

Background: Classical complement pathway activation depends on cleavage of inactive C1s by C1r.
Results: P2 Gln and P1′ Ile residues in activation loop of C1s are crucial for activation by C1r.
Conclusion: Residues at P2 and P1′ in cleavage position of C1s make important interactions with C1r active site.
Significance: Critical determinants identified for activation of the classical complement pathway.
Keywords: Complement, Molecular Dynamics, Peptidases, Protease, Serine Protease, C1r, C1s, MASP-3, Specificity, Zymogen Activation
Abstract
The serine protease, C1r, initiates activation of the classical pathway of complement, which is a crucial innate defense mechanism against pathogens and altered-self cells. C1r both autoactivates and subsequently cleaves and activates C1s. Because complement is implicated in many inflammatory diseases, an understanding of the interaction between C1r and its target substrates is required for the design of effective inhibitors of complement activation. Examination of the active site specificity of C1r using phage library technology revealed clear specificity for Gln at P2 and Ile at P1′, which are found in these positions in physiological substrates of C1r. Removal of one or both of the Gln at P2 and Ile at P1′ in the C1s substrate reduced the rate of C1r activation. Substituting a Gln residue into the P2 of the activation site of MASP-3, a protein with similar domain structure to C1s that is not normally cleaved by C1r, enabled efficient activation of this enzyme. Molecular dynamics simulations and structural modeling of the interaction of the C1s activation peptide with the active site of C1r revealed the molecular mechanisms that particularly underpin the specificity of the enzyme for the P2 Gln residue. The complement control protein domains of C1r also made important contributions to efficient activation of C1s by this enzyme, indicating that exosite interactions were also important. These data show that C1r specificity is well suited to its cleavage targets and that efficient cleavage of C1s is achieved through both active site and exosite contributions.
Introduction
Complement activation represents a crucial innate defense mechanism against invading microorganisms, providing an immediate response against microbial invasion (1). It also plays a vital role in the maintenance of immune tolerance and, because it can also target altered-self structures, is a key player in tissue homeostasis through clearance of apoptotic and necrotic cells. The complement system can be activated by the classical, lectin, or alternative pathway. The C1r3 protease is responsible for the first enzymatic events in the classical pathway of complement activation, through autoactivation and subsequent initiation of the cascade by cleaving and activating pro-enzyme C1s (2). The lectin pathway is activated by the MASP-1 and MASP-2 enzymes, whereas MASP-3, a splice variant of MASP-1, plays a presently less well characterized role in the system. The complement system is strongly implicated in many inflammatory disease states, and therefore inhibitors of the initiating proteases could be powerful anti-inflammatory agents (3). Understanding how C1r interacts with its target substrates is the key to the knowledge required to design effective inhibitors of complement activation by targeting this initiating enzyme.
It has previously been shown that C1r and C1s pro-enzymes form a heterotetrameric structure that associates with the recognition molecule, C1q, in the C1 complex (4). Binding of C1q to target ligands, such as antigen-bound antibodies, causes autoactivation of C1r by an unknown mechanism (5). It has been postulated that binding of the multiple C1q recognition sites to their ligands essentially transmits a mechanical signal to the heterotetrameric protease structure that loosens the constraints on C1r that prevent its autoactivation in the C1 complex. The activated C1r molecule is then able to cleave and activate the associated C1s enzyme, allowing it to in turn cleave its C4 and C2 physiological substrates in sequence to thus activate the complement system (6).
The C1r, C1s enzymes, and MASP enzymes are composed of six domains (Fig. 1) (7): the N-terminal CUB1-EGF-CUB2 segment of the structure contains calcium binding sites and is most likely required to bind to recognition molecules such as C1q and for homo- and heterodimerization of the enzymes. The C-terminal CCP1-CCP2-SP segment is responsible for catalysis of both a neighboring C1r molecule (autoactivation) and C1s. The CCP1 domain has been shown to play a major role in dimerization of C1r but no role in catalytic processes, whereas the CCP2 domain apparently provides an additional binding site for substrate C1r that facilitates catalysis of this substrate by the active site of the SP domain (8, 9). The precise active site specificity of C1r has never been mapped, and the relative roles played by the active site and proposed exosites in the catalysis of substrates have not previously been determined. Here we have used phage display technology to map the substrate specificity of the enzyme and then validated the data obtained using activation of the C1s protein substrate as a molecular “readout.” The data obtained and the analysis performed, including molecular dynamics simulations, indicate that the P2 Gln residue in substrates plays a vital role in catalysis by C1r, in addition to important contributions by exosite(s) most likely contained on the C1r CCP2 domain.
FIGURE 1.

C1s and MASP-3 constructs used in this study. Schematic represents the domain structures of plasma and recombinant C1r, C1s, and MASP-3. The two CUB domains, an EGF-like domain, two CCP domains, and the SP domain are indicated. The black arrow indicates the Arg-Ile cleavage site, which is required for activation of the enzyme, held together by a disulfide bond. The amino acid sequence surrounding the activation point of wild type MASP-3 and C1s are in bold type. The amino acids altered by site-directed mutagenesis are shown in red.
