

Background: Myofibroblast differentiation plays a critical role in fibrosis.
Results: Microtubule polymerization state inversely controls myofibroblast differentiation via Rho/SRF signaling. Dynamic localization of mDia2 to actin stress fibers is critical for myofibroblast differentiation and is regulated by the microtubule polymerization state.
Conclusion: Microtubule polymerization state controls myofibroblast differentiation via regulation of mDia2 localization.
Significance: This is a novel mechanism of myofibroblast differentiation and a therapeutic target.
Keywords: Actin, Cytoskeleton, Fibroblast, Microtubules, Myofibroblast, Transforming Growth Factor Beta (TGFbeta), MKL1, SRF
Abstract
Myofibroblast differentiation plays a critical role in wound healing and in the pathogenesis of fibrosis. We have previously shown that myofibroblast differentiation is mediated by the activity of serum response factor (SRF), which is tightly controlled by the actin polymerization state. In this study, we investigated the role of the microtubule cytoskeleton in modulating myofibroblast phenotype. Treatment of human lung fibroblasts with the microtubule-destabilizing agent, colchicine, resulted in a formation of numerous stress fibers and expression of myofibroblast differentiation marker proteins. These effects of colchicine were independent of Smad signaling but were mediated by Rho signaling and SRF, as they were attenuated by the Rho kinase inhibitor, Y27632, or by the SRF inhibitor, CCG-1423. TGF-β-induced myofibroblast differentiation was not accompanied by gross changes in the microtubule polymerization state. However, microtubule stabilization by paclitaxel attenuated TGF-β-induced myofibroblast differentiation. Paclitaxel had no effect on TGF-β-induced Smad activation and Smad-dependent gene transcription but inhibited actin polymerization, nuclear accumulation of megakaryoblastic leukemia-1 protein, and SRF activation. The microtubule-associated formin, mDIA2, localized to actin stress fibers upon treatment with TGF-β, and paclitaxel prevented this localization. Treatment with the formin inhibitor, SMI formin homology 2 domain, inhibited stress fiber formation and myofibroblast differentiation induced by TGF-β, without affecting Smad-phosphorylation or microtubule polymerization. Together, these data suggest that (a) TGF-β promotes association of mDia2 with actin stress fibers, which further drives stress fiber formation and myofibroblast differentiation, and (b) microtubule polymerization state controls myofibroblast differentiation through the regulation of mDia2 localization.
Introduction
Myofibroblast differentiation plays an important role in mediating the fibrotic response to tissue injury and is thought to be a key pathologic step in the pathogenesis of disorders of aberrant wound healing such as fibrosis. A highly organized actin cytoskeleton is a key morphologic feature of fully differentiated myofibroblasts, and actin dynamics participate in mediating a myriad of fibroblast functions (1), including the regulation of megakaryoblastic leukemia-1 (MKL1)3/SRF-dependent gene expression (2). An additional component of the cell cytoskeleton, the microtubule, has been implicated in a variety of cell functions directly relevant to tissue remodeling. Microtubule assembly is regulated by the application of substrate strain (3), extracellular matrix protein density (4), and matrix stiffness (5), whereas pharmacologic disruption of microtubules has been shown to result in the induction of plasminogen activator inhibitor-1 and connective tissue growth factor (CTGF) (6–8), induce increased contractility (9), and promote incorporation of soluble fibronectin into the extracellular matrix (10). Interestingly, the microtubule system can interact with the actin cytoskeleton via mechanical coupling (the tensegrity model) (11) or via direct biochemical coupling, via the microtubule-associated nucleotide exchange factor, p190RhoGEF (GEF-H1) (5, 12) or diaphanous proteins mDIA1 and mDIA2 (13–16). In light of this linkage with tissue remodeling and the actin cytoskeleton, we sought to examine whether the microtubule cytoskeleton could participate in the regulation of myofibroblast differentiation in human lung fibroblasts.
EXPERIMENTAL PROCEDURES
Isolation and Primary Culture of Human Pulmonary Fibroblasts
Human lung fibroblasts were isolated as described previously (2, 17). Briefly, tissue samples from explanted lungs from patients undergoing lung transplantation for pulmonary fibrosis were obtained and placed in DMEM with 100 units/ml streptomycin, 250 ng/ml amphotericin B, and 100 units/ml penicillin. Alveolated lung tissue was minced, washed in PBS, and plated onto 10-cm plates in growth media containing DMEM supplemented with 10% FBS, 2 mm l-glutamine, and antibiotics as above. Expanded populations of fibroblasts were subsequently subcultured after 4 to 5 days, resulting in the development of a homogenous fibroblast population. All primary cultures were used from passages 5–10 and maintained on tissue culture plastic until the time of experiments.
For all experiments, cells were subcultured by plating on collagen IV-coated tissue culture plastic at a density of 100,000/ml, followed by culture in growth media (containing 10% FBS, as above) for 24 h. Cells were then serum-starved 24 h in DMEM supplemented with 0.1% BSA and 2 mm l-glutamine, followed by treatment with the desired agonist (or vehicle) in 0.1% BSA for the desired times. Studies using paclitaxel were performed by a 30-min pretreatment with paclitaxel prior to subsequent treatment with colchicine or TGF-β for the desired times. For experiments using colchicine, cells were treated for 6 or 24 h when assessing gene expression; for experiments assessing the effect of colchicine on TGF-β effects, cells were pretreated with colchicine 30 min prior to TGF-β stimulation.
