

Abstract
Aging in the yeast Saccharomyces cerevisiae is under the control of multiple pathways. The production and accumulation of extrachromosomal rDNA circles (ERCs) is one pathway that has been proposed to bring about aging in yeast. To test this proposal, we have developed a plasmid-based model system to study the role of DNA episomes in reduction of yeast life span. Recombinant plasmids containing different replication origins, cis-acting partitioning elements, and selectable marker genes were constructed and analyzed for their effects on yeast replicative life span. Plasmids containing the ARS1 replication origin reduce life span to the greatest extent of the plasmids analyzed. This reduction in life span is partially suppressed by a CEN4 centromeric element on ARS1 plasmids. Plasmids containing a replication origin from the endogenous yeast 2 μ circle also reduce life span, but to a lesser extent than ARS1 plasmids. Consistent with this, ARS1 and 2 μ origin plasmids accumulate in ~7-generation-old cells, but ARS1/CEN4 plasmids do not. Importantly, ARS1 plasmids accumulate to higher levels in old cells than 2 μ origin plasmids, suggesting a correlation between plasmid accumulation and life span reduction. Reduction in life span is neither an indirect effect of increased ERC levels nor the result of stochastic cessation of growth. The presence of a fully functional 9.1-kb rDNA repeat on plasmids is not required for, and does not augment, reduction in life span. These findings support the view that accumulation of DNA episomes, including episomes such as ERCs, cause cell senescence in yeast.
The yeast Saccharomyces cerevisiae has proved to be a valuable model organism for investigating mechanisms of cellular aging (recently reviewed in Refs. (21, 45, and 48). Central to the biology of aging in S. cerevisiae is an asymmetric cell division process that gives rise to mother and daughter cells with different characteristics. Mother cells have a limited capacity to produce daughter cells, and the decline in this capacity with each generation is referred to as replicative aging. The limited replicative potential of yeast mother cells has been recognized since the 1950s (35). Pioneering studies in the Jazwinski and Guarente laboratories (13, 24) postulated the existence of a senescence factor/substance that accumulates in mother cells and is transmissible to daughters. Work in the Guarente laboratory identified a heritable “age” locus that regulates yeast life span (25). More recent studies have made clear that allelic variation at single genetic loci can markedly affect yeast life span, including extension of life span. This indicates that a process as complex as cellular aging is controlled by a hierarchical regulatory system. As in other model organisms, such as Drosophila melanogaster and Caenorhabditis elegans, mutations that influence yeast life span have been found to exert their effects through different physiological and genetic pathways, including those that participate in caloric restriction, gene silencing, genomic stability, growth regulation, mitochondrial function, and stress response (11, 21, 30, 45, 48).
Replicative aging is undoubtedly a complex process, even in a eukaryote as simple as S. cerevisiae, and different hypotheses have been proposed to explain yeast replicative aging. One hypothesis proposed by Sinclair and Guarente (46) posits that replicative aging is caused by progressive accumulation of extrachromosomal rDNA circles (ERCs)1 in yeast mother cells. According to this model, ERCs are produced stochastically by intrachromosomal homologous recombination at the rDNA locus and are inherited asymmetrically by mother cells, which leads to ERC accumulation and replicative senescence. The rDNA locus in S. cerevisiae consists of a tandem array of ~150 9.1-kb direct repeats, each of which encodes the four rRNAs (18, 5.8, 25, and 5 S) in precursor form. Many aspects of the ERC model have been supported experimentally. Numerous studies support the view that ERCs are produced by homologous recombination, are self-replicating, are inherited asymmetrically, and accumulate in mother cells (reviewed in Refs. 44–46).
More controversial is the role ERCs play in the aging process. Are ERCs “mediators” or “markers” of yeast aging? Certain findings link ERC production with regulation of life span and support a “mediator” role for ERCs. One of the first life span-extending mutations characterized in yeast (SIR4-42) was found to redirect Sir (silent information regulator) protein complexes to the rDNA locus and limit recombination (25, 26). Expression of SIR2, which encodes a nucleolar NAD-dependent histone deacetylase, correlates with longevity. Sir2p binds to rDNA and suppresses rDNA recombination and ERC production (14, 17, 47). Deletion of SIR2 shortens life span, whereas overexpression of SIR2 extends life span (23). FOB1 encodes a nucleolar “fork blocking” protein that binds to the replication fork barrier site in rDNA and in so doing halts DNA replication in the direction opposite of pre-35 S rRNA transcription (4, 5, 29). The replication fork barrier site and the overlapping HOT1 site promote rDNA recombination (29, 49). Mutations in or deletion of FOB1 reduces rDNA recombination, lowers ERC levels, and extends life span (12). Recombination of replication forks stalled at replication fork barrier sites is suppressed by Sir2p (2), which explains, at least in part, the role of Sir2-dependent silencing in extending life span. Also, the introduction of a plasmid carrying a stretch of rDNA, as an “artificial” ERC, was shown to reduce life span (46).
On the other hand, ERCs have been interpreted as a “marker” of aging that are a consequence, not a cause, of aging. Mutations that impair DNA replication, recombination, or repair have been observed to reduce life span without concomitant accumulation of ERCs (31, 33, 38). However, reduction in life span may be the result of the combined effects of age-dependent and age-independent processes at work in certain mutants. hrm1Δ mutants, which affect rDNA recombination, age prematurely due to a combination of the normal aging process and a G2-like cell cycle arrest (33). Similarly, sgs1 mutants exhibit a shortened life span because of the combined effects of the normal aging process and cell cycle arrest due to defective recombination (32). Some petite mutants have been shown to have elevated ERC levels (10) but extended life spans (28). However, to our knowledge, both elevated ERC levels and extended life span in petite mutants have not been demonstrated side by side in the same strain. A sir2 mutant with an extended life span was reported to have normal ERC levels (27). More generally, the effects of SIR2 on life span have been attributed to altered patterns of gene expression, including altered transcription of rDNA, which may lead to an imbalance in ribosome synthesis (19, 20). Thus, although there is agreement that the rDNA locus plays a key role in the yeast aging process, the precise role of extrachromosomal DNAs remains controversial.
To shed light on this controversy, we have developed a plasmid-based model system to investigate the role of episomal DNAs in reduction of yeast life span. Here we present the first comprehensive test of the ERC model of yeast aging proposed by Sinclair and Guarente (46). We have constructed three types of recombinant plasmid for this purpose: ARS plasmids, ARS/CEN plasmids, and 2 μ origin plasmids. ARS plasmids are most like ERCs in that they are circular DNA molecules with a replication origin but lack a cis-acting partitioning sequence. Classic pedigree analysis studies by Murray and Szostak (36) showed that ARS plasmids exhibit a strong bias to be retained in mother cells during mitosis. Thus, ARS plasmids are predicted to accumulate in mother cells like ERCs, but this has not yet been demonstrated. ARS/CEN plasmids contain a centromeric DNA region that acts in cis to attach plasmid DNA to the mitotic spindle and ensure efficient delivery to daughter cells during mitosis. ARS/CEN plasmids should not accumulate in mother cells. 2 μ origin plasmids typically contain a DNA replication origin, a cis-acting REP3/STB element, and one copy of an inverted repeat that regulates plasmid copy number (~20–40 copies/cell) (6). The REP3/STB element actively partitions plasmid DNA to daughter cells during mitosis in cir+ yeast strains (i.e. in strains that contain the endogenous 2 μ circle DNA plasmid that encodes proteins that interact in trans with REP3/STB)(6). 2 μ origin plasmids are not predicted to accumulate in mother cells, although the 2 μ plasmid partitioning machinery is not predicted to exhibit the fidelity of a centromere-based partitioning machinery. We have also constructed a series of plasmids containing functional rDNA repeat units and tested their effects on life span. This represents a significant improvement over a previously reported experiment (46), which employed a nonfunctional stretch of rDNA (i.e. rDNA incapable of being transcribed to yield full-length 35 S pre-rRNA; see “Results”).
Our studies show that yeast plasmids accumulate in mother cells and reduce replicative life span. The effect of plasmids on life span appears to be a direct effect and not an indirect effect on ERC levels in mother cells. A functional rDNA repeat unit is not required for reduction in life span, and the presence of a functional rDNA repeat does not augment reduction in life span by plasmids. Thus, plasmids containing ARS elements appear to “mimic” ERC-mediated reduction in life span. These findings provide strong evidence that replicative aging in S. cerevisiae is caused by accumulation of episomal DNA. The fact that functional rDNA sequences are not required for reduction in life span argues that expression of rDNA genes present on ERCs is not a causative process in yeast aging. This indicates that accumulation of episomal DNAs, such as ARS plasmids and ERCs, is one mechanism by which yeast life span is regulated.
EXPERIMENTAL PROCEDURES
Yeast Strains and Plasmids
W303AR5 (MATaleu2–3,112 his3–11,15 ura3–1 ade2–1 trp1–1 can1–100 RAD5 ADE2::rDNA, [cir+]) (46) was obtained from D. A. Sinclair. yAF5 and yAF6 were constructed by integrating linearized pRS305 and pRS306 (43), respectively, into the leu2–3,112 or ura3–1 loci of W303AR5, respectively, and genotypes were confirmed by Southern blotting. Plasmids were transformed into W303AR5 using a standard lithium acetate method (16). All experiments were done with freshly prepared, independently isolated, colony-purified transformants. Unless otherwise noted, yeast were grown on selective synthetic dextrose “drop in” medium (42).
