

Background: Thirty-eight NF-κB-signaling genes are analyzed for tissue expression profile and pretranscriptional mechanisms.
Results: NF-κB-signaling genes are differentially expressed and can be regulated by specific transcription factors, multiple alternative promoters/spliced isoforms, DNA methylation, and microRNAs.
Conclusion: Pretranslational regulatory mechanisms contribute to NF-κB activation and inflammatory diseases.
Significance: Pretranslational regulatory mechanisms can be used as therapeutic targets.
Keywords: Cardiovascular Disease, Cytokine, Gene Expression, NF-kappaB, Translation, NF-kappaB Activation, Cardiovascular Disease Risk Factors, Gene Expression Profile, Pretranslational Mechanisms, Proinflammatory Cytokines
Abstract
NF-κB-controlled transcriptional regulation plays a central role in inflammatory and immune responses. Currently, understanding about NF-κB activation mechanism emphasizes IκB-tethered complex inactivation in the cytoplasm. In the case of NF-κB activation, IκB phosphorylation leads to its degradation, followed by NF-κB relocation to the nucleus and trans-activation of NF-κB-targeted genes. Pretranslational mechanism mediated NF-κB activation remains poorly understood. In this study, we investigated NF-κB pretranslational regulation by performing a series of database mining analyses and using six large national experimental databases (National Center of Biotechnology Information UniGene expressed sequence tag profile database, Gene Expression Omnibus database, Transcription Element Search System database, AceView database, and Epigenomics database) and TargetScan software. We reported the following findings: 1) NF-κB-signaling genes are differentially expressed in human and mouse tissues; 2) heart and vessels are the inflammation-privileged tissues and less easy to be inflamed because lacking in key NF-κB-signaling molecular expression; 3) NF-κB-signaling genes are induced by cardiovascular disease risk factors oxidized phospholipids and proinflammatory cytokines in endothelial cells; 4) transcription factors CCAAT/enhancer-binding proteins and NF-κB have higher binding site frequencies in the promoters of proinflammatory cytokine-induced NF-κB genes; 5) most NF-κB-signaling genes have multiple alternative promoters and alternatively spliced isoforms; 6) NF-κB family genes can be regulated by DNA methylation; and 7) 27 of 38 NF-κB-signaling genes can be regulated by microRNAs. Our findings provide important insight into the mechanism of NF-κB activation, which may contribute to cardiovascular disease, inflammatory diseases, and immunological disorders.
Introduction
Nuclear factor κ light chain enhancer of activated B cells (NF-κB)3 transcription factors play critical roles in a wide range of biological processes, including immunity, stress responses, apoptosis, and maturation of various cell types, which were discovered 26 years ago (1). There are five members of the NF-κB family in mammals, including RelA (also known as p65), RelB, c-Rel (also known as Rel), and the precursor proteins NF-κB1 (p105) and NF-κB2 (p100), as shown in Fig. 1A. Classical NF-κB signaling involves at least 38 NF-κB-signaling proteins and proceeds through seven intracellular adapter family proteins, including receptor-interacting proteins (RIPKs; RIPK1/2/3) and TNF receptor-associated factors (TRAFs; TRAF2/3/5/6), which leads to the activation of inhibitor of IκB kinase kinase (IKK kinase) family protein TGFβ-activated kinase-1 (Tak1) and NF-κB-inducing kinase (NIK). In the canonical pathway, Tak1 phosphorylates IKKγ, whereas in the non-canonical pathway, NIK is required for IKKα activation and p100 phosphorylation (2). The IKK family has two catalytically active kinases, IKKα and IKKβ, and a regulatory subunit IKKγ (also known as NEMO), which phosphorylates inhibitor of κB (IκB) family protein and leads to ubiquitination and degradation of the IκBs to release and activate NF-κB (3). The IκB protein family comprises the typical IκB proteins IκBα, IκBβ, and IκBϵ, which sequester inactive NF-κB complexes in the cytoplasm of unstimulated cells, and the atypical IκB proteins IκBζ, IκBNS, and Bcl-3 (B-cell lymphoma 3), which mediate their effects in the nucleus. IKK substrates also include cyclin D1 (CCND1), nuclear receptor corepressor 2 (SMRT), CREB-binding protein (CBP), histone deacetylase 3 (HDAC3), B-cell CLL/lymphoma 10 (BCL10), and cylindromatosis turban tumor syndrome (CYLD) (4). It has been reported that additional factors can regulate NF-κB activity, such as Akirin proteins (Akirin1 and Akirin2). Mitogen-activated protein kinase-activated protein kinase 2 (MK2) has been implicated in dampening the activity of NF-κB by regulating p38. It has been suggested that at late stages of activation, nuclear RelA (p65)-containing heterodimers are removed through ubiquitylation and proteasomal degradation. NF-κB degradation can be regulated by suppressor of cytokine signaling 1 (SOCS1), which participates in the formation of elongin-B-elongin-C-cullin-2-SOCS1 (ECS) ubiquitin ligase complex; copper metabolism domain-containing 1 (COMMD1) protein, which promotes associations between RelA (p65)-containing dimers; and SOCS1, PDZ, and LIM domain protein2 (PDLIM2), which is a nuclear ubiquitin ligase and the protein inhibitor of activated STAT (PIAS) family (3).
FIGURE 1.

A, classical NF-κB signal pathway. Thirty-eight NF-κB-signaling genes are labeled by numbers. The canonical pathway is induced by TNFα, IL-1, LPS, and other stimuli, followed by sequential recruitment of the adapters of RIPK and TRAF2 to the membrane, resulting in TAK1 activation and IKK activation. Activation of IKK leads to phosphorylation of IκBα, leading to its ubiquitylation and subsequent degradation. The heterodimer p50-RelA is then released and relocated to the nucleus binding to κB sites and activates a variety of NF-κB target genes. The non-canonical pathway is induced by LPS, CD40, LMP1 (latent membrane protein-1), and other stimuli. It relies on the recruitment of TRAF6 and the heterodimer TRAF2-TRAF3 to the CD40 receptor. NIK is subsequently activated, which activates IKKα and phosphorylates the inhibitory molecule p100, part of which generates p52. The heterodimer p52-RelB is then released and relocated for NF-κB target gene trans-activation. Nuclear NF-κB subunits can be regulated by acetylases and phosphorylase (genes 30–33) and can be removed through ubiquitylation and proteasomal degradation (genes 34–38) at the late stages of activation. B, strategies of database mining and gene identification. Thirty-eight NF-κB signal genes were selected and subjected to six functional database mining tests. 1) We established tissue expression profiles of selected genes in 20 human and 19 mouse tissues by database mining using the NCBI EST database after generating an REU. Tissues were then classified based on their gene expression profiles. 2) We identified inducible NF-κB-signaling genes by querying GEO data sets based on microarray studies. 3) We identified TFs regulating NF-κB activity in the promoters of cytokine-induced NF-κB-signaling genes (1,500 bases upstream of the transcription start site) retrieved from the NIH/NCBI Entrez Gene database. Binding frequency of 10 inflammation-related TF on the promoter was analyzed using the TESS database. 4) We identified alternative promoters and spliced isoforms of NF-κB-signaling genes using the AceView database. 5) We identified DNA methylation characteristic of NF-κB family genes using the Epigenomic database. 6) We identified microRNAs potentially targeting NF-κB-signaling genes using TargetScan software by screening a microRNA library. Gene symbols and ID numbers are detailed in Table 1.