EXPERIMENTAL PROCEDURES
Construction of Recombinant Plasmids for Expression of the C1r, C1s, and MASP-3 Fragments
Recombinant C1r CCP12SP (residues Arg296–Asp705), recombinant C1r SP (residues Pro449–Asp705), recombinant C1s CCP12SP (residues Lys281–Asp688), recombinant C1s SP (residues Pro423–Asp688), and recombinant MASP-3 CCP12SP (residues Lys298–Arg728) were expressed and refolded with some modifications to previously described methods (10, 11). Briefly, genes for all recombinant proteins were synthesized (GenScript), and the DNA was cloned into the pET17b vector (EMD Biosciences). After transformation of the vector into Escherichia coli strain BL21(DE3)pLysS, cells were cultured at 37 °C in 2×TY (tryptone/yeast extract) broth with 50 μg/ml ampicillin and 34 μg/ml chloramphenicol to an A595 of 0.6, followed by induction with 1 mm isopropyl β-d-thiogalactopyranoside for 4 h. Following induction, the culture was centrifuged (27,000 × g, 20 min, 4 °C), and the cells were collected in 30 ml of 50 mm Tris-HCl, 20 mm EDTA, pH 7.4, and then frozen at −80 °C. The cells were thawed and sonicated on ice for 6 × 30 s. After centrifugation at 27,000 × g for 20 min, inclusion body pellets were sequentially washed and centrifuged with 10 ml of 50 mm Tris-HCl, 20 mm EDTA, pH 7.4. The washed pellet was resuspended in 10 ml of 8 m urea, 0.1 m Tris-HCl, 100 mm DTT, pH 8.3, at room temperature for 3 h. Refolding was initiated by rapid dilution dropwise into 50 mm Tris-HCl, 3 mm reduced glutathione, 1 mm oxidized glutathione, 5 mm EDTA, and 0.5 m arginine, pH 9.0. The renatured protein solutions were concentrated and dialyzed against 50 mm Tris-HCl, pH 9.0, and renatured proteins were purified on a 5-ml Q-Sepharose Fast Flow column (GE Healthcare). The bound protein was eluted with a linear NaCl gradient from 0 to 400 mm over 35 ml at 1 ml/min. The recombinant proteins were further purified using a Superdex 75 16/60 column (GE Healthcare) in a buffer of 50 mm Tris, 145 mm NaCl, pH 7.4, aliquoted, snap frozen, and maintained at −80 °C. The purity of the protein was confirmed by SDS-PAGE followed by Western blotting and N-terminal sequencing. Typically protein yields were between 2 and 4 mg/liter.
Western Blotting and Antibodies
Proteins were resolved by SDS-PAGE, transferred, and immunoblotted with various antibodies. The antibodies used were polyclonal C1r (Abcam), a C1s antibody directed against the unique peptide sequence CSTSVQTSRLAKSKM, and a MASP-3 antibody directed against the unique peptide sequence NPNVTDQIISSGTRT. The latter antibodies were raised in chickens as described previously (12).
Phage Display
The Novagen T7Select1-1b Phage Display system was used to generate a randomized substrate peptide library as described previously (13, 14), following the approach of Cwirla et al. (15). Amino acid peptides were displayed in low copy number (0.1–1/phage) from the T7Select1-1b vector used, making them suitable for the selection of displayed peptides that were highly susceptible to protease cleavage. As described previously (14), the substrate library was constructed by synthesizing a degenerate oligonucleotide, annealing it to complementary half-site oligonucleotides, ligating the resulting heteroduplex to vector arms and adding to a T7 phage packaging extract. The half-site oligonucleotides were 5′-GCCGCCTGGAGTGAGAG-3′ and 5′-AGCTTAGTGATGGTGATGGTGATG-3′. This library was made by using the degenerate oligonucleotide 5′-AATTCTCTCACTCCAGG-CGGC(NNK)9CATCACCATCACCATCACA-3′ (where N represents any nucleotide and K is either T or C). This added a randomized unconstrained nonameric peptide (apart from a fixed arginine residue at the fifth position) and a His6 tag to the C terminus of the 10B coat protein. The complexity of this randomized library was 7 × 106 plaque-forming units (pfu). Approximately 109 pfu of amplified phage in phage extraction buffer were bound to nickel-Sepharose beads at 4 °C. Unbound phage were removed by washing the beads with phage wash buffer (850 mm NaCl, 0.1% (w/v) Tween 20 in PBS), followed by 1 mm MgSO4 in PBS. Selection commenced by the addition of 500 nm human C1r (C1r purified from human plasma (EMD Biosciences) to the treatment tubes for rounds 1–6 of selection. Equal volumes of 1 mm MgSO4 in PBS were added to the control tube instead of protease. Both the treatment and control tubes were incubated overnight at 37 °C. Cleaved phage were recovered from the supernatant and subsequently titrated and amplified to form the sublibrary for the next round of selection. Phage that remained bound to the beads were eluted with 0.5 m imidazole and titrated to assess protease cleavage efficiency. Randomly selected individual phage plaques from round 4 were chosen for DNA sequencing. Phage DNA was amplified by PCR using dedicated primers (T7Select cloning kit; Novagen). Sequencing of PCR products using the same primers was performed using the Big-Dye 3.1 kit (GE Healthcare).
