

Abstract
GARP/LRRC32 has previously been defined as a marker of activated human regulatory T-cells (Tregs) that is responsible for surface localization of latent TGF-β1. We find that GARP and latent TGF-β1 are also found on mouse Tregs activated via TCR stimulation, but in contrast to human Tregs, GARP is also expressed at a low level on resting Tregs. The expression of GARP can be upregulated on mouse Tregs by IL-2 or IL-4 exposure in the absence of TCR signaling. GARP is expressed at a low level on Tregs within the thymus and Treg precursors from the thymus concomitantly express GARP and Foxp3 upon exposure to IL-2. The expression of GARP is independent of TGF-β1 and TGF-β1 loading into GARP and is independent of furin-mediated processing of pro-TGF-β1 to latent TGF-β1. Specific deletion of GARP in CD4+ T cells results in lack of expression of latent-TGF-β1 on activated Tregs. GARP-deficient Tregs develop normally, are present in normal numbers in peripheral tissues, and are fully competent suppressors of the activation of T conventional cells in vitro. Activated Tregs expressing GARP/latent-TGF-β1 complexes are potent inducers of Th17 differentiation in the presence of exogenous IL-6 and inducers of Treg in the presence of IL-2. Induction of both Th17 producing cells and Treg is preferentially induced by Tregs expressing the latent-TGF-β1/GARP complex on their cell surface rather than by secreted latent-TGF-β1.
Introduction
The three mammalian TGF-β genes encode a translation product consisting of an N-terminal pro-peptide (termed latency-associated peptide [LAP]) and bioactive TGF-β. This product (referred to here as pro-TGF-β) is cleaved intracellularly by furin and LAP remains non-covalently associated with TGF-β to form the small latent complex. In most cells, the small latent complex is covalently attached to latent TGF-β binding proteins (LTBP) prior to secretion. Activated Foxp3+ T regulatory cells (Treg) express a distinct latent-TGF-β binding protein termed GARP/LRRC32 (Glycoprotein A Repetitions Predominant/Leucine-rich repeat-containing protein 32) (1) that is required for surface expression of latent TGF-β1 on human Tregs as well as platelets (2–4). Recombinant latent TGF-β1 was found to directly bind to GARP by both covalent and non-covalent interactions and GARP was critical for tethering latent TGF-β1 to the cell surface. GARP was also shown to outcompete LTBP for binding to latent TGF-β1(5). Latent TGF-β does not have biological activity and the release of active TGF-β from LAP is a critical regulatory step for TGF-β function and signaling. Active TGF-β can be released from the latent-TGF-β/LTBP complex by the action of αV integrins and it has recently been reported that TGF-β is released from the latent TGF-β/GARP complex through similar mechanisms (5).
The contribution of the GARP/latent TGF-β1 complex to the suppressor function of Treg remains unclear. It was originally proposed that ectopic expression of GARP in non-Treg cells induced expression of Foxp3 and endowed the cells with partial suppressive function (1). Other studies claimed that GARP was required for the stability of the human Treg, as lentiviral mediated down-regulation of GARP expression resulted in reduced suppressor function and was associated with down-regulation of Foxp3 (6). Down-regulation of Foxp3 resulted in a concomitant down-regulation of GARP. However, more recent studies have demonstrated that Foxp3 was not essential for the expression of GARP and LAP on human Tregs, as the expression of GARP and LAP were completely normal following siRNA-mediated knocked down of Foxp3. Furthermore, transduction of GARP into Foxp3− T cells allowed for the surface expression of LAP, but no expression of Foxp3 (2). The in vitro suppressive function of Tregs with complete siRNA-mediated knock down of either GARP or TGF-β1 was only modestly reduced.
The role of GARP in Treg function has thus far been analyzed with human Treg. Here, we describe the expression of the GARP/latent TGF-β1 complex by mouse Treg. We find that GARP is expressed at low levels on resting Treg and that its expression is rapidly upregulated via TCR stimulation. Surface expression of GARP is subsequently followed by the surface expression of latent TGF-β1. Upregulation of GARP expression can also be induced by culture of Tregs in the presence of IL-2 and IL-4. Expression of GARP is not dependent upon the expression of TGF-β1, as it is retained in TGF-β1-deficient Tregs. In contrast to some of the early studies on GARP and its potential role in Treg suppressor function, GARP-deficient Tregs developed normally and were competent suppressors of T-cell proliferation in vitro. Lastly, we find that activated mouse Treg that express the GARP/latent-TGF-β1 complex on their cell surface are potent inducers of both Th17 differentiation in the presence of IL-6 and Treg differentiaton in the presence of IL-2. Induction of Th17 producing cells and Foxp3+ Treg is preferentially induced by Tregs expressing the latent-TGF-β1/GARP complex on their cell surface rather than by secreted latent-TGF-β1.
Materials and Methods
Mice
C57BL/6 and B10.A mice were purchased from DCT. Foxp3-GFP, OVA-specific TCR transgenic OT-II (CD45.1, Rag1−/−), Hy-peptide-specific TCR transgenic Marilyn (CD45.1, Rag2−/−), and PCC-Specific TCR transgenic 5CC7 (CD45.1, Rag2−/−) mice were obtained by the National Institute of Allergy and Infectious Diseases (NIAID) and were maintained by Taconic Farms (Germantown, NY) under contract by NIAID. OT-II mice were obtained from Taconic Farms and bred to Foxp3-GFP mice to generate OT-II Foxp3-GFP mice. TGF-β1fl/fl mice (7) were generously provided by Dr. Ming Li (Sloan-Kettering Memorial Institute). Furinfl/fl mice (8) were generously provided by Dr. John O’Shea. GARP (LRRC32)fl/fl mice, which have not been previously described, were generated in the laboratory of Dr. Hodaka Fujii in collaboration with Dr. Derya Unutmaz and described below. CD4-CRE mice were purchased from Taconic Farms. All animal protocols used in this study were approved by the NIAID Animal Care and Use Committee.