DNA Transfection
In all experiments, subcultured cells were grown on collagen IV-coated plates for 1 day in growth media (as above) and were serum-deprived overnight in DMEM containing 0.1% BSA and 2 mm l-glutamine prior to stimulation with desired agonists. Transient DNA transfections were performed using GenJet reagent (SignaGen Laboratories, Gaithersburg, MD) following the standard manufacturer's instructions.
Reagents
The firefly luciferase reporter driven by two copies of CArG elements (SRF-Luc) has been used previously (2, 17–20). The firefly luciferase reporter driven by four copies of Smad-binding elements was provided by Dr. Bert Vogelstein and has been used previously (2, 17). The thymidine kinase promoter-driven Renilla luciferase was from Promega (Madison, WI). Colchicine, TGF-β1, and Y27632 were from EMD Biosciences (Gibbstown, NJ). CCG-1423 was from Cayman Chemicals (Ann Arbor, MI). SMIFH2 and SB431542 were from Sigma. Antibodies against α-tubulin, β-actin, and SM-α-actin were from Sigma. CTGF, SRF, Smad4, and 14-3-3-ϵ antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Phospho-Smad2, Smad2, and phospho-Smad3, Smad3, and α/β-tubulin antibodies were from Cell Signaling (Danvers, MA). MKL-1 antibodies were from Bethyl Laboratories (Montgomery, TX). Lamin A/C antibodies were from BD Transduction Laboratories. mDIA2 antibodies were from Abcam (immunofluorescent staining) or ECM Biosciences (Western blotting, DP3491, ECM) (Versailles, KY).
Cell Lysis and Western Blotting
After stimulation of quiescent cells with desired agonists, cells were lysed in radioimmune precipitation assay buffer containing 25 mm HEPES (pH 7.5), 150 mm NaCl, 1% Triton X-100, 0.1% SDS, 2 mm EDTA, 2 mm EGTA, 10% glycerol, 1 mm NaF, 200 μm sodium orthovanadate, and protease inhibitor mixture (Sigma). Cells were scraped, sonicated for 5 s, and boiled in Laemmli buffer for 5 min. The samples were subjected to polyacrylamide gel electrophoresis followed by Western blotting with desired primary antibodies and corresponding HRP-conjugated secondary antibodies and developed by an ECL reaction (Pierce). The digital chemiluminescent images were taken by a GE LAS4000 chemiluminescence imager. Densitometry of selected blots was performed using ImageJ software.
In Vitro Isolation of Stress Fibers
Stress fibers were isolated using the method we used previously (2, 21). All of the procedures were performed on ice using the buffers containing protease inhibitor mixture (Sigma). After stimulation with desired agonists, cells were washed with PBS and then extracted with a buffer containing 2.5 mm triethanolamine (pH 8.2) for 30 min with six buffer changes, followed by extraction with 0.05% Nonidet P-40 (pH 7.2) for 5 min, and subsequent extraction with 0.5% Triton X-100 (pH 7.2) for additional 5 min. Cells were then immediately washed with cold PBS, scraped, and suspended in PBS, followed by centrifugation at 100,000 × g for 1 h. Supernatant was removed, and the pellet was sonicated in 0.5% Triton X-100, 50 mm NaCl, 20 mm Hepes (pH 7.0), 1 mm EDTA. Laemmli buffer was added, and samples were boiled for 5 min prior to further Western blot analysis as described above.
In Vitro Isolation of Soluble and Insoluble Microtubule Fractions
Soluble and insoluble fractions of microtubules were isolated using a modified method described previously (22). All procedures were performed at room temperature with warmed buffers. After stimulation with the desired agonists, cells were washed with warmed (37 °C) PBS and then extracted twice at 8 min each with a buffer containing 2 m glycerol, 0.1 m PIPES, 1 mm MgSO4, 2 mm EGTA, 0.1 mm EDTA, pH 6.75, to remove soluble proteins. The extracted cells were then lysed for 5 min with a buffer containing 25 mm Na2HPO4, 0.4 m NaCl, 0.5% SDS, pH 7.2. Cells were scraped, boiled, and centrifuged at 2000 × g for 10 min. The supernatant was then collected. Laemmli buffer was then added to both the collected extraction samples and the lysis samples, and samples were boiled for 5 min prior to further Western blot analysis.
In Vitro Isolation of Nuclear and Cytoplasmic Fractions
Preparation of nuclear and cytoplasmic fractions was performed using the NE-PER nuclear and cytoplasmic reagents (Thermo Scientific) following the manufacturer's instructions. Briefly, after stimulation with desired agonists, cells were trypsinized and washed with PBS to remove trypsin by centrifugation at 300 × g for 3 min. Cell pellets were suspended in cytoplasm extraction reagent for 10 min and pelleted again at 16,000 × g for 5 min, and the supernatant (cytoplasmic fraction) was collected. The pellets were suspended in the nuclear extraction reagent for 40 min, centrifuged at 16,000 × g for 10 min, and the supernatant (nuclear fraction) was collected. Laemmli buffer was added, and samples were boiled for 5 min prior to further Western blot analysis as described above. Immunoblotting of nuclear lamin A/C and cytoplasmic 14-3-3ϵ was performed to confirm the purity of the nuclear and cytoplasmic fractions, respectively.