The sequencing results were analyzed to determine the statistical distribution of each amino acid at each position of the nonamer (16). This analysis allowed for codon redundancy, as well as the fact that only 32 of a possible 64 codons were represented by NNK. In the following equation, Δσ indicates the difference of the observed frequency from the expected frequency in terms of standard deviations
where Obs(X) is the number of times amino acid X occurs in the selected sequences, P(X) is the theoretical probability of amino acid X occurring, and n is the total number of sequences analyzed.
Measurement of the Kinetics of Activation of Zymogen Proteases by C1r
Recombinant C1r at 100 nm was added to varying concentrations of zymogenic C1s or MASP-3 in 20 mm Tris-HCl, 100 mm NaCl, pH 7.4, and 50 μm Z-Leu-Gly-Arg-AMC (for C1s measurements (LGR-AMC)) or Z-Val-Pro-Arg-AMC (for MASP-3 measurements (VPR-AMC)) previously preincubated at 37 °C for 10 min. The appearance of fluorescence was measured using excitation and emission wavelengths of 355 and 460 nm, respectively. C1r was not active against the peptide substrates at the concentrations used, and therefore the increase in fluorescence seen was due entirely to the activity of C1s or MASP-3 activated by C1r.
The observed increase in fluorescence over time was fitted to an equation for exponential increase by nonlinear regression in GraphPad Prism: Y = Y0*exp(kobs*X). This gave a kobs value that equated to the observed rate of increase in fluorescence. The kobs values obtained at different concentrations of substrate (zymogen C1s or MASP-3) were plotted against the substrate concentrations to yield a Michaelis-Menten plot that could be fitted by nonlinear regression in GraphPad Prism to the equation: Y = Vmax*[S]/(Km + [S]). The Vmax values obtained from this analysis were converted into kcat values by taking into account the kcat of activated C1s or MASP-3 for the cognate reporter substrate used for each enzyme to yield estimates of product formation at the Vmax values obtained. The Michaelis-Menten plots could thus be used to derive Km, kcat, and kcat/Km values for the reaction of C1r with the zymogen forms of C1s and MASP-3.
Molecular Modeling and Dynamics
Missing residues (493–497) in the C1r x-ray crystal (Protein Data Bank ID code 1MD8 (7) were modeled using Modeller version 9.8 (17) The model was then superimposed onto the kallikrein chain of the kallikrein-hirustasin complex (18) using PyMOL version 1.3r2 (19). Peptides EEKQRIILG, EEKNRIILG, EEKQRAILG, EEKGRIILG, and EEKNRAILG were threaded onto the hirustasin coordinates, resulting in two C1r-peptide complexes. Each complex was then placed in a cubic unit cell with a minimum distance of 1.4 nm to the box edge and solvated in explicit SPC water (20). To neutralize the system at a physiological salt concentration of 0.1 m, Cl− and Na+ ions were randomly replaced with water molecules.
All models were then subjected to energy minimization with the conjugate gradient algorithm and a tolerance of 100 kJ mol−1·nm−1. Following the EM stage, systems were subjected to a positional restraints procedure in which a harmonic restraint was applied to all heavy atoms in C1r and bound peptides. In the procedure, the restraint was gradually decreased from 1000 to 0 kJ mol−1·nm−1 during 0.5-ns simulations. All models were then subjected to a 100-ns-long molecular dynamics simulation, each repeated three times with different random initial velocities. All simulations and trajectories analysis were conducted using the GOMACS package version 4.0.7 in conjunction with the GROMOS 53A6 united atom force field (21). During the energy minimization, the lengths of all bonds within the system were constrained using the LINCS algorithm (22). Nonbonded interactions were evaluated using a twin range cutoff scheme: interactions falling within the 0.8-nm short range cutoff were calculated every step, whereas interactions within the 1.4-nm-long cutoff were updated every three steps, together with the pair list. A reaction-field correction was applied to the electrostatic interactions beyond the long range cutoff (23), using a relative dielectric permittivity constant of ϵRF = 62 as appropriate for SPC water (24). Temperature and pressure were kept constant during simulations using the Berendsen coupling algorithm (25). Temperature was maintained at 300 K by independently coupling both protein and solvent to external temperature baths with a coupling constant of τ = 0.1 ps. The pressure was maintained at 1 bar by weakly coupling the system to an isotropic pressure bath, using an isothermal compressibility of 4.6 × 10–5 bar−1 and a coupling constant of τP = 1 ps.
The electrostatic potentials were calculated using APBS version 1.3 (26). Atomic parameters for the calculation were taken from the GROMOS 53A6 force field (21). Electrostatic potential was visualized using PyMOL version 1.3r2 (19) with positive potential in blue and negative potential in red in a range between −1 and +1 kbT/ec, where kb is the Boltzmann constant, T is the temperature (set to 300 K), and ec is electron charge.