Generation of GARP (LRRC32) Floxed Mice
To generate mice harboring a GARP allele flanked by loxP sites, a targeting vector was constructed with a 4.94 kb 5′ homology region, a 2.21 kb 3′ homology region, a neo cassette (3′-5′ orientation) flanked with FRT and loxP sites, and a fragment in which exon 1 was flanked by loxP sequences (Supplemental Fig. 1A–E). 10 μg of the targeting vector was linearlized by Not I and electroporated into iTL1 BA (CA57BL/6 × 129/SvEv) hybrid embryonic stem cells. After selection with G418 antibiotic, surviving clones were expanded for PCR and Southern blot analysis. DNA was digested with BamHI, and electrophoretically separated on a 0.8% agarose gel. After transfer to a nylon membrane, the digested DNA was hybridized with a probe targeted against the 3′ external region. DNA from C57Bl/6 (B6), 129/SvEv (129), and BA1 (C57Bl/6 × 129/SvEv) (Hybrid) mouse strains were used as wild type controls (Supplemental Fig. 1F). The expected sizes are: WT, 5.1 kbp; KO, 4.6 kbp. The probe used for Southern blot analysis was amplified by genomic PCR. The sequence of the probe (527 bp) is listed in Supplemental Figure 1G. After karyotyping, the positive clones were injected into C57BL/6 blastocysts to generate chimeric mice.
Cell isolation and flow cytometry
For purification of dendritic cells, mouse spleens were fragmented and digested for 30 min at 37°C in the presence of liberase blendzyme II (Roche) and DNase (2 μg/ml) (Roche) in complete medium (modified RPMI 1640 supplemented by 10% FBS HyClone, 50 μM 2-ME (Sigma-Aldrich), 1% sodium pyruvate, 1% nonessential amino acids, 1% HEPES, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine. They were labeled with anti-CD11c beads and purified on the AutoMACS Cell Separator (Miltenyi Biotec). T-cells from pooled lymph nodes and/or spleens were isolated using CD4-beads. CD4+Foxp3+ Treg and Foxp3− conventional T-cells (Tconv) were sorted from the pooled lymph nodes of Foxp3-GFP mice and OT-II Foxp3-GFP mice.
CD4+CD25+ Treg and CD4+CD25- Tconv were also sorted. All cell sorting was performed on FACSAria flow cytometers (BD Biosciences). Single cell suspensions were stained using the following antibodies according to the manufacturer’s protocol: Anti-mouse CD45.1 (A20), CD45.2 (104), CD4 (RM4-5), CD8 (53-6.7), Foxp3 (FLK-16s), GARP (YGIC86), IL-17A (eBio17B7), CD25 (PC61.5), RORγt (AFKJS-9), and T-bet (eBio4B10) (all from eBiosciences, San Diego, CA). Purified mouse anti-mouse LAP clone TW7-16B4(9) was generously provided by Howard Weiner (Harvard Medical School). The LAP antibody was labeled with SureLight-APC at Columbia Biosciences (Columbia, MD). Unlabeled cell-culture grade anti-LAP clone TW7-20B9 was also obtained from Biolegend (San Diego, CA). For staining of Foxp3, Tbet, or RORγt, cells were fixed and permeabilized using the Foxp3 fixation/permeabilization staining kit (eBiosciences). For staining of cytokines, cells were fixed and permeabilized using the Cytofix/Cytoperm kit from BD Biosciences.
Cytokines and other reagents
IL-4, IL-6, IL-7, IL-9, IL-15, IL-21, and TNF-α were all purchased from Peprotech and used at 10ng/ml. Recombinant human active TGF-β, latent TGF-β1, and recombinant LAP were all purchased from R&D.
In vitro T cell stimulation and differentiation
To induce Th17 differentiation, sorted Treg were activated and expanded using plate-bound anti-CD3 (1μg/well, 24-well plate) with IL-2 (100U/ml) for 2–3 days, followed by overnight culture with IL-2 alone and were generally greater than 90–95% Foxp3+. Tregs were washed and mixed 1:1 with naïve CD45.1+ OT-II cells (RAG1−/−) or CD45.1+ Marilyn cells (RAG2−/−) in the presence of recombinant mouse IL-6 (10ng/ml) and stimulated with plate-bound anti-CD3 for 4 days. In some experiments, T-cells were stimulated with DC plus soluble anti-CD3 (1μg/ml), OVA peptide (323-33), or Dby HY peptide (Anaspec, Marilyn specific). Control Th17 differentiation was performed by stimulating Foxp3− T cells in the presence of IL-6 (10ng/ml), recombinant human TGF-β1 (5ng/ml), with or without the neutralizing anti-IL-2 mAb (Clone S4B6). Differentiated T-cells were stimulated with the Cell Stimulation Cocktail and Protein Transport Inhibitors (eBioscience).
To induce Foxp3 expression in vitro, CD4+ T cells from 5CC7 (CD45.1, RAG2−) mice were stimulated with soluble anti-CD3 (1μg/ml), IL-2, TGF-β1 (5ng/ml) and irradiated T-depleted (Thy1.1) spleen cells. In other experiments, naïve cells (CD4+CD25− CD62L+CD44low) from GARP T cell conditional knockout mice were stimulated with plate-bound anti-CD3, IL-2, and TGF-β1 for 4 days.
In some experiments, pre-activated Tregs were used to induce Foxp3+ T-cells from naïve CD4+ T-cells. In these experiments, pre-activated Tregs were washed and mixed 1:1 with naïve CD45.1+ OT-II cells (RAG1−/−) in the presence of recombinant human IL-2 (100U/ml) and stimulated with soluble anti-CD3 (1μg/ml) and DCs for 4 days.
In vitro suppression assay
In vitro suppression assays were performed as previously described(10).
In vivo iTreg differentiation
In vivo iTreg differentiation was performed as previously described (11). Briefly, CD4+ cells (106) from 5CC7 (CD45.1 Rag2−/−) mice were transferred I.V. to normal B10.A recipients, which were then injected 24h later with 0.5μg of MCC peptide I.V. GARP and Foxp3 expression were assessed 12–14 days post-transfer.
IL-2 Immune Complex Treatment
C57BL/6 mice were treated with IL-2 immune complexes as previously described (12). Briefly, IL-2/anti–IL-2 mAb complexes were prepared by mixing recombinant murine IL-2 (1 μg; PeproTech) with JES6-1 (5 μg) at the optimal 1:2 molar ratio and incubated for 10 minutes at room temperature. Immune complexes were then diluted in PBS and injected I.P. on days 0, 1, and 2. Expansion of Tregs was approximately 3x in the spleen (~45% of CD4+ T-cells) by day 4.
mRNA Isolation, cDNA production and Real-Time PCR
Tregs were stimulated overnight with various γc-chain associated cytokines and then subjected to RNA extraction using TRIzol reagent. The contaminating DNA was then removed by DNase I treatment. ThermoScript RT-PCR system (Invitrogen Life Technologies) was used to generate cDNA from RNA by using oligo(dT)20 primers. Real-time PCR was conducted with the ABI Prism7900HT, using the Kapa Probe Fast Universal qPCR Kit and TaqMan Probes for GAPDH and LRRC32 (GARP).