Luciferase Reporter Assay
Cells grown on 24-well plates were co-transfected with 500 ng of desired firefly luciferase reporter plasmid and 20 ng of constitutively active thymidine kinase promoter-Renilla luciferase reporter plasmid. Cells were placed in growth media overnight and then serum-starved for 24 h, followed by stimulation with the desired agonists for the desired time points as indicated in the figure legends. Cells were then washed with PBS and lysed in protein extraction reagent (Pierce). The lysates were assayed for firefly and Renilla luciferase activity using the Dual-Luciferase assay kit (Promega). To account for differences in transfection efficiency, firefly luciferase activity of each sample was normalized to Renilla luciferase activity.
Reverse Transcription-Quantitative Real Time PCR
Total RNA was harvested using RNA STAT-60 (Tell-Test, Inc.) following the manufacturer's instructions. One microgram of total RNA was used as a template for random-primed reverse transcription using an iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer's instructions. Real time PCR analysis was performed using iTaq SYBR Green supermix with ROX (Bio-Rad) in an ABI 7500 multi-color real time PCR detection system (Applied Biosystems). PCR primers contained the following sequences: human CTGF, CGACTGGAAGACACGTTTGG (forward) and AGGCTTGGAGATTTTGGGAG (reverse); human SM-α-actin, AAAGACAGCTACGTGGGTGACGAA (forward) and TTCCATGTCGTCCCAGTTGGTGAT (reverse); and human SRF, TGAGTGCCACTGGCTTTGAAGAGA (forward) and AGAGGTGCTAGGTGCTGTTTGGAT (reverse). The 18 S transcript was used to normalize the amount and quality of the extracted RNA, using the following primer set: 18 S, GATTAAGTCCCTGCCCTTTG (forward) and GTTCACCTACGGAAACCTTG (reverse).
Indirect Immunofluorescence Microscopy
Cells were washed twice with PBS and fixed either in 4% paraformaldehyde/PBS for 15 min at room temperature followed by permeabilization with 0.2% Triton X-100/PBS for 5 min. Cells were then incubated with 2% BSA in PBS for 1 h, followed by incubation with desired primary antibodies at 4 °C overnight, five washes with PBS within 15 min, incubation with corresponding rhodamine- or FITC-conjugated secondary antibodies for 1 h at room temperature, and five washes with PBS within 15 min. The coverslips were mounted using Vectashield mounting medium containing DAPI nuclear stain (Vector Laboratories, Burlingame, CA). The immunofluorescence images were obtained using an Olympus 1X71 fluorescent microscope.
Statistical Analysis
All of the data represent the results of at least three independent experiments. Quantitative data were analyzed by the Student t test with Bonferroni's correction for multiple comparisons when appropriate. Values of p < 0.05 were considered as statistically significant.
RESULTS
Microtubule Disruption Promotes Myofibroblast Differentiation in a Rho/SRF-dependent, Smad-independent Manner
To examine whether microtubule polymerization state could influence fibroblast morphology and differentiation, we first utilized the direct pharmacologic inhibitor of microtubule polymerization, colchicine, which has been shown to fully depolymerize microtubules after 60 min (23). Treatment of human lung fibroblasts (HLF) with colchicine for 24 h resulted in a significant decrease in microtubule staining (Fig. 1A), accompanied by morphologic change, with cell widening and robust formation of actin stress fibers (Fig. 1B). Colchicine treatment also induced the expression of smooth muscle α-actin (α-SMA), which was highly enriched in the stress fibers, as assessed by phalloidin staining (Fig. 1B), suggestive of differentiation to a myofibroblast-like phenotype. Nocodazole had similar effects (data not shown). The mRNA levels of α-SMA, as well as other genes associated with myofibroblast differentiation such as CTGF and SRF were significantly up-regulated in response to colchicine treatment (Fig. 1C), demonstrating a role for microtubule dynamics in a control of gene expression. Stabilization of microtubules by 30 min of pretreatment with paclitaxel blocked colchicine-induced α-SMA expression (Fig. 1D), confirming that the effect of colchicine is mediated by microtubule disruption.
FIGURE 1.

Disruption of microtubules by colchicine promotes myofibroblast differentiation. HLF were stimulated with 1 μm colchicine (Colch) for 6 or 24 h, with or without a 30-min pretreatment with 10 μm paclitaxel, as indicated. A, immunofluorescent staining of HLF with α-tubulin antibodies. Scale bar, 200 μm. B, HLF were examined for actin stress fiber formation and α-SMA expression by phalloidin staining and immunofluorescence with α-SMA antibodies, respectively. Scale bar, 200 μm. C, levels of SRF, α-SMA, and CTGF mRNA were examined by real time PCR. D, total protein lysates from HLF were analyzed by Western blot with antibodies against α-SMA or β-actin.
Previous studies have suggested that microtubule dynamics could regulate Smad-dependent signaling in CCL64 cells, cardiac myocytes, and C2C12 myoblasts (24–26). However, microtubular disruption by colchicine in human pulmonary fibroblasts did not result in Smad nuclear translocation (Fig. 2A), or Smad-dependent gene transcription, as assessed by using Smad binding elements luciferase reporter (Fig. 2B). Furthermore, pharmacologic inhibition of TGF-β-receptor kinase (ALK5) with SB431542, which has been shown to block TGF-β-induced α-SMA expression (2), did not attenuate colchicine induced α-SMA expression (Fig. 2, C and D), suggesting that the effect of microtubule disruption was independent of TGF-β/Smad signaling. Additionally, we did not observe any induction of ERK1/2 phosphorylation by cochicine in fibroblasts (data not shown), as has been reported previously in vascular smooth muscle cells (7, 27).
