

Background: USP14 mediates protein degradation via its deubiquitination activity as well as controlling proteasome gate opening.
Results: USP14 promotes I-κB deubiquitination and degradation, therefore increasing cytokine release.
Conclusion: USP14 targets NF-κB pathway via association with RelA and regulation of I-κB stability.
Significance: This study provides a new target to mediate transcriptional activity and cytokine release.
Keywords: Cytokine, Deubiquitination, NF-κB Transcription Factor, Protein degradation, Protein Phosphorylation, USP14
Abstract
The ubiquitin-proteasome system is the major pathway of non-lysosomal intracellular protein degradation, playing an important role in a variety of cellular responses including cell division, proliferation, and apoptosis. Ubiquitin-specific protease 14 (USP14) is a component of proteasome regulatory subunit 19 S that regulates deubiquitinated proteins entering inside the proteasome core 20 S. The role of USP14 in protein degradation is still controversial. Several studies suggest that USP14 plays an inhibitory role in protein degradation. Here, in contrast, overexpression of USP14 induced I-κB degradation, which increased cytokine release in lung epithelial cells. Overexpression of HA-tagged USP14 (HA-USP14) reduced I-κB protein levels by increasing the I-κB degradation rate in mouse lung epithelial cells (MLE12). I-κB polyubiquitination was reduced in HA-USP14-overexpressed MLE12 cells, suggesting that USP14 regulates I-κB degradation by removing its ubiquitin chain, thus promoting the deubiquitinated I-κB degradation within the proteasome. Interestingly, we found that USP14 was associated with RelA, a binding partner of I-κB, suggesting that RelA is the linker between USP14 and I-κB. Lipopolysaccharide (LPS) treatment induced serine phosphorylation of USP14 as well as further reducing I-κB levels in HA-USP14-overexpressed MLE12 cells as compared with empty vector transfected cells. Further, overexpression of HA-USP14 increased the LPS-, TNFα-, or Escherichia coli-induced IL-8 release in human lung epithelial cells. This study suggests that USP14 removes the ubiquitin chain of I-κB, therefore inducing I-κB degradation and increasing cytokine release in lung epithelial cells.
Introduction
Intracellular protein degradation in the proteasome system involves covalent conjugation of ubiquitin chain(s) to the substrates that are recognized and degraded within the proteasome (1, 2). The proteasome consists of a barrel-shaped multimeric protein complex, the 20 S proteasome core particle (CP),2 the 19 S regulatory particle (RP), and numerous deubiquitinating enzymes (DUBs) (3–6). The 20 S CP contains three major proteolytic activities, including chymotrypsin-like, trypsin-like, and caspase-like activities, and functions as the catalytic center that cleaves peptides on the carboxyl side of hydrophobic, basic, and acidic amino acids (4–6). The 20 S CP is capped at each end by the 19 S RP, which controls channel opening and the remodeling of substrate proteins (4–6). DUBs disassemble ubiquitin chains to regulate protein stability. Three major DUBs, including RPN11, UCH37, and USP14, are associated with mammalian 19 S RP (4–6).
USP14, one of ubiquitin-specific peptidases, disassembles the ubiquitin chain from its substrate-distal tip (7, 8). The activity of USP14 is regulated by its interaction with 19 S RP because USP14 by itself remains relatively inactive; however, activity is greatly increased when co-incubated with 19 S RP (8). The USP14 mutant (USP14ΔUBL), which lacks the ability to bind to 19 S RP, lost its activity in the presence of the 19 S RP (8). Loss of a yeast USP14 ortholog Ubp6 rapidly reduced the free ubiquitin pool (9), suggesting a role of USP14 in regulation of ubiquitin recycling. In addition to its deubiquitination ability, USP14 regulates gate opening of 20 S CP (10). USP14 has been shown to function as an inhibitor of the proteasome because it was found to slow substrate degradation (11). The small molecule USP14 inhibitor IU1 enhanced proteasome activity and degradation of several substrates such as Tau and ataxin-3 (11).