Currently, knowledge indicates that NF-κB is present as a latent, inactive, IκB-bound protein complex in the cytoplasm of unstimulated cells. When a cell receives extracellular signals, activation of IKK complex leads to IκB phosphorylation and subsequent ubiquitination and degradation by the 26 S proteasome, followed quickly by NF-κB relocating to the nucleus and activation of NF-κB target genes. The regulatory mechanisms of the NF-κB pathway reported so far have been focused on post-translational modifications, including phosphorylation/dephosphorylation, ubiquitination, and proteasome degradation (5). However, the pretranslational regulatory mechanisms in regulating NF-κB pathways remained poorly identified. We hypothesized that in addition to post-translational mechanisms, pretranslational regulatory mechanisms play an important role in regulating NF-κB signaling. In this study, we investigated the role of tissue-specific transcription, alternative promoter/splicing, DNA methylation, and microRNA-mediated mRNA degradation and translational inhibition in NF-κB activation. Our findings provided new insights into phosphorylation- and ubiquitination-independent pretranslational mechanisms for NF-κB activation.
EXPERIMENTAL PROCEDURES
NF-κB-signaling Genes and Overall Strategy
We selected 38 NF-κB-signaling genes, including five NF-κB family genes (RelA, RelB, c-Rel, p50, and p52), six IκB family genes (IκBα, IκBβ, IκBϵ, IκBζ, IκBNS, and Bcl-3), three IKK family genes (IKKα, IKKβ, and IKKγ (NEMO)), seven adapter genes, two IKK kinase genes, six IKK substrate genes, four regulation genes, and five degradation genes (Table 1 and Fig. 1A) for the assessment of tissue expression profiles. National Center of Biotechnology Information (NCBI)/UniGene ID numbers listed in Table 1 were obtained from the National Institutes of Health (NIH)/NCBI UniGene database. The selected NF-κB-signaling genes were subjected to six functional database mining tests to 1) establish tissue expression profiles, 2) identify inducible NF-κB signaling, 3) identify transcription factor (TFs) regulating NF-κB activity in the promoters of cytokine-induced NF-κB-signaling genes, 4) identify alternative promoters and spliced isoforms of NF-κB-signaling genes, 5) identify DNA methylation characteristic of NF-κB family genes, and 6) identify potential microRNA targets on NF-κB-signaling genes by using strategies briefly summarized in Fig. 1B.
TABLE 1.
NF-κB-signaling genes in humans and mice (38 genes)
NF-κB signaling gene identifications and NCBI/UniGene ID numbers are from the NIH/NCBI UniGene database.

Data Mining and Gene Expression Profiles of NF-κB-signaling Genes in Human and Mouse Tissues (EST Analysis)
An experimental data mining strategy was applied to establish the expression profiles of mRNA transcripts of the selected genes as we described previously (6). Twenty human and 19 mouse tissues were given tissue ID numbers (Fig. 2A) and examined for mRNA expression of selected genes by mining experimentally verified human and mouse expressed sequence tag (EST) databases deposited in the NIH UniGene database. The EST database was created via cDNA cloning from various tissue cDNA libraries followed by DNA sequencing. Gene mRNA levels are described as gene transcript units per million transcripts. The gene expression profile in mouse vascular walls is not available. We generated the relative mRNA expression units (REU) of the gene by normalizing the gene transcript units per million transcripts (TPM) of the gene of interest with that of β-actin (left side of y axis in Fig. 2, B and C). In order to fairly compare gene expression across selected tissues, we further adjusted by comparison with the median REU (mREU). The mREU was determined from REU in 20 human or 19 mouse tissues selected in this study. The ratio of REU/mREU is expressed as tissue median adjusted mRNA expression units and presented in Fig. 2, B and C (right side of y axis). In order to establish a confidence interval of gene expression, we calculated the gene expression REUs and ratio of REU/mREU of randomly selected housekeeping genes in any given tissue to calculate the arbitrary units of the gene expression.
FIGURE 2.

Tissue mRNA distribution profile of NF-κB-signaling genes (EST analysis). 20 human and 19 mouse tissues were given tissue ID numbers and examined for mRNA expression by mining human and mouse EST databases on the NCBI-UniGene site (mouse vascular data were not available in the database). REU of the gene is obtained by normalizing gene transcripts per million (TPM) with that of β-actin. Tissue median-adjusted mRNA expression levels (REU/mREU) were calculated for all genes. Confidence intervals of expression of three housekeeping gene mRNAs were established. Dashed lines, upper limits of the confidence intervals of the housekeeping gene. Left and right y axes, REU and REU/mREU, respectively. A, representative tissue mRNA distribution profile of housekeeping gene PTTG1LP in humans and mice. See “Experimental Procedures” for details. B, mRNA distribution profiles of 38 NF-κB-signaling genes in 20 human tissues. C, mRNA distribution profiles of 38 NF-κB-signaling genes in 19 mouse tissues. The statistical significance was defined as when gene expression was larger than the upper limit of the confidence interval. Heart and vessel are shown as boldface columns. Gene symbols are listed in Table 1.
Housekeeping genes, including ACTB (NM_001101), ALDOA (NM_000034), ARHGDIA (NM_004309), GAPDH (NM_002046), LDHA (NM_005566), NONO (NM_001145408), PGK1 (NM_0002954), RPL11 (NM_000975), RPL19 (NM_000981), and RPS27A (NM_002954), were chosen to define confidence intervals for significant -fold change of the given genes. The confidence interval of the -fold change was generated by calculating the mean and S.D. values of the -fold change of these 10 randomly selected housekeeping genes as we described previously (6). Of note, the upper limit was the mean plus 2 × S.D.; the lower limit was the mean minus 2 × S.D. If the expression variation of a given gene in the tissues was larger than the upper limit of the confidence interval, the high expression levels of genes in the tissues was statistically significant. Any given gene transcripts, if lower than one per million, were technically presented as no expression.
Tissues were then classified based on their gene expression profiles. Three tiers of human (Table 2A) and mouse (Table 2B) tissues were categorized according to the tissue expression pattern of key components from two NF-κB signal pathways: IKKγ, IKKβ, IκBα, RelA, and p50 for canonical signaling and IKKα, RelB, and p52 for non-canonical signaling.
TABLE 2.
Classification of human and mouse tissues based on NF-κB-signaling gene expression status (EST analysis)
Three tiers of human (A) and mouse (B) tissues were categorized according to tissue expression profiles of key signaling genes: IKKα, IKKβ, IκBα, RelA, and p50 for canonical signaling and IKKα, RelB, and p52 for non-canonical signaling. Tier 1 tissue expresses both key signaling molecules and is called the “ready to go tier”; tier 2 tissue lacks key components for either canonical or non-canonical signaling and is called the “nearly ready tier”; tier 3 tissue lacks key components for both canonical and non-canonical signaling and is called the “inflammation-privileged tier” (e.g. human heart and vessel). A, tissue classification on NF-κB signaling gene expression (human). *, cardiovascular system; B, tissue classification on NF-κB signaling gene expression (mouse).