RESULTS
Analysis of the Active Site Specificity of C1r Using Phage Display Technology
The specificity of C1r for positively charged residues at the P1 position of physiological substrates was known (7), and therefore the phage library was constructed with an Arg residue fixed at the fifth position in the randomized sequence. A large concentration of the enzyme had to be used to obtain selection with the library. Six rounds of panning using cleavage by C1r were conducted, with the titer of the protease-selected sublibrary increasing at each round until round five. 94 samples were selected for sequencing, with 29 viable sequences obtained. Once adjusted statistically, the results (Fig. 2) clearly revealed that the enzyme displays considerable specificity at every position apart from P4, P3′, and P4′. The most significant results (Δσ ≥ 5) were: Gln at P2 (Δσ = 6.4), Leu at P3 (Δσ = 5.8) (Ile was nearly as high as Leu with Δσ = 4.3), Ile at P1′ (Δσ = 5.3) (Val is quite significant with Δσ = 3.9), and Tyr at P2′ (Δσ = 6.4), with Trp also significant at this position (Δσ = 4.3). The presence of Arg residues at P5 was significant (Δσ = 6.4), although it must be noted that this position is close to the phage capsid. Of the residues identified to be important at each position, it was notable that the preference for Gln residues at P2 and Ile residues at P1′ matched that of the physiological substrates for C1r, particularly those found in zymogen C1s.
FIGURE 2.

Subsite profiling of human C1r using a phage display library with a fixed P1 arginine. A library of peptides exploring the P5–P4′ positions was exposed to 500 nm human C1r over six rounds of panning. Sequences of phage cleaved by C1r were analyzed, yielding Δσ values for each subsite. The Δσ values represent the number of S.D. away from an expected “normal” to identify overrepresentation of particular amino acids at any given substrate position.
Analysis of the Importance of Cleavage Site Residues to Catalysis by C1r
Having noted that Gln and Ile residues at P2 and P1′, respectively, were important for cleavage of phage displayed substrates, we set out to confirm the importance of these residues for cleavage by C1r. A series of fluorescence-quenched peptide substrates, comprising residues from the cleavage site in C1s and some peptides found among the phage-displayed peptides, were synthesized. Unfortunately, very high concentrations of C1r were required to cleave such peptides, and erratic results were recorded for kinetic analyses. This indicated that the enzyme displayed poor activity in general against peptide substrates, and therefore another means had to be found to investigate cleavage by C1r.
We therefore decided to use a CCP1-CCP2-SP form of zymogen C1s to test for cleavage by recombinant C1r and C1r purified from human plasma. We could show that the protein substrate was efficiently cleaved by both forms of C1r, making this a better means of testing the specificity determinants for C1r cleavage (Fig. 3). We therefore constructed mutants of zymogenic C1s in which the P2 and P1′ positions were altered (Fig. 1) and tested the kinetics of cleavage of the substrates using a coupled assay in which activation of the C1s was revealed by measuring its activity against the peptide substrate, LGR-AMC (Fig. 4). The C1r had no activity against the peptide substrate at the concentrations of enzyme used. Varying the concentration of the C1s substrate allowed the effect of substrate concentration on the observed rate of activation of C1s (kobs) to be measured. The curves of fluorescence obtained could be fitted to an exponential function (Fig. 4). Plots of the kobs values obtained versus the substrate concentration could be fitted to the Michaelis-Menten equation (Fig. 4), thus allowing Km and kcat values to be estimated once the kinetics of cleavage of the peptide substrate by C1s was taken into account (Table 1).
FIGURE 3.

C1r cleavage of zymogen C1s. Recombinant wild type zymogen C1s (1 μm) was incubated with recombinant wild type C1r (10 nm) and subjected to SDS-PAGE, with subsequent immunoblot analysis using antibodies raised against a SP domain peptide of C1s (middle panel) and a polyclonal C1r antibody (bottom panel). Each panel of the experiment shows a time course (0, 1, 5, 10, and 30 min) for incubation at 37 °C (second through fifth lanes).
FIGURE 4.

Kinetics of activation of C1s and MASP-3 mutants by C1r. Left panels, progress curves of C1r (100 nm) activation of indicated concentrations of WT C1s, Q462N C1s CCP12SP, MASP-3 K448Q, and C1s SP in the presence of either 50 μm LGR-AMC (for C1s) or VPR-AMC (for MASP-3). AFU, arbitrary fluorescence units. Right panels, plots of the observed rate of activation (kobs) as a function of the concentration of substrate.
TABLE 1.
