

I. Introduction
The function of the nucleolus has been under investigation for many years, and our understanding of the role of the nucleolus in ribosome subunit assembly has become increasingly detailed [see Shaw and Jordan (1995), for cites of recent reviews and historical background]. For example, a detailed picture of the pathway for conversion of the primary pre-rRNA transcript into mature rRNAs has emerged from elegant studies done in a number of laboratories (reviewed in Sollner-Webb et al., 1996). Over the past 10 years, a great deal of progress in understanding nucleolar function has been made in the yeast Saccharomyces cerevisiae. Our understanding of pre-rRNA processing has advanced the farthest and has been reviewed (Venema and Tollervey, 1995). An aspect of nucleolar function that has lagged behind, relatively speaking, is our understanding of the macromolecular assembly events that accompany the pre-rRNA processing steps. This includes the association of ribosomal proteins with rRNAs, and the formation of intermediates such as pre-40S or pre-60S subunit assemblies within the nucleolus.
Methods for isolating subnuclear fractions enriched in nucleoli (“nucleoli”) from various cell types have been described and used to explore requirements for Pol I-dependent transcription in vitro, and to identify nucleolar proteins (Albanese and Studzinski, 1979; Beebee and Butterworth, 1977; Bombik et al., 1977; Jackowski et al., 1976; Spohn et al, 1985). Isolated nucleoli have been useful in the identification of ribosome subunit precursors in cultured cells (Kumar and Warner, 1972; Vaughan et al., 1967; Warner and Soeiro, 1967). From these studies of Warner and colleagues, the formation of 40S and 60S subunits from larger, rather than smaller, precursor ribonucleoprotein particles emerged as an important paradigm for subunit assembly. This suggested that during early processing steps a number of nonribosomal proteins associate with pre-rRNA, and are joined at later steps by ribosomal proteins recruited onto the rRNAs, which is followed by selective dissociation of nonribosomal components to yield mature ribosomal subunits that are exported to the cytosol.
Our goal in developing a method for the isolation of nucleoli from yeast is to further our understanding of subunit assembly events that accompany rRNA processing steps. Moreover, isolation of nucleoli may provide an opportunity to explore nucleolar functions not directly related to ribosome biogenesis [e.g., see Schneiter et al. (1995) and references therein]. Nucleoli are obtained from nuclei that are prepared according to a modification of a method we have published previously (Aris and Blobel, 1991). The methods for the isolation of nuclei and nucleoli are described below. The reader is also referred to methods for preparing nuclei that have been described by others (Hurt et al., 1988; Jerome and Jaehning, 1986; Lohr, 1988; Rout and Kilmartin, 1997). We wish to acknowledge that the method described herein traces its origin to the laboratory of Dr. Günter Blobel, where J.P.A. collaborated with Michael P. Rout on the nuclear fractionation studies that led to the development of this method.
II. Strains, Medium, Solutions, Buffers, and Reagents
Medium, buffers, and solutions that are sterilized are stored at 0–4°C. Ficoll and sucrose solutions, which are difficult to filter sterilize, are stored frozen at −20°C to prevent contamination. Frozen sucrose solutions should be completely mixed after thawing.
A. Strains
The protease-deficient, haploid S. cerevisiae strain BJ2168 has been used by us in the past (Aris and Blobel, 1991), and gives good yield of highly enriched nuclei. We have also used the protease-deficient yeast strain BJ5465, which contains nonrevertable alleles, with good results. Both strains are available from the Yeast Genetic Stock Center.
B. Medium
YPD, 6 liters: 1% (w/v) Bacto-yeast Extract, 2% (w/v) Bacto-peptone, 2% (w/v) glucose. Autoclave 1-liter volumes for the minimum exposure time necessary for sterilization, and begin cooling immediately after the exhaust cycle. We use Fernbach flasks.
C. Solutions and Buffers
Pretreatment buffer, 100 ml: 100 mM Tris-HCI, 50 mM DTT, 5 mM EDTA, pH 9.0. Prepare just prior to use by diluting the following stock solutions: 1 M Tris-HCI, pH 9.0; 0.5 M EDTA, pH 9.0, 1 M DTT.
Dithiothreitol stock, 1 M: prepare, filter sterilize, and store in 1-ml aliquots at −20°C. Freshly dilute into buffers that call for DTT.
Sorbitol solution, 250 ml: 50 g sorbitol (1.1 M). Autoclave to sterilize.
Cushion solution, 25 ml: 5.5 g sorbitol, 1.25 g Ficoll 400. Filter sterilize.
2 × PM buffer, 200 ml: 2.5 ml 1 M K2HPO4, 5.5 ml 1 M KH2PO4, 0.4 ml 1 M MgCl2. The pH should be 6.5. Filter sterilize.
50% Ficoll solution, 200 ml: 100.0 g Ficoll 400 (Pharmacia), 100 ml 2× PM buffer. The pH of the 50% Ficoll buffer should be 6.5.