Results
Expression of GARP and latent TGFβ-1 by mouse Treg
GARP (LRRC32) has previously been described to be a marker of activated human Treg and serves as a receptor for latent TGFβ-1 (1–4). To further analyze the expression and function of the GARP/latent TGFβ-1 complex in vitro and in vivo, we initially performed a detailed study of its expression on mouse T cells. We first analyzed unseparated CD4+ T cells from the lymph nodes of C57BL/6 mice for expression of Latency Associated Peptide (LAP) of latent TGF-β1 and GARP. Fresh CD4+Foxp3+ cells expressed a low level of GARP that increased upon stimulation with anti-CD3 and IL-2 (Fig. 1A). A small percentage of the GARP+ Tregs co-expressed LAP under resting conditions. While nearly all Foxp3+ Tregs maximally upregulated GARP expression after 24h of stimulation, co-expression of latent-TGF-β1 (LAP) was not fully gained until 48–72h of stimulation. GARP was not detected on CD4+Foxp3− cells under resting conditions, but it was detectable on a small subpopulation after long periods of stimulation in vitro. Importantly, LAP was almost exclusively expressed on GARP-expressing Foxp3+ Tregs, but was also detected on the minor population of Foxp3− T cells that expressed GARP after 72h of stimulation. GARP could also be detected on the minor Foxp3+ (~0.5%) population of CD4−CD8+ T cells.
FIGURE 1.

Expression of GARP and latent TGF-β1 by Treg. (A) GARP and LAP staining of freshly isolated lymph node cells gated on CD4+Foxp3+ and CD4+Foxp3− or CD4-enriched cells stimulated for 24, 48, or 72 h with plate-bound anti-CD3+IL-2. Quadrants are set based on background staining from isotype controls throughout. (B) Sorted CD4+Foxp3-GFP+ cells were immediately stained for GARP (Fresh) or cultured in the presence of anti-IL-2 alone or anti-IL-2 plus IL-2 (human), IL-4, IL-7, IL-9, IL-15, IL-21, or TNF-α, and GARP was measured by FACS. (C) IL-2/anti–IL-2 mAb complexes were injected I.P. on days 1–3. Splenocytes were stained for GARP/LAP on day 5. Gated on CD4+Foxp3+ cells. (D) Freshly isolated lymph node cells gated on CD4+Foxp3+ cells stained with isotype controls, surface stained for GARP/LAP, stained for GARP/LAP before and after fixation/permeabilization, or blocking of GARP/LAP with 10x unlabeled antibody prior to fixation/permeabilization, followed by intracellular staining for GARP/LAP.
In preliminary studies, we noted that culturing Tregs overnight in media alone resulted in upregulation of GARP and GARP expression was further enhanced in media supplemented with IL-2 alone (Fig. 1B). We therefore investigated whether other γc-chain signaling cytokines or TNF-α could also enhance the expression of GARP. Sorted CD4+Foxp3+ T-cells were cultured overnight in media supplemented with IL-2 (human), IL-4, IL-7, IL-9, IL-15, IL-21, or TNF-α in media supplemented with anti-mouse IL-2. Both IL-2 and IL-4 were equally capable of enhancing GARP expression, while IL-7 and IL-15 enhanced it to a lesser extent (Fig. 1B). All others cytokines had minimal, if any effect on GARP expression. Furthermore, IL-2 and IL-4 stimulation enhanced the expression on GARP mRNA (Supplemental Fig. 2A) indicating that the enhanced cell surface expression of GARP/LAP also involved de novo synthesis. To investigate if IL-2 was capable of enhancing GARP expression on Tregs in vivo, IL-2 immune complexes (JES6-1:IL-2) were administered i.p. for 3 days and GARP/LAP was measured on day 5. These complexes have previously been described to direct IL-2 to IL-2Rα-expressing cells (CD25), resulting in considerable expansion of Tregs (12). We found that these complexes not only expanded the Treg population, but also resulted in enhanced GARP/LAP expression in vivo (Fig. 1C).
As we found that culturing Tregs overnight in media alone or in IL-2/IL-4 without TCR stimulation resulted in a measurable increase in the surface expression of GARP, we investigated whether Tregs have intracellular stores of GARP. We found that staining of fresh Tregs prior to and after fixation/permeabilization resulted in significant increases in the percent positive and MFI of both GARP (MFI 152 to 327) and LAP (MFI 87.8 to 177) and increases in the percentage of LAP/GARP co-expressers, indicating that there are intracellular stores of GARP and LAP prior to activation (Fig. 1D). Pre-blocking the cell surface with unlabeled GARP and LAP antibodies prior to fixation and staining allowed us to selectively visualize the considerable contribution of intracellular GARP/LAP.
The expression of GARP was investigated in various lymphoid tissues, including the peripheral lymph nodes (pLNs), mesenteric lymph nodes (mLNs), spleen, Peyer’s patches, and thymus. We found that GARP was expressed at similar levels on resting Treg in the pLNs, mLNs, and spleen (Supplemental Fig. 2B, C). Similarly, the co-expression of LAP and GARP was similar in these locations (Supplemental Fig. 2C). In contrast, Tregs in the thymus and Peyer’s patches expressed much lower levels of GARP and GARP/LAP complexes (Supplemental Fig. 2B, C and Fig. 2A).
FIGURE 2.

Expression of GARP is gained in the thymus upon expression of Foxp3. (A) Staining of GARP on CD4+Foxp3+ cells from pooled lymph nodes (left panel) or CD4+CD8−Foxp3+ cells from the thymus of 6 week old C57BL/6 mice. (B) CD4SPFoxp3−CD25hi cells were sorted from 6–8 week old Foxp3-GFP knock-in mice, and then cultured overnight without supplementing any cytokines or supplementing IL-2 (100U/ml). (C) Staining for GARP gating on CD4+Foxp3− and CD4+Foxp3+ cells from IL-2 cultured CD4SPFoxp3−CD25hi thymocytes from (B).
Expression of GARP is acquired in the thymus simultaneously with Foxp3
Although GARP and the GARP/LAP complex were expressed at very low levels on thymic Tregs, we determined at what developmental stage thymic Tregs acquired GARP expression. We sorted CD4+CD8−Foxp3−CD25hi Treg precursors from thymi of Foxp3-GFP knock in mice. As reported previously (15), culture of these precursors with IL-2 resulted in induction of Foxp3 expression in a substantial proportion of these cells. GARP expression was also induced by culture in IL-2 and was selectively expressed on the Foxp3+ subpopulation (Fig. 2B, C).