Kinetic parameters for cleavage of wild type and mutant forms of C1s and MASP-3 by C1r CCP12SP and C1r SP enzymes
Cleavage of the wild type and mutant forms of C1s and MASP-3 was followed by monitoring the appearance of their activity using fluorescent substrates, and data were fitted to allow the determination of the kinetic parameters shown below.
| Substrate | Km | kcat | kcat/Km |
|---|---|---|---|
| nm | s−1 | m−1s−1 | |
| Substrate with C1r CCP1-CCP2SP | |||
| Wild type C1s CCP1-CCP2SP | 22.0 | 6.45 × 10−2 | 2.9 × 106 |
| C1s CCP1-CCP2SP Q462N | 148 | 6.3 × 10−3 | 4.2 × 104 |
| C1s CCP1-CCP2SP Q462G | 42.6 | 3.9 × 10−2 | 9.1 × 105 |
| C1s CCP1-CCP2SP I464A | 38.8 | 4.75 × 10−3 | 1.2 × 105 |
| C1s CCP1-CCP2SP Q462N and I464A | 445 | NDa | ND |
| C1s SP | 45.4 | 3.25 × 10−2 | 7.2 × 105 |
| Wild type MASP-3 CCP1-CCP2SP | 237 | ND | ND |
| MASP-3 K448Q | 62.2 | 2.1 × 10−2 | 3.4 × 105 |
| Substrate with C1r SP | |||
| Wild type C1s CCP1-CCP2SP | 191 | 7.4 × 10−3 | 3.9 × 104 |
| C1s SP | 1730 | 3.3 × 10−2 | 1.9 × 104 |
a ND, not determined because rates of cleavage were too low to allow determination of these parameters.
The data obtained showed that wild type zymogen C1s was very efficiently cleaved by C1r, with a very low Km value of 22 nm and an overall kcat/Km value of 2.9 × 106 m−1·s−1. Substitution of the P2 Gln residue of zymogen C1s by a chemically similar Asn residue (Q462N) resulted in a 69-fold decrease in the kcat/Km value for the reaction, strongly influenced by a 7-fold increase in the Km value. Interestingly, substitution of the P2 Gln by a Gly residue (Q462G) brought about a much smaller 3-fold decrease in the kcat/Km value, indicating that the substitution by the Asn residue was especially detrimental at this position.
Altering the P1′ Ile residue to an Ala residue (I464A) also had a strong effect on the kcat/Km value for the reaction, decreasing it 24-fold, mainly due to a decrease in the kcat value for the reaction (14-fold). Interestingly, the activated C1s with an Ala residue at the new N terminus was still active against the peptide substrate, with the kcat/Km value of 1.2 × 103 m−1·s−1 for the mutant only 6-fold lower than that for wild type C1s (7.1 × 103 m−1·s−1) (Fig. 5), indicating that the Ala residue was able to substitute for Ile at this crucial position. It is worth noting that substituting the Ile at the new N terminus of activated trypsin with an Ala residue resulted in a similar reduction in activity against most peptide substrates tested (27). Alteration of both the P2 and P1′ residues simultaneously resulted in a form of C1s which C1r was only able to cleave very weakly, such that kcat residues could not be estimated and the Km value was increased >20-fold.
FIGURE 5.

Dependence of initial velocity on substrate concentration for wild type C1s and C1s I464A. Initial velocity values for cleavage of Z-LGR-AMC (0–500 μm) by 100 nm wild type C1s (A) and C1s I464A (B), to the substrate are shown. Data were fitted by nonlinear regression to the Michaelis-Menten equation.
These data indicated that the Gln residue found at the P2 position of the physiological substrates was of high importance for efficient cleavage by C1r. To further verify this, we substituted the Lys residue found at the P2 position of zymogen MASP-3 with a Gln residue (K448Q) (Fig. 1) and investigated whether C1r, which was essentially unable to cleave wild type MASP-3 (Fig. 5), could efficiently activate this protease with a similar domain structure to C1s. Interestingly, the mutated MASP-3 was efficiently activated by C1r (Fig. 6), with a kcat/Km value only 8.5-fold lower than that for wild type C1s zymogen (Fig. 4 and Table 1). The kcat value was comparable with that found for C1s, whereas the Km value was nearly 3-fold higher.
FIGURE 6.

C1r cleavage of wild type and mutant MASP-3. Top panel, zymogen forms of wild type MASP-3 and K448Q MASP-3 were incubated with or without C1r at 37 °C for 60 or 300 min, as indicated, and samples were analyzed by SDS-PAGE. Bottom panel, cleavage of the K448Q mutant after 300 min was further analyzed by immunoblotting using antibodies raised against a MASP-3 SP domain peptide.
Investigation of the Relative Effect of CCP Domains on Cleavage by C1r
These data indicate that the residues found in the activation loop of the zymogens capable of being activated by C1r play a major role in recognition of the active site of C1r and in turn validate the results demonstrated using phage display technology. It appears that there was still some activation of C1s occurring even when both the important P2 and P1′ residues were altered, however, indicating that other parts of the enzyme might be playing a role in recognizing cognate physiological substrates of the enzyme. It has previously been demonstrated that the CCP domains of C1r play an important role in the recognition of substrate C1r molecules in the autoactivation reaction (8). We therefore set out to determine the importance of such exosites on the body of the protease by examining the effect of eliminating the CCP domains from the C1s substrate, reasoning that these domains may be playing an important role in the recognition of the substrate protein. We found that the C1s SP domain alone was activated with a 4-fold lower kcat/Km value than that found for the CCP1-CCP2-SP form of the substrate, due to changes in both the Km and kcat values for the reaction (Fig. 4 and Table 1). Removal of the CCP domains from C1r had a much stronger effect on the interaction, decreasing the kcat/Km value 74-fold. Removal of the CCP domains from the C1s substrate resulted in only a further 2-fold decrease in the kcat/Km value, indicating that the CCP domains of C1r played the dominant role in recognition of the substrate protein.