Descriptions of plasmids are provided in Table I. A 200-bp fragment containing ARS1 was amplified by PCR with primers 5′-GGAAGCTTCCAAATGATTTAGCATTATC-3′ and 5′-CCGAATTCTGTGGAGACAAATGGTG-3′ using template YRp17. A 200-bp fragment containing the rDNA ARS was amplified by PCR with primers 5′-CCAAGCTTGTGGACAGAGGAAAAGG-3′ and 5′-GGGAATTCATAACAGGAAAGTAACATCC-3′ using template pJPA102 (rDNA repeat with AhdI end points in pCR4, see below). A 753-bp fragment containing CEN4 was amplified by PCR with primers 5′-GCGGATCCCCTAGGTTATCTATGCTG-3′ and 5′-GGGAATTCCTAGGTACCTAAATCCTC-3′ using template YCp50. A 1346-bp region of 2 μ circle DNA, containing the REP3/STB cis-acting stability element and a single 599-bp repeat region, was amplified by PCR with primers 5′-CCGGATCCAACGAAGCATCTGTGCTTC-3′ and 5′-CCAAGCTTTATGATCCAATATCAAAGG-3′ using pRS424 as template. rDNA repeats were amplified by PCR using as template size-selected (8–10 kb), genomic DNA that was digested with the appropriate enzyme (AhdI, PsiI, or XmaI). The following primer pairs were used: AhdI end points, 5′-GGGATCCATGTCGGCGGCAGTATTG-3′ and 5′-CCTGCAGCTGTCCCACATACTAAATCTCTTC-3′; PsiI end points, 5′-GGGATCCTAATATACGATGAGGATGATAGTG-3′ and 5′-CCTGCAGTAATAGATATATACAATACATGTTTTTACC-3′; XmaI end points, 5′-CCCGGGGCACCTGTCACTTTGG-3′ and 5′-CCCGGGTAAACCCAGTTCCTCACTAT-3′. PCR was performed for 20 cycles with 15-s denaturation and annealing times using Pfu Turbo DNA polymerase (Stratagene). PCR products were purified (Qiagen), digested with restriction enzymes, and ligated directly into recipient vectors, or cloned into pCR4-TOPO (Invitrogen), excised, gel-purified, and ligated into recipient vectors (see Table I). ARS elements were cloned between HindIII and EcoRI sites. CEN4 was cloned between EcoRI and BamHI sites. The 2 μ origin was cloned between HindIII and BamHI sites. rDNA inserts were cloned between Pst I and BamHI sites in pAF-15, which is derived from pRS424 and contains LoxP sites that were inserted at EcoRI and SpeI sites using annealed primer pairs: 5′-AATTATAACTTCGTATAATGTATGCTATACGAAGTTAT-3′ and 5′-AATTATAACTTCGTATAGCATACATTATACGAAGTTAT-3′ (EcoRI); 5′-CTAGATAACTTCGTATAATGTATGCTATACGAAGTTAT-3′ and 5′-CTAGATAACTTCGTATAGCATACATTATACGAAGTTAT-3′ (SpeI). All cloned inserts were sequenced in their entirety. Plasmids pJPA105, pJPA106, and pJPA107 (that contain rDNA inserts) were propagated in Escherichia coli DH5α grown in LB medium with 25 μg/ml carbenicillin at 30 °C to avoid insert instability.
Table I.
Plasmids used in this study
| Plasmid | Origin, Insert | Marker | Backbone |
|---|---|---|---|
| pJPA105 | 2 μ, rDNA repeat (XmaI end points) | TRP1 | pAF15a |
| pJPA106 | 2 μ, rDNA repeat (AhdI end points) | TRP1 | pAF15 |
| pJPA107 | 2 μ, rDNA repeat (PsiI end points) | TRP1 | pAF15 |
| pJPA113 | ARS1 | URA3 | pRS306 (43) |
| pJPA114 | rDNA ARS | URA3 | pRS306 |
| pJPA116 | ARS1, CEN4 | URA3 | pRS306 |
| pJPA117 | rDNA ARS, CEN4 | URA3 | pRS306 |
| pJPA138 | 2 μ | URA3 | pRS306 |
| pJPA133 | ARS1 | LEU2 | pRS305 (43) |
| pJPA136 | ARS1, CEN4 | LEU2 | pRS305 |
| pJPA148 | 2 μ | LEU2 | pRS305 |
Derived from pRS424 (8), see “Experimental Procedures.”
Replicative Life Span Determinations
Replicative life span determinations were done essentially as described (39) with a few modifications. Six transformants were streaked individually on one side of an SD agar plate, and 10 virgin mother cells from each (n = 60) were positioned in an orthogonal grid pattern. Virgin mothers that failed to give rise to five daughters were not included in the data set. Due to the low mitotic stability of ARS-plasmids, it was necessary to start with ~250 virgin mother cells from ARS1-plasmid transformants to obtain n = 50–60 for life span determinations. A Zeiss Tetrad microscope equipped with ×16 eyepieces was used for micromanipulations as described (42). SD agar plates were weighed at the beginning of each experiment and sterile water was pipetted into four small notches at the edge of each plate on a daily basis to compensate for evaporation and prevent increases in osmolality, which could potentially affect results (22). During life span experiments, plates were incubated at 30 °C during the daytime and stored overnight (~12 h) at 14 °C. We found that extended periods (≥24 h) at 14 °C reduced life spans of transformants and control strains (data not shown). At the end of a life span experiment, mother cells not having divided for 2 days were transferred to nonselective SD or YPD medium, and cells that resumed mitosis were excluded from the data set. This allowed us to exclude data from mother cells that stopped dividing due to plasmid loss rather than cell senescence. Data were entered into an Excel spreadsheet template file (available on request) that automatically calculated relevant life span data values and performed Wilcoxon two-sample paired signed rank tests. Images of terminal cells were collected using a Spot-2 CCD camera (Diagnostic Imaging) affixed to a Zeiss Tetrad microscope and “terminal” cell morphology analysis was done as described (32).
Mitotic Stability and ADE2 Marker Loss
For each plasmid, five transformants were grown in selective SD liquid medium for 2 days at 30 °C to saturation (A600 = 1.1–1.5; 0.5–1 × 107 colony-forming units/ml; growth to late log gave results similar to stationary phase). Approximately 200–250 colony-forming units of each transformant were plated on nonselective SD medium, grown for 2–3 days at 30 °C, replica-plated onto selective and nonselective agar media, and grown for 3–4 days at 30 °C. For ADE2 marker loss determinations, saturated liquid cultures were prepared from five transformants, diluted, and spread on 15-cm selective SD agar plates containing 5 μg/ml adenine hemisulfate and 5 μg/ml histidine to enhance red color production.
Southern Blot Analysis and Quantitation
DNA was extracted from yeast cells using a glass bead/phenol method, digested with restriction enzymes according to the supplier (New England Biolabs), separated on agarose gels, and capillary-transferred to positively charged nylon membrane under alkaline conditions using standard methods (50). For each plasmid copy number and ERC monomer level determination, five plasmid transformants were analyzed in parallel. Digestion with BamHI or PstI yielded single plasmid-specific or genome-specific bands of different sizes that hybridized to 32P-labeled probe generated by random-primed labeling (New England Biolabs). PstI and BamHI do not cleave rDNA. Genomic bands were used as internal standards for measurements of plasmid levels. Chromosomal rDNA bands were used as internal standards for measurements of ERC monomer levels. Blots were hybridized first to URA3 or LEU2 probe, followed by stripping and hybridization to rDNA probe. Data from the same blots were used to prepare Figs. 2 and 3B. Southern data were acquired with a Typhoon PhosphorImager and analyzed using ImageQuant software (Amersham Biosciences).
Fig. 2. Plasmid inheritance studies.
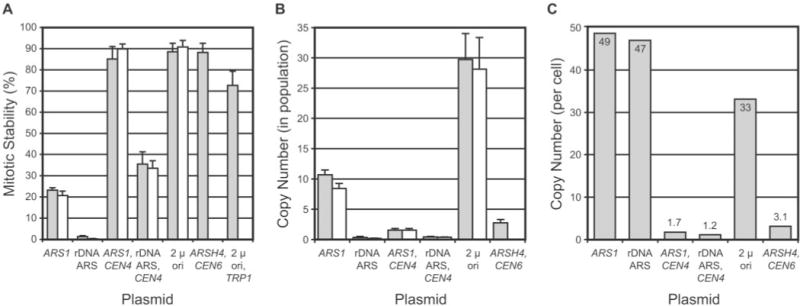
Plasmids are denoted by cis-acting element(s). See Table I for plasmid descriptions. pRS316 (ARSH4, CEN6)(43) and pRS424 (2 μ ori, TRP1) (8) are included for comparison purposes. A, mitotic stability determinations. Mitotic stability is defined as the percentage of colony-forming units in a culture grown under selective conditions that contains plasmid-borne selectable marker. Side-by-side bars are determinations from separate experiments. Average and S.D. values are plotted. B, plasmid copy number in toto for cell population. Average and S.D. values from Southern blots of genomic DNA digested with BamHI(filled bars) and PstI(open bars) are shown. C, plasmid copy number on a per cell basis. Values were calculated by dividing copy number values from B by mitotic stability values from A (average of both experiments). The variances in copy number values were determined, assuming a log normal distribution of values. Variances for all values were near 1.0, with the exception of the rDNA ARS plasmid copy number, which had a variance of 3.0, which is indicative of a higher level of error in this measurement.