GARP is expressed by in vitro and In vivo generated iTregs
iTregs were induced in vitro from TCR transgenic 5CC7 cells on a RAG−/− background by activation with soluble anti-CD3 and irradiated T-cell depleted spleen cells in the presence of TGF-β1 and IL-2. A high percentage of the cells expressed Foxp3 (Fig. 3A). Surprisingly, both Foxp3+ and Foxp3− cells expressed similar levels of GARP and LAP. This result differs markedly from our previous studies on human T cells induced to express Foxp3 in vitro under similar culture conditions that were GARP and LAP negative (2).
FIGURE 3.
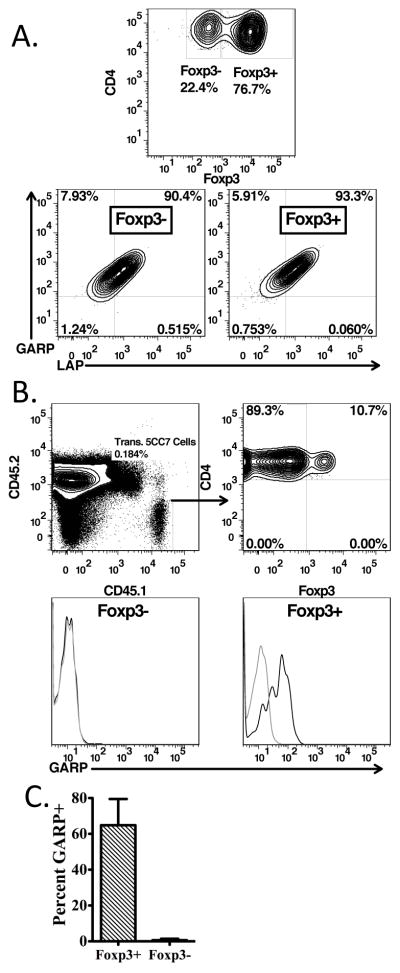
GARP is expressed by in vivo and in vitro generated iTregs. (A) CD4+ Rag2−/− 5CC7 T-cells were cultured with soluble anti-CD3 (1μg/ml), IL-2 (100U/ml), and human recombinant TGF-β1 with irradiated T-depleted splenocytes for 4 days, then stained for CD4, GARP, LAP, and Foxp3. Left panel is gated on CD4+ cells, while center and right panels are gated on the CD4+Foxp3− and CD4+Foxp3+ populations, respectively. (B, C) 106 CD4+CD45.1+Rag2−/− 5CC7 T cells were transferred into B10.A mice that were then injected i.v. with 0.5μg MCC peptide the following day. (B) 12–14 days post-transfer, CD4-enriched cells from lymph nodes and spleen were stained for CD4, CD45.1, CD45.2, Foxp3, and GARP (upper panels). GARP expression on the gated Foxp3− and Foxp3+ cells is shown in the lower panels. (C) Average percent GARP+ of transferred CD4+CD45.1+ from 5 mice. Error bars represent of the mean of 5 mice +/− SD.
In order to examine the expression of GARP/LAP on Tregs induced in vivo, we used a previously established system in which pigeon cytochrome-C (PCC) specific transgenic (5CC7) T-cells on a RAG2−/− background are transferred into B10.A mice, followed by a low dose of MCC peptide(11). Approximately 2 weeks post-transfer, 5CC7 cells were assessed for their expression on Foxp3 and GARP. 10–30% of 5CC7 cells gained expression of Foxp3, and the majority of Foxp3+ population expressed GARP (Fig. 3B, C). This result differs from the studies on in vitro generated iTregs where both the Foxp3+ and Foxp3− populations expressed GARP/LAP. It remains possible that activation in the presence of IL-2 and TGF-β1 may be sufficient to induce the expression of GARP in vitro, whereas the expression of Foxp3 appears to be required to stably express GARP in vivo.
Expression of GARP is independent of TGF-β1 and loading of TGF-β1 is independent of furin processing
As GARP is expressed concomitantly with Latent-TGF-β1, we investigated the dependence of GARP expression on the expression of TGF-β1. We crossed TGF-β1fl/fl mice to CD4-CRE (7) mice. Unstimulated CD4+Foxp3+ cells from homozygous floxed CRE+ mice expressed GARP at levels similar to CRE− littermates, but did not express latent TGF-β1 (Fig. 4A, left panels). Furthermore, stimulation of the TGF-β1−/− CD4+ T-cells with anti-CD3 and IL-2 in serum-free media resulted in upregulation of GARP to levels similar to those of the CRE− littermates, but no expression of latent TGF-β1 (Fig. 4A, right panels). To determine if TGF-β1−/− CD4+ T-cells could bind free latent TGF-β1, unfractionated CD4+ T-cells were activated for 48h, incubated with human latent TGF-β1, washed and then stained for human LAP (Fig. 4B). Expression of LAP could readily be detected on Foxp3+ T cells, but not Foxp3− T cells.
FIGURE 4.

Expression of GARP/LAP on TGF-β1-, Furin-, and GARP-deficient Tregs. Staining of GARP/LAP CD4+Foxp3+ gated cells from lymph nodes or on CD4-enriched cells after activation with plate-bound anti-CD3 and IL-2 in serum-free media. Cells are from (A.) tgfb1fl/fl x CD4-CRE, (C) furinfl/fl x CD4-CRE, and (D) lrrc32fl/fl x CD4-CRE (GARP deficient). (B.) CD4+ T-cells from tgfb1fl/fl x CD4-CRE were stimulated as in (A) for 48 hours, then incubated in fresh media with (black lines) or without (grey lines) recombinant human latent TGF-β1 for 30′ at 37°C, then thoroughly washed and stained with anti-human LAP. Gating on CD4+Foxp3- cells (left panel) or CD4+Foxp3+ cells (right panel).
TGF-β1 is produced as an inactive precursor polypeptide (pro-TGF-β) that is proteolytically processed by the pre-protein convertase, furin, in order to produce latent TGF-β1, which must then be further processed in order to produce biologically active TGF-β1 (13). To determine if furin processing of pro-TGF-β1 is required for binding to GARP, furinfl/fl mice were crossed to the CD4-CRE mice (8, 14). CRE recombinase mediated deletion of furin exon 1 in CD4+ T-cells was confirmed by PCR (data not shown). Unstimulated CD4+Foxp3+ cells from the homozygous furinfl/fl-CRE+ mice co-expressed GARP and latent-TGF-β1 to at levels similar to the CRE− littermates (Fig. 4C, left panels). Furthermore, stimulation of furinfl/fl T cells from Cre+ mice with anti-CD3 and IL-2 in serum-free media resulted in upregulation of the GARP/latent-TGF-β1 complex to levels similar to those seen on the CRE− littermates (Fig. 4C, right panels). Therefore, furin processing of pro-TGF-β1 to latent TGF-β1 is not required for loading onto GARP (5).