It is essential that the 50% Ficoll buffer be prepared accurately. A common source of poor results in our experience is hasty preparation of this solution. We suggest the following protocol: Add 100 ml of 2× PM buffer and 30 ml ddH2O to a 400-ml beaker with a petri dish cover and a stir bar. Heat to almost boiling with a microwave oven or a hotplate stirrer. Add 100 g of Ficoll 400 slowly with continuous, rapid stirring for 5–10 min. Keep solution hot, but avoid boiling (e.g., microwave intermittently) and keep lid on beaker. Condensation on beaker walls will wash down undissolved Ficoll. Approximately 1 hr of moderate stirring and gentle heating will dissolve the Ficoll. Cool to room temperature, and transfer to a 250-ml glass cylinder using several rinses with ddH2O to transfer all of the Ficoll solution. Wait for bubbles to rise to the surface. Add ddH2O to final volume (200 ml). Mix for 30–60 min (e.g., on rotator) until completely homogeneous. Store at −20°C in five aliquots in sterile, plastic 50-ml tubes.
Lysis (20% Ficoll) solution, 75 ml: 30.0 ml 50% Ficoll, 22.5 ml 2× PM buffer, ddH2O to 75 ml.
30% Ficoll solution, 30 ml: 18.0 ml 50% Ficoll, 6.0 ml 2× PM buffer, ddH2O to 30 ml.
40% Ficoll solution, 30 ml: 24.0 ml 50% Ficoll, 3.0 ml 2× PM buffer, ddH2O to 30 mls.
Protease inhibitor cocktails (PICs):
PIC-D, 10 ml: 0.88 g PMSF (0.5 M, Sigma, P7626), 10 mg pepstatin A (Boehringer, 1359 053), 10 mg chymostatin (Boehringer, 1004 638); dissolve in DMSO and store at −20°C in 1-ml aliquots.
PIC-W, 10 ml: 1.57 g benzamidine (1 M, Sigma, B6506), 5 mg leupeptin (Sigma, L2884), 5 mg bestatin (Sigma, B8385), ddH2O to 10 ml; dissolve in ddH2O and store at −20°C in 1-ml aliquots.
Dilute both protease inhibitor cocktails 1/1000 immediately prior to use.
Add PIC-D with rapid stirring below the surface of buffer or solution.
PSM1 buffer, 200 ml: 1.6 ml 1 M K2HPO4, 2.4 ml 1 M KH2PO4, 17.12 g ultrapure sucrose (250 mM), 0.2 ml 1 M MgCl2; filter sterilize.
Heparin solution, 1 ml: Prepare 10 mg/ml solution of Sigma grade I-A sodium salt (H3393) in 50% glycerol/ddH2O; store at −20°C
DNase I solution: Prepare a 10 mg/ml stock of DNase I (Sigma Type II, D4527) in 50% glycerol, 20 mM KPi, pH 7, 1 mM MgCl2, 1 mM DTT; freeze as 10-μl aliquots at −70°C.
10 × BTM buffer, 100 ml: 2.09 g bisTris (100 mM), 1 ml 1M MgCl2; adjust with HCl to pH 6.5; filter sterilize; dilute to make 1× BTM.
2.5 M sucrose/BTM, 200 ml: 165 g ultrapure sucrose; add warm (~37°C) 1 × BTM to 180 ml; mix and dissolve; cool to room temperature; adjust refractive index to 1.4540.
2.25 M sucrose/BTM, 40 ml: 36 ml 2.5 M sucrose/BTM, 4 ml 1× BTM; adjust refractive index to 1.4417.
2.0 M sucrose/BTM, 40 ml: 32 ml 2.5 M sucrose/BTM, 8 ml 1× BTM; adjust refractive index to 1.4298.
1.75 M sucrose/BTM, 40 ml: 28 ml 2.5 M sucrose/BTM, 12 ml 1× BTM; adjust refractive index to 1.4178.
1.5 M sucrose/BTM, 40 ml: 24 ml 2.5 M sucrose/BTM, 16 ml 1× BTM; adjust refractive index to 1.4059.
D. Reagents
Glusulase (NEN, DuPont). Use directly without dilution, filtration, or centrifugation.
Zymolyase 100T (ICN). Freshly prepare 10 mg/ml Zymolyase 100T in ddH2O. Use a sonifier to disperse enzyme (but do not allow to warm above 30°C). Store on ice until use; 1 ml is usually sufficient for the average yield of yeast (~3 × 1011 cells). We have also used yeast lytic enzyme (100,000 U/g; from ICN) in place of Zymolyase 100T.