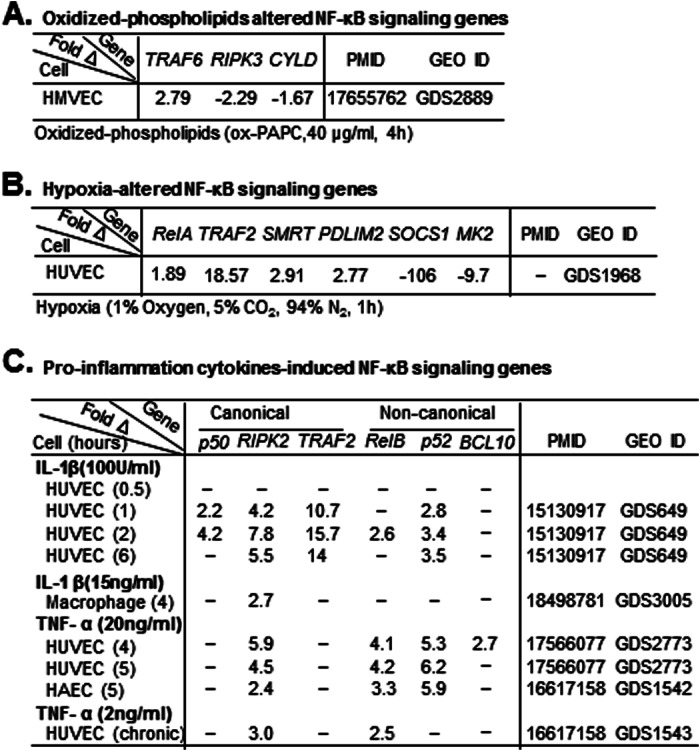
Identification of NF-κB-signaling Genes Induced by Oxidized Phospholipids, Hypoxia, and Proinflammatory Cytokines in Human Endothelial Cells and Macrophages (Gene Expression Omnibus (GEO) Analysis)
To analyze NF-κB-signaling gene expression in response to cardiovascular diseases risk factors, we examined data sets in the Gene Expression Omnibus repository, which was established based on microarray studies (Fig. 1B). We selected seven data sets (Table 3): GDS2889 from human microvascular endothelial cells treated with oxidized phospholipid (40 μg/ml, 4 h); GDS1968 from human umbilical vein endothelial cells (HUVEC) with hypoxia stimulation (1% oxygen, 5% CO2, and 94% N2, 1 h); GDS649 from HUVEC with proinflammatory cytokine interleukin-1 (IL-1) stimulation (100 units/ml, 0.5, 1, 2, and 6 h); GDS3005 from monocyte-derived macrophages stimulated with IL-1 (15 ng/ml, 4 h); and GDS2773/GDS1542/GDS1543 from HUVEC/human aortic endothelial cells acutely activated by soluble TNF-α (20 ng/ml, 4 or 5 h) or chronically activated by expression of an uncleavable form of the trans-membrane TNF precursor.
TABLE 3.
Identification of NF-κB-signaling genes induced by oxidized phospholipids, hypoxia, and proinflammatory cytokines in human endothelial cells and macrophages (GEO analysis)
NF-κB-signaling genes induced by oxidized phospholipids, hypoxia, IL-1, or TNF-α in human EC and macrophage were identified by data mining in GEO data sets based on microarray studies. A, oxidized phospholipid-altered genes. Three genes were identified. ox-PAPC, oxidized 1-palmitoyl-2-arachidonyl-sn-glycero-3-phosphorylcholine; 40 μg/ml, 4 h. B, hypoxia-altered genes. Six genes were identified. C, cytokine-induced genes. IL-1 induced five genes. TNF-α induced four genes. HMVEC, human microvascular endothelial cells. B, hypoxia-altered NF-κB-signaling genes. C, proinflammation cytokine-induced NF-κB-signaling genes.
Analysis of TF Binding Frequencies of Cytokine-induced NF-κB-signaling Genes (TESS Analysis)
The promoter sequences (1500 bp upstream of the transcription start site) of six cytokine-induced NF-κB-signaling genes identified in Table 3C (RelB, p50, p52, TRAF2, RIPK2, and Bcl-10) and three randomly chosen housekeeping genes (ACTB, GAPDH, and ARHGDIA) were retrieved from the NIH/NCBI Entrez Gene database and analyzed for binding sites of 10 inflammation-related TFs (AP-1 (activator protein 1), C/EBPs, Elk1 (E twenty-six (ETS)-like transcription factor 1), glucocorticoid receptor (GR), heat shock factor protein 1 (HSF-1), interferon regulatory factor 1 (IRF-1), Mef2 (myocyte enhancer factor-2), NF-κB, p53 (tumor protein 53), and Pu.1 (hematopoietic transcription factor Pu.1)) identified from our previous study using the TESS database (7). TF binding frequency denotes the number of the binding sites for each TF in the promoter region. The confidence interval was set by using the mean ± 2 × S.D. of the TF binding frequencies in the promoter of three housekeeping genes. The binding frequency higher than the uppermost confidence interval (p < 0.05) is considered significant.
Identification of Alternative Spliced Isoform and Alternative Promoter of NF-κB Family Genes (AceView Database Analysis)
The presence and features of alternative promoters and spliced isoforms of five NF-κB family genes (RelA, RelB, c-Rel, p50, and p52) were examined with the AceView database as we previously reported (7, 8).
Identify DNA Methylation of NF-κB Family Genes (Epigenomics Database Analysis)
DNA methylation of five NF-κB family genes (RelA, RelB, c-Rel, p50, and p52) were examined by using the Epigenomics database. The DNA methylation status of promoter regions and genomic DNA sequences were identified from experimental data and deposited in the Epigenomics database.
Prediction of MicroRNAs That Potentially Target the List of NF-κB-signaling Genes (TESS Analysis)
We employed a computational prediction strategy to identify candidate microRNAs that potentially target 3′-untranslated regions (UTRs) of 38 NF-κB-signaling genes listed in Table 1 by screening a microRNA library and using a microRNA prediction software program, TargetScan, developed at the Massachusetts Institute of Technology (Cambridge, MA). This software generates a context value and context percentage for microRNA-gene interaction. We then judged each microRNA-gene reaction based on confidence intervals we established recently (9). MicroRNA-gene interactions with a context value ≤−0.22 and context percentage ≥70 were accepted.
RESULTS
The NF-κB-signaling Genes Are Differentially Expressed in Human and Mouse Tissues
The expression profiles of 38 NF-κB-signaling genes in 20 human tissues and 19 mouse tissues are established and shown in Fig. 2, B and C. Note that the mouse vascular tissue is not examined because it is not available in the database. Statistical significance is defined as when a gene expression in given tissues is higher than the upper limit of the confidence interval. To emphasize the vascular system, the columns for heart and vasculature are shown in boldface type. In contrast to the housekeeping genes, NF-κB-signaling genes are differentially expressed in human and mouse tissue. NF-κB family genes have lower expression levels in most human tissues compared with that in mouse tissues. RelA and p50 are highly expressed in thymus. Fourteen other NF-κB-signaling genes are highly expressed in lymph node, including IκB proteins (IκBβ and IκBϵ), IKK complex (IKKα) and adapters (TRAF3, TRAF5, TRAF6, and RIPK1), IKK kinase (TAK1), IKK substrates (SMRT, CBP, Bcl-10, and CYLD), and regulation and degradation factors (AKIRIN2 and SOCS1). In addition, some IκB and IKK substrate members and degradation genes are highly expressed in muscle and pituitary gland.