Fig. 3. ERC formation in yeast transformants.
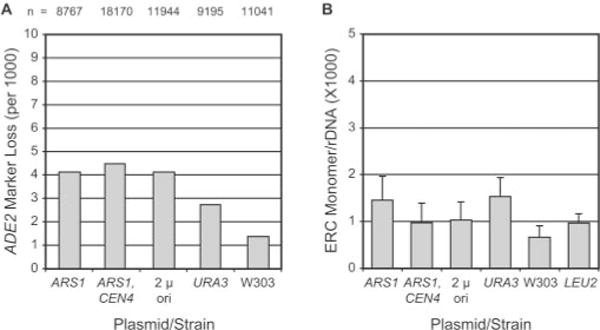
Plasmids are denoted by cis-acting element(s). See Table I for plasmid descriptions. Control strains W 303 AR 5 (W303), yAF5 (LEU2), and yAF6 (URA3) did not contain plasmid. A, ADE2 marker loss assay. The number of halfred sectored colonies on minimal selective medium per total colony number defines the per (first) cell division rate of loss of the ADE2 marker from the rDNA repeat in W303AR5. The total number (n) of colonies scored is shown. B, ERC monomer levels. Southern blotting analyses of DNA from transformed and control strains grown on selective media were done to quantify chromosomal rDNA and ERC monomer band levels (see “Experimental Procedures”). ERC monomer band intensity was divided by chromosomal rDNA band intensity to give a normalized ERC monomer/chromosomal rDNA ratio. Average and S.D. values are plotted.
Cell Sorting
Old yeast cells were collected following biotinylation and sorting with magnetic beads as described (46). Transformants were grown to midlog phase (A600 < 0.5) in SD liquid medium under selection prior to biotinylation. Following biotinylation, cells were grown overnight in selective SD medium, from which both young and old cells were obtained.
RESULTS
Roles for Different Cis-acting Plasmid Sequences in Reduction of Yeast Replicative Life Span
To study the effects of plasmids on yeast replicative life span, we generated two series of plasmids based on commonly used integrating vectors pRS306 and pRS305 (43). In each plasmid, we inserted ARS1, or ARS1 and CEN4, or the 2 μ circle origin (see “Experimental Procedures”). ARS1(autonomous replicating sequence 1) is a nuclear genomic DNA replication origin whose function and domain organization have been studied in detail (reviewed in Ref. 3). Centromeric DNA from chromosome IV (CEN4) has been mapped and functionally dissected (reviewed in Ref. 7). The region of the 2 μ circle plasmid extending from REP3 through the adjacent 599-bp repeat functions as a replication origin as well as a cis-acting plasmid partitioning element (reviewed in Refs. 6 and 41). The plasmids used in this study are summarized in Table I.
To evaluate effects on life span, plasmids were transformed into strain W303AR5 (46). For each plasmid, six independently isolated transformants were analyzed in parallel, and each life span curve reflects their collective behavior. Selection for the plasmid was maintained during life span analyses. Virgin mother cells unable to give rise to five daughters were discarded to exclude contributions from mother cells without plasmid. To identify mother cells that stopped dividing due to plasmid loss, rather than senescence, cells that had not divided in 2 days were transferred to nonselective medium and monitored for cell division and colony formation. A low percentage (< 10%) of mother cells were found to give rise to colonies and were excluded from the life span data set. Life span plates were incubated during the daytime at 30 °C but placed overnight (~12 h) at 14 °C, which gave a slightly but significantly (p < 0.01) longer life span than observed on plates stored overnight at 4 °C (Fig. 1A).
Fig. 1. Life span analysis of plasmid-transformed yeast.
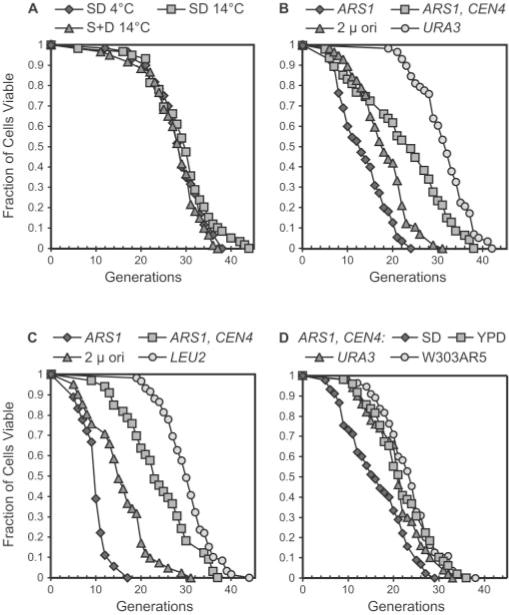
The number of daughter cells (generations) produced per mother cell are plotted as a function of mother cell viability. A, life span curves of strain W303AR5 (46) grown on SD (synthetic dextrose) and S + D (dextrose added after autoclaving) media at 30 °C during the daytime and stored overnight (~12 h) at 4 °C or 14 °C. The number (n) of mother cells analyzed per curve is as follows: SD4 °C,n = 60; SD 14 °C,n = 59; S + D 14 °C, n = 60. B, life span curves of W303AR5 transformed with plasmid pJPA113 (ARS1), pJPA116 (ARS1, CEN4), or pJPA138 (2 μ ori) and control strain yAF6 (URA3) (n = 55, 47, 57, and 58, respectively). C, life span curves of W303AR5 transformed with plasmids pJPA133 (ARS1), pJPA136 (ARS1, CEN4), pJPA148 (2 μ ori), and control strain yAF5 (LEU2) (n = 38, 33, 41, and 59, respectively). D, life span curves of W303AR5 transformed with pJPA116 (ARS1, CEN4) determined on SD and YPD (n = 45 and 49, respectively). Life spans of control strains yAF6 and W303AR5 were determined on YPD (n = 50 and 55, respectively). Plasmids are described in Table I.
Interestingly, transformants harboring pJPA113 (ARS1) showed dramatic reductions in both average and maximum life span compared with the Ura+ control strain yAF6 (Fig. 1B). yAF6 differs from pJPA113 transformants only in terms of plasmid DNA topology (i.e. integrated in yAF6 and episomal in transformants). Transformants containing pJPA116 (ARS1, CEN4) have a reduced average life span compared with yAF6 but exhibit a maximum life span similar to yAF6 (Fig. 1B). Thus, the addition of a CEN4 element to an ARS1 plasmid suppresses reduction in maximum life span but does not completely compensate for or protect against effects on average life span. Plasmids containing the 2 μ circle origin of replication were also constructed and analyzed. Yeast cells harboring pJPA138 (2 μ ori) show a reduction in both average and maximum life span (Fig. 1B). Generally speaking, the extent of reduction in average and maximum life span in pJPA138 (2 μ ori) transformants is intermediate between that observed in pJPA113 (ARS1) and pJPA116 (ARS1, CEN4) transformants (Fig. 1B). The results from multiple life span experiments are summarized in Table II.
Table II.
Life span data summary
| Plasmid/Strain | Average life span | Maximum life span | n* |
|---|---|---|---|
| pJPA113 | 12.4 ± 1.8 | 21.8 ± 2.2 | 4 |
| pJPA116 | 23 ± 1.4 | 39 ± 2.7 | 4 |
| pJPA138 | 16.3 ± 1.8 | 31.3 ± 0.6 | 3 |
| yAF6 | 33.2 ± 3.0 | 42 ± 1 | 3 |
Number of separate life span experiments.
The results reported above were obtained with plasmids carrying a URA3 selectable marker. To eliminate the possibility that effects of plasmids on life span were due to URA3 or medium lacking uracil, we constructed plasmids with a LEU2 selectable marker (Table I) and conducted life span experiments on medium lacking leucine. The results obtained with the LEU2 plasmid series were very similar to results obtained with the URA3 plasmid series (Fig. 1C). pJPA133 (ARS1) caused dramatic reductions in average (9.9 generations) and maximum (17 generations) life spans compared with the Leu+ control strain yAF5. yAF5 yielded an average (30.3 generations) and a maximum (44 generations) life span very similar to the average and maximum life span for yAF6 (Table II). Transformants containing pJPA136 (ARS1, CEN4) yielded a maximum life span of 38 generations but an average life span of 24 generations, similar to what was observed for the URA3 plasmid pJPA116 (ARS1, CEN4). Plasmid pJPA148 (2 μ ori) reduced the average (15.5 generations) and maximum (31 generations) life span to an extent intermediate between pJPA133 (ARS1) and pJPA136 (ARS1, CEN4) (Fig. 1C), similar to what was observed with the URA3 plasmid pJPA138 (2 μ ori) (Fig. 1B).
The reduction in average life span by ARS1, CEN4 plasmids pJPA116 and pJPA136 was unexpected. A similar plasmid had previously been reported to have no effect on life span when grown on YPD medium (46). One possible explanation for this difference was that ARS1, CEN4 plasmids are occasionally lost from mother cells, causing them to cease division on selective medium prior to senescence, which would result in a reduction in average life span. To test this, ARS1, CEN4 plasmid transformants were analyzed on nonselective YPD medium as done previously (46). On YPD, transformants carrying pJPA116 (ARS1, CEN4) were as long lived as control strains yAF6 (URA3) and W303AR5 (Fig. 1D). pJPA116 transformants analyzed in parallel on selective SD medium showed a reduction in average life span (Fig. 1D), as expected. These findings support the interpretation that ARS1, CEN4 plasmids, which are present at near unit copy number in transformants (see below), are occasionally lost from mother cells, rendering them unable to divide at a point in their life span prior to normal senescence.