FIGURE 2.
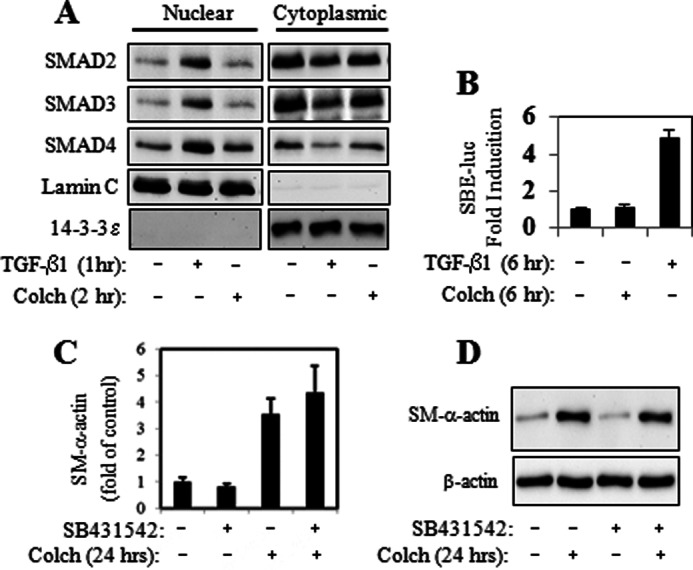
Colchicine-induced myofibroblast differentiation is not mediated by Smad activation. HLF were stimulated with 1 ng/ml of TGF-β1 or 1 μm colchicine (Colch) in the presence or absence of 10 μm SB431542 (added 30 min prior to stimulation) for the times indicated. Cells were examined for the nuclear localization of Smad2/3/4 by in vitro fractionation (A), for activation of Smad-binding element-luciferase reporter (SBE-luc) (B), and for the expression of α-SMA mRNA (C) or protein (D).
In contrast, colchicine stimulated phosphorylation of cofilin (Fig. 3A), which is known to occur in a Rho kinase/LIM kinase-dependent manner (28) and has been used as a marker for Rho activation (2). Both colchicine-induced actin stress fiber formation and α-SMA expression were dependent on Rho signaling, as the Rho kinase inhibitor, Y27632, fully blocked this induction (Fig. 3B). Finally, colchicine-induced α-SMA expression was attenuated by pharmacological inhibition of SRF (Fig. 3C), which has been previously shown by us to be activated by TGF-β through the Rho/stress fiber pathway (2) and play a critical role in TGF-β-induced myofibroblast differentiation (17).
FIGURE 3.

Colchicine-induced myofibroblast differentiation is mediated by ROCK and SRF activities. HLF were pretreated for 30 min with or without 10 μm Y27632 or 10 μm CCG-1423 as indicated, followed by stimulation with 1 μm colchicine (Colch) for 24 h. A, colchicine stimulates phosphorylation of cofilin, as assessed by Western blotting with phosphor-Ser-3-cofilin antibodies. B, Rho kinase inhibition by Y27632 attenuates colchicine-induced actin stress fiber formation and α-SMA protein expression. C, the SRF inhibitor, CCG-1423, blocked colchicine-induced α-SMA protein expression. Densitometry results are shown below the respective blots.
Stabilization of Microtubules Attenuates TGF-β-induced Myofibroblast Differentiation through Inhibition of Actin/SRF Signaling
Given the well established role of TGF-β1 in inducing myofibroblast differentiation (29) and the observed induction of a myofibroblast phenotype by microtubule disruption, we sought to examine whether (a) TGF-β1 affects microtubule stability and (b) whether microtubule polymerization could influence TGF-β1-induced myofibroblast differentiation. As shown in Fig. 4A, TGF-β1 treatment for 24 h did not result in any appreciable alteration in microtubule configuration, as assessed by immunofluorescent staining. Furthermore, subcellular fractionation of insoluble (polymerized) and soluble (depolymerized) tubulin also revealed that TGF-β1, applied either for 1 or 24 h, does not affect global tubulin polymerization. In contrast, treatment with paclitaxel (microtubule stabilizer) or colchicine (microtubule disruptor) for either 90 min (Fig. 4B) or 24 h (Fig. 4C) redistributed tubulin to the insoluble and soluble fractions, respectively, which is consistent with previously published observations (23). Despite the lack of effect TGF-β on microtubule dynamics, stabilization of microtubules by paclitaxel resulted in inhibition of TGF-β1-induced expression of myofibroblast-associated genes, including α-SMA, CTGF, and SRF, both at mRNA and protein levels (Fig. 5), suggesting that microtubule dynamics can potentially interact with TGF-β1-dependent signaling.
FIGURE 4.