Growing evidence suggests that USP14 regulates various biological responses including neural function (12, 13), chemokine signaling (14), and tumorigenesis (15, 16). The ataxia mutation (axJ) mice, a loss of Usp14 function strain, exhibited a reduced growth rate, ataxia, and hindlimb muscle wasting (12, 17). The neuron-specific overexpression of Usp14 restored viability and motor system function to the axJ mice (13, 17). USP14 deubiquitinated chemokine receptor CXCR4, whereas both overexpression and RNAi-induced knockdown of USP14 blocked ligand-induced CXCR4 degradation (14). Further, knockdown or inhibition of USP14 by siRNA or inhibitor (IU1) reduced murine norovirus-1 (MNV-1) nonstructural gene expression in RAW cells (18). These data indicate that USP14 is implicated in chemokine receptor signaling, virus infection, and immune responses. A recent study using an inhibitor of USP14, b-AP15, revealed that inhibition of USP14 resulted in accumulation of polyubiquitin, tumor cell apoptosis, a lower tumor progression rate, and organ infiltration in an acute myeloid leukemia model (16). These studies suggest that USP14 is a new drug target in anticancer and anti-infection.
The role of USP14 in the regulation of lung inflammation has not yet been studied. Here, we show that USP14 regulates I-κB stability and cytokine release in lung epithelial cells. This is the first study demonstrating that overexpression of USP14 reduces I-κB ubiquitination as well as its protein levels, thus increasing interleukin-8 (IL-8) release. Further, we demonstrate that USP14 interacts with phosphorylated RelA. This study will provide a basis for developing a new therapeutic strategy to treat lung inflammatory diseases.
MATERIALS AND METHODS
Cell Culture and Reagents
Murine lung epithelial (MLE12) cells (ATCC, Manassas, VA) were cultured with medium supplemented with hydrocortisone, insulin, transferrin, estrogen, selenium (HITES), 10% fetal bovine serum (FBS), and antibiotics at 37 °C in 5% CO2 incubator. Human lung epithelial (Beas2B) cells (ATCC, Manassas, MA) were cultured in BEGM medium with 10% FBS and antibiotics. HA tag, FLAG tag, I-κB, RelA, phospho-JNK, and ubiquitin antibodies were obtained from Cell Signaling (Danvers, MA). Cycloheximide, USP14 shRNA, and β-actin antibody were obtained from Sigma. USP14 antibody, immobilized protein A/G beads, and control IgG were from Santa Cruz Biotechnology (Santa Cruz, CA). HA-USP14 plasmid was kindly provided by Wade Harper (Harvard Medical School). FLAG-RelA was kindly provided by Thomas Gilman (Boston University). Escherichia coli isolated from chronic obstructive pulmonary disease patients were kindly provided by Rama Mallampalli (University of Pittsburgh). All materials used in the experiments are in the highest grades and are commercially available.
Plasmid Transfection
MLE12 cells were nucleofected with plasmids. 1 × 106 MLE cells were suspended in 120 μl of nucleofection buffer and well mixed with 3 μg of plasmid DNA in an electroporation cuvette. Electroporation was performed in the NucleofectionTM II system (Lonza, Gaithersburg, MD), and the cells were cultured in 2 ml of complete HITES (hydrocortisone, insulin, transferrin, estrogen, selenium) medium for 48 h. Beas2B cells were transfected with FuGENE HD transfection reagent (Roche Applied Science). Briefly, 2 μg of plasmid were mixed with 3 μl of FuGENE HD reagent in BEBM blank medium for 10 min. The mixture was added into Beas2B cell culture medium to grow for a further 2 days.
Preparation of Cell Lysates, Immunoprecipitation, and Western Blotting
Cell lysates in 120 μl of lysis buffer (20 mm Tris-HCl (pH 7.4), 150 mm NaCl, 2 mm EGTA, 5 mm β-glycerophosphate, 1 mm MgCl2, 1% Triton X-100, 1 mm sodium orthovanadate, 10 μg/ml protease inhibitors, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 μg/ml pepstatin) were sonicated on ice for 12 s and centrifuged at 500 × g for 5 min at 4 °C in a microcentrifuge. For immunoprecipitation, equal amounts of cell lysates (1 mg) were incubated with 2 μg/ml specific primary antibodies overnight at 4 °C followed by the addition of 40 μl of protein A/G-agarose for 2 h at 4 °C. For Western blotting, equal amounts of supernatant (20 μg) were subjected to 10% SDS-PAGE gels, transferred to polyvinylidene difluoride membranes, blocked with 5% (w/v) nonfat milk in TBST (25 mm Tris-HCl, pH 7.4, 137 mm NaCl, and 0.1% Tween 20) for 1 h, and incubated with primary antibodies in 5% (w/v) BSA in TBST for 1–2 h. The membranes were washed at least three times with TBST at 10-min intervals followed by a 1-h incubation with mouse, rabbit, or goat horseradish peroxidase-conjugated secondary antibody (1: 2,000). The membranes were developed with an enhanced chemiluminescence detection system according to manufacturer's instructions.
