

Background: Feedback regulatory processes controlling iNOS expression are incompletely defined.
Results: Induction of iNOS expression was attenuated by 1) inhibition of iNOS activity, 2) prevention of Ras S-nitrosylation, and 3) inhibition of PI3K and mTOR activity.
Conclusion: iNOS-derived NO amplifies iNOS expression through S-nitrosylation of Ras and activation of PI3K and mTOR.
Significance: We have defined a previously unrecognized positive feedback pathway that amplifies human iNOS expression.
Keywords: Nitric Oxide, Nitric-oxide Synthase, Ras, S-Nitrosylation, Signal Transduction
Abstract
The production of nitric oxide (NO) by inducible NO synthase (iNOS) regulates many aspects of physiology and pathology. The expression of iNOS needs to be tightly regulated to balance the broad ranging properties of NO. We have investigated the feedback regulation of cytokine-induced iNOS expression by NO in human cells. The pharmacological inhibition of iNOS activity reduced iNOS protein levels in response to cytokine stimulation in a human epithelial cell line (A549 cells) as well as in primary human astrocytes and bronchial epithelial cells. The addition of exogenous NO using a NO donor prevented the reduction in iNOS levels caused by blockade of iNOS activity. Examination of signaling pathways affected by iNOS indicated that NO S-nitrosylated Ras. Transfection of cells with a S-nitrosylation-resistant Ras mutant reduced iNOS protein levels, indicating a role for this Ras modification in the amplification of iNOS levels. Further, the induction of iNOS protein levels correlated with the late activation of the phosphatidylinositol 3-kinase/Akt and mammalian target of rapamycin (mTOR) pathways, and inhibition of these signaling molecules reduced iNOS levels. Altogether, our findings reveal a previously unknown regulatory pathway that amplifies iNOS expression in human cells.
Introduction
Nitric oxide (NO) is a bioactive gas involved in many aspects of physiology and pathology (1). It is produced from the conversion of l-arginine to l-citrulline and NO by a family of nitric-oxide synthases (NOSs), of which there are three isoforms: neuronal NOS (nNOS),3 inducible NOS (iNOS), and endothelial NOS (eNOS). nNOS and eNOS are expressed constitutively in neurons and endothelial cells, respectively, and iNOS is induced in several cell types (2–7). The induction of iNOS is involved in the regulation of immunity, the clearance of pathogens, and the pathogenesis of several conditions (8, 9).
Cytokines are one of the main inducers of iNOS expression. In mice, the proinflammatory cytokines TNF, IL-1, and IFNγ act together to induce very high levels of iNOS in many cell types (10). Pathogen-derived molecules, such as lipopolysaccharide, also stimulate iNOS expression (11). In contrast to mice, expression of iNOS in humans is relatively restricted to certain cell types, and controversy exists regarding differences between the regulation of human and mouse expression of this enzyme (12). iNOS expression has been documented in macrophages from patients with Mycobacterium tuberculosis infection, although neither bacterial components nor the combination of TNF, IL-1, and IFNγ induces substantial levels of iNOS expression in cultured human macrophages (13–15). Also, evaluation of human iNOS gene promoter activity in transgenic mice expressing human iNOS promoter enhanced GFP has shown extensive species-specific distinctions in expression patterns of iNOS under both basal and inflammatory settings (16). These findings highlight the importance of defining mechanisms regulating iNOS expression specifically in human cells to provide insight into human physiology and pathology.
Inducible expression of iNOS has been defined in only a handful of primary human cell types such as astrocytes, hepatocytes, and bronchial epithelial cells (17, 18). Certain human cell lines have also been instrumental in defining the role of signaling pathways in the regulation of human iNOS expression. The cytokines TNF, IL-1, and IFNγ act in synergy to induce human iNOS gene expression through the coordinated activation of NF-κB, JAK-STAT1, ERK1/2, and p38 MAPK pathways (19–22). In contrast, phosphatidylinositol 3-kinase (PI3K) inhibits cytokine-induced human iNOS gene promoter activity, although the effect of this signaling pathway on post-transcriptional regulation of iNOS levels has not been examined (23). Activation of Ras is also involved in the induction of iNOS in some cell types, although how Ras interacts with other signaling pathways in this setting is incompletely defined (24).
The cellular effects of NO are mediated largely by the activation of soluble guanylate cyclase (sGC) and by protein S-nitrosylation. Activation of sGC by NO initiates cGMP signaling pathways by converting GTP to cGMP (25). NO also directly modifies proteins by S-nitrosylation, which is a post-translational modification that involves the covalent addition of a nitroso moiety onto cysteines to form S-nitrosocysteine. This type of protein modification regulates the enzymatic properties of a large number of proteins and has recently been suggested to have similar biological significance as protein phosphorylation (26). Lander et al. (27, 28) have shown that Ras is S-nitrosylated on cysteine 118 and that this leads to Ras activation. However, very little is known about how this modification of Ras affects cell biology.
The mammalian target of rapamycin (mTOR) is a 289-kDa serine threonine protein kinase that regulates cell size, proliferation, survival, and metabolism (29). In mammalian cells, mTOR forms two complexes, mTOR complex 1 (mTORC1) and mTORC2. Activation of mTORC1 supports cell growth by increasing protein translation and lipid biosynthesis through activation of p70 S6 kinase and the translation initiation factor eIF-4E BP1. mTORC2 promotes proliferation and survival, at least in part, by inhibiting activity of the transcription factors Foxo1 and Foxo3a (30). The Akt and mTOR pathways are tightly linked as Akt can activate mTORC1 and this kinase is activated downstream of mTORC2 (31, 32).
iNOS expression is tightly controlled by feedback mechanisms that fine-tune its expression. The nature of NO-mediated feedback regulation of iNOS expression is cell- and context-dependent. NO, either provided exogenously by NO donors or synthesized endogenously, has been reported to act as part of a negative feedback loop to down-regulate iNOS expression (33, 34). iNOS-derived NO can also inhibit iNOS enzymatic function through post-translational modifications (35, 36). In contrast to negative regulation, NO positively regulates iNOS expression in certain rodent cell types and in human cells, but the mechanisms by which this positive feedback occurs is poorly understood (37–40).
We showed previously that iNOS-derived NO augments human allogeneic immune responses (41, 42) and, in the current study, we define a positive feedback mechanism by which iNOS-derived NO amplifies iNOS protein expression in human cells. Pharmacological inhibition of iNOS activity attenuated iNOS protein expression, and the addition of exogenous NO was sufficient to rescue iNOS levels. Potentiation of iNOS expression by NO was due to S-nitrosylation of Ras, which increased iNOS levels through supporting the sustained activation of Akt. This NO-mediated augmentation of iNOS protein levels also involved activation of the mTOR pathway, as demonstrated by the attenuation of iNOS protein levels by rapamycin. Altogether, our findings highlight an important regulatory process controlling the expression of iNOS in human cells and identify a novel role of Ras S-nitrosylation in this process.