In mouse tissues (Fig. 2C), 22 of the 38 NF-κB-signaling genes are highly expressed in lymph node, including three NF-κB family genes (RelA, p50, and p52), three IκB proteins (IκBβ, IκBϵ, and IκBζ), two IKK complexes (Ikkα and Ikkβ), five adapters in the NF-κB-signaling pathway (Traf2, Traf3, Traf5, Traf6, and Ripk1), IKK kinase (Tak1), three IKK substrates (Hdac3, Bcl-10, and Cyld), and two regulators (Akirin1 and Mk2) and three inhibitors (Socs1, Commd1, and Pdlim2) of NF-κB. Fourteen genes are highly expressed in pituitary gland (RelB, Ikkβ, Ikkγ, Traf6, Ccnd1, Smrt, Cbp, Hdac3, Cyld, Akirin2, p38, Commd1, Pias1, and Pias4); nine genes are highly expressed in spleen (RelA, c-Rel, NF-κB1, Bcl-3, Ikkβ, Traf2, Ripk2, Bcl-10, and Socs1).
Interestingly, in addition to immune defense tissues (lymph node and spleen) where NF-κB components were highly expressed, most of the NF-κB-signaling genes (36 of 38) have low level expression in heart and vascular tissues except for SOCS1 and COMMD1, which are highly expressed in human heart, and Bcl-10, which is highly expressed in human vessels.
Heart and Vessel Lack Key Components for both Canonical and Non-canonical Signaling and Are Defined as Tier 3 Inflammation-privileged Tissues
Based on the tissue expression pattern of key components of NF-κB signal pathways (IKKγ, IKKβ, IκBα, RelA, and p50 for canonical signaling and IKKα, RelB, and p52 for non-canonical signaling), human (Table 2A) and mouse (Table 2B) tissues are categorized as three tiers. The tier 1 tissues express all key molecules in both signaling pathways and are defined as the “ready to go tier.” Brain, embryonic tissue, kidney, lung, pancreas, spleen, and thymus are tier 1 tissues in both human and mice. Tier 2 tissue lacks key components for one of the pathways and is called the “nearly ready tier.” Bone, connective tissue, liver, muscle, ovary, and testis are tier 2a tissues and lack of one or two molecules from the non-canonical signaling in both humans and mice. Bone marrow is the tier 2b tissue in human, missing three components of the canonical pathway, but belongs to tier 1 in mice. The tier 3 tissue lacks a key component for both canonical and non-canonical signal pathways and is the “inflammation-privileged tier.” Heart, vasculature, and pituitary gland are tier 3 tissues in humans. Eye, lymph node, and pituitary gland are tier 3 tissues in mice.
The compositions of the three tiers in mice are mostly similar to those of humans for most tissues. There are some differences. Heart is a tier 3 tissue in human but tier 1 in mice. In contrast, lymph node and eye are tier 1 tissues in human but tier 3 in mice. Bone marrow is tier 2b in human but tier 1 in mice. Blood is tier 1 in human and tier 2 in mice.
CVD Risk Factors Affect NF-κB Signaling by Directly Altering Gene Expression or Indirectly via the Action of Proinflammatory Cytokines in Vascular Cells
It was suggested that endothelial cell-specific NF-κB signaling plays a critical role in promoting chronic inflammatory atherosclerosis (10). We hypothesized that chronic stimulation with CVD risk factors could induce the NF-κB-signaling gene directly or indirectly via inflammatory cytokines in endothelial cells. We tested this hypothesis by examining the effect of chronic treatment of CVD risk factors on NF-κB-signaling gene expression in vascular cells by using a data mining approach against the GEO data sets, which have been established based on microarray studies. As shown in Table 3, we found that oxidized phospholipids (40 μg/ml, 4 h) induced TRAF6 and reduced RIPK2 and CYLD levels in cultured human microvascular endothelial cells (Table 3A). Hypoxia (1% oxygen, 1 h) induced RelA, TRAF2, SMRT, and PDLIM2 and reduced SOCS1 and MK2 in cultured HUVEC (Table 3B). Interestingly, oxidized phospholipids and hypoxia reduced the expressions of inhibitory molecules of NF-κB pathways CYLD and SOCS1 in endothelial cells. As shown in Table 3C, proinflammatory cytokine IL-1β induced NF-κB-signaling genes in both signal pathways in endothelial cells and macrophages, whereas TNF-α induced more non-canonical pathway components than canonical pathway genes in endothelial cells.
Proinflammatory TF C/EBPs and NF-κB Have Higher Binding Site Frequency in IL-1β- and TNF-α-induced NF-κB-signaling Gene Promoters
TFs are master genes controlling the expressions of other genes. It is well accepted that multiple binding sites for a given TF in a promoter increase the likelihood of actual binding and activity (11). Data in Table 3C suggest that proinflammatory cytokines may induce the NF-κB-signaling gene via an inflammation-related transactivation mechanism. We therefore examined binding frequency of inflammation-related TF in the promoters of proinflammatory cytokine- induced NF-κB-signaling gene identified in Table 3C. Promoter sequences (1,500 bases upstream of the gene transcription start site) of cytokine-induced genes (RelB, p52, p50, TRAF2, and RIPK2) and housekeeping genes (ACTB, GAPDH, and ARHGDIA) are retrieved from the NCBI Entrez database. TF binding frequency of 10 inflammation-related TFs (AP-1, C/EBPs, Elk1, GR, HSF-1, IRF-1, Mef2, NF-κB, p53, and Pu.1) identified from our previous study (7) were analyzed using TF database Transcription Element Search System (TESS) (Fig. 3A). We found that TFs NF-κB and CCAAT/enhancer-binding proteins (C/EBPs) have higher binding frequency in the promoters of cytokine-induced NF-κB-signaling genes compared with that in housekeeping genes (Fig. 3B). The other eight inflammation-related TFs have similar binding frequency in the promoters of genes from both groups.
FIGURE 3.

Transcription factor binding frequencies of cytokine-induced NF-κB-signaling genes (TESS analysis). A, strategic schematics. The promoter sequences (1,500 bp upstream of the transcription start site) of six cytokine-induced NF-κB-signaling genes identified in Table 3C (RelB, p50, p52, TRAF2, RIPK2, and Bcl-10) and three randomly chosen housekeeping genes (ACTB, GAPDH, and ARHGDIA) were retrieved from the NIH/NCBI Entrez Gene database and analyzed for binding sites of 10 inflammation-related TFs (AP-1, C/EBPs, Elk1, GR, HSF-1, IRF-1, Mef2, NF-κB, p53, and Pu.1) identified from our previous study (7) using the TESS database. B, TF binding frequency. Binding frequency denotes the number of the binding sites for each TF on the promoter region. The confidence interval was set by using the mean ± 2 × S.D. (error bars) of the TF binding frequencies in the promoter of three housekeeping genes. Binding frequency higher than the uppermost confidence interval (p < 0.05) is considered significant. Compared with the control group, the promoter regions of cytokine-induced NF-κB family/signaling genes (RelB, p50, p52, TRAF2, RIPK2, and Bcl-10) have significantly higher frequencies of the binding sites for C/EBPs and NF-κB.