Immunofluorescence Staining
MLE12 cells grown on glass bottom dishes were fixed with 3.7% formaldehyde for 20 min, immunostained with USP14, HA tag, or RelA antibody, washed three times, and incubated with fluorescent conjugated second antibodies. Images were captured by Nikon ECLIPSE TE 300 inverted microscope.
Cytokine ELISA Measurement
Cell culture media were replaced with blank media before LPS treatment. At the end of the experiment, cell supernatants were collected, centrifuged at 1,000 × g for 5 min at 4 °C, and frozen at −80 °C for later analysis of IL-8 by ELISA kit according to the manufacturer's instructions.
Statistical Analyses
All results were subjected to statistical analysis using one-way analysis of variance and, wherever appropriate, the data were also analyzed by Student-Newman-Keuls test and expressed as mean ± S.D. Data were collected from at least three independent experiments, and p < 0.05 was considered significant.
RESULTS AND DISCUSSION
Overexpression of USP14 Reduces I-κB Expression
The nuclear factor NF-κB pathway has long been considered a proinflammatory signaling pathway that regulates the expression of proinflammatory genes including cytokines and chemokines (19). To investigate the role of USP14 in inflammatory lung diseases, we first tested whether USP14 regulates I-κB stability, which has been implicated in NF-κB activity (19). MLE12 cells were overexpressed with an HA-tagged human USP14 (HA-USP14) plasmid for 48 h. Cell lysates were analyzed for I-κB and IKKα expression. As shown in Fig. 1A, overexpression of HA-USP14 reduced I-κB level without altering IKKα and β-actin expression. Consistently, overexpression of HA-USP14 decreased levels of I-κB in a dose-dependent manner (Fig. 1B). The overexpressed HA-USP14 was confirmed with antibodies to USP14 and HA tag (Fig. 1, A and B). To further determine the role of USP14 in I-κB stability, I-κB levels were examined in USP14-overexpressed and USP14 knockdown MLE cells in the presence of cycloheximide (20 μg/ml), a protein synthesis inhibitor. Cycloheximide-mediated I-κB degradation was attenuated by USP14 shRNA-mediated knockdown of USP14, whereas overexpression of HA-USP14 promotes the I-κB degradation (Fig. 1C). Transfection of USP14 shRNA significantly reduced USP14 level (Fig. 1C). USP14 has been considered as an inhibitor of the proteasome because USP14 stabilized ubiquitinated cyclin B protein in vitro (11) and Tau and TDP-43 proteins in Usp14−/− murine embryonic fibroblasts (11). Nagai et al. (20) have shown that overexpression of USP14 inhibited the endoplasmic reticulum-associated degradation pathway, and knockdown of USP14 reversed the effect. In contrast to these findings, Peth et al. (10) found that USP14 controls 20 S gate opening, thus activating the proteasome, in a deubiquitination enzyme activity-independent manner. In addition, loss of USP14 function increased GABAA receptor levels in the Purkinje cells from the axJ mice (17). The present study provides new evidence that USP14 promotes certain protein degradation, such as that of I-κB. Given that USP14 is the deubiquitination enzyme, we hypothesize that USP14 is responsible for the removal of the ubiquitin chain bound to I-κB. To further investigate whether USP14 regulates deubiquitination of I-κB, ubiquitinated proteins were immunoprecipitated from proteasome inhibitor (MG-132)-treated vector alone or HA-USP14-overexpressed MLE12 cell lysates followed by I-κB immunoblotting. In addition, I-κB was immunoprecipitated followed by immunoblotting analysis with a ubiquitin antibody. Fig. 1D shows that MG-132-induced accumulation of polyubiquitinated I-κB was reduced by overexpression of HA-USP14. The data reveal that USP14 reduces I-κB polyubiquitination. Sun et al. (21) had reported that USP11 targets I-κB for its deubiquitination. Here, we report that USP14 mediates I-κB deubiquitination and stability. USP14 is a component of proteasome 19 S RP and controls ubiquitin recycling and proteasome gate opening (8–10). Deubiquitination of I-κB by USP11 increases I-κB stability (21), whereas ubiquitin removal by USP14 promotes I-κB entrance into the 20 S CP for degradation. USP14 has been known to regulate gate opening of the 20 S CP (10). A future study will investigate whether the gate opening function of USP14 contributes to the regulation of I-κB stability.