To confirm previously published results on human T cells that GARP is required for the surface expression of latent-TGF-β1 in the murine system, we crossed GARPfl/fl mice to CD4-CRE mice. As these mice have not been previously described, we examined Treg and Tconv development. The percentages of thymocyte subpopulations (Supplemental Fig. 3A, upper panels) including the percentages of Foxp3+ cells within the CD4 single positive population (lower panels) were identical in CRE+ and CRE− littermates. The percentages of CD4+Foxp3− and CD4+Foxp3+ T cells in lymph nodes and spleen were also identical in CRE+ and CRE− littermates (Supplemental Fig. 3B). In addition, the level of CD25 expression was similar on Foxp3+ T cells from CRE+ and CRE− littermates (Supplemental Fig. 3C). Therefore, it does not appear that expression of GARP is required for the normal development of Tregs in the thymus or for their maintenance in the periphery. Fresh CD4+Foxp3+ cells from GARPfl/fl- CRE+ mice did not express GARP, nor did they have any surface latent TGF-β1 expression, as compared to the CRE− littermates (Fig. 4D, left panels). Furthermore, stimulation with anti-CD3 and IL-2 in serum free media resulted in the upregulation of GARP and latent TGF-β1 only in CRE− littermates (Fig. 4D, right panels), thus confirming previous reports (2–4) that GARP is required for surface expression of TGF-β1 on Tregs.
In order to determine if GARP is required for in vitro differentiation of iTregs, naïve CD4+CD25−CD62LhiCD44low T cells from GARPfl/fl− CRE+ and CRE− littermates were stimulated with plate-bound anti-CD3 for four days in the presence of TGF-β1 and IL-2. No difference was observed in the induction of Foxp3+cells in the presence and absence of GARP expression (Supplemental Fig. 3D).
Expression of GARP or TGF-β1 is not required for Treg suppression in vitro
To determine the ability of GARP−/− Tregs to act as suppressor cells in vitro, we sorted CD4+CD25hi cells from GARPfl/fl CD4-CRE+ and CD4-CRE− mice. Wild type and GARP−/− Tregs were equally effective in suppressing the proliferation of CD4+CD25− responder cells (Fig. 5A). Similarly, Tregs from TGF-β1−/− mice were as effective as Tregs from wild type mice in suppressing responder cell proliferation (Fig. 5B), as reported previously (9). Furthermore, Treg from GARP−/− mice were similar to Treg from wild type mice in that they were nonresponsive to TCR stimulation alone, but proliferated vigorously to stimulation with anti-CD3 and IL-2 (Fig. 5C). Thus, neither cell surface associated nor secreted TGF-β1 plays a major role in the standard in vitro assays for Treg function.
FIGURE 5.

Neither GARP nor TGF-β1 contributes to Treg suppression. (A) Wild type T effector (CD4+CD25−, 5x104) cells were activated with soluble anti-CD3 and irradiated T-cell depleted splenocytes in the presence or absence of wild type or GARP−/− Tregs from lrrc32fl/fl x CD4-CRE mice at an effector to Treg ratio of 16:1 to 1:2 for 3 days. Cultures were pulsed with [3H]-thymidine for the last 6 hours of culture. (B) As in (A), except using wild type or TGF-β1−/− Tregs from tgfb1fl/fl x CD4-CRE mice. (C) Wild type or GARP−/− Tregs from lrrc32fl/fl x CD4-CRE were stimulated with soluble anti-CD3 and irradiated T-cell depleted splenocytes in the presence or absence of IL-2. Cultures were pulsed as in (A) after 3 days.
Treg-mediated Th17 differentiation and Treg-mediated Foxp3 induction are primarily mediated by GARP-associated TGF-β1
It has previously been reported that in the presence of IL-6, activated Tregs are capable of driving differentiation of naïve CD4+ T-cells to Th17 cells(15). To directly determine the contributions of secreted versus cell associated TGF-β1 to the induction of Th17 cells, we activated sorted Tregs from Foxp3-GFP mice for 72h so that they would express high levels of the GARP/LAP complex on the cell surface. The activated Tregs were then co-cultured with congenically marked naïve T-cells in the presence of IL-6, and the co-cultures were restimulated with plate-bound anti-CD3 for 4 days. Upon restimulation, approximately ~40% of the naïve cells expressed IL-17A upon restimulation with PMA and ionomycin (Fig. 6A). Surprisingly, this level of IL-17 induction was similar to that seen when the naïve responder cells were stimulated in the presence of TGF-β1, IL-6, and anti-IL-2, conditions we have previously shown to be optimal for induction of Th17 cells (16). Furthermore, these cells lacked the ability to produce IL-10, similar to those stimulated in the presence of TGF-β1, IL-6, and anti-IL-2 and in contrast to those stimulated in the presence of TGF-β1 and IL-6 (Supplemental Fig. 4A). Additionally, these cells expressed RORγt, while lacking the expression of Foxp3 and T-bet. This is in contrast to those stimulated in the presence of TGF-β1 and IL-6, which not only expressed RORγt, but also expressed considerable Foxp3 (Supplemental Fig. 4B).
FIGURE 6.

Treg-mediated Th17 differentiation requires activation of latent TGF-β1. Naïve CD45.1+ OT-II cells from Rag1−/− mice were activated with plate-bound anti-CD3 in the presence of IL-6 (10ng/ml), TGF-β1 (5ng/ml), with (left) or without (right) anti-IL-2, or IL-6 and pre-activated CD4+Foxp3+ T-cells for 4 days. Cells were reactivated with Cell Stimulation Cocktail and Protein Transport Inhibitors from eBioscience, then stained for CD4, CD45.1, CD45.2, and IL-17A. Percents indicate the percentage IL-17A+cells derived from the CD4+CD45.1+CD45.2− cells in the culture. (B) Th17 cells were generated as in (A) with pre-activated CD4+Foxp3+ T-cells and IL-6 in the presence of antibodies against GARP, LAP (TW7-16B4), TGF-β, or with recombinant human LAP. (C) As in (B), except comparing the ability of two different anti-LAP antibody clones (TW7-16B4 and TW7-20B9) to block Th17 differentiation (Left and Right Panels). Right panel shows averages of an experiment performed in triplicate ± SD.