EXPERIMENTAL PROCEDURES
Cell Culture
Human lung epithelial cells (A549) were purchased from ATCC and cultured in F-12K medium containing 10% FBS at 37 °C in a humidified atmosphere of 95% air and 5% CO2. Primary human astrocytes were purchased from Lonza, cultured as above in ABM medium (Lonza, Basel, Switzerland) supplemented with the AGM SingleQuot Kit (Lonza), and used between passages 4 and 12 for experiments. Primary bronchial epithelial cells were also purchased from Lonza, cultured in BEGM with the associated supplements, and used between passages 2 and 6. RAW 264.7 cells were cultured in DMEM + 10% FCS. All experiments were performed at least three times.
For all experiments using A549 cells, confluent cells were incubated in basal medium in the absence of serum for 24 h prior to cytokine treatment. Primary cells were not serum-starved. To induce iNOS, cells were treated with a cytokine mixture (CM) of recombinant human IL-1α (PeproTech, Rocky Hill, NJ), recombinant human TNFα (PeproTech), and recombinant human IFNγ (Invitrogen) for the indicated times. iNOS activity was inhibited by incubating cells with 1400W (50 μm unless indicated otherwise; Sigma-Aldrich) or l-NAME (Cayman Chemicals, Ann Arbor, MI) 1 h prior to cytokine stimulation. Nitric oxide was added using the slow release NO donor NOC-18 (Calbiochem). Inhibition of signaling pathways was achieved by incubating cells with the pharmacological inhibitors FPT inhibitor II (100 μm; Calbiochem), LY294002 (50 μm; Calbiochem), Akt inhibitor VII (50 μm, Calbiochem), PD98059 (50 μm; Cayman Chemicals), epidermal growth factor receptor (EGFR) inhibitor (20 μm; Calbiochem), and PP2 (20 μm; Calbiochem). sGC was inhibited using 1H-[1,2,4]oxadiazole[4,3-a]quinoxaline-1-one (ODQ; Cayman Chemicals), and the mTOR pathway was inhibited with rapamycin (50 nm; LC Laboratories, Woburn, MA).
SDS-PAGE and Immunoblotting
Cells were lysed as described previously (43). Briefly, cells were washed two times with Dulbecco's phosphate-buffered saline and then harvested in 1× radioimmune precipitation buffer (1% (v/v) Triton X-100, 1%(w/v) sodium deoxycholate, 0.1% SDS, 150 mm NaCl, 50 mm Tris·HCl, 2 mm EDTA supplemented with fresh 1 mm sodium orthovanadate, 1 mm PMSF, 50 mm NaF, 10 mm β-glycerophosphate) using a rubber scraper. The lysate was then centrifuged at 17,000 × g for 10 min to remove insoluble material.
Immunoblot was performed as described previously (43). Primary antibodies used were: polyclonal anti-iNOS (sc-651; Santa Cruz Biotechnology), monoclonal anti-phospho-Thr-308-Akt (2965S; Cell Signaling Technology), monoclonal anti-phospho-ERK1/2 (4370S; Cell Signaling Technology), monoclonal anti-phospho-p70 S6 kinase (9234S; Cell Signaling Technology), monoclonal anti-p70 S6 kinase (2708S; Cell Signaling Technology), monoclonal anti-β-actin (A1978, Sigma-Aldrich), polyclonal anti-Ras (3965S, Cell Signaling Technology), and monoclonal anti-Ras (OP23, Calbiochem).
RNA Isolation and Quantitative RT-PCR Analysis
Total RNA was isolated from cells using an RNeasy mini kit as per the manufacturer's instructions (Qiagen), and TaqMan quantitative RT-PCR was performed as described (44) using validated primer/probe sets for iNOS and GAPDH (Applied Biosystems). Data were acquired on an ABI 7900HT fast real-time PCR system (Applied Biosystems).
Quantification of NO Production
NO production was measured by quantifying the accumulation of nitrite in culture supernatants as described previously (41) using a NO-specific chemiluminescence analyzer (Sievers, Boulder, CO).
Transfection of Cells
A549 cells were harvested by trypsinization, and 1 × 106 cells were transfected with 6 μg of wild type or 6 μg of C118S Ras plasmid using an Amaxa nucleofector as per the manufacturer's instructions (Lonza) and as described previously (45). The wild-type (WT) and C118S Ras plasmids were kindly provided by Dr. Kenneth Tang (Cornell University). Following transfection, cells were rested in 6-well tissue culture plates for 24 h and then used for experiments.
Detection of S-Nitrosylation
The biotin switch method to measure S-nitrosylation was performed using the S-nitrosylation protein detection assay kit (Cayman Chemicals) as per the manufacturer's instructions. Following the biotin switch assay, Ras was immunoprecipitated using a monoclonal Ras antibody (Cell Signaling Technology), and the immunoprecipitate was immunoblotted for biotin using an HRP-conjugated monoclonal biotin antibody (Cell Signaling Technology) and a rabbit polyclonal Ras antibody (EMD Millipore).
Statistical Analyses
Immunoblot images were quantified by densitometry using the National Institutes of Health ImageJ software, and protein expression data were normalized to β-actin levels. Unless otherwise indicated, data are expressed as mean ± S.E. obtained in at least three independent experiments. Statistical comparisons were made with a Student's t test or with analysis of variance followed by Tukey's honestly significant difference test. Statistically significant differences were defined as p < 0.05.
RESULTS
iNOS-derived NO Amplifies iNOS Protein Expression
To study the impact of iNOS-derived NO on the feedback regulation of human iNOS expression, A549 cells were stimulated with CM composed of TNFα (10 ng/ml), IL-1α (10 ng/ml), and IFNγ (50 ng/ml). This stimulus is known to optimally induce the expression of iNOS (19). The induction of iNOS expression was evident in CM-treated cells as early as 3–4 h after stimulation and continued to increase until 6 h after stimulation, which was the last time point examined (Fig. 1A). CM also induced the production of NO that was concomitant with the induction of iNOS expression. NO levels were significantly increased at 4 h after stimulation and continued to increase thereafter (Fig. 1B). Also, NO production in response to CM was completely prevented by the iNOS inhibitor 1400W (data not shown).
FIGURE 1.

iNOS-derived NO amplifies iNOS protein expression. A, A549 cells were stimulated with CM for the indicated time points, and iNOS expression was determined by immunoblot. The graph on the bottom is the mean ± S.E. of the percentage of iNOS expression over a series of five separate experiments. *, p < 0.01 as compared with t = 0 h. B, A549 cells were stimulated with CM, and supernatant was collected at the indicated time points. NO production was determined by quantifying nitrite accumulation in cell culture supernatants using a Siever NO analyzer. Data presented are the mean ± S.E. of the -fold increase in nitrite levels over a series of five separate experiments. *, p < 0.05 as compared with t = 0 h. C, A549 cells were untreated or treated with CM, and iNOS or total NOS activity was inhibited by adding the indicated concentrations of 1400W or l-NAME, respectively. Cells were lysed at 5 h after stimulation, and iNOS expression was determined by immunoblot. The effect of 1400W on iNOS expression was quantified over a series of independent experiments, and the graph on the right is the mean ± S.E. of the percentage of iNOS expression relative to cells stimulated with CM that were not treated with 1400W. *, p < 0.01 as compared with cells stimulated with CM in the absence of 1400W. D, A549 cells were untreated (UNT) or treated with CM. iNOS was inhibited by the addition of 1400W (50 μm), and NO was added to some 1400W-treated cells using the NO donor NOC-18 at the indicated concentrations. Cells were lysed at 5 h after stimulation, and iNOS expression was determined by immunoblot. The effect of NOC-18 on iNOS expression was quantified over a series of independent experiments, and the graph on the bottom is the mean ± S.E. of the percentage of iNOS expression relative to cells stimulated with CM that were not treated with 1400W or NOC-18. *, p < 0.01 as compared with cells treated with CM that were not treated with 1400W; ns, not significant. E, A549 cells were untreated or stimulated with CM. iNOS activity was inhibited by the addition of 1400W (50 μm), and NO was provided by the addition of NOC-18 (10 μm). At 5 h after stimulation, the cells were harvested, RNA was isolated, and iNOS mRNA levels were quantified by quantitative RT-PCR. F, the mouse macrophage cell line, RAW 264.7, was unstimulated or stimulated with LPS + IFNγ. iNOS was inhibited in stimulated cells by the addition of 1400W. Cells were lysed, and iNOS expression was determined by immunoblot.