Molecular Dynamics Simulations of the Interaction between C1r and Zymogen C1s
Both phage display libraries and kinetic studies have shown the importance of Gln and Ile residues at position P2 and P1′, respectively, for substrate cleavage by C1r. To gain a structural and dynamic insight into C1r-substrate interactions, we modeled a series of five nona-peptides bound to C1r and subjected them to molecular dynamics simulations. Snapshots after 100 ns of simulations indicate that whereas the WT C1s peptide maintains the modeled canonical and stable conformation in the active site (Fig. 7A), other peptides, representing mutations at P2 and P1′, undergo rapid conformational fluctuations, resulting in the loss of most protein-peptide interactions (Fig. 7, B–F, and supplemental Movies S1 and S2). The calculated root mean square deviation for the last 90 ns of simulation for the peptide similar to WT C1s was 0.22 ± 0.05 nm (EEKQRIILG), significantly lower than the root mean square deviation calculated for the other peptides: 0.62 ± 0.09 nm (EEKNRIILG), 0.52 ± 0.07 nm (EEKQRAILG), 0.56 ± 0.09 nm (EEKGRIILG), and 0.53 ± 0.1 nm (EEKNRAILG). The simulations provide a simple and physically straightforward molecular insight into the differences in measured Km and kcat. The mutation Ile→Ala at P1′ causes a weakening of its interactions at S1′. This is also the case for the mutation Gln→Gly at position P2, in which an amide group is removed, resulting in a loss of interactions between P2 and S2. In contrast, the mutation Gln→Asn at P2 results in conformational change of the side chain and subsequent loss of interactions with S2 (Fig. 8). Modeling and molecular dynamics simulations suggest that the shortening of the side chain and different side chain rotamer preferences of Asn compared with Gln may result in the introduction of its polar amide group into the nonpolar portion of the S2 binding pocket, destabilizing the C1r-peptide complex.
FIGURE 7.

Structural models of complexes between C1r protease and bound peptides. A–E, snapshot after 100 ns of simulation of C1r protease, represented as a gray scheme, with bound EEKQRIILG (A), EEKNRIILG (B), EEKQRAILG (C), EEKGRIILG (D), and EEKNRAILG (E) represented as sticks. F, root mean square deviation (RMSD) of backbone atoms of the EEKQRIILG (black), EEKNRIILG (red), EEKQRAILG (blue), EEKGRIILG (green), and EEKNRAILG (orange) peptides calculated after structural fitting of C1r backbone atoms.
FIGURE 8.
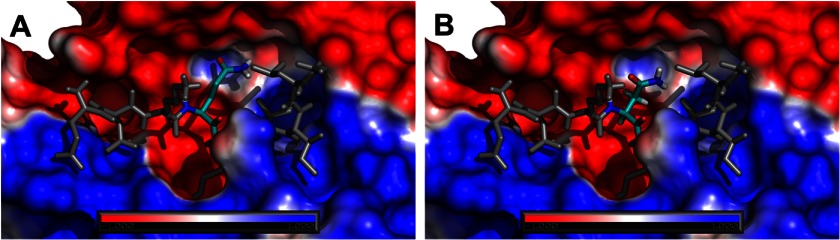
Electrostatic potential of C1r protease with bound EEKQRIILG (A) and EEKNRIILG (B) represented as sticks mapped on its solvent accessible surface. Color coding is according to the electrostatic potential gradient, where positively and negatively charged areas are represented in blue and red (iso-values from +1 kbT/ec to −1 kbT/ec), respectively.
DISCUSSION
The complement system is vital for the proper function of the immune system, but also contributes to inflammatory diseases, therefore understanding initiating events in the pathways controlling activation is crucial to the design of inhibitors that can precisely target them to alleviate diseases in which complement is involved (3). Here we have provided evidence that indicates that the recognition of residues in the cleavage site by the active site of C1r, the initiating protease of the classical pathway, is equally important to recognition of substrate C1s via exosites found on the CCP domains of the activating C1r enzyme.
Analysis of the specificity of C1r using phage display technology revealed that the enzyme displayed strong specificity for residues at the P2, P1′, and P2′ of substrates. The strong preference displayed for Gln residues at P2 and Ile residues at P1′ matched those found in physiological substrates of C1r, i.e. itself and zymogen C1s. The lack of convincing activity of C1r for peptide substrates meant that entire protein substrates had to be used to map the importance of the cleavage site residues for recognition by C1r. These analyses confirmed the importance of the P2 Gln residue in particular and introduction of this residue alone into MASP-3, a lectin pathway protease with similar domain structure (6), but different cleavage site residues at the nonprime side in particular, were sufficient to render this protease efficiently activated by C1r.