III. Isolation of Nuclei and Nucleoli
A. Growth of Yeast and Preparation of Spheroplasts
Yeast strain differences and/or differences in culture medium result in a range of efficiencies of spheroplasting with the enzyme preparations we use. Pretreatment has allowed us to employ the conditions described below with different strain backgrounds grown in different media. Because we have made changes to our previously reported method for preparing yeast nuclei (Aris and Blobel, 1991), which serves as the starting material for the isolation of nucleoli, we present the entire method for nuclei here.
Grow yeast in YPD as follows. Day −4: Grow a fresh patch of cells (from permanent culture at −70°C) on a YPD plate at 30°C. Day −2: Grow a 5-ml YPD culture overnight at 30°C. Day −1: Measure the OD600 of the overnight culture. Dilute overnight culture into 50 ml of YPD at 30°C such that the OD600 value will be 0.5–1 at 5 PM. The doubling time of BJ5465 or BJ2168 in YPD at 30°C is typically ~ 110 min in our experience. Calculate the volume from the 50-ml starter culture to be added to each of the six 1-liter volumes of YPD to give an OD600 = 0.6–0.7 after an overnight growing period. Add starter culture to the 1-liter volumes of YPD equilibrated to 30°C, and agitate at 200–225 rpm.
Harvest yeast at OD600 = 0.6–0.7. Do not exceed OD600 = 0.8. Pool and stir culture in an ice-water bath while collecting cells in the next step. Because light scattering estimates from spectrophotometric optical density measurements are not very precise, different spectrophotometers will give different estimates of cell density. In our laboratory an OD600 of 0.6 = ~5 × 107 cells/ml.
Centrifuge in 250-ml bottles for 5 min at 4°C in a Sorvall GSA rotor at 5000 RPM (~4000 gmax).
Resuspend cell pellets in ice-cold water, and pool into a single, tared 250-ml centrifuge bottle. Centrifuge as in step 3. Keep pellets and suspension on ice.
Pour off and aspirate to remove all supernatant. Determine wet weight of cell pellet.
For each 1 g of wet weight, resuspend the cell pellet in 4 ml of freshly prepared pretreatment buffer. Swirl intermittently for 10 min at room temperature.
Fill centrifuge bottle to neck with ice-cold water and mix. Dilute 5 μl of cell suspension in 495 μl of water for counting cell number using a hemacytometer. Assume the centrifuge bottle holds 275 ml. The total yield will usually be ~3 × 1011 cells (70 cells per 1/16 square of the corner grid of a hemacytometer is 3 × 1011 cells).
Centrifuge cells as in step 3. Do this while calculating yield. Pour off and aspirate to remove all supernatant.
Resuspend the cell pellet in ice-cold sorbitol solution. Centrifuge cells as in step 3. Pour off and aspirate to remove all supernatant.
Set up digestion. For every 1 × 1011 cells, disperse the wet cell pellet in 25 ml sorbitol solution. For every 1 × 1011 cells, add 1.5 ml Glusulase and 0.3 ml 10 mg/ml Zymolyase 100T. Digest at 30°C for 1.5–2.5 hr with gentle swirling. Cap bottle loosely. Check progress of digestion every 15–20 min (using at least 400× magnification). By about 1 hr of digestion spheroplasts will clump, but will separate by about 2 hr. The digestion is complete when spheroplasts appear round and individual, with no clumps. Clumps of cells will not be lysed effectively, which will result in a lower yield of nuclei.
For BJ2168, we find that there are ~1.6 × 1010 cells per gram wet weight of cell pellet. For other strains that we have used, this value varies from (1.2–1.7) × 1010 cell per gram wet weight. For convenience in routine nuclei preparations from BJ2168, digestions are set up on a per gram wet cell weight basis, as follows: 4 ml sorbitol solution, 0.25 ml Glusulase, 50 μl 10 mg/ml Zymolyase 100T. If nuclei will be used for in vitro transcription, or other functional studies (e.g., see Bednarek et al., 1995), the digestion step should be followed by a recovery step in which spheroplasts are incubated in osmotically stabilized medium. For example, 30 min of gentle swirling in 1 M sorbitol, 1% Bacto-peptone, 1% glucose, 0.5% Bacto-yeast extract, 20 mM KPi, pH 6.5, can be used. Optimum recovery conditions should be determined empirically.
B. Isolation of Nuclei-Enriched Fraction
Set up the Ficoll gradients during the spheroplast digestion step. Plan for one gradient per 1 × 1011 cells. Typically, three gradients will be needed. Pour the step gradients as follows: (A) warm the 50, 40, and 30% Ficoll solutions to ~25°C; (B) add PICs to 20 ml of each of the Ficoll solutions in 50-ml tubes and vortex to mix; (C) pipete 6.50 g of the 50% Ficoll solution into the bottom of a Beckman SW28 ultraclear tube, place the tube on ice, and prepare additional tubes as needed; (D) pipet 6.5 g of the 40% and 6.5 g of the 30% Ficoll solutions into the SW28 tubes; (E) chill gradients on ice at least 10 min.