The role of iNOS activity in regulating iNOS protein levels was studied by treating cells with the iNOS inhibitor 1400W or the pan-NOS inhibitor l-NAME followed by examination of iNOS protein expression by immunoblot 6 h later. The inhibition of iNOS activity using either inhibitor significantly reduced CM-induced iNOS protein levels (Fig. 1C). To confirm that NO produced by iNOS was involved in the augmentation of iNOS protein levels, the NO donor NOC-18 was added to cells at the same time as iNOS inhibition with 1400W. The addition of exogenous NO in this manner prevented the reduction in iNOS expression caused by pharmacological inhibition, indicating that iNOS-derived NO stimulates a positive feedback pathway that amplifies iNOS protein levels (Fig. 1D). Interestingly, inhibition of iNOS activity did not affect iNOS mRNA levels, indicating a specific effect of NO in the amplification of protein levels through a post-transcriptional mechanism (Fig. 1E).
Other studies have shown in mouse cells that iNOS-derived NO can inhibit iNOS expression (33, 34). It is possible that NO has pleiotropic effects on cell signaling and that the effect of iNOS-derived NO may be different in humans and mice (12). As such, the effect of iNOS activity on the feedback regulation of iNOS expression in a mouse cell line was examined. Stimulation of RAW 264.7 cells with LPS + IFNγ induced iNOS expression and, in contrast to our observation in human cells but consistent with previous studies examining mouse macrophages (33, 34), inhibition of iNOS activity with 1400W increased iNOS levels (Fig. 1F).
The role of iNOS activity in regulating iNOS protein levels was also examined in primary cells. Primary human astrocytes stimulated with CM express iNOS with maximal levels apparent at 24 h after stimulation (Fig. 2A). Consistent with our findings in A549 cells, inhibition of iNOS activity with 1400W dose-dependently inhibited CM-induced iNOS protein levels in astrocytes (Fig. 2B). A similar effect of 1400W was also observed in experiments with primary human bronchial epithelial cells (Fig. 2C).
FIGURE 2.

iNOS activity amplifies iNOS protein expression in primary human cells. A, primary human astrocytes were unstimulated or stimulated with CM. Cells were lysed at the indicated time points, and iNOS expression was determined by immunoblot. B, astrocytes were unstimulated or stimulated with CM in the absence or presence of the indicated concentrations of 1400W. Cells were lysed at 24 h after stimulation, and iNOS expression was determined by immunoblot. The effect of 1400W on iNOS expression in astrocytes was quantified over a series of experiments, and the graph on the bottom shows the mean ± S.E. of the percentage of iNOS expression relative to cells stimulated with CM that were not treated with 1400W. *, p < 0.02 as compared with cells stimulated with CM in the absence of 1400W. C, primary human bronchial epithelial cells were unstimulated or stimulated with CM in the absence or presence of 1400W. Cells were lysed at 18 h after stimulation, and iNOS expression was determined by immunoblot.
S-Nitrosylation of Ras by iNOS-derived NO
NO regulates signaling pathways largely through the activation of sGC and by protein modification through S-nitrosylation (25). Therefore, the role of these processes in positive feedback regulation of iNOS expression was evaluated. To determine the role of sGC, A549 cells were treated with the sGC inhibitor ODQ prior to CM stimulation, and iNOS expression was examined. There was no effect of sGC inhibition on CM-induced iNOS expression, indicating that the feedback regulation of iNOS expression occurs through an sGC-independent mechanism (Fig. 3A).
FIGURE 3.
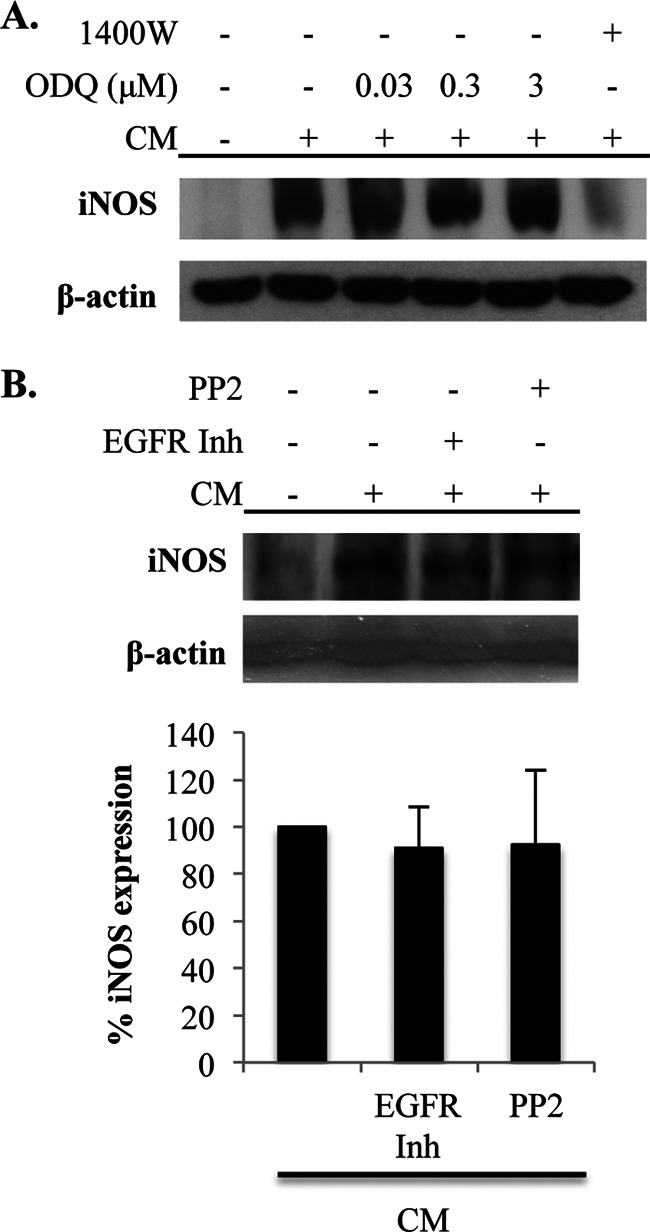
iNOS-mediated amplification of iNOS levels does not depend on sGC, EGFR, or Src activation. A, A549 cells were unstimulated or stimulated with CM. sGC was inhibited by the addition of the indicated concentrations of ODQ. Cells were lysed at 5 h after stimulation, and iNOS expression was determined by immunoblot. B, A549 cells were unstimulated or stimulated with CM in the absence or presence of an EGFR inhibitor (EGFR Inh) or the Src inhibitor PP2. Cells were lysed at 5 h after stimulation, and iNOS expression was determined by immunoblot. The graph on the bottom is the mean ± S.E. of the percentage of iNOS expression relative to cells stimulated with CM that were not treated with the inhibitors.