TGF-β1 does not regulate gross microtubule polymerization state. A, immunofluorescent staining of HLF with α-tubulin antibodies following 24-h stimulation with TGF-β1. Scale bar, 100 μm. B and C, HLF were pretreated with either vehicle, 10 μm paclitaxel, or 1 μm colchicine (Colch) for 30 min, followed by stimulation with 1 ng/ml TGF-β1 for either 1 (B) or 24 h (C). Microtubule and soluble tubulin fractions were isolated and examined by Western blotting with α-tubulin antibodies. Densitometry results are shown below the respective blots.
FIGURE 5.

Polymerization of microtubules by paclitaxel attenuates TGF-β1-induced myofibroblast differentiation. HLF were pretreated with indicated concentration of paclitaxel for 0.5 h, followed by stimulation with 1 ng/ml TGF-β1 for 24 h. Cell extracts were examined for the expression of SRF, CTGF, and α-SMA mRNA by real time quantitative PCR (A) or of protein levels by Western blotting (B).
Given our recent observations establishing the role of stress fiber formation and SRF activation in mediating TGF-β-induced gene expression (2), we examined whether microtubule stabilization by paclitaxel affected this axis of TGF-β signaling. Treatment with paclitaxel resulted in the inhibition of TGF-β1-induced actin stress fiber formation (Fig. 6, A and B). We have previously shown that stress fiber formation leads to the nuclear accumulation of the transcription co-activator, MKL1, activating SRF-dependent gene transcription in response to TGF-β1 (2). Additionally, under treatment with TGF-β1, MKL1 expression increases in an SRF-dependent manner (2). In the present study, we observed that paclitaxel attenuates both TGF-β1-induced MKL1 expression and nuclear accumulation (Fig. 6C), as well as SRF-dependent gene transcription (Fig. 6D). We did not observe significant effects of paclitaxel on TGF-β1-induced Smad2/3 phosphorylation, Smad nuclear translocation, or Smad-dependent gene expression in HLF (supplemental Fig. S1). Together, these data suggest that regulation of TGF-β1-induced myofibroblast differentiation by paclitaxel occurs through inhibition of the stress fiber/MKL1/SRF axis but not through immediate Smad signaling.
FIGURE 6.

Paclitaxel regulates TGF-β1-induced stress fiber formation, MKL1 nuclear accumulation, and SRF activation. A, HLF were pretreated with 10 μm paclitaxel for 0.5 h, followed by stimulation with 1 ng/ml TGF-β1 for 24 h. Cells were analyzed for stress fiber formation by fluorescent phalloidin staining. Scale bar, 200 μm. Under these treatment conditions, HLF were also assessed for total α-SMA expression and stress fiber formation by in vitro fractionation (B), for nuclear accumulation of MKL1 by in vitro fractionation (C), or for SRF-luciferase reporter activation (D).
TGF-β1 Promotes Association of mDia2 with Actin Stress Fibers, which Is Controlled by Microtubule Polymerization State
Diaphanous-related formins belong to a family of multifunctional proteins known to independently promote both microtubule stabilization and actin polymerization (13–16, 30). In vitro studies have shown that microtubules inhibit the ability of the family member, mDia2 to promote actin polymerization (14). We have observed that under basal conditions, mDia2 co-localized with microtubules in quiescent fibroblasts, as assessed by immunofluorescent microscopy (Fig. 7, A and B), which is consistent with others having shown association of mDia2 with microtubules in vitro (13) and in NIH3T3 cells (31). In vitro fractionation of stress fibers from HLF cells showed little association of mDia with the stress fiber fraction under the basal conditions. However, upon TGF-β1 or colchicine treatment, the levels of mDia2 were significantly increased in the stress fiber fraction (Fig. 7C). Furthermore, stabilization of microtubules with paclitaxel abrogated TGF-β1-induced accumulation of mDIA2 in the stress fiber fraction (Fig. 7C).
FIGURE 7.

TGF-β1-induced accumulation of mDia2 with stress fibers and regulation by microtubule dynamics. Co-localization of mDia2 with microtubules in quiescent fibroblasts, as assessed by immunofluorescent microscopy are shown (A and B). A, shown are the images of HLF stained with antibodies against α-tubulin, mDia2, or merged (counter stained with DAPI) as indicated, taken at 600× magnification. Scale bar, 50 μm. B, the same fields as in A were imaged at 1000× magnification, and the selected areas (dashed lines) were magnified using Adobe Photoshop. Scale bar, 10 μm. C, HLF were pretreated with or without 10 μm paclitaxel for 0.5 h, followed by stimulation with 1 ng/ml TGF-β1 for 24 h, as indicated. mDia2 levels in stress fiber fraction as well as total levels of mDia2 were examined by Western blotting with mDia2 antibodies. Densitometry results are shown below the respective blots. Colch, colchicine.
Given the inverse relation between microtubule polymerization state and localization of mDia2 to stress fibers, we then examined the role of mDia in TGF-β1-induced stress fiber formation and myofibroblast differentiation using the mDia inhibitor SMIFH2 (32). As shown in Fig. 8A, pretreatment of fibroblasts with SMIFH2 attenuated TGF-β1-induced α-SMA expression at the doses previously shown to be effective in mDia inhibition (32, 33). Importantly, SMIFH2 attenuated actin stress fiber formation in response to TGF-β1 (Fig. 8B). Control experiments showed that SMIFH2 had no effect on TGF-β1-induced Smad phosphorylation (Fig. 8C), as well as on gross microtubule polymerization state, modulated by paclitaxel or colchicine (Fig. 8D). Taken together, these data suggest that (a) TGF-β1 promotes association of mDia2 with actin stress fibers, which further drives stress fiber formation and myofibroblast differentiation, and (b) microtubule polymerization inhibits myofibroblast differentiation through the regulation of mDia2 localization and/or activity.
