma gondii inspired by structural analysis. Journal of medicinal chemistry. 2007;50(5):940–950. doi: 10.1021/jm061027h. [DOI] [PubMed] [Google Scholar]
- 12.Riley HAG, AR . Organic Syntheses. Vol. 2. Wiley & Sons; New York, NY: 1943. General procedure for synthesis of glyoxal derivatives; pp. 509–510. [Google Scholar]
- 13.Corelli F, Summa V, Brogi A, Monteagudo E, Botta M. Chiral Azole Derivatives2. Synthesis of Enantiomerically Pure 1-Alkylimidazoles. J Org Chem. 1995;60(7):2008–2015. [Google Scholar]
- 14.Sabbah M, Soulere L, Reverchon S, Queneau Y, Doutheau A. LuxR dependent quorum sensing inhibition by N,N′-disubstituted imidazolium salts. Bioorganic & medicinal chemistry. 2011;19(16):4868–4875. doi: 10.1016/j.bmc.2011.06.075. [DOI] [PubMed] [Google Scholar]
- 15.Xi ZX, Liu FH, Zhou YB, Chen WZ. CuI/L (L = pyridine-functionalized 1,3-diketones) catalyzed C-N coupling reactions of aryl halides with NH-containing heterocycles. Tetrahedron. 2008;64(19):4254–4259. [Google Scholar]
- 16.Lu Y, Li CM, Wang Z, Ross CR, 2nd, Chen J, Dalton JT, Li W, Miller DD. Discovery of 4-substituted methoxybenzoyl-aryl-thiazole as novel anticancer agents: synthesis, biological evaluation, and structure-activity relationships. Journal of medicinal chemistry. 2009;52(6):1701–1711. doi: 10.1021/jm801449a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leonessa F, Green D, Licht T, Wright A, Wingate-Legette K, Lippman J, Gottesman MM, Clarke R. MDA435/LCC6 and MDA435/LCC6MDR1: ascites models of human breast cancer. British journal of cancer. 1996;73(2):154–161. doi: 10.1038/bjc.1996.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vredenburg MR, Ojima I, Veith J, Pera P, Kee K, Cabral F, Sharma A, Kanter P, Greco WR, Bernacki RJ. Effects of orally active taxanes on P-glycoprotein modulation and colon and breast carcinoma drug resistance. Journal of the National Cancer Institute. 2001;93(16):1234–1245. doi: 10.1093/jnci/93.16.1234. [DOI] [PubMed] [Google Scholar]
- 19.Zhang S, Morris ME. Effects of the flavonoids biochanin A, morin, phloretin, and silymarin on P-glycoprotein-mediated transport. The Journal of pharmacology and experimental therapeutics. 2003;304(3):1258–1267. doi: 10.1124/jpet.102.044412. [DOI] [PubMed] [Google Scholar]
- 20.Dong X, Mattingly CA, Tseng MT, Cho MJ, Liu Y, Adams VR, Mumper RJ. Doxorubicin and paclitaxel-loaded lipid-based nanoparticles overcome multidrug resistance by inhibiting P-glycoprotein and depleting ATP. Cancer research. 2009;69(9):3918–3926. doi: 10.1158/0008-5472.CAN-08-2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barbier P, Dorleans A, Devred F, Sanz L, Allegro D, Alfonso C, Knossow M, Peyrot V, Andreu JM. Stathmin and interfacial microtubule inhibitors recognize a naturally curved conformation of tubulin dimers. The Journal of biological chemistry. 2010;285(41):31672–31681. doi: 10.1074/jbc.M110.141929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dorleans A, Gigant B, Ravelli RB, Mailliet P, Mikol V, Knossow M. Variations in the colchicine-binding domain provide insight into the structural switch of tubulin. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(33):13775–13779. doi: 10.1073/pnas.0904223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.