NF-κB-signaling Genes Have Multiple Alternative Promoters and Alternatively Spliced Isoforms
Recent findings suggest that alternative splicing of RNA transcripts may either enhance or antagonize the NF-κB pathway components (12) and that alternative promoters may mediate differential gene induction in response to different stimuli (13). Alternative splicing probably regulates about 50% human and mouse gene RNA transcripts in whole transcriptomes (14). Because NF-κB-signaling genes are induced differentially (Table 3), we examined alternative promoters and splicing isoforms of NF-κB-signaling genes by using the AceView-NCBI database, which is the most comprehensive database of alternative promoters and alternatively spliced gene isoforms, generated from the experimental data of cDNA sequencing from various tissue mRNA transcriptomes under non-stimulatory physiological conditions (15). As shown in Table 4A, we found that the RNA transcripts of all human NF-κB-signaling genes encode multiple open reading frames and have alternatively spliced isoforms. Further, most of the human NF-κB-signaling genes have alternative promoters. For example, RelA has 18 potential open reading frame isoforms and two alternative promoters. P50 has 15 potential open reading frame isoforms and six alternative promoters. Interestingly, IκBa and RIPK3 have secreted isoforms. Most of NF-κB-signaling genes exhibit proinflammaiton function, except for NF-κB degradation proteins and IKK substrates CYLD, which display anti-inflammation function.
TABLE 4.
Alternative splicing isoforms and promoters in NF-κB family genes (AceView database analysis)
The presence and features of alternative promoters and spliced isoforms of NF-κB-signaling genes were examined with the AceView database. A, human NF-κB-signaling gene RNA transcripts encoded multiple open reading frame isoforms. Most of the genes have more than one probable alternative promoter. Most NF-κB-signaling genes are proinflammation genes except for CYLD and NF-κB degradation genes. n/a, information not available in the AceView-NCBI database. B, list of the tissue expressions of alternatively splicing isoform/ORF-encoded domain/interaction with other proteins of five NF-κB family genes. RHD, Rel homology domain; TAD, transactivation domain; DD, death domain; CCS, coiled-coil stretch; ANK, ankyrin repeats; PXD, peroxisomal domain; SPXD, second peroximal domain. n/a, no domain retained.
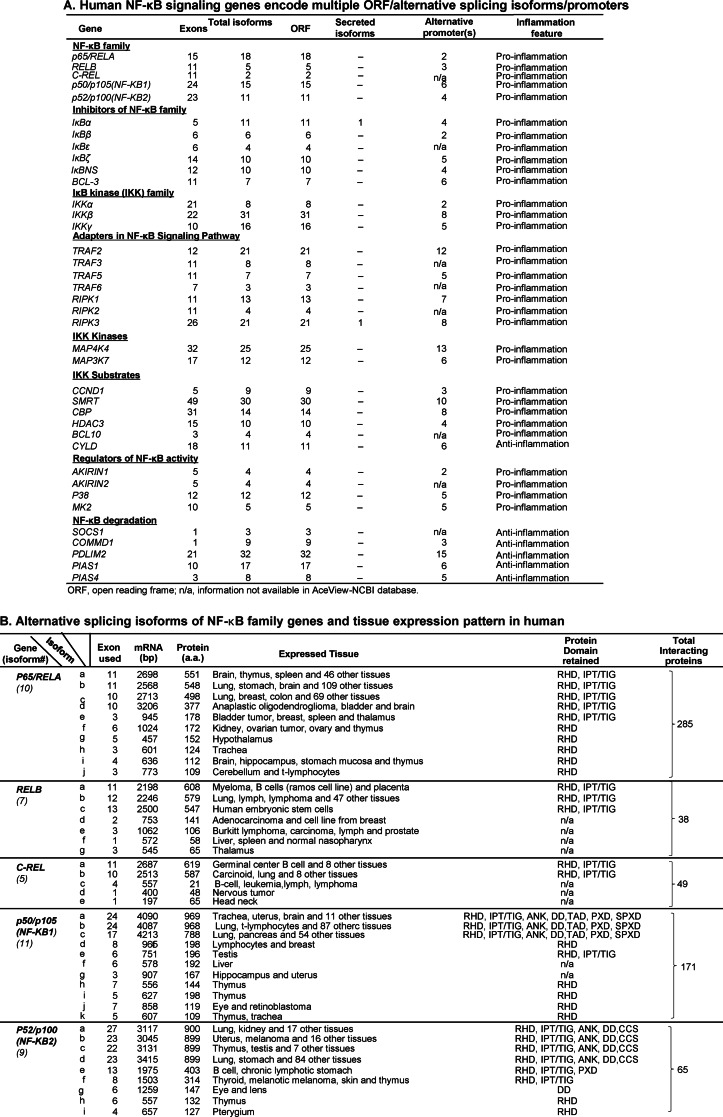
Because alternative splicing and promoters may alter protein domains and interacting proteins, we next examined isoforms of five NF-κB family genes expressed in human tissue, their protein binding domain retained, potential interacting proteins, and tissue expression pattern using the AceView database. As shown in Table 4B, we found that all five NF-κB family genes have multiple isoforms expressed in different tissues, which retain different protein domains and have multiple potential interacting proteins. We name the expressed isoforms in α, β order based on their protein size. For example, RelA has 10 isoforms (from a to j) expressed in humans. RelA isoform a (BelA-a) is encoded by 11 exons and a 2,685-bp mRNA transcript, is 551 amino acids in length, and express in the brain, thymus, spleen, and 46 other tissues. At the same time, we identified an ORF-encoded domain, which showed that NF-κB family proteins have many interaction partners to fulfill their temporally and spatially well orchestrated functions. RelA isoforms contain either one or two protein domains (RHD and IPT/TIG or only RHD) and interact with as many as 285 other proteins. In addition, we found that larger isoforms have broader tissue expression patterns than the shorter ones because RelA-a/b/c are expressed in multiple tissues, ranging from 46 to 112 different tissues, and RelA-d/e/f/g/h/i/j isoforms are expressed only in as few as 1–4 tissues.
DNA Methylation Regulates NF-κB Family Gene Expression
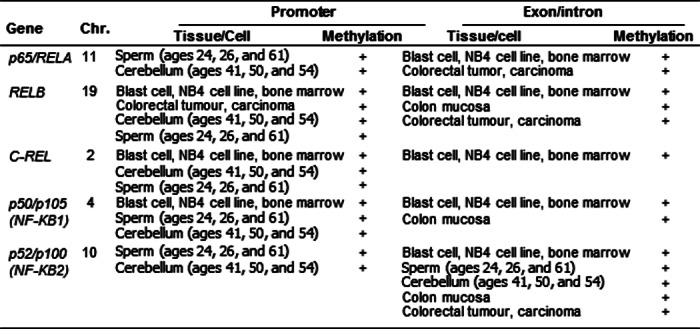
Because DNA hypomethylation is the best characterized epigenetic regulation process in autoimmune disease (16), we assessed methylation status in the promoters and genomic DNA of 5 NF-κB family genes by searching the NCBI-Epigenomics database. DNA methylation of promoter regions and genomic DNA sequences was identified from experimental data deposited in the Epigenomics database. As shown in Table 5, we found that all five NF-κB family genes are characterized for DNA methylation in both promoter and exon/intron regions, suggesting that these genes are potentially subjected to DNA methylation-based epigenetic regulation.