In addition to sGC, NO also activates cell signaling pathways through the modification of proteins by S-nitrosylation. In fact, this protein modification has been reported to induce iNOS expression through activation of EGFR and Src (46, 47). To determine the involvement of EGFR and Src in NO-mediated amplification of iNOS expression in our experiments, their activity was inhibited pharmacologically. However, neither inhibition of EGFR nor inhibition of Src had any effect on iNOS expression, indicating that the NO-induced amplification of iNOS protein levels in our studies is independent of these signaling pathways (Fig. 3B).
The activity of Ras is also known to be regulated by S-nitrosylation (27). Due to the proximal location of Ras in several signaling pathways implicated in the regulation of iNOS expression, we examined whether Ras activity was required for the NO-mediated amplification of iNOS expression. Inhibition of Ras activity using a farnesyltransferase inhibitor reduced iNOS expression in response to CM stimulation (Fig. 4A). S-Nitrosylation of Ras was then examined using the biotin switch assay. Ras was S-nitrosylated in CM-stimulated cells (Fig. 4B). The elimination of CM-induced Ras S-nitrosylation by 1400W and lack of detection of S-nitrosylation when the biotin labeling step was omitted during the biotin switch procedure demonstrated the specificity of the assay for this NO-induced protein modification (Fig. 4B and data not shown).
FIGURE 4.

S-Nitrosylation of Ras amplifies iNOS protein levels. A, A549 cells were unstimulated or stimulated with CM in the absence or presence of a farnesyltransferase inhibitor (FPT Inh. II), which prevents Ras activation. Cells were lysed at 5 h after stimulation, and iNOS expression was determined by immunoblot. B, A549 cells were unstimulated or stimulated with CM in the absence or presence of 1400W. Cells were harvested, and the biotin switch assay was performed to detect S-nitrosylation. Ras was immunoprecipitated (IP) from the lysates, and S-nitrosylation (SNO) of Ras was detected by immunoblotting for biotin. Total Ras levels in the immunoprecipitate were also examined. C, cells were transfected with an empty vector plasmid or plasmid encoding WT Ras. Transfected cells were untreated or stimulated with CM, and iNOS levels were determined by immunoblot 5 h later. D, A549 cells were transfected with WT Ras or C118S Ras and then left untreated (UNT) or stimulated with CM. Cells were lysed at 5 h after stimulation, and iNOS and Ras expression was determined by immunoblot. iNOS expression was quantified in a series of independent experiments, and the graph depicts the mean ± S.E. of the percentage of iNOS expression relative to cells transfected with WT Ras and stimulated with CM. *, p < 0.01 as compared with cells transfected with WT Ras and stimulated with CM.
Ras S-Nitrosylation Is Required for Positive Feedback Regulation of iNOS Expression
Ras is S-nitrosylated on cysteine 118 (28). To study the role of Ras S-nitrosylation in the regulation of iNOS expression, cells were transfected with WT H-Ras or modified H-Ras in which cysteine 118 is mutated to serine (C118S). C118S Ras cannot be activated by NO but has GTPase activity comparable with WT Ras in response to canonical stimuli (48). Moreover, expression of C118S Ras in human cells has been shown to inhibit activation of endogenous Ras by NO (28). A549 cells express abundant levels of Ras, but endogenous expression of the H-Ras isoform was not detected (data not shown). Transfection of cells with WT Ras did not affect the induction of iNOS by CM (Fig. 4C). Equivalent amounts of WT or C118S H-Ras were detected after transfection with the respective vectors. Remarkably, transfection of cells with C118S Ras reduced the levels of iNOS protein in response to CM stimulation in a manner similar to that observed when iNOS activity was inhibited, indicating that S-nitrosylation of Ras participates in the positive feedback regulation of iNOS expression (Fig. 4D).
Positive Feedback Regulation of iNOS by NO Requires Activation of PI3K and Akt
Two signaling pathways activated by Ras are the MAPK and PI3K-Akt pathways (49–51). To examine the potential role of these pathways in controlling the NO-mediated amplification of iNOS levels, we first characterized their activation in response to CM by visualizing Thr-308 phosphorylation of Akt and phosphorylation of ERK 1/2. In response to CM, there was rapid initial phosphorylation of Akt that reached maximal levels at 15 min after stimulation. Interestingly, there was a second phase of Akt phosphorylation that coincided with the expression of iNOS beginning at 3 h after stimulation and reaching maximal levels at 4–5 h after stimulation. Similar kinetics of ERK1/2 phosphorylation were observed, although late phase ERK1/2 phosphorylation was evident slightly earlier than Akt phosphorylation (Fig. 5A).
FIGURE 5.

Activation of the PI3K-Akt pathway by Ras S-nitrosylation amplifies iNOS levels. A, A549 cells were stimulated with CM, and cell lysates were prepared at the indicated time points. iNOS expression, Akt phosphorylation (pAkt) (on Thr-308), and ERK1/2 phosphorylation (pERK1/2) were examined by immunoblot. B, cells were unstimulated or stimulated with CM, and iNOS was inhibited in CM-treated cells with 1400W. In some 1400W-treated cells, NO was added using the indicated concentrations of NOC-18. Cells were lysed 5 h after stimulation, and phosphorylation of Akt and ERK1/2 was determined by immunoblot. C, cells were transfected with WT or C118S Ras and then left unstimulated or stimulated with CM. Phosphorylation of Akt and ERK1/2 and levels of H-Ras were determined by immunoblot. D and E, A549 cells were unstimulated or stimulated with CM. PI3K, Akt, and ERK1/2 activation was inhibited by the addition of LY294002 or Akt inhibitor VII (Akt Inh) (D) or PD98059 (E), respectively, at 2 h after stimulation with CM. Cells were lysed at 5 h after stimulation, and iNOS expression was determined by immunoblot.
The association between the kinetics of iNOS expression and activation of Akt and ERK1/2 suggested a potential role of iNOS in activation of these kinase pathways. To examine this, A549 cells were treated with 1400W prior to stimulation with CM, and Akt and ERK1/2 phosphorylation was examined. Inhibition of iNOS activity had no effect on the initial activation of Akt (data not shown) but diminished the phosphorylation of Akt at 5 h after stimulation (Fig. 5B). The addition of exogenous NO with NOC-18 rescued Akt activation in the presence of 1400W, indicating a role for iNOS-derived NO in this process (Fig. 5B). No clear effect of iNOS inhibition on the activation of ERK1/2 was observed.