After digestion, transfer the spheroplasts to Oakridge polycarbonate centrifuge tubes and place on ice for 10 min. Use one 50-ml Oakridge tube for every 1 × 1011 cells.
Centrifuge for 5 min at 4°C in a Sorvall HB-4 rotor at 5000 RPM (~4000 gmax).
Resuspend spheroplast pellets with 25 ml of ice-cold sorbitol solution. Centrifuge again as in the previous step.
Resuspend spheroplasts in 20 ml of ice-cold sorbitol solution. Freshly add PICs to the cushion buffer. Pipet 6 ml of cushion buffer onto the bottoms of the tubes.
Centrifuge spheroplasts through cushion for 10 min at 4°C in Sorvall HB-4 rotor at 5000 RPM. Aspirate off supernatant and keep wet spheroplast pellets on ice until lysis.
Lyse spheroplasts. Freshly add PICs to lysis buffer. To each pellet in an Oakridge tube, add 25 ml of lysis buffer at room temperature. Quickly repipet with 25-ml pipette to disperse pellet. Immediately transfer to Potter-Elvehjem homogenizer. Lyse with five strokes and fast-spinning pestle using a motorized drive. After lysis, transfer lysate back into a 50-ml Oakridge centrifuge tube submerged to neck in ice. Work quickly to complete this step in 3–5 min for each tube.
Chill spheroplast lysate in tube submerged to neck in ice for 10–15 min. Check lysate with microscope (400×). Spheroplasts should be completely disrupted. Nuclei are small, dark, and round in appearance. Vacuoles are large and refractile.
Centrifuge lysate for 5 min at 2°C in a precooled Sorvall HB-4 rotor at 9000 RPM (~13,000 gmax). Pour supernatant into a second centrifuge tube. Do not transfer any of the loose pellet.
Centrifuge for 10 min at 2°C in HB-4 rotor at 9000 RPM. A small tight pellet should be observed.
Using a 25-ml pipet, carefully transfer the supernatant to the step gradients on ice. Do not transfer any of the pellet. If necessary, top off the SW28 tubes with cold 1 × PM buffer containing PICs. Hold the loaded step gradients submerged to tops of the tubes in ice for at least 10 min to completely chill the load zone and step gradient.
Centrifuge the gradients for 60 min at 2°C in a Beckman SW28 rotor at 18,000 RPM (58,400 gmax). Precool rotor and buckets.
Nuclei occur in the 40% layer and at the 30–40% and 40–50% interfaces. Bands are usually visible at interfaces, but the 40% layer will also contain nuclei. A white film layer should be visible at the top of the gradient. A thick band should be visible at the 20–30% interface. A small pellet, with a halo appearance, should occur at the bottom. Using an aspirator, carefully remove the white film layer, and some of the 20% Ficoll load. Alternatively, these fractions may be collected.
Collect nuclei with a 20-ml syringe and a 16-gauge needle. Carefully insert needle below the 40–50% interface near the seam at the bottom of the ultraclear tube. Position the needle with the beveled edge up in the center of the tube near the 40–50% interface. Move the needle back and forth smoothly to collect the 40% layer. Slowly remove ~8 ml per gradient, which corresponds to the 40% layer plus some of the 50% layer. This will require 2–3 min per gradient because the cold Ficoll suspension is viscous. Avoid the 30% layer because it contains membranes (along with some nuclei). Remove the needle from the syringe and expel nuclei into a 50 ml-tube on ice. Mix nuclei to create a uniform suspension. Note: If nuclei will not be used to prepare nucleoli, one may elect to perform only one Ficoll step gradient (omit the next step), and harvest less nuclei (~6 ml) at this step to achieve a reasonable level of enrichment of nuclei. However, do not attempt to prepare nucleoli starting with nuclei isolated from only one Ficoll step gradient.
Repeat the Ficoll step gradient using two Ficoll gradients. Set up gradients as above except use 5.5 g per step. Dilute nuclei with ice-cold 1 × PM buffer plus PICs and load the second gradients. Perform centrifugation as described above. Collect up to 10 ml of nuclei as indicated in step 14. The second Ficoll step gradient has a reduced accumulation at the 20–30% interface and a smaller pellet, which permits all of the 40% layer, and some of the 30% and 50% layers, of the gradient to be collected. Pool collected nuclei and mix to create a homogeneous suspension. Do protein determination, or save ~100 μl of nuclei at −20° for this purpose. Aliquot nuclei in 10-mg amounts, if protein concentration has been determined, or in amounts equal to the volume collected per gradient. Nuclei stored at −70°C for several months may be used to prepare nucleoli without significantly compromising yield or enrichment.