FIGURE 1.
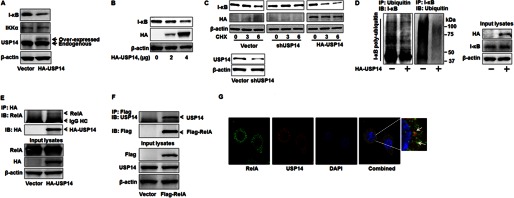
Overexpression of USP14 reduces I-κB levels and ubiquitination via association with RelA. A, MLE12 cells were transfected with an HA-USP14 plasmid (2 μg) for 48 h. Cell lysates were subjected to immunoblotting with antibodies to I-κB, IKKα, USP14, and β-actin. B, MLE12 cells were transfected with an HA-USP14 plasmid (2 and 4 μg) for 48 h. Cell lysates were subjected to I-κB, HA tag, and β-actin. C, MLE12 cells were transfected with an empty vector or an HA-USP14 plasmid for 48 h or with USP14 shRNA (shUSP14) for 72 h. Cell lysates were subjected to immunoblotting with antibodies to I-κB, HA tag, and β-actin (upper panel). Cell lysates from vector or shUSP14 transfected cells were analyzed with USP14 and β-actin antibodies (lower panel). CHX, cycloheximide. D, MLE12 cells were transfected with an empty vector or an HA-USP14 plasmid (3 μg) for 48 h prior to MG-132 (20 μm, 4 h) treatment. Cell lysates were subjected to immunoprecipitation (IP) with a ubiquitin antibody followed by I-κB immunoblotting (IB, left panel). Cell lysates were subjected to immunoprecipitation with an I-κB ubiquitin antibody followed by ubiquitin immunoblotting (IB, middle panel). Input lysates were subjected to immunoblotting with antibodies to HA tag, I-κB, and β-actin (right panel). E, MLE12 cells were transfected with an empty vector or an HA-USP14 plasmid (3 μg) for 48 h. Cell lysates were subjected to immunoprecipitation with an HA tag antibody followed by immunoblottings with antibodies to RelA and HA tag. Input lysates were subjected to immunoblotting with antibodies to RelA, HA tag, and β-actin. F, MLE12 cells were transfected with an empty vector or a FLAG-RelA plasmid (3 μg) for 48 h. Cell lysates were subjected to immunoprecipitation with a FLAG tag antibody followed by immunoblottings with antibodies to USP14 and FLAG tag. Input lysates were subjected to immunoblotting with antibodies to USP14, FLAG tag, and β-actin. G, MLE12 cells grown on glass bottom dishes were fixed and immunostained with antibodies to RelA (shown in green) and USP14 (shown in red). Nuclei were stained with DAPI (blue). Shown are representative images from three independent experiments.
USP14 Interacts with RelA
DUBs remove the ubiquitin chain via direct interaction with its ubiquitinated substrates or the ubiquitin chain (22). For instance, USP22 directly interacts with and mediates deubiquitination of Sirt1 (23). To investigate the mechanism by which USP14 regulates I-κB stability, we examined whether USP14 directly interacts with I-κB or its binding partner RelA. Cell lysates from MLE12 cells transfected with an empty vector alone or HA-USP14 plasmid were subjected to immunoprecipitation with an HA tag antibody followed by immunoblotting with antibodies to RelA or HA tag. As shown in Fig. 1E, HA-USP14 was associated with RelA. Further, cells were transfected with the FLAG-RelA plasmid. The overexpressed FLAG-RelA was immunoprecipitated followed by USP14 immunoblotting analysis. Consistent with the data from Fig. 1E, USP14 was associated with overexpressed FLAG-RelA. Co-immunoprecipitation has failed to detect the association between USP14 and I-κB (data not shown). The association between endogenous USP14 and RelA was confirmed by co-immunofluorescence staining with antibodies to USP14 and RelA. As shown in Fig. 1G, both USP14 and RelA were localized in the cytoplasm. A portion of the co-localized USP14 and RelA was detected. The ubiquitinated I-κB degradation in the proteasome has been well studied, whereas the detailed understanding of precisely how the NF-κB·I-κB complex interacts with the proteasome remains unclear. The present study indicates that RelA might play a novel role in recruiting the RelA·I-κB complex to USP14 on the proteasome 19 S RP, therefore regulating I-κB remodeling and promoting its entrance through the 20 S gate for degradation. The RelA binding domain within USP14 will need to be identified in future studies.