The involvement of Ras S-nitrosylation in the activation of Akt and ERK1/2 was then determined. A549 cells were transfected with WT or C118S Ras prior to stimulation with CM. Cells were lysed at 5 h after stimulation, and phosphorylation of Akt and ERK1/2 was determined. Phosphorylation of both Akt and ERK1/2 was observed in cells transfected with WT Ras. However, the presence of C118S Ras prevented the phosphorylation of Akt without affecting phosphorylation of ERK1/2 (Fig. 5C). To specifically address the role of the late activation of the PI3K-Akt pathway in the positive feedback regulation of iNOS expression, A549 cells were CM-stimulated and then treated with the PI3K inhibitor LY294002 or an Akt inhibitor at 2 h after stimulation. This 2-h time point precedes the second phase of Akt activation observed after CM stimulation but follows down-regulation of the initial activation of Akt in response to CM (Fig. 5A). Therefore, the addition of LY294002 and the Akt inhibitor at this time point does not affect early processes controlled by the initial activation of this pathway but specifically inhibits those affected by the second phase of PI3K-Akt activation. Inhibition of the second phase of PI3K-Akt activation reduced iNOS protein levels (Fig. 5D). The MEK1/2 inhibitor PD98059 was also added at 2 h after stimulation, but this did not affect iNOS levels (Fig. 5E). These findings establish a role for the PI3K-Akt pathway in mediating the positive feedback regulation of iNOS expression by Ras S-nitrosylation.
Positive Feedback Regulation of iNOS Is Regulated by mTOR
The signaling mechanisms downstream of PI3K-Akt involved in the positive feedback regulation of iNOS protein levels by NO were investigated. mTOR is a metabolic sensor that is activated by Akt and regulates protein synthesis (29). We considered a role for this pathway in the positive feedback regulation of iNOS because of our observation that iNOS mRNA levels are not affected by iNOS inhibition (Fig. 1E), which suggested the involvement of post-transcriptional mechanisms. Activation of the mTOR pathway in response to CM was first examined. Stimulation of A549 cells with CM resulted in the phosphorylation of p70S6 kinase, an event that is downstream of mTOR activation and that drives increased protein translation, at time points concomitant with the induction of iNOS (Fig. 6A).
FIGURE 6.

iNOS-derived NO amplifies iNOS protein levels through activation of mTOR. A, A549 cells were stimulated with CM, and lysates were prepared at the indicated time points. Activation of the mTOR pathway was determined by immunoblot for phosphorylated p70S6K (p-p70S6K), which is downstream of mTOR activation. Total p70S6K levels were also examined. B, cells were unstimulated or stimulated with CM, and iNOS activity was inhibited by the addition of 1400W. Cells were lysed at 5 h after stimulation, and phosphorylation of p70S6K was determined by immunoblot. C, cells were unstimulated or stimulated with CM. mTOR activity was inhibited by adding rapamycin (Rapa; 50 nm) at 2 h after stimulation. Lysates were prepared at 5 h after stimulation, and iNOS expression and p70S6K phosphorylation were determined by immunoblot. iNOS expression was quantified in a series of independent experiments, and the graph on the bottom is the mean ± S.E. of the percentage of iNOS expression relative to cells stimulated with CM in the absence of rapamycin. *, p < 0.02 as compared with cells stimulated with CM in the absence of rapamycin. D, A549 cells were unstimulated or stimulated with CM. mTOR and Akt activity was prevented in CM-stimulated cells by the addition of rapamycin or Akt inhibitor VII (Akt Inh.). RNA was isolated at 5 h after stimulation, and the iNOS mRNA expression was quantified by quantitative RT-PCR.
The effect of iNOS activity on activation of the mTOR pathway was then examined. Inhibition of iNOS activity with 1400W inhibited phosphorylation of p70S6 kinase at late time points (Fig. 6B). Also, treatment of cells with the mTOR inhibitor rapamycin at 2 h after stimulation with CM also reduced iNOS protein levels (Fig. 6C). Finally, in support of a role for NO-mediated activation of Akt and mTOR in amplifying iNOS expression through a post-transcriptional mechanism, inhibition of Akt and mTOR activity did not reduce iNOS mRNA expression in response to CM stimulation (Fig. 6D). Taken together, these findings indicate that iNOS-derived NO augments iNOS protein levels through the activation of mTOR.
DISCUSSION
Studying the regulation of human iNOS expression has implications for understanding the pathogenesis of several diseases. We have identified a previously unrecognized positive feedback mechanism controlling the expression of iNOS by endogenously produced NO in human cells. Specifically, our data show that NO produced by iNOS acts to S-nitrosylate Ras. This leads to the activation of the PI3K-Akt and mTOR pathways, which amplify iNOS protein levels.
The expression of iNOS and resultant production of NO are complexly regulated. Factors such as species, NO levels, and the cellular redox state affect the outcome of NO on biological processes and signaling. Some studies, mostly performed in mouse cells, have shown that iNOS-derived NO down-regulates iNOS expression (33, 34). The inhibition of iNOS expression by NO may involve post-translational modification of transcription factors or related regulatory factors (52, 53). In addition to regulating expression, NO is also able to attenuate iNOS activity by destabilizing iNOS dimers through effects on the heme prosthetic group or by S-nitrosylating iNOS directly (35). In contrast to negative regulation of iNOS by NO, studies have also established that NO can augment iNOS expression (37–39). Our findings support a role for iNOS-derived NO in the amplification of iNOS levels in human cells. The paradoxical effect of NO on inhibiting and augmenting iNOS expression likely depends on the levels of available gas within cells, with high levels being inhibitory and lower levels being stimulatory (40). Indeed, iNOS expression and production of NO have been suggested to be lower in cultured human as compared with mouse macrophages (54). Moreover, the human cells used in our studies produce 10–50-fold less NO in response to cytokine stimulation than do mouse macrophage cell lines (data not shown).
Although we have focused our studies on iNOS, NO also regulates the expression of eNOS and nNOS. Specifically, the addition of exogenous NO increases eNOS levels and the inhibition of NOS activity decreases basal eNOS levels in endothelial cells, indicative of a positive feedback process controlling the basal expression of this NOS isoform (55). In contrast, NO production by eNOS can down-regulate the induction of eNOS expression that occurs in response to shear stress (56, 57). nNOS expression is also inhibited by NO in certain pathological settings (58). The mechanisms by which either positive or negative feedback regulation of eNOS and nNOS is controlled remain poorly understood, but these complex feedback mechanisms are likely in place to precisely control the broad ranging biological effects of NO.
Ras has been defined to undergo S-nitrosylation at cysteine 118, and this modification controls cellular processes such as progress through the cell cycle, cell death, T cell activation, and responses to ischemia (27, 59–62). Several signaling pathways are activated downstream of Ras, including PI3K-Akt and MAPK. Despite the clear role of Ras S-nitrosylation in amplifying iNOS expression in our experiments, we were not able to observe any differences in global Ras activation, as defined by Raf1 binding, within whole cell lysates after the inhibition of iNOS (data not shown). However, it is known that S-nitrosylation of Ras also regulates the intracellular localization of this GTPase and that Ras activates Raf1-independent signaling pathways (63, 64). Indeed, Ras S-nitrosylation triggers activation of the PI3K pathway by increasing the interaction of this GTPase with PI3K at cellular membranes, and it also regulates the interaction of Ras with membrane surfaces (65, 66).