Protein Determination
The preparation of nucleoli requires an accurate determination of protein concentration, which we have found to be more reliable than OD260 measurements for determining yield of nuclei. The Bradford dye binding assay and the Lowry method work equally well, using either BSA or bovine gamma globulin as standards (Ausubel et al., 1996). It is important to remove Ficoll from the sample, however. For this, dilute nuclei in 10 volumes of PSM1 buffer (without DTT or PICs), mix very well, and centrifuge at 12,000 g for 30 min at 4°C. Nuclei may be resuspended in ddH2O or 0.1% SDS in TE buffer (10 mM Tris-HCl, pH 8, 1 mM EDTA). We routinely dilute ~50 μl of nuclei, centrifuge, resuspend in 50 μl of TE/SDS, and use a modified Lowry procedure (Ausubel et al., 1996).
C. Isolation of Nucleoli-Enriched Fraction
Dilute a volume of yeast nuclei equivalent to 10 mg of nuclear protein into five volumes of ice-cold PSM1 buffer containing freshly added PICs and freshly added 1 mM DTT. Use a 50-ml Oakridge polycarbonate tube. Vortex to disperse nuclei completely in PSM1 buffer. Placing frozen nuclei in an ice-water bath will hasten thawing. Plan for 10 mg of nuclei for each step gradient (see step 8 below). Use of an entire nuclear prep will result in two to four gradients, depending on the yield of nuclei. The use of an oversized 50-ml tube in this step results in a thin pellet in step 3. Too thick a nuclear pellet in step 3 causes isolated nucleoli to be more aggregated.
Centrifuge for 10 min at 2°C in a Sorvall HB-4 rotor at 8000 RPM (~10,000 gmax). Begin pouring the sucrose step gradients at this point (see step 8 below). The gradients must be on ice by step 7.
Carefully remove all of the supernatant by aspiration. The pellet should form a thin, opalescent layer. Keep pellets on ice.
Resuspend each nuclear pellet in 1 ml ice-cold PSM1 buffer containing freshly added PICs and 1 mM DTT. Disperse pellet as follows: repipet using a 1-ml micropipettor for 1 min on ice, vortex for 15 sec, add 5 μl DNase I solution, and vortex again for 15 sec. Each 50-ml Oakride tube should contain one pellet from 10 mg of nuclear protein, 1 ml of PSM1 buffer, and 5 μl (50 μg) of DNase I.
Place tube at room temperature for 4 min following resuspension.
Add 25 μl (250 μg) of the heparin solution. Mix by gentle vortexing for 15 sec.
Hold at room temperature for 45 sec (1 min at room temperature after the addition of heparin). Chill on ice for 10–15 min before loading onto the gradient. The step gradients should also be on ice at this time.
-
Set up the sucrose step gradient in an Ultraclear SW50.1 centrifuge tube as follows:
(A) warm the sucrose solutions to room temperature and add PICs and 1 mM DTT;
(B) add 1 ml of 2.5 M sucrose solution;
(C) add 1.5 ml of 2.0 M sucrose solution;
(D) add 1 ml of 1.75 M sucrose solution;
(E) add 0.75 ml of 1.5 M sucrose solution.
Place gradient on ice for 10 min prior to spin. Tube capacity is ~5.2 ml.
Load digested nuclei (~1 ml) on gradient. If necessary, top off with PSM1 buffer.
Centrifuge for 3 hr at 2°C in the SW50.1 rotor at 45,000 RPM (~243,000 gmax).
After centrifugation, a band at each interface should be visible. Nucleoli are present at the 2.0 M/2.5 M interface, and can be collected with a syringe and 20-gauge needle by inserting the needle bevel-up just below the interface and removing ~1 ml from the gradient. Do this carefully, and sacrifice some of the 2.0 M step to avoid collecting the 1.75 M/2.0 M interface. If other fractions are desired, collect each interface with a 1-ml pipet tip, starting with the load zone. Collect each step and the band beneath it in one fraction. Pipet slowly from the surface of liquid, slowly rotating tube, with the tip at side wall of tube. Nucleoli may be stored frozen at −70°C.
To analyze nucleoli and other fractions from the step gradient by SDS-PAGE, it is convenient to precipitate proteins with TCA using Triton X-100 detergent as a carrier. For this, we add 1/100 volume of 20% Triton X-100 to a sample, adjust the TCA concentration to 10% (w/v) and the sucrose concentration to < 0.5 M, mix well, and place on ice for at least 1 hr. After centrifugation, the pellet is washed with acetone to remove the Triton X-100, and is resuspended and boiled in SDS sample buffer.
IV. Optimization of the Method for the Isolation of Nucleoli
During the development of this method for nucleoli a number of steps in the procedure were optimized by comparison of different conditions. For the sake of brevity, the results of these comparisons are summarized here, without presentation of the data from the corresponding experiments.