FIGURE 8.

mDIA activity is required for TGF-β1-induced actin stress fiber formation and myofibroblast differentiation. A–D, HLF were pretreated with indicated concentrations of SMFH2 for 30 min, followed by treatment with 1 ng/ml TGF-β1, 10 μm paclitaxel, or 1 μm colchicine (Colch) for the times indicated. Cell extracts were examined by Western blotting for the expression of α-SMA (A), stress fiber formation (B), phosphorylation of Smad2 (C), or microtubule polymerization state (D). Densitometry results are shown below the respective blots.
DISCUSSION
This study describes four major findings: (i) gross microtubule polymerization state inversely affects myofibroblast differentiation by controlling the actin stress fiber/MKL1/SRF signaling, but not Smad-dependent gene transcription; (ii) polymerization of microtubules prevents myofibroblast differentiation in response to TGF-β1, again, through the regulation of stress fiber/MKL1/SRF, but not Smad signaling; (iii) TGF-β1 promotes accumulation of mDia2 in the actin stress fiber fraction - a process controlled by microtubule polymerization state; and (iv) mDia activity is critical for stress fiber formation and myofibroblast differentiation in response to TGF-β1.
Our results indicate that microtubule depolymerization alone is sufficient to induce myofibroblast-like morphologic and, importantly, gene expression changes (Fig. 1). In contrast to TGF-β1-induced myofibroblast differentiation, these effects of microtubule disruption were independent of Smad signaling, as colchicine had no effect on Smad phosphorylation, nuclear localization or Smad-dependent gene transcription; and its effects were not associated with activation of TGF-β receptor (Fig. 2). Previous studies performed in mink lung epithelial cells, cardiac myocytes and C2C12 myoblasts have implicated microtubules in regulating Smad2/3 cytosolic localization via binding to β-tubulin (24, 25). These reports demonstrated that disruption of microtubules by nocodazole or colchicine for 18 h resulted in the induction of both basal and TGF-β-induced Smad-phosphorylation and Smad-dependent gene transcription. The differences between their and our results may represent cell-specific regulation of Smads by microtubule dynamics and suggest that at least in human lung fibroblasts, microtubule disruptors have no effect on Smad-dependent gene transcription, which is also consistent with the results of others obtained on human renal fibroblasts (8). Furthermore, these previous reports showed that microtubule stabilization by paclitaxel had no effect on TGF-β-induced Smad2 phosphorylation or Smad reporter activity in C2C12, Mv1Lu, RI14, and HepG2 cells (24, 26), which is consistent with our results obtained with human lung fibroblasts (supplemental Fig. S1).
Our data show that microtubule disruption results in Rho activation and stress fiber formation (Fig. 3), consistent with previous findings in fibroblasts (8, 10). However, to our knowledge, this is the first report linking microtubule disruption to the expression of the myofibroblast marker gene, α-SMA, through this mechanism. Furthermore, we show for the first time that even though TGF-β1 does not affect overall microtubule polymerization state (Fig. 4), its ability to induce myofibroblast differentiation is dramatically reduced by the microtubule-polymerizing agent paclitaxel (Fig. 5). Importantly, paclitaxel elicits its effect through the regulation of the stress fiber/MKL1/SRF axis (Fig. 6) without affecting TGF-β1-induced Smad signaling (supplemental Fig. S1). This suggests that myofibroblast differentiation could be targeted independently of TGF-β proximal signaling, via pharmacologic modification of the microtubule polymerization state. This intriguing possibility for treating the fibrotic response has begun to be explored, with reports demonstrating that treatment with paclitaxel attenuates the fibrotic response in several models of experimental fibrosis (34–37). The therapeutic potential of this strategy for treatment of pulmonary fibrosis is an area of current investigation in our laboratory.
The other novel basic finding of this study relates to a dynamic localization of mDia2 to stress fibers and its role in TGF-β1-induced myofibroblast differentiation. Diaphanous proteins are evolutionarily conserved multi-domain proteins that are able to interact with the actin cytoskeleton, via its formin homology 2 (FH2) domain, promoting actin polymerization (15, 16). The mammalian diaphanous family member, mDia2 also has the ability to interact and stabilize microtubules (13). Our results show that (a) mDIA2 is recruited to the actin stress fibers in response to TGF-β1 or colchicine treatment, (b) this recruitment can be regulated by microtubule polymerization (Fig. 7C), and (c) pharmacological inhibition of mDia attenuates stress fiber formation and α-SMA expression (Fig. 8). This suggests a model wherein the initial recruitment of mDia2 to stress fibers further promotes actin polymerization to drive myofibroblast differentiation.
Mechanistically, the fact that paclitaxel inhibited TGF-β1-induced accumulation of mDia2 at stress fibers (Fig. 7C), suggests that microtubules may sequester mDia2 from stress fibers. However, this does not explain TGF-β1-induced mDia translocation to stress fibers, which is not accompanied by disruption of microtubules (Fig. 4). However, TGF-β1 induces Rho signaling in fibroblasts (2) and Rho kinase has been shown to phosphorylate mDia2 and increase its actin-polymerizing activity (38). Thus, it is possible that mDia translocation to stress fibers in response to TGFβ is also mediated through this mechanism, which will be investigated in the future.