Our findings also suggest a complex role of the PI3K pathway in the regulation of iNOS expression. Analysis of human iNOS promoter activity has shown that PI3K activation attenuates iNOS gene transcription through the recruitment and binding of Foxo3a to the iNOS gene promoter (23). Our data show that the augmentation of iNOS levels by NO occurs at a post-transcriptional level because NO does not effect iNOS mRNA levels. We also show that inhibition of PI3K and mTOR, at a time point after the induction of gene transcription but immediately prior to the detectable induction of iNOS protein, reduces the accumulation of iNOS protein levels. As such, there may be at least two opposing effects of PI3K on iNOS regulation that act together to fine-tune expression of this enzyme, namely attenuation of gene transcription and post-transcriptional amplification of iNOS protein levels.
Augmentation of iNOS protein levels by NO involves the activation of mTOR because treatment of cells with the mTOR inhibitor rapamycin attenuated iNOS protein levels. mTOR is known to increase protein translation (67). In addition to regulating protein translation, the activity of mTOR may also regulate iNOS expression through effects on protein stability (68). The specific post-transcriptional processes modulated by NO that augment iNOS protein levels require further study.
In conclusion, we have shown that NO from iNOS triggers a positive feedback loop that amplifies iNOS protein levels through the S-nitrosylation of Ras in human cells. Our findings also show that this biochemical modification of Ras preferentially activates the PI3K and mTOR pathways. The precise control of iNOS expression by feedback mechanisms may be important in defining the biological actions of this enzyme. The mechanisms regulating iNOS expression are likely to affect various aspects of physiology and pathology.
Acknowledgments
We are grateful to Dr. Kenneth Teng for providing the WT and C118S Ras plasmids and to Winnie Enns for laboratory support.
This work was funded by a Natural Sciences and Engineering Research Council Discovery Grant (to J. C. C.).
- nNOS
- neuronal nitric oxide synthase
- eNOS
- endothelial nitric oxide synthase
- iNOS
- inducible nitric oxide synthase
- CM
- cytokine mixture
- l-NAME
- l-NG-nitroarginine methyl ester
- mTOR
- mammalian target of rapamycin
- mTORC1
- mammalian target of rapamycin complex 1
- mTORC2
- mammalian target of rapamycin complex 2
- ODQ
- 1H-[1,2,4]oxadiazole[4,3-a]quinoxaline-1-one
- sGC
- soluble guanylate cyclase
- EGFR
- epidermal growth factor receptor.
REFERENCES
- 1. Moncada S., Higgs E. A. (2006) The discovery of nitric oxide and its role in vascular biology. Br. J. Pharmacol. 147, S193–S201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bredt D. S., Hwang P. M., Glatt C. E., Lowenstein C., Reed R. R., Snyder S. H. (1991) Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature 351, 714–718 [DOI] [PubMed] [Google Scholar]
- 3. Sessa W. C., Harrison J. K., Barber C. M., Zeng D., Durieux M. E., D'Angelo D. D., Lynch K. R., Peach M. J. (1992) Molecular cloning and expression of a cDNA encoding endothelial cell nitric oxide synthase. J. Biol. Chem. 267, 15274–15276 [PubMed] [Google Scholar]
- 4. Nishida K., Harrison D. G., Navas J. P., Fisher A. A., Dockery S. P., Uematsu M., Nerem R. M., Alexander R. W., Murphy T. J. (1992) Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J. Clin. Invest. 90, 2092–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marsden P. A., Schappert K. T., Chen H. S., Flowers M., Sundell C. L., Wilcox J. N., Lamas S., Michel T. (1992) Molecular cloning and characterization of human endothelial nitric oxide synthase. FEBS Lett. 307, 287–293 [DOI] [PubMed] [Google Scholar]
- 6. Xie Q. W., Cho H. J., Calaycay J., Mumford R. A., Swiderek K. M., Lee T. D., Ding A., Troso T., Nathan C. (1992) Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science 256, 225–228 [DOI] [PubMed] [Google Scholar]
- 7. Lowenstein C. J., Glatt C. S., Bredt D. S., Snyder S. H. (1992) Cloned and expressed macrophage nitric oxide synthase contrasts with the brain enzyme. Proc. Natl. Acad. Sci. U.S.A. 89, 6711–6715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bogdan C., Röllinghoff M., Diefenbach A. (2000) The role of nitric oxide in innate immunity. Immunol. Rev. 173, 17–26 [DOI] [PubMed] [Google Scholar]
- 9. Nathan C. (2011) Is iNOS beginning to smoke? Cell 147, 257–258 [DOI] [PubMed] [Google Scholar]
- 10. Stuehr D. J., Cho H. J., Kwon N. S., Weise M. F., Nathan C. F. (1991) Purification and characterization of the cytokine-induced macrophage nitric oxide synthase: an FAD- and FMN-containing flavoprotein. Proc. Natl. Acad. Sci. U.S.A. 88, 7773–7777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xie Q. W., Whisnant R., Nathan C. (1993) Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon γ and bacterial lipopolysaccharide. J. Exp. Med. 177, 1779–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schneemann M., Schoedon G. (2002) Species differences in macrophage NO production are important. Nat. Immunol. 3, 102. [DOI] [PubMed] [Google Scholar]
- 13. Nicholson S., Bonecini-Almeida Mda G., Lapa e Silva J. R., Nathan C., Xie Q. W., Mumford R., Weidner J. R., Calaycay J., Geng J., Boechat N., Linhares C., Rom W., Ho J. L. (1996) Inducible nitric oxide synthase in pulmonary alveolar macrophages from patients with tuberculosis. J. Exp. Med. 183, 2293–2302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schneemann M., Schoedon G., Hofer S., Blau N., Guerrero L., Schaffner A. (1993) Nitric oxide synthase is not a constituent of the antimicrobial armature of human mononuclear phagocytes. J. Infect. Dis. 167, 1358–1363 [DOI] [PubMed] [Google Scholar]
- 15. Padgett E. L., Pruett S. B. (1992) Evaluation of nitrite production by human monocyte-derived macrophages. Biochem. Biophys. Res. Commun. 186, 775–781 [DOI] [PubMed] [Google Scholar]
- 16. Yu Z., Xia X., Kone B. C. (2005) Expression profile of a human inducible nitric oxide synthase promoter reporter in transgenic mice during endotoxemia. Am. J. Physiol. Renal. Physiol. 288, F214–F220 [DOI] [PubMed] [Google Scholar]
- 17. Lee S. C., Brosnan C. F. (1996) Cytokine regulation of iNOS expression in human glial cells. Methods 10, 31–37 [DOI] [PubMed] [Google Scholar]