Molecular dynamics simulations confirmed that the Gln residue found at P2 was indeed highly important for recognition by the active site of C1r. These studies particularly explained why substituting an Asn residue at the P2 site was so much more detrimental than altering this residue to a Gly. Reduction of the side chain group of the P2 residue by one carbon most likely brings the polar head groups of the Asn residue into contact with hydrophobic residues surrounding the S2 pocket of C1r, which is clearly highly detrimental for binding, as demonstrated in the molecular dynamics simulations. Alteration of the P2 Gln to a Gly residue eliminates the binding interaction predicted to occur at S2 for the polar head group of the Gln residue, but the clash between the polar group on Asn and the hydrophobic surrounds of the S2 pocket is not found for the Gly residue, thus explaining its lower effect on the kcat/Km value for C1r activation of C1s.
Our results indicate that the CCP domains of C1r also play an important role in the activation of C1s by C1r, whereas the CCP domains of C1s play a lesser role in recognition of the activating protease. The crystal structure of C1r (7) shows the protease in a head to tail dimer, with the CCP domains of the protease mediating strong contacts with the dimer partner. C1r and C1s apparently form a tetramer in the C1 complex, and it has been postulated that the head to tail dimer of C1r forms the core of the tetrameric structure. Such an arrangement would indeed facilitate contact between the active site of one C1r molecule with the cleavage loop of the other partner, albeit that a rearrangement would still be required over that found in the crystal structure to allow these regions to form the intimate contacts required for activation (28). Our results indicate that a substantial change must occur in the C1 complex following autoactivation of C1r to allow the C1r CCP domains to mediate contacts with the C1s molecule. Such exosite interactions would then be in addition to the contacts formed between active site of C1r and the cleavage loop of C1s. Our results have therefore provided important insights into the contacts required between the active site of C1r and the cleavage loop of C1s, with the P2 Gln residue being of particular importance in this regard. These data will facilitate the development of inhibitors of C1r for the treatment of inflammatory diseases.
Supplementary Material
Acknowledgments
We thank Antony Matthews for supplying the phage display library used in this study, Mrs. Usha Koul for excellent technical assistance, and Prof. J. A. Huntington (University of Cambridge) for useful discussions.
This work was supported by a program grant from the National Health and Medical Research Council of Australia.

This article contains supplemental Movies S1 and S2 and an additional reference.
- C1r
- complement subcomponent 1r
- AMC
- 7-amino-4-methyl coumarin
- C1
- complement component 1
- C1q
- complement subcomponent 1q
- C1s
- complement subcomponent 1s
- CCP
- complement control protein
- CUB
- module originally found in complement C1r/C1s, Uegf, and bone morphogenetic protein
- MASP
- mannose-binding lectin-associated serine protease
- SP
- serine protease.
REFERENCES
- 1. Frank M. M., Fries L. F. (1991) The role of complement in inflammation and phagocytosis. Immunol. Today 12, 322–326 [DOI] [PubMed] [Google Scholar]
- 2. Duncan R. C., Wijeyewickrema L. C., Pike R. N. (2008) The initiating proteases of the complement system: controlling the cleavage. Biochimie 90, 387–395 [DOI] [PubMed] [Google Scholar]
- 3. de Cordoba S. R., Tortajada A., Harris C. L., Morgan B. P. (2012) Complement dysregulation and disease: from genes and proteins to diagnostics and drugs. Immunobiology 217, 1034–1046 [DOI] [PubMed] [Google Scholar]
- 4. Gaboriaud C., Teillet F., Gregory L. A., Thielens N. M., Arlaud G. J. (2007) Assembly of C1 and the MBL- and ficolin-MASP complexes: structural insights. Immunobiology 212, 279–288 [DOI] [PubMed] [Google Scholar]
- 5. Arlaud G. J., Gaboriaud C., Thielens N. M., Budayova-Spano M., Rossi V., Fontecilla-Camps J. C. (2002) Structural biology of the C1 complex of complement unveils the mechanisms of its activation and proteolytic activity. Mol. Immunol. 39, 383–394 [DOI] [PubMed] [Google Scholar]
- 6. Gál P., Dobó J., Závodszky P., Sim R. B. (2009) Early complement proteases: C1r, C1s and MASPs. A structural insight into activation and functions. Mol. Immunol. 46, 2745–2752 [DOI] [PubMed] [Google Scholar]
- 7. Budayova-Spano M., Grabarse W., Thielens N. M., Hillen H., Lacroix M., Schmidt M., Fontecilla-Camps J. C., Arlaud G. J., Gaboriaud C. (2002) Monomeric structures of the zymogen and active catalytic domain of complement protease C1r: further insights into the C1 activation mechanism. Structure 10, 1509–1519 [DOI] [PubMed] [Google Scholar]