TABLE 5.
DNA methylation characteristic of NF-κB family genes (Epigenomics database analysis)
DNA methylation of five NF-κB family genes (RelA, RelB, c-Rel, p50, and p52) was examined by using the Epigenomics database. DNA methylation of promoter regions and genomic DNA sequences was identified from experimental data deposited in the Epigenomics database. All five NF-κB family genes have DNA methylation in the promoter and exon-intron regions. Chr., chromosome.
MicroRNA Targets Most of the NF-κB Signal Gene's mRNA 3′-UTRs
MicroRNA (also known as miRNA or miR) is a type of epigenetic regulation of gene expression (17), which predominantly targets the 3′-UTR of mRNA, leads to mRNA degradation or inhibition of mRNA translation, and contributes to various biological processes, including cell growth, differentiation, proliferation, and apoptosis (18, 19).
Recently, we examined the reliability of computationally identified microRNA interactions by comparing 45 interactions between 28 experimentally verified human microRNAs and 36 identified with Tarbase (version 5.0), an online database of experimentally verified miRNAs (20). We established microRNA interaction confidence intervals for the context score as mean ± 2 × S.D. = −0.25 ± 0.12 and for context percentage, 76.07 ± 19.07 (9). Based on these confidence intervals, we identified a group of microRNAs that potentially target 27 (71%) of 38 NF-κB-signaling genes, as listed in Table 6A. Several of these microRNAs have been experimentally confirmed to regulate NF-κB-signaling expression, such as miR-200 for RelA, miR-9/181b and Let-7 for p50, miR-125a/b for IκBα, miR-125b for Bcl-3, miR-31 for IKKα, miR200c and miR-199 for IKKβ, miR-31 for NIK, miR-10 for Tak1, miR-16 for SMRT, and miR-19 for CYLD, respectively (21–24).
TABLE 6.
MicroRNAs targeting NF-κB-signaling genes (TESS analysis)
The potential microRNA targets of NF-κB signaling genes were examined with microRNA target prediction software by screening a microRNA library and analyzing the context score and context score percentage, based on the confidence interval we established previously (9). A, potential microRNA targeting on NF-κB signaling genes. All miRNA interactions with a context value of ≤−0.22 and context percentage of ≥70 were accepted. These confidence intervals were generated from 45 interactions between 28 experimentally verified human microRNAs and 36 genes found in Tarbase, an online database of experimentally verified microRNAs (for details, see “Experimental Procedures”). *, microRNA involved in the pathway confirmed by experiment. B, pro- and anti-inflammation microRNAs targeting on NF-κB- signaling genes. MicroRNAs were classified based on their inflammation feature. C, anti-inflammation microRNA reduction leads to NF-κB activation; identification of anti-inflammation microRNA down-regulated by inflammatory stimuli, leading to NF-κB activation.

Based on the target gene function, microRNAs potentially targeting NF-κB components are classified into two groups, anti-inflammatory microRNAs and proinflammatory microRNAs (Table 6B). Of note, the five anti-inflammatory microRNAs, including Let-7, miR-7, miR-15a, miR-29a, and miR-143, were also found in our previous report of anti-inflammatory/anti-atherogenic microRNAs (9). In addition, proinflammatory stimuli, such as TNF-α, IL-1β, and lipopolysaccharide (LPS), induce RelB and IKKα via down-regulating microRNA-7 and Let-7 (Table 6C), respectively.
DISCUSSION
The NF-κB family is one of the best characterized transcription factor families, first discovered about 26 years ago. The regulatory mechanisms of the NF-κB pathway reported so far have been focused on post-translational modifications, including phosphorylation/dephosphorylation, ubiquitination, and proteasome degradation (5). However, pretranslational regulatory mechanisms, which are phosphorylation- and ubiquitination-independent, remained poorly identified. In this study, we extensively examined pretranslational mechanisms of NF-κB-signaling gene regulation, including tissue-specific transcription, alternative promoter/splicing, DNA methylation, and microRNA-mediated mRNA degradation and translational inhibition, by using several experimental results-based NIH/NCBI databases and other software.
We reported here that mRNAs of NF-κB family and signaling genes are differentially expressed in human and mouse tissues. This result suggests that the NF-κB pathway may be regulated via a tissue-specific manner. NF-κB is activated primarily by two pathways: the canonical pathway and the non-canonical pathway. The canonical pathway mediates inflammatory responses. The non-canonical pathway is involved in immune cell differentiation and maturation and secondary lymphoid organogenesis. The expression of the essential molecules of NF-κB pathways in embryonic tissues suggests the role of these pathways in developmental process in addition to its roles in promoting cell survival and inflammation.
We defined human cardiovascular tissue as inflammation-privileged tissues, based on the low or lack of expression of key NF-κB-signaling genes for both canonical and non-canonical signal pathways. This finding suggests that NF-κB activation requires induction of both NF-κB-signaling pathways in the human cardiovascular system. Therefore, human cardiovascular tissues are less likely to be inflamed than other tissues in response to stimulation and are relatively protected against acute low threshold inflammation damage. Of note, most risk factors cause cardiovascular inflammation and diseases in a chronic manner. This situation may result from the requirement for chronic induction of NF-κB-signaling genes in these inflammation-privileged tissues. We previously proposed to term heart and vessel, for the first time, as inflammation-privileged tissue because of the lack of expression of some components of the inflammation initiation protein complex termed as the inflammasome (6). Our current finding is consistent with and supports the notion of defining human heart and vasculature as inflammation-privileged tissue. This new concept provides novel insights into our understanding of inflammatory regulation in cardiovascular tissues, which will benefit future strategies to identify therapeutic targets for CVD.
We noticed that immune privilege sites, ovary, testis, and pituitary gland, stayed in the second tier and third tier. This feature is conserved through the evolution from mice to humans. These results suggest that 1) human tissues have different inflammation potential; 2) the reproductive tissues (ovary and testis) enjoy both immune privilege and inflammation privilege; and 3) human heart and vasculature are inflammation-privileged sites and less easy to be inflamed in response to NF-κB-signaling stimuli.
We also found that CVD risk factors, including oxidized phospholipids and hypoxia, directly induce NF-κB signal gene expression or indirectly via actions of proinflammatory cytokines in vascular cells. Different risk factors that activate the NF-κB signal may do so through a synergistic mechanism. This result suggests that pretranslational mechanisms that are the focus of this study are pathophysiologically relevant.