Supplementary Material
This work was supported by National Institutes of Health Grants K08 HL093367 (to N. S.), R01 GM85058 (to N. O. D), and T32HL007237 (to J. K.). This work was also supported by American Lung Association Dalsemer Award DA-161895-N (to N. S.) and American Heart Association Fellowship 10PRE4190120 (to J. K.).

This article contains supplemental Figs. S1 and S2.
- MKL1
- megakaryoblastic leukemia-1
- SRF
- serum response factor
- HLF
- human lung fibroblast(s)
- CTGF
- connective tissue growth factor
- α-SMA
- smooth muscle α-actin.
REFERENCES
- 1. Sandbo N., Dulin N. (2011) Actin cytoskeleton in myofibroblast differentiation: ultrastructure defining form and driving function. Transl. Res. 158, 181–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sandbo N., Lau A., Kach J., Ngam C., Yau D., Dulin N.O. (2011) Delayed stress fiber formation mediates pulmonary myofibroblast differentiation in response to TGF-β. Am. J. Physiol. Lung Cell Mol. Physiol. 301, L656–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Putnam A. J., Schultz K., Mooney D. J. (2001) Control of microtubule assembly by extracellular matrix and externally applied strain. Am. J. Physiol. Cell Physiol. 280, C556–564 [DOI] [PubMed] [Google Scholar]
- 4. Mooney D. J., Hansen L. K., Langer R., Vacanti J. P., Ingber D. E. (1994) Extracellular matrix controls tubulin monomer levels in hepatocytes by regulating protein turnover. Mol. Biol. Cell. 5, 1281–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heck J. N., Ponik S. M., Garcia-Mendoza M. G., Pehlke C. A., Inman D. R., Eliceiri K. W., Keely P. J. (2012) Microtubules regulate GEF-H1 in response to extracellular matrix stiffness. Mol. Biol. Cell. 23, 2583–2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Samarakoon R., Goppelt-Struebe M., Higgins P. J. (2010) Linking cell structure to gene regulation: signaling events and expression controls on the model genes PAI-1 and CTGF. Cell Signal. 22, 1413–1419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Samarakoon R., Higgins C. E., Higgins S. P., Higgins P. J. (2009) Differential requirement for MEK/ERK and SMAD signaling in PAI-1 and CTGF expression in response to microtubule disruption. Cell Signal. 21, 986–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ott C., Iwanciw D., Graness A., Giehl K., Goppelt-Struebe M. (2003) Modulation of the expression of connective tissue growth factor by alterations of the cytoskeleton. The J. Biol. Chem. 278, 44305–44311 [DOI] [PubMed] [Google Scholar]
- 9. Danowski B. A. (1989) Fibroblast contractility and actin organization are stimulated by microtubule inhibitors. J. Cell Sci. 93, 255–266 [DOI] [PubMed] [Google Scholar]
- 10. Zhang Q., Magnusson M. K., Mosher D. F. (1997) Lysophosphatidic acid and microtubule-destabilizing agents stimulate fibronectin matrix assembly through Rho-dependent actin stress fiber formation and cell contraction. Mol. Biol. Cell. 8, 1415–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ingber D. E. (2003) Tensegrity I. Cell structure and hierarchical systems biology. J. Cell Sci. 116, 1157–1173 [DOI] [PubMed] [Google Scholar]
- 12. Krendel M., Zenke F. T., Bokoch G. M. (2002) Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 4, 294–301 [DOI] [PubMed] [Google Scholar]
- 13. Bartolini F., Moseley J. B., Schmoranzer J., Cassimeris L., Goode B. L., Gundersen G. G. (2008) The formin mDia2 stabilizes microtubules independently of its actin nucleation activity. J. Cell Biol. 181, 523–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gaillard J., Ramabhadran V., Neumanne E., Gurel P., Blanchoin L., Vantard M., Higgs H. N. (2011) Differential interactions of the formins INF2, mDia1, and mDia2 with microtubules. Mol. Biol. Cell. 22, 4575–4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chesarone M. A., DuPage A. G., Goode B. L. (2010) Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat. Rev. Mol. Cell Biol. 11, 62–74 [DOI] [PubMed] [Google Scholar]
- 16. Goode B. L., Eck M. J. (2007) Mechanism and function of formins in the control of actin assembly. Annu. Rev. Biochem. 76, 593–627 [DOI] [PubMed] [Google Scholar]
- 17. Sandbo N., Kregel S., Taurin S., Bhorade S., Dulin N. O. (2009) Critical role of serum response factor in pulmonary myofibroblast differentiation induced by TGF-β. Am. J. Respir. Cell Mol. Biol. 41, 332–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Davis A., Hogarth K., Fernandes D., Solway J., Niu J., Kolenko V., Browning D., Miano J. M., Orlov S. N., Dulin N. O. (2003) Functional significance of protein kinase A activation by endothelin-1 and ATP: negative regulation of SRF-dependent gene expression by PKA. Cell Signal. 15, 597–604 [DOI] [PubMed] [Google Scholar]