USP14 Promotes Cytokine Release
The role of USP14 in cytokine release has not yet been studied. First, we examined whether LPS treatment induced post-translational modification, such as phosphorylation, of USP14. MLE12 cells were transfected with an empty vector alone or the HA-USP14 plasmid for 48h prior to LPS treatment followed by immunoprecipitation with an antibody to phospho-serine and immunoblotting with an HA tag antibody. Interestingly, the LPS treatment increased serine phosphorylation of USP14 at 15 min (Fig. 2A). The post-translational modification of DUBs has not yet been well demonstrated. A recent study from Zhang et al. (24) reports that USP4 is phosphorylated by AKT, which mediates its stability. Growing evidence suggests that phosphorylation of the components of the 19 S RP regulates proteasome activity. Subunits of proteasomes including α subunits C8 (α7), C9 (α3), and ATPase subunit S4 (rpt2) have been shown to be modified by phosphorylation (25). Djakovic et al. (26) reported that phosphorylation of the 19 S ATPase subunit, Rpt6, by the calcium/calmodulin-dependent protein kinase II α (CaMKIIα) may promote the tethering of proteasomes to scaffolds and cytoskeletal components. This study is the first to demonstrate a post-translational modification of USP14. The phosphorylation site and role of the serine phosphorylation in the regulation of USP14 activity will need to be further investigated. Further, we found that overexpression of USP14 promoted LPS-induced degradation of I-κB without altering phosphorylation of JNK1/2 (Fig. 2B), suggesting that USP14 is involved in LPS-induced NF-κB pathway, but not JNK signaling. To investigate the involvement of USP14 in LPS-induced cytokine release, human lung epithelial cells (Beas2B) were transfected with HA-USP14 plasmid prior to LPS treatment. The IL-8 levels in cell culture media were measured by a human IL-8 ELISA kit. Consistently, LPS treatment significantly increased IL-8 production. Interestingly, overexpression of HA-USP14 promoted LPS-induced IL-8 release (Fig. 2C). Further, overexpression of HA-USP14 increased TNFα- or E. coli (isolated from chronic obstructive pulmonary disease patients)-induced IL-8 secretion (Fig. 2D). The data demonstrate that USP14 promotes antigen-induced I-κB degradation as well as cytokine release in lung epithelial cells.
FIGURE 2.

Overexpression of USP14 promotes LPS-induced IL-8 release. A, MLE12 cells were transfected with an empty vector or an HA-USP14 plasmid (3 μg) for 48 h. Cell lysates were subjected to immunoprecipitation (IP) with a phospho-serine (p-serine) antibody followed by immunoblottings (IB) with an antibody to HA tag. Input lysates were subjected to immunoblotting with antibodies to HA tag and β-actin. B, MLE12 cells were transfected with an empty vector or an HA-USP14 plasmid (3 μg) for 48 h prior to LPS treatment (10 μg/ml, 30 min). Cell lysates were subjected to immunoblotting with antibodies to I-κB, phospho-JNK (p-JNK), HA tag, and β-actin. C, Beas2B cells were transfected with an empty vector or an HA-USP14 plasmid (3 μg) for 48 h prior to LPS treatment (10 μg/ml, 3 h). Media were collected, and IL-8 release was measured by IL-8 ELISA kit. *, p < 0.01 as compared with cells treated with LPS alone. D, Beas2B cells were transfected with an empty vector or an HA-USP14 plasmid (3 μg) for 48 h prior to TNFα (10 μg/ml) treatment or E. coli infection (10 multiplicity of infection) for 3 h. Media were collected, and IL-8 release was measured by IL-8 ELISA kit. *, p < 0.01 as compared with cells treated with TNFα alone; **, p < 0.01 as compared with cells treated with E. coli alone.