We reported here that transcription factors C/EBPs and NF-κB have higher binding site frequencies in the promoters of proinflammatory cytokine-induced NF-κB genes. This result suggests that C/EBPs may mediate the NF-κB signal gene induced by IL-1β and TNF-α and that the NF-κB signal gene mediates IL-1β- and TNF-α-induced self-amplification of the NF-κB pathway via a positive feedback manner. It was reported that C/EBPs mediate the induction of several target genes, including preprotachykinin, choline acetyltransferase, cytokines (IL-6, IL-1β, and TNF-α), and cyclooxygenase-2 (Cox2) (25). Taken together, we propose that C/EBPs mediate IL-1β and TNF-α-induced NF-κB-signaling gene expressions, leading to self-amplification of NF-κB activation via a transcription-dependent manner.
Interestingly, we found that most NF-κB family/signaling genes have multiple alternative promoters and alternatively spliced isoforms, which may regulate the expressions, structures, and functions of these components. We found that alternative splicing regulates gene expression and protein structures of NF-κB signaling with rates (100%) significantly higher than that (50%) of general human genes. Many alternative spliced isoforms have significantly shortened mRNA transcripts and proteins (Table 4B), which could make protein-protein interaction less efficient or make it inhibitory due to the dominant negative effects. Because potential binding differences of signaling components to their adapter proteins/kinases/substrates determine the function of the signaling, alternative splicing could regulate NF-κB signaling. Further, we identified that IκBa and RIPK3 have secreted isoforms. The functional significance of the secreted isoforms remains to be identified. It was suggested that ∼75% of alternatively spliced protein isoforms are involving in signaling regulation (26). Future work is needed to map alternative promoters and recognition sequence for alternative splicing responsible for tissue-specific and/or stimulation-specific NF-κB activation.
We demonstrated here that the expression of NF-κB-signaling genes is subject to epigenetic regulation, including DNA methylation and microRNA regulation. These data suggest that environmental and disease risk factors could alter NF-κB activity via DNA methylations (27). We found that most of the NF-κB-signaling gene's mRNA 3′-UTRs can be regulated by microRNAs with a binding quality equivalent to what has been approved experimentally. Moreover, some NF-κB-signaling genes are targets of multiple microRNAs, such as RelA, p50, iκBζ, and IKKα/β. Some microRNAs or microRNA clusters can regulate a few NF-κB-signaling genes. However, most microRNAs are not shared, suggesting that microRNA epigenetic regulation may be responsible for tissue-/cell-specific, spatially and temporally regulated NF-κB-signaling gene expression.
It is worthwhile to point out that the tissue expression profile was assessed by a database mining approach utilizing the NCBI/UniGene database, in which mRNA levels were calculated based on the copy number of EST cDNA clone sequencing, which is more accurate than traditional hybridization- and primer annealing-based approaches, such as Northern blots and RT-PCR analysis, as we discussed previously (7). More importantly, these studies involve using multiple large NCBI databases established with experimental data, including the NCBI-GEO repository database for microarray data from endothelial cells, the NCBI-AceView database for alternative splicing and alternative promoter data, and the NCBI-Epigenomics database for DNA methylation data. In the last 10 years, our laboratories have developed a successful experimental data mining strategy, which has led to generation of numerous novel working models, including stimulation-responsive splicing in generation of autoantigens (28) and the three-tier model and inflammation privilege for inflammation initiation (6). As more high throughput experimental data are deposited in the NCBI and other databases, data mining is becoming a powerful tool for screening potential mechanisms and identifying novel drug targets.
Taking the results together, we propose a novel working model of NF-κB activation (Fig. 4). Using CVD as an example, we elucidated that CVD risk factors initiate NF-κB signaling by binding to pathogen-associated molecular pattern receptors, such as Toll-like receptors (29), and then induce NF-κB gene transcription via four pretranslational regulatory mechanisms, including TF C/EBPs and NF-κB, DNA methylation, alternative promoters and splicing, and anti-inflammation related microRNAs. In response to CVD risk factors, NF-κB-signaling genes can be induced by TF C/EBPs and NF-κB-mediated transactivation, by positive feedback of proinflammatory cytokine induction via an autocrine manner, by using alternative promoters and splicing for efficient expression of fully active spliced isoforms, and by DNA demethylation in cytokine-inducible NF-κB-signaling gene promoters. Finally, CVD risk factors may suppress NF-κB-targeted anti-inflamamtory microRNA expression, leading to increased NF-κB gene expression. In conclusion, our findings on pretranslational mechanisms provide new insight into NF-κB activation related to inflammatory diseases, immunological diseases, and cancers.
FIGURE 4.
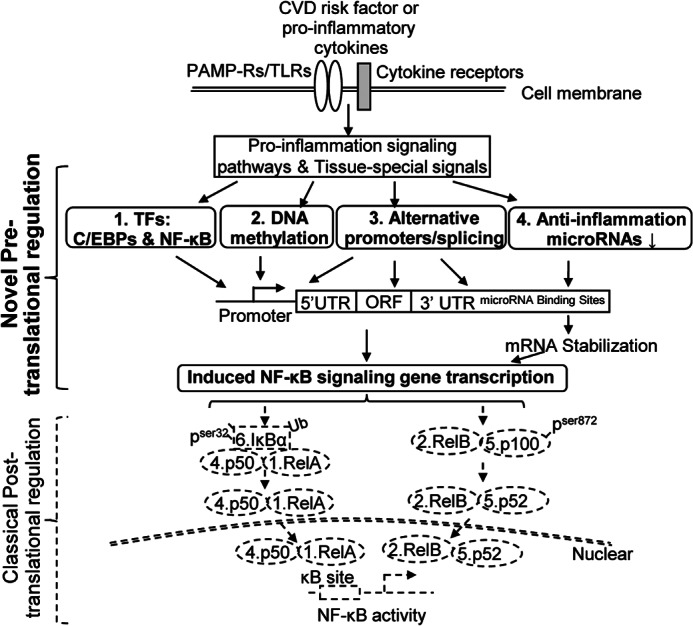
Working model. Four novel pretranslational regulatory mechanisms responsible for NF-κB activation are identified in this study. In addition to classical post-translational regulatory pathways (detailed in Fig. 2), NF-κB signaling can be activated by four pretranslational regulatory mechanisms, leading to induced NF-κB-signaling gene transcription. These include 1) transactivation via C/EBPs and NF-κB consensus sites; 2) promoter DNA methylation; 3) using alternative promoters and alternative spliced mRNA transcripts for efficient expression of fully active spliced isoforms; and 4) reducing anti-inflammatory microRNA, which in turn stabilizes NF-κB mRNAs. Dashed lines, classical NF-κB activation signaling. Solid lines, the newly identified NF-κB activation mechanism identified in this study.
This work was supported, in whole or in part, by National Institutes of Health Grants HL67033, HL77288 HL82774, HL110764, and HL117654 (to H. W.) and HL094451 and HL108910 (to X. F. Y.).
- NF-κB
- nuclear factor κ light chain enhancer of activated B cells
- CVD
- cardiovascular disease
- RIPK
- receptor-interacting protein kinase
- TRAF
- TNF receptor-associated factor
- IKK
- IκB kinase
- NIK
- NF-κB-inducing kinase
- CBP
- CREB-binding protein
- CREB
- cAMP-response element-binding protein
- CYLD
- cylindromatosis turban tumor syndrome
- EST
- expressed sequence tag
- REU
- relative mRNA expression unit
- mREU
- median REU
- HUVEC
- human umbilical vein endothelial cell(s)
- TF
- transcription factor
- C/EBP
- CCAAT/enhancer-binding protein
- GR
- glucocorticoid receptor
- IPT/TIG
- immunoglobulin, plexins, transcription factors-like transcription factor immunoglobulin.