- 19. Hogarth D. K., Sandbo N., Taurin S., Kolenko V., Miano J. M., Dulin N. O. (2004) Dual role of PKA in phenotypic modulation of vascular smooth muscle cells by extracellular ATP. Am. J. Physiol. Cell Physiol. 287, C449–456 [DOI] [PubMed] [Google Scholar]
- 20. Taurin S., Sandbo N., Yau D. M., Sethakorn N., Kach J., Dulin N. O. (2009) Phosphorylation of myocardin by extracellular signal-regulated kinase. J. Biol. Chem. 284, 33789–33794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Katoh K., Kano Y., Fujiwara K. (2000) Isolation and in vitro contraction of stress fibers. Methods Enzymol. 325, 369–380 [DOI] [PubMed] [Google Scholar]
- 22. Yu J. Z., Dave R. H., Allen J. A., Sarma T., Rasenick M. M. (2009) Cytosolic Gαs acts as an intracellular messenger to increase microtubule dynamics and promote neurite outgrowth. J. Biol. Chem. 284, 10462–10472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Platts S. H., Falcone J. C., Holton W. T., Hill M. A., Meininger G. A. (1999) Alteration of microtubule polymerization modulates arteriolar vasomotor tone. Am. J. Physiol. 277, H100–106 [DOI] [PubMed] [Google Scholar]
- 24. Dong C., Li Z., Alvarez R., Jr., Feng X. H., Goldschmidt-Clermont P. J. (2000) Microtubule binding to Smads may regulate TGF beta activity. Mol. Cell. 5, 27–34 [DOI] [PubMed] [Google Scholar]
- 25. Dai P., Nakagami T., Tanaka H., Hitomi T., Takamatsu T. (2007) Cx43 mediates TGF-β signaling through competitive Smads binding to microtubules. Mol. Biol. Cell. 18, 2264–2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhu S., Goldschmidt-Clermont P. J., Dong C. (2004) Transforming growth factor-β-induced inhibition of myogenesis is mediated through Smad pathway and is modulated by microtubule dynamic stability. Circ. Res. 94, 617–625 [DOI] [PubMed] [Google Scholar]
- 27. Samarakoon R., Higgins P. J. (2002) MEK/ERK pathway mediates cell shape-dependent plasminogen activator inhibitor type 1 gene expression upon drug-induced disruption of the microfilament and microtubule networks. J. Cell Sci. 115, 3093–3103 [DOI] [PubMed] [Google Scholar]
- 28. Maekawa M., Ishizaki T., Boku S., Watanabe N., Fujita A., Iwamatsu A., Obinata T., Ohashi K., Mizuno K., Narumiya S. (1999) Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285, 895–898 [DOI] [PubMed] [Google Scholar]
- 29. Desmoulière A., Geinoz A., Gabbiani F., Gabbiani G. (1993) Transforming growth factor-β 1 induces α-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 122, 103–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wen Y., Eng C. H., Schmoranzer J., Cabrera-Poch N., Morris E. J., Chen M., Wallar B. J., Alberts A. S., Gundersen G. G. (2004) EB1 and APC bind to mDia to stabilize microtubules downstream of Rho and promote cell migration. Nat. Cell Biol. 6, 820–830 [DOI] [PubMed] [Google Scholar]
- 31. Palazzo A. F., Cook T. A., Alberts A. S., Gundersen G. G. (2001) mDia mediates Rho-regulated formation and orientation of stable microtubules. Nat. Cell Biol. 3, 723–729 [DOI] [PubMed] [Google Scholar]
- 32. Rizvi S. A., Neidt E. M., Cui J., Feiger Z., Skau C. T., Gardel M. L., Kozmin S. A., Kovar D. R. (2009) Identification and characterization of a small molecule inhibitor of formin-mediated actin assembly. Chem. Biol. 16, 1158–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oakes P. W., Beckham Y., Stricker J., Gardel M. L. (2012) Tension is required but not sufficient for focal adhesion maturation without a stress fiber template. J. Cell Biol. 196, 363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brahn E., Tang C., Banquerigo M. L. (1994) Regression of collagen-induced arthritis with taxol, a microtubule stabilizer. Arthritis Rheum. 37, 839–845 [DOI] [PubMed] [Google Scholar]
- 35. Liu X., Zhu S., Wang T., Hummers L., Wigley F. M., Goldschmidt-Clermont P. J., Dong C. (2005) Paclitaxel modulates TGFβ signaling in scleroderma skin grafts in immunodeficient mice. PLoS Med. 2, e354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang D., Sun L., Xian W., Liu F., Ling G., Xiao L., Liu Y., Peng Y., Haruna Y., Kanwar Y. S. (2010) Low-dose paclitaxel ameliorates renal fibrosis in rat UUO model by inhibition of TGF-β/Smad activity. Lab. Invest. 90, 436–447 [DOI] [PubMed] [Google Scholar]
- 37. Sun L., Zhang D., Liu F., Xiang X., Ling G., Xiao L., Liu Y., Zhu X., Zhan M., Yang Y., Kondeti V. K., Kanwar Y. S. (2011) Low-dose paclitaxel ameliorates fibrosis in the remnant kidney model by down-regulating miR-192. J. Pathol. 225, 364–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Staus D. P., Taylor J. M., Mack C. P. (2011) Enhancement of mDia2 activity by Rho-kinase-dependent phosphorylation of the diaphanous autoregulatory domain. Biochem. J. 439, 57–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.