This study is the first demonstrating that USP14 regulates cytokine release via modulating I-κB stability. This reveals a new mechanism by which the NF-κB pathway is regulated by USP14 in lung epithelial cells and suggests that the association between USP14 and RelA is the link between the NF-κB complex and the proteasome. The present study will provide a new therapeutic target to diminish lung inflammation. Several inhibitors have been developed to inhibit USP14 activity (11, 16, 18); however, their specificity remains unclear. The implications of these inhibitors in lung inflammation will likely be examined in future studies. In addition, an isoform of USP14, the short form (USP14SF), has been reported (11). Its expression in the lung cells and its involvement in the NF-κB pathway and inflammatory responses will be investigated.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 HL091916 and HL112791 (to Y. Z.). This work was also supported by American Heart Association Award 12SDG9050005 (to J. Z.).
- CP
- core particle
- RP
- regulatory particle
- DUB
- deubiquitinating enzyme
- IKKα
- IκB kinase α
- TBST
- Tris-buffered saline with Tween
- MLE
- mouse lung epithelial cells.
REFERENCES
- 1. Hershko A., Ciechanover A. (1992) The ubiquitin system for protein degradation. Annu. Rev. Biochem. 61, 761–807 [DOI] [PubMed] [Google Scholar]
- 2. Schwartz A. L., Ciechanover A. (1992) Ubiquitin-mediated protein modification and degradation. Am. J. Respir. Cell Mol. Biol. 7, 463–468 [DOI] [PubMed] [Google Scholar]
- 3. Ugai S., Tamura T., Tanahashi N., Takai S., Komi N., Chung C. H., Tanaka K., Ichihara A. (1993) Purification and characterization of the 26S proteasome complex catalyzing ATP-dependent breakdown of ubiquitin-ligated proteins from rat liver. J. Biochem. 113, 754–768 [DOI] [PubMed] [Google Scholar]
- 4. Saeki Y., Tanaka K. (2012) Assembly and function of the proteasome. Methods Mol. Biol. 832, 315–337 [DOI] [PubMed] [Google Scholar]
- 5. Kim H. M., Yu Y., Cheng Y. (2011) Structure characterization of the 26S proteasome. Biochim. Biophys. Acta 1809, 67–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Adams J. (2003) The proteasome: structure, function, and role in the cell. Cancer Treat. Rev. 29, Suppl. 1, 3–9 [DOI] [PubMed] [Google Scholar]
- 7. Lee M. J., Lee B. H., Hanna J., King R. W., Finley D. (2011) Trimming of ubiquitin chains by proteasome-associated deubiquitinating enzymes. Mol. Cell. Proteomics 10, R110.003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hu M., Li P., Song L., Jeffrey P. D., Chenova T. A., Wilkinson K. D., Cohen R. E., Shi Y. (2005) Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 24, 3747–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hanna J., Hathaway N. A., Tone Y., Crosas B., Elsasser S., Kirkpatrick D. S., Leggett D. S., Gygi S. P., King R. W., Finley D. (2006) Deubiquitinating enzyme Ubp6 functions noncatalytically to delay proteasomal degradation. Cell 127, 99–111 [DOI] [PubMed] [Google Scholar]
- 10. Peth A., Besche H. C., Goldberg A. L. (2009) Ubiquitinated proteins activate the proteasome by binding to Usp14/Ubp6, which causes 20S gate opening. Mol. Cell 36, 794–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee B. H., Lee M. J., Park S., Oh D. C., Elsasser S., Chen P. C., Gartner C., Dimova N., Hanna J., Gygi S. P., Wilson S. M., King R. W., Finley D. (2010) Enhancement of proteasome activity by a small-molecule inhibitor of USP14. Nature 467, 179–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Crimmins S., Jin Y., Wheeler C., Huffman A. K., Chapman C., Dobrunz L. E., Levey A., Roth K. A., Wilson J. A., Wilson S. M. (2006) Transgenic rescue of ataxia mice with neuronal-specific expression of ubiquitin-specific protease 14. J. Neurosci. 