REFERENCES
- 1. Baltimore D. (2011) NF-κB is 25. Nat Immunol. 12, 683–685 [DOI] [PubMed] [Google Scholar]
- 2. Hayden M. S., Ghosh S. (2008) Shared principles in NF-κB signaling. Cell 132, 344–362 [DOI] [PubMed] [Google Scholar]
- 3. Ghosh S., Hayden M. S. (2008) New regulators of NF-κB in inflammation. Nat. Rev. Immunol. 8, 837–848 [DOI] [PubMed] [Google Scholar]
- 4. Perkins N. D. (2007) Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 8, 49–62 [DOI] [PubMed] [Google Scholar]
- 5. Vallabhapurapu S., Karin M. (2009) Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 27, 693–733 [DOI] [PubMed] [Google Scholar]
- 6. Yin Y., Yan Y., Jiang X., Mai J., Chen N. C., Wang H., Yang X. F. (2009) Inflammasomes are differentially expressed in cardiovascular and other tissues. Int. J. Immunopathol. Pharmacol. 22, 311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li X., Mai J., Virtue A., Yin Y., Gong R., Sha X., Gutchigian S., Frisch A., Hodge I., Jiang X., Wang H., Yang X. F. (2012) IL-35 is a novel responsive anti-inflammatory cytokine. A new system of categorizing anti-inflammatory cytokines. PLoS One 7, e33628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ng B., Yang F., Huston D. P., Yan Y., Yang Y., Xiong Z., Peterson L. E., Wang H., Yang X. F. (2004) Increased noncanonical splicing of autoantigen transcripts provides the structural basis for expression of untolerized epitopes. J. Allergy Clin. Immunol. 114, 1463–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Virtue A., Mai J., Yin Y., Meng S., Tran T., Jiang X., Wang H., Yang X. F. (2011) Structural evidence of anti-atherogenic microRNAs. Front Biosci. 16, 3133–3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gareus R., Kotsaki E., Xanthoulea S., van der Made I., Gijbels M. J., Kardakaris R., Polykratis A., Kollias G., de Winther M. P., Pasparakis M. (2008) Endothelial cell-specific NF-κB inhibition protects mice from atherosclerosis. Cell Metab. 8, 372–383 [DOI] [PubMed] [Google Scholar]
- 11. Holloway D. T., Kon M., DeLisi C. (2005) Integrating genomic data to predict transcription factor binding. Genome Inform. 16, 83–94 [PubMed] [Google Scholar]
- 12. Leeman J. R., Gilmore T. D. (2008) Alternative splicing in the NF-κB signaling pathway. Gene 423, 97–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davuluri R. V., Suzuki Y., Sugano S., Plass C., Huang T. H. (2008) The functional consequences of alternative promoter use in mammalian genomes. Trends Genet. 24, 167–177 [DOI] [PubMed] [Google Scholar]
- 14. Su Z., Wang J., Yu J., Huang X., Gu X. (2006) Evolution of alternative splicing after gene duplication. Genome Res. 16, 182–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thierry-Mieg D., Thierry-Mieg J. (2006) AceView. A comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 7, Suppl. 1, S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lopez-Pedrera C., Pérez-Sánchez C., Ramos-Casals M., Santos-Gonzalez M., Rodriguez-Ariza A., Cuadrado M. J. (2012) Cardiovascular risk in systemic autoimmune diseases. Epigenetic mechanisms of immune regulatory functions. Clin. Dev. Immunol. 2012, 974648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bartel D. P. (2009) MicroRNAs. Target recognition and regulatory functions. Cell 136, 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang C. (2008) MicroRNAs. Role in cardiovascular biology and disease. Clin. Sci. 114, 699–706 [DOI] [PubMed] [Google Scholar]
- 19. Naeem H., Küffner R., Csaba G., Zimmer R. (2010) miRSel. Automated extraction of associations between microRNAs and genes from the biomedical literature. BMC Bioinformatics 11, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sethupathy P., Corda B., Hatzigeorgiou A. G. (2006) TarBase. A comprehensive database of experimentally supported animal microRNA targets. RNA 12, 192–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun X., Icli B., Wara A. K., Belkin N., He S., Kobzik L., Hunninghake G. M., Vera M. P., MICU Registry, Blackwell T. S., Baron R. M., Feinberg M. W. (2012) MicroRNA-181b regulates NF-κB-mediated vascular inflammation. J. Clin. Invest. 122, 1973–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Qi J., Qiao Y., Wang P., Li S., Zhao W., Gao C. (2012) MicroRNA-210 negatively regulates LPS-induced production of proinflammatory cytokines by targeting NF-κB1 in murine macrophages. FEBS Lett. 586, 1201–1207 [DOI] [PubMed] [Google Scholar]
- 23. Kim S. W., Ramasamy K., Bouamar H., Lin A. P., Jiang D., Aguiar R. C. (2012) MicroRNAs miR-125a and miR-125b constitutively activate the NF-κB pathway by targeting the tumor necrosis factor α-induced protein 3 (TNFAIP3, A20). Proc. Natl. Acad. Sci. U.S.A. 109, 7865–7870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yamagishi M., Nakano K., Miyake A., Yamochi T., Kagami Y., Tsutsumi A., Matsuda Y., Sato-Otsubo A., Muto S., Utsunomiya A., Yamaguchi K., Uchimaru K., Ogawa S., Watanabe T. (2012) Polycomb-mediated loss of miR-31 activates NIK-dependent NF-κB pathway in adult T cell leukemia and other cancers. Cancer Cell 21, 121–135 [DOI] [PubMed] [Google Scholar]
- 25. Takata Y., Kitami Y., Yang Z. H., Nakamura M., Okura T., Hiwada K. (2002) Vascular inflammation is negatively autoregulated by interaction between CCAAT/enhancer-binding protein-δ and peroxisome proliferator-activated receptor-γ. Circ. Res. 91, 427–433 [DOI] [PubMed] [Google Scholar]
- 26. Modrek B., Lee C. (2002) A genomic view of alternative splicing. Nat. Genet. 30, 13–19 [DOI] [PubMed] [Google Scholar]
- 27. Maunakea A. K., Chepelev I., Zhao K. (2010) Epigenome mapping in normal and disease States. Circ. Res. 107, 327–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang F., Chen I. H., Xiong Z., Yan Y., Wang H., Yang X. F. (2006) Model of stimulation-responsive splicing and strategies in identification of immunogenic isoforms of tumor antigens and autoantigens. Clin. Immunol. 121, 121–133 [DOI] [PubMed] [Google Scholar]
- 29. Yang X. F., Yin Y., Wang H. (2008) Vascular inflammation and Atherogenesis are activated via receptors for PAMPs and suppressed by regulatory T cells. Drug Discov. Today Ther. Strateg. 5, 125–142 [DOI] [PMC free article] [PubMed] [Google Scholar]