Induction of Th17 producing cells in the presence of activated Tregs was completely inhibited by the pan-neutralizing TGF-β antibody 11D1 (Fig. 6B) and substantially by recombinant human LAP. Th17 induction was also inhibited by 60–70% by anti-mouse LAP (clone TW7-16B4), but not by a second anti-LAP mAb (TW7-2089) that recognizes an epitope distinct from that recognized by TW7-16B4 (9) (Fig. 6C). Anti-GARP (clone YGIC86) (Fig. 6C) or two different anti-GARP mAbs generated in house had no inhibitory effects on Treg-mediated Th17 induction (data not shown).
These studies suggest that Treg-mediated Th17 differentiation requires their production of TGF-β1 and likely involves conversion of latent TGF-β1 to the active form. It remains possible that inhibitory effects of anti-LAP mAb TW-16B4 are mediated by interfering with conversion of latent to active TGF-β1. In order to determine if the source of the active TGF-β1 needed for Treg-mediated Th17 differentiation is the cell surface associated GARP/Latent-TGF-β1 complex or secreted latent-TGF-β1, we compared the capacity of activated CD4+CD25hi cells from T cell conditional (CD4-CRE+) TGF-β1−/−, Furin −/− and GARP−/− mice to their wild type counterparts (CD4-CRE−) to drive Th17 differentiation. Induction of Th17 cells was completely dependent on T cell produced TGF-β1, as Tregs from TGF-β1−/− mice were unable to drive Th17 differentiation (Fig. 7A). Furthermore, Tregs from Furin−/− mice were also completely unable to drive Th17 differentiation (Fig. 7B), most likely because the TGF-β1 produced by furin−/− Tregs remains in the proprotein form. The induction of Th17 cells in the presence of GARP−/− Tregs was reduced by ~75% demonstrating that the majority of the activated TGF-β1 mediating Th17 induction was derived from the cell surface associated GARP/Latent TGF-β1 complex (Fig. 7C).
FIGURE 7.

Treg-mediated Th17 differentiation and Treg induction require Treg derived TGF-β1 and presentation via GARP. Naïve CD45.1+ OT-II cells from Rag1−/− mice were activated with soluble anti-CD3 (1μg/ml) and dendritic cells in the presence of IL-6 (10ng/ml) and pre-activated CD4+CD25hi Tregs from tgfb1fl/fl x CD4-CRE (A), furinfl/fl x CD4-CRE (B), and lrrc32fl/fl x CD4-CRE (C) mice as in Figure 6. Cells from CRE-negative littermates were used as controls. Percents indicate the percentage IL-17A+ from the CD4+CD45.1+CD45.2− cells within the culture. Bar graphs on right panels indicate the average of duplicates within the experiment ± SD. (D) Naïve CD45.1+ OT-II cells from Rag1−/− mice were activated with soluble anti-CD3 (1μg/ml) and dendritic cells in the presence of IL-2 (100U/ml) and pre-activated CD4+CD25hi Tregs from lrrc32fl/fl x CD4-CRE.
Previously, our group and others described the ability of activated Tregs to drive differentiation of naïve T-cells to express Foxp3 when activated in the presence of IL-2 (17), and that this was dependent upon TGF-β1 produced by Tregs. In order to determine if induction of Foxp3 expression was dependent upon the GARP/Latent TGF-β1 complex, naive T-cells were activated with anti-CD3 in the presence of activated Tregs from CD4-conditional knockouts of GARP or their wildtype counterparts (CD4-CRE−) and IL-2. GARP −/− Tregs were unable to efficiently induce Foxp3-expression in naïve T-cells in comparison to GARP-sufficient Tregs (Fig. 7D).
Treg-mediated Th17 differentiation requires TCR-reactivation
Thus far, both the activated Tregs and the naïve responder cells were simultaneously activated via the TCR for induction of IL-17 producing cells. To determine if TCR reactivation of the activated Tregs was required for induction of Th17 cells, activated polyclonal Tregs were cultured with naïve OT-II CD4+ T cells in the presence of dendritic cells, IL-6, and either OVA323-329, or soluble anti-CD3. No induction of Th17 cells was seen in the presence of OVA323-329 and polyclonal Tregs (Fig. 8A). In contrast, Th17 cells were induced with activated polyclonal Tregs in the presence of anti-CD3. Thus, even though activated Tregs express high levels of the GARP/Latent TGF-β1 complex, they must be reactivated via the TCR to initiate release and activation of latent TGF-β1 required for induction of Th17 cells.
FIGURE 8.

Treg-mediated Th17 differentiation requires TCR-reactivation. (A) Naïve CD45.1+ OT-II cells from Rag1−/− mice were cultured with IL-6 (10ng/ml), dendritic cells, and pre-activated CD4+Foxp3+ T-cells in the presence of soluble anti-CD3 (left), or OVA peptide (323-339) for 4 days. (B) Naïve CD45.1+ Marilyn cells from Rag2−/− mice were cultured with dendritic cells with OVA and HY peptides either with IL-6 alone (left panel) or with IL-6 and pre-activated OT-II CD4+Foxp3+ Tregs (center panels). The right panel was cultured with IL-6, dendritic cells, soluble anti-CD3, and pre-activated OT-II CD4+Foxp3+ Tregs.
To determine if activated Tregs could mediate induction of Th17 cells by a bystander pathway, we cultured activated OT-II Tregs and naïve T cells from congenically marked Marilyn mice that are specific for an HY peptide (Dby) in the presence of both OVA323-329 and Dby (Fig. 8B). As expected, culture of Marilyn T cells with IL-6 alone did not result in Th17 differentiation, whereas culture with IL-6 and OT-II Tregs in combination drove Th17 differentiation. Therefore, Tregs responding to different antigens presented by the same antigen-presenting cells can drive Th17 differentiation from naïve T-cells.
Discussion
Here, we report an analysis of the expression of GARP and the GARP/latent-TGF-β1 complex on mouse Treg and highlight potential functions of the complex on Treg. Resting mouse Treg express low levels of GARP and much of this constitutive expression is not complexed with latent-TGF-β1. The expression of GARP is upregulated by culture of Tregs in media alone in the absence of cytokines and can be further upregulated by addition of IL-2 or IL-4. TCR stimulation induces an initial upregulation of GARP expression that is rapidly followed by detection of the GARP/latent-TGF-β1 complex. A large proportion of this initial “burst” in surface expression of GARP may be due to its release from intracellular stores. We observed that the baseline surface expression of GARP/latent-TGF-β1 varies in different tissues tested, which may be a reflection of the cytokine milieu in different sites or the activation status of the Tregs (Fig. 2, Supplemental Fig. 2). Thymic Treg in general expressed a low, but reproducible, level of GARP consistent with their resting state. When Treg precursors were induced to express Foxp3 by culture in IL-2, the induction of Foxp3 expression was accompanied by GARP expression.