26, 11423–11431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen P. C., Qin L. N., Li X. M., Walters B. J., Wilson J. A., Mei L., Wilson S. M. (2009) The proteasome-associated deubiquitinating enzyme Usp14 is essential for the maintenance of synaptic ubiquitin levels and the development of neuromuscular junctions. J. Neurosci. 29, 10909–10919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mines M. A., Goodwin J. S., Limbird L. E., Cui F. F., Fan G. H. (2009) Deubiquitination of CXCR4 by USP14 is critical for both CXCL12-induced CXCR4 degradation and chemotaxis but not ERK activation. J. Biol. Chem. 284, 5742–5752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shinji S., Naito Z., Ishiwata S., Ishiwata T., Tanaka N., Furukawa K., Suzuki H., Seya T., Matsuda A., Katsuta M., Tajiri T. (2006) Ubiquitin-specific protease 14 expression in colorectal cancer is associated with liver and lymph node metastases. Oncol. Rep. 15, 539–543 [PubMed] [Google Scholar]
- 16. D'Arcy P., Brnjic S., Olofsson M. H., Fryknäs M., Lindsten K., De Cesare M., Perego P., Sadeghi B., Hassan M., Larsson R., Linder S. (2011) Inhibition of proteasome deubiquitinating activity as a new cancer therapy. Nat. Med. 17, 1636–1640 [DOI] [PubMed] [Google Scholar]
- 17. Lappe-Siefke C., Loebrich S., Hevers W., Waidmann O. B., Schweizer M., Fehr S., Fritschy J. M., Dikic I., Eilers J., Wilson S. M., Kneussel M. (2009) The ataxia (axJ) mutation causes abnormal GABAA receptor turnover in mice. PLoS Genet. 5, e1000631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perry J. W., Ahmed M., Chang K. O., Donato N. J., Showalter H. D., Wobus C. E. (2012) Antiviral activity of a small molecule deubiquitinase inhibitor occurs via induction of the unfolded protein response. PLoS Pathog. 8, e1002783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tak P. P., Firestein G. S. (2001) NF-κB: a key role in inflammatory diseases. J. Clin. Invest. 107, 7–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nagai A., Kadowaki H., Maruyama T., Takeda K., Nishitoh H., Ichijo H. (2009) USP14 inhibits ER-associated degradation via interaction with IRE1α. Biochem. Biophys. Res. Commun. 379, 995–1000 [DOI] [PubMed] [Google Scholar]
- 21. Sun W., Tan X., Shi Y., Xu G., Mao R., Gu X., Fan Y., Yu Y., Burlingame S., Zhang H., Rednam S. P., Lu X., Zhang T., Fu S., Cao G., Qin J., Yang J. (2010) USP11 negatively regulates TNFα-induced NF-κB activation by targeting on IκBα. Cell Signal 22, 386–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reyes-Turcu F. E., Ventii K. H., Wilkinson K. D. (2009) Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annual review of biochemistry 78, 363–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin Z., Yang H., Kong Q., Li J., Lee S. M., Gao B., Dong H., Wei J., Song J., Zhang D. D., Fang D. (2012) USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol. Cell 46, 484–494 [DOI] [PubMed] [Google Scholar]
- 24. Zhang L., Zhou F., Drabsch Y., Gao R., Snaar-Jagalska B. E., Mickanin C., Huang H., Sheppard K. A., Porter J. A., Lu C. X., ten Dijke P. (2012) USP4 is regulated by AKT phosphorylation and directly deubiquitylates TGF-β type I receptor. Nat. Cell Biol. 14, 717–726 [DOI] [PubMed] [Google Scholar]
- 25. Rivett A. J., Bose S., Brooks P., Broadfoot K. I. (2001) Regulation of proteasome complexes by γ-interferon and phosphorylation. Biochimie 83, 363–366 [DOI] [PubMed] [Google Scholar]
- 26. Djakovic S. N., Marquez-Lona E. M., Jakawich S. K., Wright R., Chu C., Sutton M. A., Patrick G. N. (2012) Phosphorylation of Rpt6 regulates synaptic strength in hippocampal neurons. J. Neurosci. 32, 5126–5131 [DOI] [PMC free article] [PubMed] [Google Scholar]