- 18. Nussler A. K., Di Silvio M., Billiar T. R., Hoffman R. A., Geller D. A., Selby R., Madariaga J., Simmons R. L. (1992) Stimulation of the nitric oxide synthase pathway in human hepatocytes by cytokines and endotoxin. J. Exp. Med. 176, 261–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taylor B. S., de Vera M. E., Ganster R. W., Wang Q., Shapiro R. A., Morris S. M., Jr., Billiar T. R., Geller D. A. (1998) Multiple NF-κB enhancer elements regulate cytokine induction of the human inducible nitric oxide synthase gene. J. Biol. Chem. 273, 15148–15156 [DOI] [PubMed] [Google Scholar]
- 20. Ganster R. W., Taylor B. S., Shao L., Geller D. A. (2001) Complex regulation of human inducible nitric oxide synthase gene transcription by Stat 1 and NF-κB. Proc. Natl. Acad. Sci. U.S.A. 98, 8638–8643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Poljakovic M., Nygren J. M., Persson K. (2003) Signalling pathways regulating inducible nitric oxide synthase expression in human kidney epithelial cells. Eur. J. Pharmacol. 469, 21–28 [DOI] [PubMed] [Google Scholar]
- 22. Kristof A. S., Marks-Konczalik J., Moss J. (2001) Mitogen-activated protein kinases mediate activator protein-1-dependent human inducible nitric-oxide synthase promoter activation. J. Biol. Chem. 276, 8445–8452 [DOI] [PubMed] [Google Scholar]
- 23. Kristof A. S., Fielhaber J., Triantafillopoulos A., Nemoto S., Moss J. (2006) Phosphatidylinositol 3-kinase-dependent suppression of the human inducible nitric-oxide synthase promoter is mediated by FKHRL1. J. Biol. Chem. 281, 23958–23968 [DOI] [PubMed] [Google Scholar]
- 24. Pahan K., Liu X., McKinney M. J., Wood C., Sheikh F. G., Raymond J. R. (2000) Expression of a dominant-negative mutant of p21ras inhibits induction of nitric oxide synthase and activation of nuclear factor-κB in primary astrocytes. J. Neurochem. 74, 2288–2295 [DOI] [PubMed] [Google Scholar]
- 25. Tennyson A. G., Lippard S. J. (2011) Generation, translocation, and action of nitric oxide in living systems. Chem. Biol. 18, 1211–1220 [DOI] [PubMed] [Google Scholar]
- 26. Hess D. T., Matsumoto A., Kim S. O., Marshall H. E., Stamler J. S. (2005) Protein S-nitrosylation: purview and parameters. Nat. Rev. Mol. Cell Biol. 6, 150–166 [DOI] [PubMed] [Google Scholar]
- 27. Lander H. M., Ogiste J. S., Pearce S. F., Levi R., Novogrodsky A. (1995) Nitric oxide-stimulated guanine nucleotide exchange on p21ras. J. Biol. Chem. 270, 7017–7020 [DOI] [PubMed] [Google Scholar]
- 28. Lander H. M., Hajjar D. P., Hempstead B. L., Mirza U. A., Chait B. T., Campbell S., Quilliam L. A. (1997) A molecular redox switch on p21ras: structural basis for the nitric oxide-p21ras interaction. J. Biol. Chem. 272, 4323–4326 [DOI] [PubMed] [Google Scholar]
- 29. Powell J. D., Delgoffe G. M. (2010) The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity 33, 301–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laplante M., Sabatini D. M. (2009) mTOR signaling at a glance. J. Cell Sci. 122, 3589–3594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Inoki K., Li Y., Zhu T., Wu J., Guan K. L. (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 4, 648–657 [DOI] [PubMed] [Google Scholar]
- 32. Sarbassov D. D., Ali S. M., Sengupta S., Sheen J. H., Hsu P. P., Bagley A. F., Markhard A. L., Sabatini D. M. (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 22, 159–168 [DOI] [PubMed] [Google Scholar]
- 33. Griscavage J. M., Rogers N. E., Sherman M. P., Ignarro L. J. (1993) Inducible nitric oxide synthase from a rat alveolar macrophage cell line is inhibited by nitric oxide. J. Immunol. 151, 6329–6337 [PubMed] [Google Scholar]
- 34. Hinz B., Brune K., Pahl A. (2000) Nitric oxide inhibits inducible nitric oxide synthase mRNA expression in RAW 264.7 macrophages. Biochem. Biophys. Res. Commun. 271, 353–357 [DOI] [PubMed] [Google Scholar]
- 35. Albakri Q. A., Stuehr D. J. (1996) Intracellular assembly of inducible NO synthase is limited by nitric oxide-mediated changes in heme insertion and availability. J. Biol. Chem. 271, 5414–5421 [DOI] [PubMed] [Google Scholar]
- 36. Galijasevic S., Saed G. M., Diamond M. P., Abu-Soud H. M. (2003) Myeloperoxidase up-regulates the catalytic activity of inducible nitric oxide synthase by preventing nitric oxide feedback inhibition. Proc. Natl. Acad. Sci. U.S.A. 100, 14766–14771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mühl H., Pfeilschifter J. (1995) Amplification of nitric oxide synthase expression by nitric oxide in interleukin 1β-stimulated rat mesangial cells. J. Clin. Invest. 95, 1941–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Boese M., Busse R., Mülsch A., Schini-Kerth V. (1996) Effect of cyclic GMP-dependent vasodilators on the expression of inducible nitric oxide synthase in vascular smooth muscle cells: role of cyclic AMP. Br. J. Pharmacol. 119, 707–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pérez-Sala D., Cernuda-Morollón E., Díaz-Cazorla M., Rodríguez-Pascual F., Lamas S. (2001) Posttranscriptional regulation of human iNOS by the NO/cGMP pathway. Am. J. Physiol. Renal Physiol. 280, F466–F473 [DOI] [PubMed] [Google Scholar]
- 40. Sheffler L. A., Wink D. A., Melillo G., Cox G. W. (1995) Exogenous nitric oxide regulates IFN-γ plus lipopolysaccharide-induced nitric oxide synthase expression in mouse macrophages. J. Immunol. 155, 886–894 [PubMed] [Google Scholar]
- 41. Choy J. C., Wang Y., Tellides G., Pober J. S. (2007) Induction of inducible NO synthase in bystander human T cells increases allogeneic responses in the vasculature. Proc. Natl. Acad. Sci. U.S.A. 104, 1313–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Choy J. C., Pober J. S. (2009) Generation of NO by bystander human CD8 T cells augments allogeneic responses by inhibiting cytokine deprivation-induced cell death. Am. J. Transplant. 9, 2281–2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. von Rossum A., Krall R., Escalante N. K., Choy J. C. (2011) Inflammatory cytokines determine the susceptibility of human CD8 T cells to Fas-mediated activation-induced cell death through modulation of FasL and c-FLIPS expression. J. Biol. Chem. 286, 21137–21144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Escalante N. K., von Rossum A., Lee M., Choy J. C. (2011) CD155 on human vascular endothelial cells attenuates the acquisition of effector functions in CD8 T cells. Arterioscler. Thromb. Vasc. Biol. 31, 1177–1184 [DOI] [PubMed] [Google Scholar]