We have also examined the effects of two well known plasmids that carry the TRP1-selectable marker. pTV3 carries the 2 μ origin, whereas pRS314 carries ARSH4 and CEN6(41, 43). Life spans of transformants containing each plasmid were analyzed on medium lacking tryptophan. pTV3 transformants had an average life span of 18.7 and a maximum life span of 32, both values of which are in good agreement with corresponding values for the 2 μ origin plasmids pJPA138 and pJPA148 (see above and Table II). pRS314 had average and maximum life spans of 21 and 41, respectively, which are in good agreement with values obtained with the ARS1/CEN4 plasmids pJPA116 and pJPA136 (see above and Table II). These data allow us to exclude a specific role for ARS1 and CEN4 in life span reductions presented above (Fig. 1).
Plasmid Inheritance Correlates with Reduction in Yeast Life Span
The plasmids used in this study were constructed to explore relationships between plasmid inheritance and effects on life span. Mitotic stability and plasmid copy number are widely used measures of plasmid DNA inheritance. Mitotic stability is defined as the proportion of a population of cells grown under selection that contains plasmid. We determined the mitotic stability and plasmid copy number of the plasmids used in life span experiments. Included in our studies were plasmids containing the rDNA ARS. rDNA repeats contain a single, relatively weak ARS (34). pJPA114 and pJPA117 contain the rDNA ARS at the same position as ARS1 in pJPA113 and pJPA116, respectively (see Table I and “Experimental Procedures”).
Plasmid pJPA113 (ARS1) was found to have a mitotic stability of ~20% (Fig. 2A), which is typical of yeast replicating plasmids containing ARS1, which exhibit a mother cell partitioning bias (36). pJPA116 (ARS1, CEN4) exhibited a much higher mitotic stability, ~90%, which is consistent with the presence of CEN4 centromeric DNA, and agrees with the mitotic stability of pRS316 (ARSH4, CEN6) (Fig. 2A). pJPA138 (2 μ ori) showed a high degree of mitotic stability, ~90% (Fig. 2A). The 2 μ origin plasmid pRS424 had a somewhat lower mitotic stability by comparison (Fig. 2A). pJPA114 (rDNA ARS) has a very low mitotic stability, < 1% (Fig. 2A). The presence of CEN4 with the rDNA ARS in pJPA117 improves mitotic stability to ~35% (Fig. 2A). These results with pJPA114 and pJPA117 are consistent with the low efficiency of the rDNA ARS (34). Not surprisingly, it was impractical for us to carry out life span analyses of transformants containing pJPA114.
Plasmid copy number was determined using Southern blot analysis. Copy number determinations using two different restriction enzymes gave comparable results (Fig. 2B). pJPA113 (ARS1) exhibited the highest plasmid copy number (Fig. 2C). Plasmids pJPA116 (ARS1, CEN4) and pJPA117 (rDNA ARS, CEN4) exhibited near unit copy number values (Fig. 2C), which is typical of centromeric plasmids (7), such as pRS316 (ARSH4, CEN6)(43). pJPA138 (2 μ ori) exhibited a copy number of ~33 (Fig. 2C), which is in the range of copy number values reported for other 2 μ origin plasmid vectors (41). The high copy number of pJPA113 is primarily due to the asymmetric inheritance of this plasmid and its accumulation in mother cells rather than ARS strength per se. We reach this conclusion because pJPA114, which contains a weak (rDNA ARS) replication origin, achieves a copy number almost as great as pJPA113, which contains a strong (ARS1) replication origin (Fig. 2C). Thus, pJPA113 demonstrates a correlation between extent of reduction of transformant life span (Fig. 1B) and tendency to be inherited asymmetrically and attain a high copy in yeast cells (Fig. 2C).
Plasmids Do Not Significantly Increase ERC Levels
The results presented above suggest that reduction in life span by the ARS1 plasmid pJPA113 is due to asymmetric inheritance and accumulation in mother cells. An alternative explanation is that pJPA113 increases ERC levels in transformed cells and thereby reduces life span indirectly. To address this possibility, we measured recombination at the rDNA locus using an ADE2 marker loss assay and measured ERC levels in transformed cells by Southern blotting.
To analyze the frequency of recombination at the rDNA locus, we took advantage of the fact that W303AR5 contains ADE2 integrated at the rDNA locus (46). Recombination between flanking rDNA repeats results in loss of ADE2 and a change in colony color. The frequency of half-red sectored colonies is a measure of rDNA recombination rate (events per cell division). Transformation of yeast with plasmid results in a small increase in rDNA recombination as measured by ADE2 marker loss. For W303AR5, we find that ADE2 marker loss occurs at a frequency of ~1.3 per thousand cell doublings (Fig. 3A), which is in good agreement with frequencies reported by others (23, 31, 33). The rate of ADE2 marker loss from yAF6 (URA3) occurs at ~2.7 per thousand (Fig. 3A). Transformants containing the three plasmids used in this study, pJPA113 (ARS1), pJPA116 (ARS1, CEN4), and pJPA138 (2 μ ori), exhibited marker loss rates of 4.1, 4.5, and 4.1 per thousand cell doublings, respectively. The differences between transformants and yAF6 represent increases of less than 2-fold. Higher levels of ADE2 marker loss are typically observed in strains with reduced life spans. For example, short lived sir2Δ mutants exhibit ADE2 marker loss rates > 10-fold higher than isogenic SIR2 strains (23).
To directly compare ERC levels, yeast transformants and control strains were analyzed by Southern blotting, and ERC monomer bands were quantitated (see “Experimental Procedures”). ERC monomers consist of a single 9.1-kb rDNA repeat and were chosen for purposes of quantitation because they are well resolved from chromosomal rDNA and other ERC bands on Southern blots. ERC monomer levels in transformants were not significantly different from ERC monomer levels in control strains. Control strains W303AR5 and yAF6 (URA3) have ~0.0007 and 0.0015 ERC monomers per total chromosomal rDNA, respectively (Fig. 3B). Transformants bearing pJPA113 (ARS1), pJPA116 (ARS1, CEN4), and pJPA138 (2 μ ori) have ERC monomer levels of 0.0014, 0.001, and 0.001, respectively (Fig. 3B). These values are within the error of measurements and are not significantly different (Fig. 3). For comparison, we examined yAF5 (LEU2), which contains a copy of pRS305 integrated at the leu2–113 locus, and found that the ERC monomer level was 0.001, which is intermediate between W303AR5 and yAF6 (Fig. 3B). Quantitation of slower migrating ERC multimer bands did not reveal significant differences in levels between transformant and control strains (data not shown). We conclude that plasmids do not have a significant effect on ERC levels.
Plasmid Accumulation Correlates with Reduction in Life Span
If plasmids reduce life span in a manner analogous to ERCs, then plasmid DNAs should accumulate in old mother cells. To test this prediction, we used a biotinylation and magnetic sorting approach to isolate ~7-generation-old yeast cells (see “Experimental Procedures”). Plasmid DNA levels in young and old cells were measured by quantitative Southern blotting.
The ages of old and young (unsorted) cells were determined by counting bud scars stained with Calcofluor (40). From single sort experiments, the average ages of yeast transformed with pJPA113, pJPA116, pJPA138, and yAF6 were 6.9, 7.0, 6.1, and 6.2 generations, respectively (Fig. 4A). Young cells from the same cultures were an average of 1.5, 1.4, 1.1, and 1.1 generations old, respectively (Fig. 4A). Inspection of the Southern blot clearly reveals increases in relative amounts of pJPA113 (ARS1) and pJPA138 (2 μ ori) in old cells (Fig. 4B). pJPA116 (ARS1, CEN4) did not accumulate in old cells, and it yields bands similar in their intensities to corresponding bands from yAF6 (Fig. 4B). In a striking illustration of the accumulation of pJPA113 and pJPA138 in old cells, the linearized plasmid DNA bands can be observed by ethidium bromide staining (Fig. 4D). ERC levels in young and old cells were also analyzed by Southern blotting. Hybridization to rDNA probe revealed ERC bands and a broad band corresponding to the rDNA locus on chromosome XII (Fig. 4C). We note that all old cell preparations contained increased numbers of both monomeric and slower migrating ERC species (Fig. 4C). The ERC and rDNA repeat bands collapse to a single 9.1-kb band following digestion with KpnI, which cuts rDNA once (data not shown).
Fig. 4. Plasmid DNA and ERC levels in young and old cells.