Both in vivo and in vitro generated iTregs expressed the GARP/LAP complex. This result should be contrasted to results of studies on human T cells, which failed to express GARP or the GARP/latent-TGF-β1 complex (2) following in vitro induction of Foxp3 expression by TCR stimulation in the presence of TGF-β1. However, it should be noted that the human Foxp3+ T cells induced in vitro lacked Treg suppressor function and produced IL-2. We also observed that a substantial proportion of mouse T cells in the iTreg generation cultures that did not express Foxp3 expressed the GARP/latent-TGF-β1 complex. In contrast, in our in vivo Treg induction model, GARP/latent-TGF-β1 complexes could only be detected on cells that expressed Foxp3. It is likely that expression of Foxp3 is required for stable expression of GARP/LAP, however in the artificial in vitro situation the presence of TGF-β1 and IL-2 are sufficient to induce their expression. We could only detect very low percentages of Foxp3− GARP/latent-TGF-β1 expressing cells in any lymphoid population studied consistent with other studies. Thus, expression of GARP represents an excellent marker of bona fide Foxp3+ Treg in the mouse although it does not distinguish between thymic-derived from peripherally induced Treg.
Expression of GARP was not dependent upon the expression of latent-TGF-β1, as cells from T cell conditional TGF-β1−/− mice expressed levels of GARP equivalent to those detected on wild type Treg both before and after T cell activation. This result confirms studies in which activated human T cells continued to express GARP after siRNA-mediated knock down of TGF-β1 (2). As was observed with human Tregs, we could reconstitute LAP expression on activated mouse Tregs from TGF-β1−/− mice by incubation with human recombinant latent TGF-β1. We could not detect binding of latent-TGF-β1 to activated Foxp3− T cells. Thus, the GARP/latent-TGF-β1 complex can be generated intracellularly, as well as extracellularly. It remains to be determined what fraction of the GARP/latent-TGF-β1 complex is created by these two mechanisms during T cell activation. It is also possible that free GARP molecules on the cell surface may compete with LTBP for binding of secreted latent-TGF-β1. We could readily detect the GARP/latent-TGF-β1 complex on the surface of activated Tregs from T cell conditional furin deficient mice indicating that both Prolatent-TGF-β1 and furin-processed latent-TGF-β1 could interact with GARP.
Using CD4 conditional knockouts of GARP, we confirm siRNA-mediated knock down studies on human Treg that GARP is absolutely required for the expression of latent TGF-β1 on the cell surface. The contribution of cell surface expressed latent-TGF-β1 in mediating Treg suppression of responder T cell activation has been controversial (1, 2, 4, 6). Following siRNA-mediated knock down of human GARP, a modest reduction of suppressor capacity was observed. However, GARP−/− mouse Tregs were as suppressive as wild type Tregs in mediating suppression in vitro and also maintained their anergic phenotype in that they failed to proliferate in response to stimulation with anti-CD3 alone. Although GARP−/− Tregs can still secrete latent-TGF-β1 as measured by ELISA (data not shown), it is unlikely that this secreted latent-TGF-β1 plays any role in Treg suppression in vitro as the suppressive ability of TGF-β1−/− Tregs is unimpaired (7).
As previous studies suggested that activated Treg could also induce Th17 cells in the presence of IL-6 (15), we explored in depth the role of the GARP/latent-TGF-β1 complex in this in vitro assay. Activation of co-cultures of activated Tregs and naïve responder T cells in the presence of IL-6 resulted in a high efficiency of induction of IL-17 producing cells approximating the number of IL-17 producers observed in cultures of naïve responder cells in the presence of TGF-β1 and IL-6. The induction of IL-17 producing cells was markedly inhibited by recombinant LAP, certain mAbs to LAP, and by anti-TGF-β indicating that activated Tregs released latent-TGF-β1 following reactivation and that the latent-TGF-β1 was then processed to active TGF-β1. Activated Tregs from T cell conditional knockouts of TGF-β1 or of furin failed to induce IL-17 cells indicating that that Treg-derived TGF-β1 was the major source of TGF-β1 in this system. To distinguish whether secreted latent-TGF-β1 or the GARP/latent-TGF-β1 complex was the source of the active TGF-β1 for induction of Th17 cells, we compared wild type and GARP−/− Tregs for their capacity to induce IL-17 producing cells. Surprisingly, wild type Tregs induced 3–4 times more IL-17 producing cells that Tregs from T cell conditional GARP−/− mice. This result strongly suggests that one major function of the GARP/latent-TGF-β1 complex may be to act as a source of active TGF-β1 for induction of Th17 production.
We had previously demonstrated that activation of co-cultures of activated Tregs and naïve T cells resulted in induction of Foxp3 expression and Treg function in a low percentage of responder T cells in a TGF-β1-dependent manner (17). The Tregs induced in this process of infectious tolerance were suppressive in vitro and in vivo and this pathway may represent a mechanism by which Treg maintain tolerance. The major source of TGF-β1 in this system also appears to be from the GARP/latent-TGF-β1 complex, as activated Treg from GARP−/− mice were poor inducers of Foxp3 expression in naïve responder cells. It is likely that in vivo Treg and antigen-specific T cells are stimulated on the surface of the APC in the form of a three cell interaction. Stimulation of the Treg would induce the GARP/latent-TGF-β1 complex which depending on the local milieu (+/−IL-6) would result in induction of Treg or Th17 cells.
Lastly, one must question why Foxp3+ Treg, the most potent suppressor T cell population, would play an important role in the induction of potentially pathogenic Th17 cells. There is little doubt that TGF-β1 is required for induction of pathogenic Th17 cells in vivo as local, but not systemic administration of anti-TGF-β inhibits Th17 cell generation (18) and mice with a T cell specific deletion of TGF-β1 fail to generate Th17 cells are resistant to experimental autoimmune encephalomyelitis (EAE) (7). However, mice with a Treg cell-specific deletion of TGF-β1generated Th17 cells and were susceptible to EAE (19). Conditional deletion of TGF-β1 in Tregs resulted in increased numbers of Tregs in certain lymphoid tissues, but not others, raising the possibility that TGF-β1 produced by Treg cells is specifically required for inhibiting Treg cell proliferation and controlling the size of the Treg pool. One study has suggested that Tregs are critically important for the induction of Th17 differentiation (20), but this study proposed that Treg promote Th17 production by a TGF-β1-independent mechanism related to their capacity to consume IL-2. Although Th17 cells are thought to be pathogenic, some studies demonstrate the existence of non-pathogenic IL-17 producing Th17 cells (21, 22) and it remains possible that Treg-induced Th17 cells are actually non-pathogenic. Furthermore, the development of pathogenic versus non-pathogenic Th17 cells may be contingent on the type of TGF-β present during the initial stage of Th17 differentiation and studies from the Kuchroo laboratory have demonstrated that TGF-β3, but not TGF-β1, is the critical TGF-β isoform required for induction of pathogenic Th17 cells (23). Intriguingly, latent human TGF-β1 and -β2, but not -β3, were capable of forming complexes with human GARP (2) raising the possibility that latent-TGF-β1/GARP complexes may play a unique role in the differentiation of a subset of Th17 cells.