A. DNase I Treatment
The digestion of nuclei with DNase I fragments chromosomal DNA and allows treatment with heparin to release nucleosomes. The efficacy of this step was evaluated by agarose gel electrophoresis of resuspended nuclei treated with DNase I and heparin, or not. The use of the conditions described above results in production of DNA fragments of less than 500 bp in length. The use of up to 10-fold more DNase I did not yield a visible change in DNA fragment size in agarose gels, or improve release of nucleosomes as judged by relative amounts of histones visualized by Coomassie staining of fractions from the step gradient analyzed by SDS-PAGE. It is possible that adequate digestion of nuclei may be achieved with less DNase I.
B. Heparin/Nuclear Protein Ratio
The ratio of heparin to nuclear protein was also examined over a range of 2.5–250 μg heparin per 1 mg of nuclear protein (25 μg/mg protein is used above), but little change in the protein profile across the step gradient fractions was observed. We note that the presence of 0.1% Triton X-100 detergent in the step gradient augments the action of heparin, and reduces the yield of nucleoli dramatically.
We have previously described a method for subfractionating isolated nuclei (Aris and Blobel, 1991), which stipulated the use of buffers lacking Mg2+ and containing EDTA for the purpose of dissociating histones from chromatin digested with DNase I. In the method described herein, brief exposure to a low concentration of heparin in the presence of Mg2+ is used to facilitate dissociation of histones and other chromatin-associated proteins. Heparin has been used previously for this purpose by Courvalin and co-workers (1982), as well as by Rout and Kilmartin (1990). A concentration of 1 mM Mg2+ has been chosen because it approximates the intracellular value (see Loukin and Kung, 1995), and increases the likelihood that nucleoli will support run-on transcription by RNA polymerase I, and other nucleolar functions.
C. Buffer Ionic Strength
The presence of NaCl in buffers at concentrations of 50–300 mM was examined in order to ascertain if increased ionic strength may improve release of chromatin-associated proteins prior to the sucrose step gradient. Low salt concentrations (50–150 mM) did not improve histone release, and high salt concentrations (200–300 mM) reduced the yield of nucleoli and increased the amount of Noplp detected in upper step gradient fractions.
D. Sedimentation Conditions
The isolation method calls for loading of the DNase digestion mixture directly onto the sucrose step gradient. Sedimentation of digested nuclei, followed by gentle resuspension, prior to loading on the sucrose step gradient was also tried, but resulted in the presence of aggregates of nucleoli at the 2.0/2.5 M interface, as judged by light microscopy. Centrifugation times of 2, 4, 8, and 20 hr (at 243,000 gmax) were compared with different step gradient sucrose concentrations. Gradient composition was adjusted to give optimal results with a 2-hr centrifugation time for purposes of convenience as well as to retain biologic activity in isolated nucleoli.
V. Results
A. Characterization of the Nucleolus-Enriched Fraction
The nucleolar fraction obtained with this method has been characterized by SDS-PAGE, immunoblot analysis, and light and electron microscopy. Histones are present predominantly in the load, and 1.5 M sucrose fractions at the top of the gradient, indicating that DNase I digestion and heparin treatment have released these proteins from chromatin (Fig. 1). Immunoblotting shows that nucleoli are highly enriched in the nucleolar protein Noplp (Fig. 1, lane 2.0). The nucleolar proteins Nop2p and Nsrlp are distributed across fractions from the step gradient almost identically to Noplp (data not shown). However, the nuclear polyadenylated RNA binding protein Nab3p is prominent in the top gradient fractions and depleted from nucleoli (Fig. 1). The 2.5 M fraction contains little protein, even though any material that pellets is collected with this fraction (Fig. 1). Thus, nucleoli do not sediment through the 2.5 M step. From comparisons of immunoblotting results from multiple experiments, we estimate that the yield of nucleoli is approximately one-third of the starting amount of nuclei.
Fig. 1.
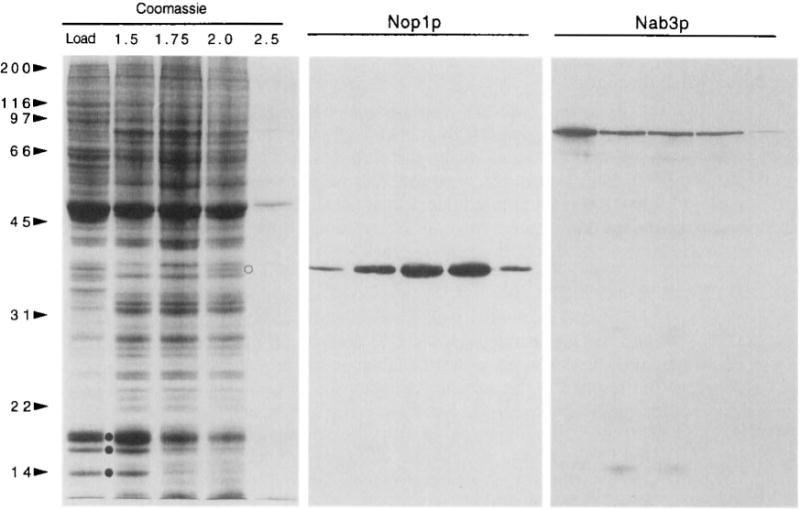
Characterization of sucrose step gradient fractions. Proteins from equal volumes of the five steps were TCA precipitated, electrophoresed on a 10.5% Polyacrylamide gel, and stained with Commassie blue (de Beus et al., 1994). Each step was collected with the interface below it, and with a small amount of the next step. “Load” designates the top fraction, and other fractions are designated by the molarity of sucrose in the step (1.5, 1.75, 2.0, 2.5). Nucleoli are harvested from the 2.0 M sucrose fraction and interface. Histones (•) and the nucleolar protein Noplp (o) are indicated. Positions of molecular weight standards are shown (sizes in kilodaltons). Immunoblots were prepared in parallel and probed with mAb A66 against the 36-kDa nucleolar protein Noplp (Aris and Blobel, 1988), or with mAb 2F12 against the 90-kDa nuclear poly(A)+ binding protein Nab3p (Wilson et al., 1994).