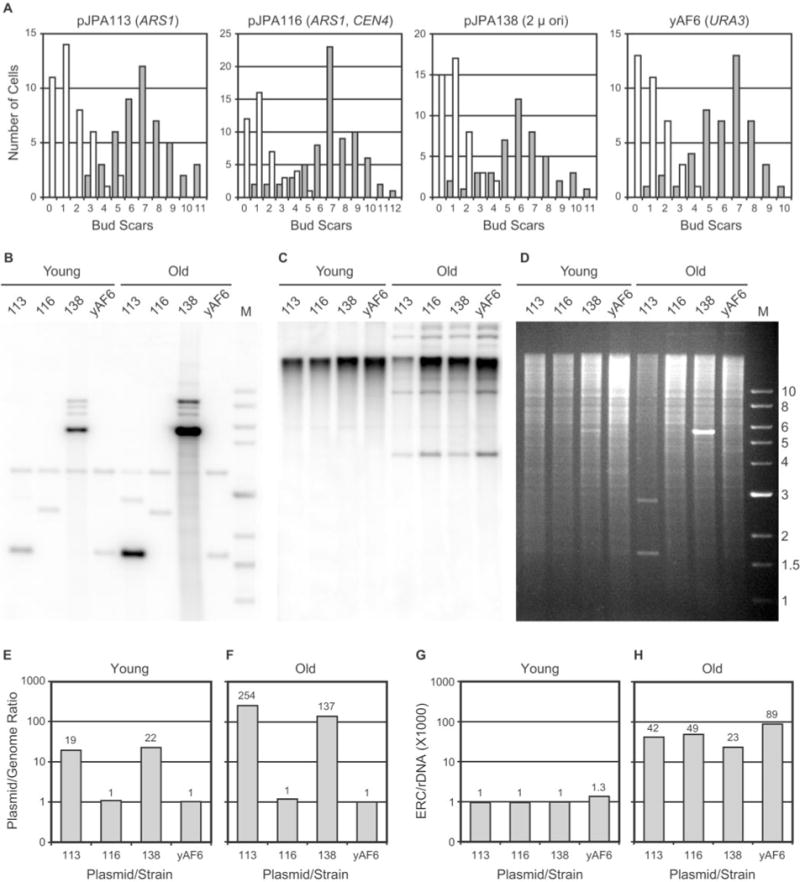
A conveys the cis-acting elements present in each plasmid (see also Table I). Plasmids are abbreviated by numbers in B–H. All plasmids carry URA3. Control strain yAF6 (URA3) did not contain plasmid. Old cells were harvested using a biotinylation and magnetic sorting approach (see “Experimental Procedures”). A, age profile histograms of young and old cells. The number of cells is plotted as a function of the number of bud scars (n > 40 for each histogram). B, Southern blot of plasmid DNAs. PstI-digested genomic DNA yields a 3.67-kb URA3 band. Other bands are plasmid-derived. Genomic URA3DNA in lane Old 113 migrated as two bands due to partial overdigestion of this sample. C, Southern blot of ERCs. D, ethidium bromide-stained agarose gel corresponding to the blot in B and C. DNA marker sizes (in kb) are shown. E and F, plasmid levels in young and old cells (quantitation of data presented in B). G and H, ERC monomer levels in young and old cells (quantitation of data presented in C). E–H, ratios of episome (plasmid or ERC monomer) band intensity divided by chromosomal rDNA band intensity (×1000) are plotted (on a semilog scale). See Fig. 1B for corresponding life span data. Comparable results were obtained from similar cell sorting and Southern blotting experiments and are discussed under “Results.”
Chromosomal and plasmid band intensities were quantitated using a PhosphorImager. Consistent with our determinations in Fig. 2, pJPA113 (ARS1) and pJPA138 (2 μ ori) are present at high copy number in young cells, but pJPA116 (ARS1, CEN4) is not (Fig. 4E). In ~7-generation-old transformants, the plasmid copy numbers for pJPA113 and pJPA138 are dramatically increased, reaching values of 254 and 137, respectively (Fig. 4F). This represents a difference in copy number between young and old cells of ~13-fold for pJPA113 and ~6-fold for pJPA138. By comparison, 7-generation-old pJPA116 transformants show no significant increase in plasmid copy number (Fig. 4F).
In a separate experiment with pJPA113 and pJPA116 transformants, in which genomic DNA was digested with BamHI instead of PstI, quantitative analysis revealed that young cells contained 27 and 1.5 plasmids/cell, respectively, whereas old cells contained 283 and 1.2 plasmids/cell, respectively (data not shown). This corresponds to a ~10-fold increase in plasmid copy number for pJPA113 in ~7-generation old cells and no significant increase in pJPA116 copy number, which agrees with findings presented in Fig. 4E and F.
ERC monomer levels were also quantitated in young and ~7-generation old transformants and yAF6. ERC monomer levels in young cells were equal or close to 0.001 (Fig. 4G), which agrees with measurements presented above (Fig. 3B). In old cells, however, ERC monomer levels were appreciably higher and exhibited increases between ~20- and ~70-fold (Fig. 4H). The levels of ERCs we observe in ~7-generation-old cells appear comparable with ERC levels in sorted cells of similar age reported by others (e.g. Refs. 23 and 46), although quantitative analysis of ERC levels in young and old yeast cells is not commonly reported in the literature.
In the experiment shown in Fig. 4, ERC monomer levels in yAF6 (URA3) are higher than in transformants (Fig. 4H). This raises the question of whether the presence of plasmid reduces ERC levels. In a separate experiment, ERC monomer levels in young cells were equal or close to 0.001 (ERC monomer/chromosomal rDNA), and ERC monomer levels in old transformants containing pJPA113, pJPA116, and pJPA138 and in old yAF6 cells were determined to be 0.083, 0.075, 0.045, and 0.081, respectively (data not shown). The similar ERC levels in yAF6 and transformants in this experiment suggest that plasmid vectors do not appreciably affect ERC monomer levels (see also Fig. 5).
Fig. 5. Plasmid DNA and ERC levels in young and old cells.
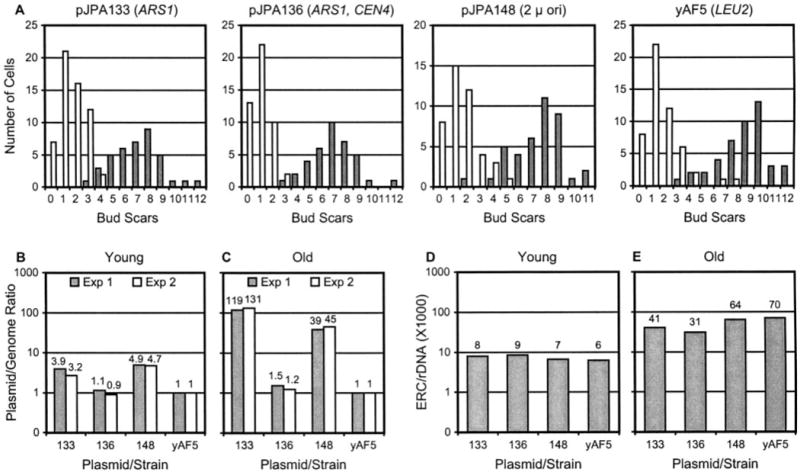
Plasmids are abbreviated by numbers in B–E. A conveys the cis-acting elements present in each plasmid (see also Table I). All plasmids carry LEU2. Control strain yAF5 (LEU2) did not contain plasmid. Data were collected as described in the legend to Fig. 4. A, age profile histograms of young and old cells. B and C, plasmid levels in young and old cells (semilog plot). Data from two Southern blotting experiments are shown (Exp 1 and Exp 2). D and E, ERC monomer levels in young and old cells (semilog plot). See Fig. 1C for corresponding life span data.
Does the extent of ERC accumulation in old cells in Fig. 4 agree with predictions based on our estimates of rates of recombination within the rDNA locus (see above and Fig. 3)? If we assume that extrachromosomal rDNA repeats are generated at a rate of 0.5/cell/generation and that ERCs are retained in mother cells, then 6–7 generations should yield an increase of between 32- and 64-fold, which is similar to the observed range of increase from 20- to 70-fold (Fig. 4G and H).
We have also quantitated the relative amount of all ERCs (i.e. monomers, multimers, and concatemers) found in old transformants containing pJPA113, pJPA116, and pJPA138 and in old yAF6 cells. We found levels of 0.140, 0.136, 0.086, and 0.238, respectively (extrachromosomal rDNA/chromosomal rDNA; data not shown). These values mirror levels of accumulation of ERC monomers presented in Fig. 4H. Thus, ERC monomers comprise about one-fourth to one-third of all extrachromosomal rDNA repeats and are present at similar levels relative to all ERCs in old transformed and untransformed cells.
To extend these studies, yeast sorting experiments were done with transformants containing the LEU2 plasmids pJPA133 (ARS1), pJP136 (ARS1, CEN4), and pJPA148 (2 μ ori) and with the LEU2 strain yAF5. The average ages of sorted yeast transformed with pJPA133, pJPA136, pJPA148, and yAF5 are 7.1, 7.0, 7.6, and 7.9 generations, respectively (Fig. 5A). Young cells from the same cultures were an average of 1.7, 1.0, 1.6, and 1.6 generations, respectively (Fig. 5A). pJPA133 and pJPA148 attain copy number levels of 119 and 39, respectively, in ~7-generation-old cells (Fig. 5C). This represents an increase in copy number between young and old cells of ~30-and ~8-fold for pJPA133 and pJPA148, respectively. pJPA136 did not show a significant increase in old cells (Fig. 5C). In comparison with pJPA113 and pJPA138, pJPA133 and pJPA148 reached lower absolute levels of plasmid in ~7-generation-old cells. However, pJPA133 and pJPA148 accumulated to similar extents in terms of -fold increase. To resolve whether this difference in absolute levels of plasmids in old cells was due to experimental error, sorting experiments with transformants and the control strain were repeated, followed by Southern analyses. The repeat experiment gave results very similar to first experiment, in terms of both absolute level of plasmid in young and old cells and -fold increase in young and old cells (Fig. 5B and C). This indicates that plasmids with identical ARS1 origins and CEN4 elements, but with different backbones and selectable markers, are maintained at different absolute copy number levels in young and old cells. Nevertheless, similar -fold differences in plasmid levels are observed between young and ~7-generation-old cells. This indicates that ASR1 and CEN4 elements present on plasmids functionally determine patterns of plasmid inheritance and accumulation during yeast mother cell replication.