Supplementary Material
Acknowledgments
We would like to thank Julie Edwards, Tom Moyer, and Elina Stregevsky in the National Institute of Allergy and Infectious Diseases Flow Cytometry Section for sorting our cells. We thank Howard Weiner for providing the anti-mouse LAP antibody TW7-16B4 and John O’Shea for providing Furin floxed mice.
Abbreviations used in this article
- GARP/LRRC32
Glycoprotein A Repetitions Predominant/Leucine-rich repeat-containing protein 32
- LAP
Latency Associated Peptide
- Treg
Regulatory T-cell
- Treg
Regulatory T-cell
- iTreg
Induced Regulatory T-cell
- MCC
Moth Cytochrome C
- LTBP
latent TGF-β binding proteins
Footnotes
Supported by funds from the Intramural Program of the National Institute of Allergy and Infectious Diseases Diseases (E.M.S and Grant-in-Aid for Scientific Research (C) (#23590569) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, Osaka Clinical Immunology Foundation, Takeda Science Foundation, Senshin Medical Research Foundation, Japan Rheumatism Foundation, Itoh Chubei Foundation (H.F.).
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Wang R, Wan Q, Kozhaya L, Fujii H, Unutmaz D. Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PLoS One. 2008;3:e2705. doi: 10.1371/journal.pone.0002705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A. 2009;106:13445–13450. doi: 10.1073/pnas.0901944106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang R, Kozhaya L, Mercer F, Khaitan A, Fujii H, Unutmaz D. Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A. 2009;106:13439–13444. doi: 10.1073/pnas.0901965106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stockis J, Colau D, Coulie PG, Lucas S. Membrane protein GARP is a receptor for latent TGF-beta on the surface of activated human Treg. Eur J Immunol. 2009;39:3315–3322. doi: 10.1002/eji.200939684. [DOI] [PubMed] [Google Scholar]
- 5.Wang R, Zhu J, Dong X, Shi M, Lu C, Springer TA. GARP regulates the bioavailability and activation of TGFbeta. Mol Biol Cell. 23:1129–1139. doi: 10.1091/mbc.E11-12-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Probst-Kepper M, Geffers R, Kroger A, Viegas N, Erck C, Hecht HJ, Lunsdorf H, Roubin R, Moharregh-Khiabani D, Wagner K, Ocklenburg F, Jeron A, Garritsen H, Arstila TP, Kekalainen E, Balling R, Hauser H, Buer J, Weiss S. GARP: a key receptor controlling FOXP3 in human regulatory T cells. J Cell Mol Med. 2009;13:3343–3357. doi: 10.1111/j.1582-4934.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 2007;26:579–591. doi: 10.1016/j.immuni.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 8.Roebroek AJ, Taylor NA, Louagie E, Pauli I, Smeijers L, Snellinx A, Lauwers A, Van de Ven WJ, Hartmann D, Creemers JW. Limited redundancy of the proprotein convertase furin in mouse liver. J Biol Chem. 2004;279:53442–53450. doi: 10.1074/jbc.M407152200. [DOI] [PubMed] [Google Scholar]
- 9.Oida T, Weiner HL. TGF-beta induces surface LAP expression on murine CD4 T cells independent of Foxp3 induction. PLoS One. 5:e15523. doi: 10.1371/journal.pone.0015523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188:287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gottschalk RA, Corse E, Allison JP. TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J Exp Med. 207:1701–1711. doi: 10.1084/jem.20091999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311:1924–1927. doi: 10.1126/science.1122927. [DOI] [PubMed] [Google Scholar]
- 13.Dubois CM, Blanchette F, Laprise MH, Leduc R, Grondin F, Seidah NG. Evidence that furin is an authentic transforming growth factor-beta1-converting enzyme. Am J Pathol. 2001;158:305–316. doi: 10.1016/s0002-9440(10)63970-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pesu M, Watford WT, Wei L, Xu L, Fuss I, Strober W, Andersson J, Shevach EM, Quezado M, Bouladoux N, Roebroek A, Belkaid Y, Creemers J, O’Shea JJ. T-cell-expressed proprotein convertase furin is essential for maintenance of peripheral immune tolerance. Nature. 2008;455:246–250. doi: 10.1038/nature07210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178:6725–6729. doi: 10.4049/jimmunol.178.11.6725. [DOI] [PubMed] [Google Scholar]
- 16.Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O’Shea J. Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 17.Andersson J, Tran DQ, Pesu M, Davidson TS, Ramsey H, O’Shea JJ, Shevach EM. CD4+ FoxP3+ regulatory T cells confer infectious tolerance in a TGF-beta-dependent manner. J Exp Med. 2008;205:1975–1981. doi: 10.1084/jem.20080308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veldhoen M, Hocking RJ, Flavell RA, Stockinger B. Signals mediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat Immunol. 2006;7:1151–1156. doi: 10.1038/ni1391. [DOI] [PubMed] [Google Scholar]
- 19.Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO. Autocrine transforming growth factor-beta1 promotes in vivo Th17 cell differentiation. Immunity. 34:396–408. doi: 10.1016/j.immuni.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y, Haines CJ, Gutcher I, Hochweller K, Blumenschein WM, McClanahan T, Hammerling G, Li MO, Cua DJ, McGeachy MJ. Foxp3(+) regulatory T cells promote T helper 17 cell development in vivo through regulation of interleukin-2. Immunity. 34:409–421. doi: 10.1016/j.immuni.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 21.Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY, O’Connor W, Jr, Rongvaux A, Van Rooijen N, Haberman AM, Iwakura Y, Kuchroo VK, Kolls JK, Bluestone JA, Herold KC, Flavell RA. Control of TH17 cells occurs in the small intestine. Nature. 475:514–518. doi: 10.1038/nature10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Y, Weiner J, Liu Y, Smith AJ, Huss DJ, Winger R, Peng H, Cravens PD, Racke MK, Lovett-Racke AE. T-bet is essential for encephalitogenicity of both Th1 and Th17 cells. J Exp Med. 2009;206:1549–1564. doi: 10.1084/jem.20082584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, Kuchroo VK. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 13:991–999. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.