All fractions from the step gradient have been examined by light and electron microscopy. The results with the nucleolar fraction and the fraction just above it on the gradient are shown in Fig. 2. The nucleolar fraction viewed by light microscopy reveals particles that are approximately 1 μm in diameter, relatively uniform in size distribution, and dark by phase contrast (Fig. 2, A and B). Irregular phase-dense material may be seen at the edge of particles, and is likely to correspond to nuclear envelope and/or endoplasmic reticulum. Nucleoli viewed by electron microscopy retain the associated nuclear envelope (Fig. 2, C and D). Electron microscopy also shows heavily stained fragments, consistent with the loss of structural integrity of some nucleoli. Although some Noplp is found in lighter fractions from the sucrose gradient (Fig. 1), these fractions contain an abundance of membranes, and are not highly enriched in nucleoli (Fig. 2E).
Fig. 2.
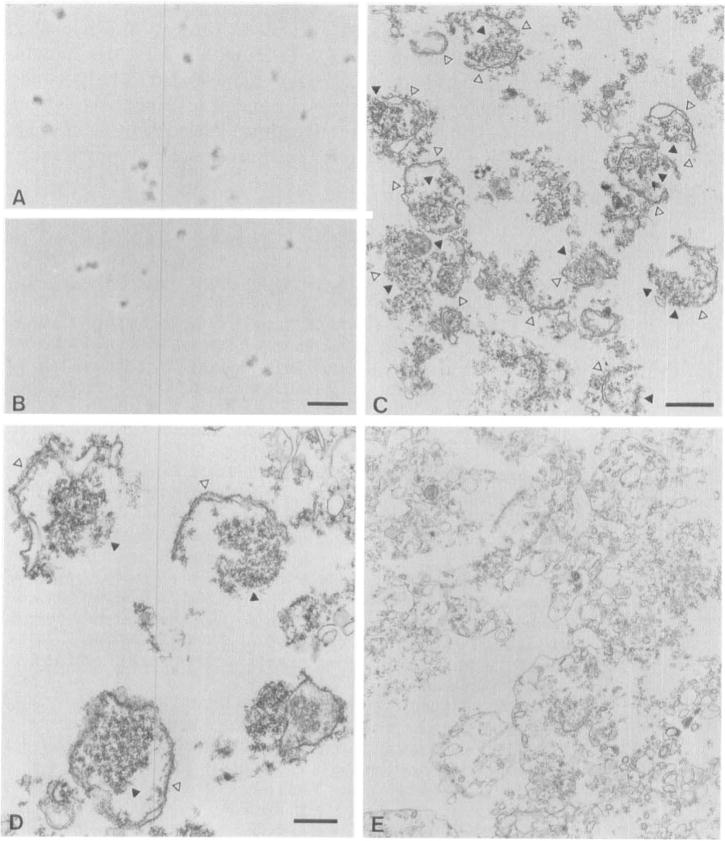
Characterization of enriched nucleoli by light and electron microscopy. (A and B) Nucleoli in sucrose from two different step gradients were viewed by phase-contrast microscopy; scale bar, 10 μm. (C and D) Micrographs of nucleoli are shown at two magnifications. The nuclear envelope (△) and the nucleolus (▲) are marked. A micrograph of the fraction immediately above the nucleoli (corresponding to lane 1.75 in Fig. 1) is also shown (E). (C) Scale bar, 1 μm; (D) scale bar, 0.5 μm. Magnification is the same in C and E. For EM, nucleoli were fixed, embedded in Spurr’s resin, poststained, and viewed using standard methods, as described previously (de Beus et al., 1994).