Next, ERC monomer levels in transformants containing pJPA133, pJPA136, and pJPA148 and in strain yAF5 were quantitated. In young cells, ERC monomers were detected at relatively high levels (Fig. 5C). However, ERC monomer levels in ~7-generation-old cells were similar to levels observed above for pJPA113, pJPA116, and pJPA138 transformants (compare Figs. 4H and 5D). Thus, ERCs in the Leu+ transformants showed accumulation over a range of ~3- to ~12-fold between young and old transformants. This range of -fold increase is ~7-fold lower than the ~20- to ~70-fold increase in ERC levels between young and old Ura+ transformants. This suggests that the rate of ERC accumulation during the aging process is regulated so that old cells of similar ages contain similar levels of ERCs despite differences in initial levels of ERCs in.
An important trend emerges from our studies of plasmid accumulation in old cells. Plasmids that accumulate to the greatest degree in old cells (Figs. 4 and 5) exert the most profound effect on life span (Fig. 1). ARS1 plasmids attain the highest copy numbers in old cells and have the most pronounced effect on life span. ARS1/CEN4 plasmids maintain a copy number near unity in young and old cells and have a small effect on maximum life span and a moderate effect on average life span. Plasmids with 2 μ origins attain a copy number in old cells roughly half that of ARS1 plasmids and reduce life span roughly half as much as ARS1 plasmids. This suggests the existence of an inverse relationship between plasmid accumulation in old cells and reduction in yeast life span.
Terminal Cell Morphology
Currently, in the field of yeast aging, there are few approaches available to directly address the senescent phenotype in old nondividing cells. To address this issue indirectly, we scrutinized the “terminal” morphology of cells at the end of their life span. The rationale for this approach is that cell morphology is a phenotypic indicator of cell cycle stage and can serve as a basis to compare senescent cells (32). If cell morphology in terminal transformed cells is very different from the morphology of terminal wild type cells, this would imply that different mechanisms may bring about the senescent phenotype in transformed and untransformed cells.
To examine terminal yeast cells, images of terminal cells were collected from three different life span experiments. Three different cell morphologies were scored: unbudded cells, single-budded cells with small buds, and single-budding cells with large buds (32). Bud emergence in S. cerevisiae correlates with entrance into S phase, and small buds are indicative of early S phase, whereas large buds are indicative of late S/G2 or mitotic arrest. Unbudded cells are in G1 phase. Between 10 and 15% of the terminal cells, transformed or untransformed, had multiple buds (data not shown) and were omitted from this comparison. For pJPA113 (ARS1) and pJPA116 (ARS1, CEN4) transformants and W303AR5, more than 50% of terminal cells were unbudded (Fig. 6). Typically, between 50 and 60% of senescent yeast cells have been found to be unbudded (32, 33). pJPA116 transformant cells consistently yielded the highest proportion (~65%) of unbudded cells (Fig. 6). yAF6 (URA3) and pJPA138 (2 μ ori) transformants ceased dividing with a predominance, yet a lower percentage, of unbudded cells (Fig. 6). Thus, the majority of pJPA113 transformants, like W303AR5 cells, senesced in G1, as expected. In addition, similar proportions of small budded and large budded terminal cells in senescent pJPA113 transformants and W303AR5 cells (Fig. 6) indicate that similar proportions of these cells arrested in similar phases (S or G2/M) of the cell cycle. Thus, this analysis supports the interpretation that pJPA113 (ARS1) reduces life span by a normal aging process.
Fig. 6. Terminal morphology of senescent cells.
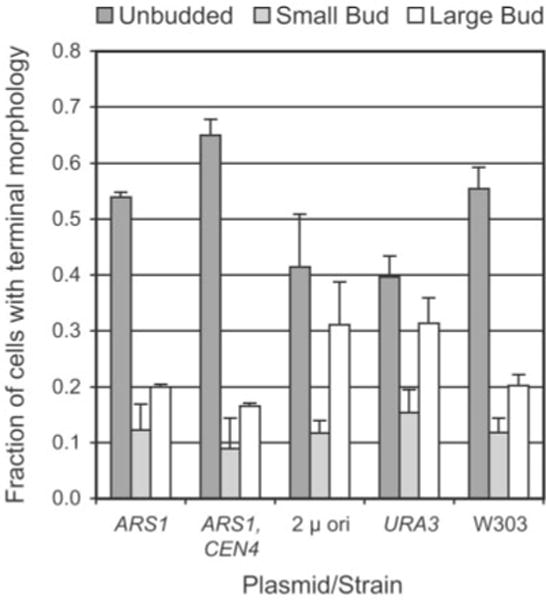
Cells at the end of life span experiments were classified according to budding pattern as described (32). Small buds were defined as having a diameter less than 25% of the diameter of the mother cell. All other buds were classified as large. Average and S.D. values from three independent experiments are shown (n > 40 for each transformant or control strain in each experiment).
Do Functional rDNA Transcription Units Play a Role in Reduction of Life Span?
Although plasmids without rDNA sequences reduce yeast life span, it is important to consider a potential role for rDNA sequences in life span reduction. It is possible that ERCs reduce life span in a manner that is mechanistically more complex than the manner in which plasmid episomes reduce life span. There are significant differences in coding potential between plasmids and ERCs. The 9.1-kb rDNA repeat carries genes for rRNA precursors as well as the gene TAR1, which lies on the strand opposite the 25 S rRNA and encodes a mitochondrial protein (9). One way to address this issue is to ask whether or not a plasmid vector carrying an rDNA repeat unit has a more pronounced effect on life span than plasmid vector alone. It is important to note this issue was not completely addressed in a previous study employing the rDNA-containing plasmid pDS163 (46). Plasmid pDS163 does not contain a functional 9.1-kb rDNA repeat unit. The rDNA on pDS163 consists of a 12.1-kb insert extending from an EcoRI site within the coding sequence of 5.8 S rRNA to the 5′-most EcoRI site in the 25 S rRNA coding region (data not shown). The 12.1-kb fragment does not carry a full-length 35 S pre-rRNA transcription unit and is capable of producing only a truncated 35 S pre-rRNA transcript, which if processed would be incapable of yielding mature 25 S rRNA.
To determine whether an episomal rDNA repeat influences life span, we constructed three plasmids containing 9.1-kb rDNA repeats and used them in life span experiments. The three plasmids, pJPA105, pJPA106, and pJPA107, contain 9.1-kb repeats with different endpoints in the plasmid pAF15, which contains a 2 μ origin (see “Experimental Procedures” and Table I). Plasmid pJPA105 contains a repeat with XmaI end points, which has been shown by Nomura and colleagues to functionally complement an rDNA deletion in vivo(37). pJPA106 and pJPA107 contain repeats with AhdI and PsiI end points, respectively, which should not interfere with rDNA gene expression. A 2 μ origin plasmid was used, because plasmids constructed with rDNA inserts whose replication relied solely on the rDNA ARS were found to integrate into the chromosomal rDNA locus with high frequency (as determined by Southern blot analysis; data not shown). Life span determinations of W303AR5 transformants containing pAF15, pJPA105, pJPA106, and pJPA107 were done as described above (see Fig. 1). pJPA105, pJPA106, pJPA107, and pAF15 transformants gave very similar life span curves, indicating that the presence of a functional rDNA repeat does not have a dramatic effect on life span (Fig. 7). All four plasmids affect life span to an extent similar to the 2 μ origin plasmids pJPA138 and pJPA148 (Fig. 1B and C), although the average life spans for pJPA105, pJPA106, pJPA107, and pAF15 (13.3, 11.8, 11.7, and 12.2 generations, respectively) are lower than the average life spans for pJPA138 and pJPA148 transformants (15.5 and 16.3 generations, respectively; Fig. 1 and Table II). Life span curves for pJPA106 and pJPA107 transformants did not show a statistically significant difference from pAF15 transformants based on the Wilcoxon signed pair rank test (p > 0.05) Only transformants carrying pJPA105 and pAF15 exhibited a statistically significant difference (p < 0.05), but this represents a small increase in life span of transformants carrying pJPA105. These findings support the conclusion that the presence of a full-length rDNA repeat per se is not required for, and does not necessarily augment, reduction in yeast life span.
Fig. 7. Life span analysis of yeast transformed with plasmids containing rDNA repeats.