- 45. Liu A. C., Lee M., McManus B. M., Choy J. C. (2012) Induction of endothelial nitric oxide synthase expression by IL-17 in human vascular endothelial cells: implications for vascular remodeling in transplant vasculopathy. J. Immunol. 188, 1544–1550 [DOI] [PubMed] [Google Scholar]
- 46. Rahman M. A., Senga T., Ito S., Hyodo T., Hasegawa H., Hamaguchi M. (2010) S-Nitrosylation at cysteine 498 of c-Src tyrosine kinase regulates nitric oxide-mediated cell invasion. J. Biol. Chem. 285, 3806–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Switzer C. H., Glynn S. A., Cheng R. Y., Ridnour L. A., Green J. E., Ambs S., Wink D. A. (2012) S-Nitrosylation of EGFR and Src activates an oncogenic signaling network in human basal-like breast cancer. Mol. Cancer Res. 10, 1203–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mott H. R., Carpenter J. W., Campbell S. L. (1997) Structural and functional analysis of a mutant Ras protein that is insensitive to nitric oxide activation. Biochemistry 36, 3640–3644 [DOI] [PubMed] [Google Scholar]
- 49. Thomas S. M., DeMarco M., D'Arcangelo G., Halegoua S., Brugge J. S. (1992) Ras is essential for nerve growth factor- and phorbol ester-induced tyrosine phosphorylation of MAP kinases. Cell 68, 1031–1040 [DOI] [PubMed] [Google Scholar]
- 50. Rodriguez-Viciana P., Warne P. H., Dhand R., Vanhaesebroeck B., Gout I., Fry M. J., Waterfield M. D., Downward J. (1994) Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 370, 527–532 [DOI] [PubMed] [Google Scholar]
- 51. Rodriguez-Viciana P., Warne P. H., Vanhaesebroeck B., Waterfield M. D., Downward J. (1996) Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 15, 2442–2451 [PMC free article] [PubMed] [Google Scholar]
- 52. Yu Z., Kuncewicz T., Dubinsky W. P., Kone B. C. (2006) Nitric oxide-dependent negative feedback of PARP-1 trans-activation of the inducible nitric-oxide synthase gene. J. Biol. Chem. 281, 9101–9109 [DOI] [PubMed] [Google Scholar]
- 53. Chang K., Lee S. J., Cheong I., Billiar T. R., Chung H. T., Han J. A., Kwon Y. G., Ha K. S., Kim Y. M. (2004) Nitric oxide suppresses inducible nitric oxide synthase expression by inhibiting post-translational modification of IκB. Exp. Mol. Med. 36, 311–324 [DOI] [PubMed] [Google Scholar]
- 54. Weinberg J. B., Misukonis M. A., Shami P. J., Mason S. N., Sauls D. L., Dittman W. A., Wood E. R., Smith G. K., McDonald B., Bachus K. E., et al. (1995) Human mononuclear phagocyte inducible nitric oxide synthase (iNOS): analysis of iNOS mRNA, iNOS protein, biopterin, and nitric oxide production by blood monocytes and peritoneal macrophages. Blood 86, 1184–1195 [PubMed] [Google Scholar]
- 55. Yuhanna I. S., MacRitchie A. N., Lantin-Hermoso R. L., Wells L. B., Shaul P. W. (1999) Nitric oxide (NO) upregulates NO synthase expression in fetal intrapulmonary artery endothelial cells. Am. J. Respir. Cell Mol. Biol. 21, 629–636 [DOI] [PubMed] [Google Scholar]
- 56. Grumbach I. M., Chen W., Mertens S. A., Harrison D. G. (2005) A negative feedback mechanism involving nitric oxide and nuclear factor κ-B modulates endothelial nitric oxide synthase transcription. J. Mol. Cell. Cardiol. 39, 595–603 [DOI] [PubMed] [Google Scholar]
- 57. Vaziri N. D., Wang X. Q. (1999) cGMP-mediated negative-feedback regulation of endothelial nitric oxide synthase expression by nitric oxide. Hypertension 34, 1237–1241 [DOI] [PubMed] [Google Scholar]
- 58. De Alba J., Cárdenas A., Moro M. A., Leza J. C., Lorenzo P., Boscá L., Lizasoain I. (1999) Down-regulation of neuronal nitric oxide synthase by nitric oxide after oxygen-glucose deprivation in rat forebrain slices. J. Neurochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 59. Oliveira C. J., Curcio M. F., Moraes M. S., Tsujita M., Travassos L. R., Stern A., Monteiro H. P. (2008) The low molecular weight S-nitrosothiol, S-nitroso-N-acetylpenicillamine, promotes cell cycle progression in rabbit aortic endothelial cells. Nitric Oxide 18, 241–255 [DOI] [PubMed] [Google Scholar]
- 60. Teng K. K., Esposito D. K., Schwartz G. D., Lander H. M., Hempstead B. L. (1999) Activation of c-Ha-Ras by nitric oxide modulates survival responsiveness in neuronal PC12 cells. J. Biol. Chem. 274, 37315–37320 [DOI] [PubMed] [Google Scholar]
- 61. Ibiza S., Pérez-Rodríguez A., Ortega A., Martínez-Ruiz A., Barreiro O., García-Domínguez C. A., Víctor V. M., Esplugues J. V., Rojas J. M., Sánchez-Madrid F., Serrador J. M. (2008) Endothelial nitric oxide synthase regulates N-Ras activation on the Golgi complex of antigen-stimulated T cells. Proc. Natl. Acad. Sci. U.S.A. 105, 10507–10512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen S. C., Huang B., Liu Y. C., Shyu K. G., Lin P. Y., Wang D. L. (2008) Acute hypoxia enhances proteins' S-nitrosylation in endothelial cells. Biochem. Biophys. Res. Commun. 377, 1274–1278 [DOI] [PubMed] [Google Scholar]
- 63. Deora A. A., Hajjar D. P., Lander H. M. (2000) Recruitment and activation of Raf-1 kinase by nitric oxide-activated Ras. Biochemistry 39, 9901–9908 [DOI] [PubMed] [Google Scholar]
- 64. Williams J. G., Pappu K., Campbell S. L. (2003) Structural and biochemical studies of p21Ras S-nitrosylation and nitric oxide-mediated guanine nucleotide exchange. Proc. Natl. Acad. Sci. U.S.A. 100, 6376–6381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Deora A. A., Win T., Vanhaesebroeck B., Lander H. M. (1998) A redox-triggered Ras-effector interaction: Recruitment of phosphatidylinositol 3′-kinase to Ras by redox stress. J. Biol. Chem. 273, 29923–29928 [DOI] [PubMed] [Google Scholar]
- 66. Shanshiashvili L., Narmania N., Barbakadze T., Zhuravliova E., Natsvlishvili N., Ramsden J., Mikeladze D. G. (2011) S-Nitrosylation decreases the adsorption of H-Ras in lipid bilayer and changes intrinsic catalytic activity. Cell Biochem. Biophys. 59, 191–199 [DOI] [PubMed] [Google Scholar]
- 67. Thoreen C. C., Chantranupong L., Keys H. R., Wang T., Gray N. S., Sabatini D. M. (2012) A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jin H. K., Ahn S. H., Yoon J. W., Park J. W., Lee E. K., Yoo J. S., Lee J. C., Choi W. S., Han J. W. (2009) Rapamycin down-regulates inducible nitric oxide synthase by inducing proteasomal degradation. Biol. Pharm. Bull. 32, 988–992 [DOI] [PubMed] [Google Scholar]