B. Application: Monoclonal Antibody Preparation
We have used isolated nucleoli to prepare monoclonal antibodies against nucleolar proteins (S. Chen, J. E. Dove, and J. P. Aris, unpublished results). Six monoclonal antibodies have been characterized by indirect immunofluorescence localization and Western blotting, and have been used to screen yeast expression libraries. At this stage, putative, novel nucleolar proteins are being expressed in yeast with an epitope tag for the purpose of confirming their subcellular localization.
References
- Albanese EA, Studzinski GP. J Cell Physiol. 1979;99(1):55–65. doi: 10.1002/jcp.1040990108. [DOI] [PubMed] [Google Scholar]
- Aris JP, Blobel G. J Cell Biol. 1988;107:17–31. doi: 10.1083/jcb.107.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aris JP, Blobel G. In: Methods in Enzymology. Guthrie C, Fink GR, editors. Vol. 194. Academic Press; San Diego, CA.: 1991. pp. 735–749. [DOI] [PubMed] [Google Scholar]
- Ausubel FA, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current Protocols in Molecular Biology. Vol. 3. Greene Publishing and Wiley-Interscience; New York: 1996. [Google Scholar]
- Bednarek SY, Ravazzola M, Hosobuchi M, Amherdt M, Perrelet A, Schekman R, Orci L. Cell (Cambridge, Mass) 1995;83(7):1183–1196. doi: 10.1016/0092-8674(95)90144-2. [DOI] [PubMed] [Google Scholar]
- Beebee TJ, Butterworth PH. Eur J Biochem. 1977;77(2):341–348. doi: 10.1111/j.1432-1033.1977.tb11673.x. [DOI] [PubMed] [Google Scholar]
- Bombik BM, Huang CH, Baserga R. Proc Natl Acad Sci USA. 1977;74(1):69–73. doi: 10.1073/pnas.74.1.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courvalin JC, Dumontier M, Bornens M. J Biol Chem. 1982;257(1):456–463. [PubMed] [Google Scholar]
- de Beus E, Brockenbrough JS, Hong B, Aris JP. J Cell Biol. 1994;127(6):1799–1813. doi: 10.1083/jcb.127.6.1799. Part 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurt EC, McDowall A, Schimmang T. Eur J Cell Biol. 1988;46(3):554–563. [PubMed] [Google Scholar]
- Jackowski G, Suria D, Liew CC. Can J Biochem. 1976;54(1):9–14. doi: 10.1139/o76-002. [DOI] [PubMed] [Google Scholar]
- Jerome JF, Jaehning JA. Mol Cell Biol. 1986;6(5):1633–1639. doi: 10.1128/mcb.6.5.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Warner JR. J Mol Biol. 1972;63(2):233–246. doi: 10.1016/0022-2836(72)90372-5. [DOI] [PubMed] [Google Scholar]
- Lohr D. In: Yeast A Practical Approach. Campbell I, Duffus JH, editors. IRL Press; Oxford: 1988. pp. 125–145. [Google Scholar]
- Loukin S, Kung C. J Cell Biol. 1995;131:1025–1037. doi: 10.1083/jcb.131.4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout MP, Kilmartin JV. J Cell Biol. 1990;111:1913–1927. doi: 10.1083/jcb.111.5.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout MP, Kilmartin JV. In: Cell Biology: A Laboratory Handbook. Celis JE, editor. Academic Press; San Diego, CA: 1997. (in press) [Google Scholar]
- Schneiter R, Kadowaki T, Tartakoff AM. Mol Biol Cell. 1995;6(4):357–370. doi: 10.1091/mbc.6.4.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ, Jordan EG. Annu Rev Cell Dev Biol. 1995;11:93–121. doi: 10.1146/annurev.cb.11.110195.000521. [DOI] [PubMed] [Google Scholar]
- Sollner-Webb B, Tycowski KT, Steitz JA. In: Ribosomal RNA: Structure, Evolution, Processing, and Function in Protein Synthesis. Zimmermann RA, Dahlberg AE, editors. CRC Press; Boca Raton, FL: 1996. pp. 469–490. [Google Scholar]
- Spohn WH, Ahn YS, Busch RK, Busch H. Cancer Invest. 1985;3(4):307–320. doi: 10.3109/07357908509039793. [DOI] [PubMed] [Google Scholar]
- Vaughan MH, Warner JR, Darnell JE. J Mol Biol. 1967;25(2):235–251. doi: 10.1016/0022-2836(67)90140-4. [DOI] [PubMed] [Google Scholar]
- Venema J, Tollervey D. Yeast. 1995;11(16):1629–1650. doi: 10.1002/yea.320111607. [DOI] [PubMed] [Google Scholar]
- Warner JR, Soeiro R. Proc Natl Acad Sci USA. 1967;58(5):1984–1990. doi: 10.1073/pnas.58.5.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SM, Datar KV, Paddy MR, Swedlow JR, Swanson MS. J Cell Biol. 1994;127(5):1173–1184. doi: 10.1083/jcb.127.5.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]