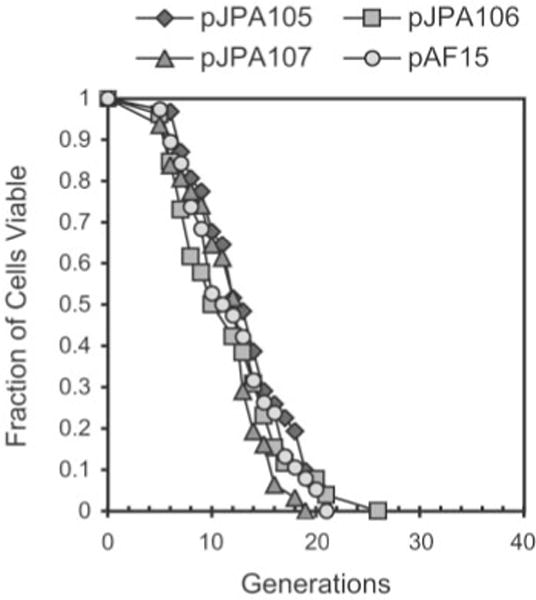
Number of daughter cells (generations) produced per mother cell are plotted as a function of mother cell viability. Life span analysis was done as described in the legend to Fig. 1 using W303AR5 carrying plasmids pJPA105 (n = 45), pJPA106 (n = 43), or pJPA107 (n = 46) and control plasmid pAF15 (n = 46). Plasmids pJPA105, pJPA106, and pJPA107 contain full-length (9.1-kb) rDNA repeats with different endpoints (see Table I and “Experimental Procedures”). pJPA105 contains an rDNA insert with XmaI endpoints, which has been shown to be functional in vivo(37).
DISCUSSION
Budding yeast is an excellent system in which to study cell-autonomous mechanisms of aging. Mechanisms linked to genome stability, metabolic damage, and metabolic regulation have been found to regulate yeast replicative life span (1, 11, 21, 30, 45, 48). Sinclair and Guarente (46) have proposed that a key regulator of life span is the cellular level of ERCs. To study this proposal, we have used plasmids to model ERC inheritance and accumulation, two processes that govern ERC levels in yeast cells. Our work shows that plasmid DNAs bring about significant reductions in yeast life span. We find that ARS1 and 2 μ origin plasmids specifically accumulate in old yeast cells and that the level of accumulation of ARS1 and 2 μ origin plasmids in old cells correlates with the extent of reduction in life span. This is the first demonstration to our knowledge of an inverse relationship between DNA episome level in old cells and reduction in life span. We find that plasmids have a direct effect on life span and do not indirectly reduce life span by increasing recombination at the rDNA locus and increasing ERC levels in transformed cells. Analysis of the “terminal” morphology of senescent cells indicates that plasmids do not cause a stochastic arrest in the cell cycle, which is consistent with a normal aging process. Reduction in life span does not require that plasmids carry rDNA repeat sequences, and the presence of a full-length, functional 9.1-kb rDNA repeat on a plasmid does not augment reduction in life span. These findings confirm the work of Sinclair and Guarente (46) and provide significant new support for their ERC model by directly demonstrating a relationship between plasmid inheritance, plasmid accumulation, and reduction in life span. Our studies also highlight the value of plasmids as tools to investigate properties of ERCs that are relevant to the aging process in yeast.
Why do ARS plasmids accumulate in mother cells? It has long been appreciated that ARS plasmids are inherited asymmetrically and accumulate in mother cells (36). This accounts for the high copy number and low mitotic stability of ARS plasmids. However, accumulation of ARS plasmids in cells that are multiple generations old has not been directly demonstrated. Our studies are the first to directly demonstrate that ARS1-containing plasmids accumulate to high levels in old yeast cells. Although ARS1 plasmid partitioning bias is well known, little is understood about its underlying mechanism. One possibility is that plasmid partitioning bias is due to the nature of cell and nuclear division in budding yeast. During closed mitosis in yeast, an intact nucleus elongates along the axis of the mitotic spindle and adopts an elongated “dumbbell” shape due to constriction of the nucleus at the bud neck. Chromosomes pass though the constriction at the bud neck by virtue of their attachment to the mitotic spindle, which is able to exert force on chromosomes. In the absence of spindle attachment, passage of DNA molecules through the constriction at the bud neck may be limited. Consistent with this notion, the relatively small (1.45-kb) TRP RI plasmid has been shown to be inherited efficiently and to exhibit high mitotic stability (51). The small size of the TRP RI plasmid may allow it to readily distribute between mother and daughter cells through the bud neck constriction. Commonly used yeast recombinant DNA vectors are typically larger than the TRP RI plasmid and require cis-acting sequences and trans-acting factors to be stably inherited.
Why do budding yeast exhibit a mother cell plasmid segregation bias? One possibility is that mother cell segregation bias is a mechanism to protect progeny cells from potential “parasitic” effects of episomal DNAs acquired from the environment. The 2 μ circle is a “commensal” episomal DNA that Futcher et al. (15) have depicted as a sexually transmitted selfish DNA. The 2 μ circle depends on its capacity to overcome mother cell segregation bias (see below) in order to survive in a host population in the absence of any selective value. Another possibility is that mother cell segregation bias is a mechanism to increase the longevity of progeny cells by limiting transmission of ERCs.
Why do ARS1 plasmids bring about cellular senescence more rapidly than do ERCs? One possibility is that virgin mothers contain more plasmids than ERCs at the start of life span experiments. Virgin mothers must contain at least one ARS plasmid but probably contain on average ~0.5 ERC/cell. The difference in origin strength between ARS1 and the rDNA ARS may also be important. ARS1 is a relatively “strong” ARS and capable of supporting rapid plasmid accumulation in mother cells. ERCs contain a comparatively “weak” ARS that is likely to support only relatively slow accumulation in mother cells. The rDNA ARS contains an ACS (ARS consensus sequence) that departs from the consensus at position 1, a change that has been shown to reduce ARS function, primarily by limiting DNA unwinding (34). This difference in strength could explain why ARS1 plasmids bring about senescence in mother cells more rapidly than do ERCs. ARS1 plasmids are replicated more efficiently than ERCs, which increases the rate of ARS1 plasmid accumulation in mother cells compared with ERCs.
Do cis-acting sequences that counteract mother cell segregation bias suppress reduction in life span by ARS1 plasmids? Yes, ARS1/CEN4 plasmids reduce life span to a lesser extent than ARS1 plasmids, which is consistent with results of Sinclair and Guarente (46). However, inclusion of CEN4 on ARS1 plasmids suppresses the reduction in maximum life span by ARS1 plasmids but does not fully suppress the reduction in average life span. Our studies also directly show that ARS1/CEN4 plasmids do not accumulate in ~7-generation-old mother cells. The reduction in life span is not specific for the combination of ARS1 and CEN4. The combination of ARSH4 and CEN6 (in pRS314) (43) reduces average life span with a minimal effect on maximum life span. The fact that centromeric DNA elements suppress reduction in maximum life span supports the conclusion that ARS1 plasmids exert their effect by accumulation in mother cells, as discussed above.
Why do 2 μ origin plasmids reduce life span? One clue emerges from the observation that endogenous 2 μ circles do not accumulate in old cells (data not shown). Although both 2 μ origin plasmids and 2 μ circles contain the REP3/STB cis-acting stability element, 2 μ origin plasmids contain a single 599-bp segment, whereas 2 μ circles contain two 599-bp segments arranged as an inverted repeat (6, 41). More efficient autoregulation of 2 μ circle copy number and inheritance is likely to prevent accumulation in old cells. It is important to note that 2 μ circles can be toxic to cells when present at high copy number. Constitutive expression of the 2 μ amplification machinery results in high copy number and has deleterious effects on cell growth (6). Similarly, mutations in NIB1/ULP1 result in unusually high levels of 2 μ circles, formation of large inviable or mitotically arrested cells, and clonal lethality (18). Studies by Dobson and co-workers2 indicate that a modified form of Rep2p, a 2 μ circle-encoded plasmid partitioning protein, accumulates in ulp1 mutants, suggesting that ULP1 is involved in partitioning of 2 μ circles during mitosis. This suggests that high levels of 2 μ circles in nib1/ulp1 mutants may result from asymmetric inheritance. In this sense, phenotypes associated with nib1/ulp1 defects may share mechanistic underpinnings with senescent phenotypes associated with asymmetric inheritance of plasmids and ERCs.
Why do 2 μ origin plasmids have an intermediate effect on life span? Although 2 μ origin plasmids accumulate in ~7-generation-old mother cells, they attain levels approximately half that observed with ARS1 plasmids. As mentioned above, comparison of results with 2 μ origin and ARS1 plasmids supports an important inverse relationship; the extent of plasmid accumulation in old cells correlates with the extent of reduction in life span.
Our findings raise the question of by what mechanism(s) plasmids, and by implication ERCs, reduce life span in yeast. It is clear that asymmetric inheritance of plasmid DNAs has the potential to burden mother cells with high DNA content. If we assume that a 5-kb plasmid is replicated once each S phase and uniformly inherited by the mother cell during M phase, then 12 doublings will yield a plasmid DNA content in excess of the nuclear genomic DNA content (5 × 212 = 20.5 Mb of plasmid DNA content > ~13 Mb of nuclear genomic DNA content). Of course, this example is an oversimplification and omits factors such as origin firing frequency and segregation efficiency. However, we note that after 12 generations, 90% of pJPA113 (5.7 kb of ARS1 plasmid) transformants were senescent, and after 20 generations, 90% of pJPA133 (4.8 kb of ARS1 plasmid) transformants were senescent. The fact that significant percentages of senescent mother cells arise between 10 and 20 generations is consistent with the accumulation of plasmid DNA content to a level that approaches or exceeds nuclear genomic DNA content. Similarly, Sinclair and Guarente (46) have estimated that the ERC content of old cells exceeds the content of the linear genome. One possible mechanism for reduction of life span is that accumulated plasmid DNAs interfere with cellular DNA metabolism. Another possibility is that plasmid DNAs bind regulatory proteins, such as transcription factors, which could alter genome-wide patterns of gene expression. Thus, plasmids are likely to be useful tools for exploring possible mechanisms by which ERCs reduce life span. Also, studies with plasmids should improve our understanding of mechanisms by which episomal DNAs accumulate during yeast aging. This, in turn, should shed additional light on the role ERCs play in the yeast aging process.