- 8. Kardos J., Gál P., Szilágyi L., Thielens N. M., Szilágyi K., Lõrincz Z., Kulcsár P., Gráf L., Arlaud G. J., Závodszky P. (2001) The role of the individual domains in the structure and function of the catalytic region of a modular serine protease, C1r. J. Immunol. 167, 5202–5208 [DOI] [PubMed] [Google Scholar]
- 9. Lacroix M., Ebel C., Kardos J., Dobó J., Gál P., Závodszky P., Arlaud G. J., Thielens N. M. (2001) Assembly and enzymatic properties of the catalytic domain of human complement protease C1r. J. Biol. Chem. 276, 36233–36240 [DOI] [PubMed] [Google Scholar]
- 10. Duncan R. C., Bergström F., Coetzer T. H., Blom A. M., Wijeyewickrema L. C., Pike R. N. (2012) Multiple domains of MASP-2, an initiating complement protease, are required for interaction with its substrate C4. Mol. Immunol. 49, 593–600 [DOI] [PubMed] [Google Scholar]
- 11. Duncan R. C., Mohlin F., Taleski D., Coetzer T. H., Huntington J. A., Payne R. J., Blom A. M., Pike R. N., Wijeyewickrema L. C. (2012) Identification of a catalytic exosite for complement component C4 on the serine protease domain of C1s. J. Immunol. 189, 2365–2373 [DOI] [PubMed] [Google Scholar]
- 12. Goldring J. P., Thobakgale C., Hiltunen T., Coetzer T. H. (2005) Raising antibodies in chickens against primaquine, pyrimethamine, dapsone, tetracycline, and doxycycline. Immunol. Invest. 34, 101–114 [PubMed] [Google Scholar]
- 13. Kerr F. K., Thomas A. R., Wijeyewickrema L. C., Whisstock J. C., Boyd S. E., Kaiserman D., Matthews A. Y., Bird P. I., Thielens N. M., Rossi V., Pike R. N. (2008) Elucidation of the substrate specificity of the MASP-2 protease of the lectin complement pathway and identification of the enzyme as a major physiological target of the serpin, C1-inhibitor. Mol. Immunol. 45, 670–677 [DOI] [PubMed] [Google Scholar]
- 14. Kaiserman D., Bird C. H., Sun J., Matthews A., Ung K., Whisstock J. C., Thompson P. E., Trapani J. A., Bird P. I. (2006) The major human and mouse granzymes are structurally and functionally divergent. J. Cell Biol. 175, 619–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cwirla S. E., Peters E. A., Barrett R. W., Dower W. J. (1990) Peptides on phage: a vast library of peptides for identifying ligands. Proc. Natl. Acad. Sci. U.S.A. 87, 6378–6382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matthews D. J., Goodman L. J., Gorman C. M., Wells J. A. (1994) A survey of furin substrate specificity using substrate phage display. Protein Sci. 3, 1197–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eswar N, Marti-Renom M. A, Webb B, Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., Sali A. (2006) Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 5, 5.6.1–5.6.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mittl P. R., Di Marco S., Fendrich G., Pohlig G., Heim J., Sommerhoff C., Fritz H., Priestle J. P., Grütter M. G. (1997) A new structural class of serine protease inhibitors revealed by the structure of the hirustasin-kallikrein complex. Structure 5, 253–264 [DOI] [PubMed] [Google Scholar]
- 19. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.3r1, Schrödinger, LLC, New York [Google Scholar]
- 20. Berendsen H. J., Postma J. P., van Gunsteren W. F. (1981) in Intermolecular Forces (Pullman B., ed) pp. 331–342, Reidel, Dordrecht [Google Scholar]
- 21. Oostenbrink C., Villa A., Mark A. E., van Gunsteren W. F. (2004) A biomolecular force field based on the free enthalpy of hydration and solvation: the GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 25, 1656–1676 [DOI] [PubMed] [Google Scholar]
- 22. Hess B., Berendsen H. J., Fraaije J. G. (1997) LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 [Google Scholar]
- 23. Tironi S. R., Smith P. E., van Gunsteren W. F. (1995) A generalized reaction field method for molecular-dynamics simulations. J. Chem. Phys. 102, 5451–5459 [Google Scholar]
- 24. Hünenberger H. T., van Gunsteren W. F. (2001) Comparison of four methods to compute the dielectric permittivity of liquids from molecular dynamics simulations. J. Chem. Phys. 115, 1125–1136 [Google Scholar]
- 25. Berendsen H. J., van Gunsteren J. P., Dinola A., Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 26. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hedstrom L., Lin T. Y., Fast W. (1996) Hydrophobic interactions control zymogen activation in the trypsin family of serine proteases. Biochemistry 35, 4515–4523 [DOI] [PubMed] [Google Scholar]
- 28. Wallis R., Mitchell D. A., Schmid R., Schwaeble W. J., Keeble A. H. (2010) Paths reunited: initiation of the classical and lectin pathways of complement activation. Immunobiology 215, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.