Acknowledgments
We are grateful to Carl Feldherr, Michal Jazwinski, David Sinclair, and Maury Swanson for comments on the manuscript. We thank Fernando Castro and Raj Mehta for assistance with sequencing rDNA repeats.
Footnotes
This project was supported in part by funding from the Ellison Medical Foundation. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: ERC, extrachromosomal rDNA circle; rDNA, ribosomal DNA; rRNA, ribosomal RNA.
M. Dobson, personal communication.
References
- 1.Aguilaniu H, Gustafsson L, Rigoulet M, Nystrom T. Science. 2003;299:1751–1753. doi: 10.1126/science.1080418. [DOI] [PubMed] [Google Scholar]
- 2.Benguria A, Hernandez P, Krimer DB, Schvartzman JB. Nucleic Acids Res. 2003;31:893–898. doi: 10.1093/nar/gkg188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bielinsky AK, Gerbi SA. J Cell Sci. 2001;114:643–651. doi: 10.1242/jcs.114.4.643. [DOI] [PubMed] [Google Scholar]
- 4.Brewer BJ, Fangman WL. Cell. 1988;55:637–643. doi: 10.1016/0092-8674(88)90222-x. [DOI] [PubMed] [Google Scholar]
- 5.Brewer BJ, Lockshon D, Fangman WL. Cell. 1992;71:267–276. doi: 10.1016/0092-8674(92)90355-g. [DOI] [PubMed] [Google Scholar]
- 6.Broach JR, Volkert FC. In: The Molecular and Cellular Biology of the Yeast Saccharomyces. Pringle JR, editor. Vol. 1. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1991. pp. 297–331. [Google Scholar]
- 7.Carbon J, Clarke L. New Biol. 1990;2:10–19. [PubMed] [Google Scholar]
- 8.Christianson TW, Sikorski RS, Dante M, Shero JH, Hieter P. Gene (Amst) 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- 9.Coelho PS, Bryan AC, Kumar A, Shadel GS, Snyder M. Genes Dev. 2002;16:2755–2760. doi: 10.1101/gad.1035002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Conrad-Webb H, Butow RA. Mol Cell Biol. 1995;15:2420–2428. doi: 10.1128/mcb.15.5.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Costa V, Moradas-Ferreira P. Mol Aspects Med. 2001;22:217–246. doi: 10.1016/s0098-2997(01)00012-7. [DOI] [PubMed] [Google Scholar]
- 12.Defossez PA, Prusty R, Kaeberlein M, Lin SJ, Ferrigno P, Silver PA, Keil RL, Guarente L. Mol Cell. 1999;3:447–455. doi: 10.1016/s1097-2765(00)80472-4. [DOI] [PubMed] [Google Scholar]
- 13.Egilmez NK, Jazwinski SM. J Bacteriol. 1989;171:37–42. doi: 10.1128/jb.171.1.37-42.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fritze CE, Verschueren K, Strich R, Easton Esposito R. EMBO J. 1997;16:6495–6509. doi: 10.1093/emboj/16.21.6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Futcher B, Reid E, Hickey DA. Genetics. 1988;118:411–415. doi: 10.1093/genetics/118.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gietz RD, Woods RA. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- 17.Gotta M, Strahlbolsinger S, Renauld H, Laroche T, Kennedy BK, Grunstein M, Gasser SM. EMBO J. 1997;16:3243–3255. doi: 10.1093/emboj/16.11.3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holm C. Cell. 1982;29:585–594. doi: 10.1016/0092-8674(82)90174-x. [DOI] [PubMed] [Google Scholar]
- 19.Jazwinski SM. Trends Genet. 2000;16:506–511. doi: 10.1016/s0168-9525(00)02119-3. [DOI] [PubMed] [Google Scholar]
- 20.Jazwinski SM. Ann N Y Acad Sci. 2000;908:21–30. doi: 10.1111/j.1749-6632.2000.tb06632.x. [DOI] [PubMed] [Google Scholar]
- 21.Jazwinski SM. Annu Rev Microbiol. 2002;56:769–792. doi: 10.1146/annurev.micro.56.012302.160830. [DOI] [PubMed] [Google Scholar]
- 22.Kaeberlein M, Andalis AA, Fink GR, Guarente L. Mol Cell Biol. 2002;22:8056–8066. doi: 10.1128/MCB.22.22.8056-8066.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaeberlein M, McVey M, Guarente L. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kennedy BK, Austriaco NR, Jr, Guarente L. J Cell Biol. 1994;127:1985–1993. doi: 10.1083/jcb.127.6.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kennedy BK, Austriaco NR, Jr, Zhang J, Guarente L. Cell. 1995;80:485–496. doi: 10.1016/0092-8674(95)90499-9. [DOI] [PubMed] [Google Scholar]
- 26.Kennedy BK, Gotta M, Sinclair DA, Mills K, McNabb DS, Murthy M, Pak SM, Laroche T, Gasser SM, Guarente L. Cell. 1997;89:381–391. doi: 10.1016/s0092-8674(00)80219-6. [DOI] [PubMed] [Google Scholar]
- 27.Kim S, Benguria A, Lai CY, Jazwinski SM. Mol Biol Cell. 1999;10:3125–3136. doi: 10.1091/mbc.10.10.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kirchman PA, Kim S, Lai CY, Jazwinski SM. Genetics. 1999;152:179–190. doi: 10.1093/genetics/152.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobayashi T, Horiuchi T. Genes Cells. 1996;1:465–474. doi: 10.1046/j.1365-2443.1996.d01-256.x. [DOI] [PubMed] [Google Scholar]
- 30.Mandavilli BS, Santos JH, Van Houten B. Mutat Res. 2002;509:127–151. doi: 10.1016/s0027-5107(02)00220-8. [DOI] [PubMed] [Google Scholar]
- 31.Mays Hoopes LL, Budd M, Choe W, Weitao T, Campbell JL. Mol Cell Biol. 2002;22:4136–4146. doi: 10.1128/MCB.22.12.4136-4146.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McVey M, Kaeberlein M, Tissenbaum HA, Guarente L. Genetics. 2001;157:1531–1542. doi: 10.1093/genetics/157.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merker RJ, Klein HL. Mol Cell Biol. 2002;22:421–429. doi: 10.1128/MCB.22.2.421-429.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller CA, Umek RM, Kowalski D. Nucleic Acids Res. 1999;27:3921–3930. doi: 10.1093/nar/27.19.3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mortimer RK, Johnson JR. Nature. 1959;183:1751–1752. doi: 10.1038/1831751a0. [DOI] [PubMed] [Google Scholar]
- 36.Murray AW, Szostak JW. Cell. 1983;34:961–970. doi: 10.1016/0092-8674(83)90553-6. [DOI] [PubMed] [Google Scholar]
- 37.Oakes M, Aris JP, Brockenbrough JS, Wai H, Vu L, Nomura M. J Cell Biol. 1998;143:23–34. doi: 10.1083/jcb.143.1.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park PU, Defossez PA, Guarente L. Mol Cell Biol. 1999;19:3848–3856. doi: 10.1128/mcb.19.5.3848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park PU, McVey M, Guarente L. Methods Enzymol. 2002;351:468–477. doi: 10.1016/s0076-6879(02)51865-6. [DOI] [PubMed] [Google Scholar]
- 40.Pringle JR. Methods Enzymol. 1991;194:732–735. doi: 10.1016/0076-6879(91)94055-h. [DOI] [PubMed] [Google Scholar]
- 41.Rose AB, Broach JR. Methods Enzymol. 1990;185:234–279. doi: 10.1016/0076-6879(90)85024-i. [DOI] [PubMed] [Google Scholar]
- 42.Sherman F. Methods Enzymol. 2002;350:3–41. doi: 10.1016/s0076-6879(02)50954-x. [DOI] [PubMed] [Google Scholar]
- 43.Sikorski RS, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sinclair D, Mills K, Guarente L. Annu Rev Microbiol. 1998;52:533–560. doi: 10.1146/annurev.micro.52.1.533. [DOI] [PubMed] [Google Scholar]
- 45.Sinclair DA. Mech Ageing Dev. 2002;123:857–867. doi: 10.1016/s0047-6374(02)00023-4. [DOI] [PubMed] [Google Scholar]
- 46.Sinclair DA, Guarente L. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- 47.Smith JS, Brachmann CB, Pillus L, Boeke JD. Genetics. 1998;149:1205–1219. doi: 10.1093/genetics/149.3.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tissenbaum HA, Guarente L. Dev Cell. 2002;2:9–19. doi: 10.1016/s1534-5807(01)00098-3. [DOI] [PubMed] [Google Scholar]
- 49.Ward TR, Hoang ML, Prusty R, Lau CK, Keil RL, Fangman WL, Brewer BJ. Mol Cell Biol. 2000;20:4948–4957. doi: 10.1128/mcb.20.13.4948-4957.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu P, Brockenbrough JS, Metcalfe AC, Chen S, Aris JP. J Biol Chem. 1998;273:16453–16463. doi: 10.1074/jbc.273.26.16453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zakian VA, Scott JF. Mol Cell Biol. 1982;2:221–232. doi: 10.1128/mcb.2.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]