

Background: Cannabinoids regulate serotonin signaling in prefrontal cortex.
Results: Cannabinoid-induced up-regulation and enhanced activity of serotonin 2A (5-HT2A) receptors are regulated by G-protein receptor kinase 5 (GRK5) in neuronal cells.
Conclusion: Cannabinoids differentially regulate expression of GRKs, which could contribute to the modulation of CB2 receptor signaling.
Significance: Cannabinoid-induced up-regulation of 5-HT2A receptors could represent an adverse effect of repeated exposure to cannabinoids.
Keywords: Arrestin, Cannabinoid Receptors, Cannabinoids, Molecular Pharmacology, Receptor Regulation, 5-HT2A Receptor, β-Arrestin, CB2 Receptor, GRK Proteins
Abstract
We have recently reported that cannabinoid agonists can up-regulate and enhance the activity of serotonin 2A (5-HT2A) receptors in the prefrontal cortex (PFCx). Increased expression and activity of cortical 5-HT2A receptors has been associated with neuropsychiatric disorders, such as anxiety and schizophrenia. Here we report that repeated CP55940 exposure selectively up-regulates GRK5 proteins in rat PFCx and in a neuronal cell culture model. We sought to examine the mechanism underlying the regulation of GRK5 and to identify the role of GRK5 in the cannabinoid agonist-induced up-regulation and enhanced activity of 5-HT2A receptors. Interestingly, we found that cannabinoid agonist-induced up-regulation of GRK5 involves CB2 receptors, β-arrestin 2, and ERK1/2 signaling because treatment with CB2 shRNA lentiviral particles, β-arrestin 2 shRNA lentiviral particles, or ERK1/2 inhibitor prevented the cannabinoid agonist-induced up-regulation of GRK5. Most importantly, we found that GRK5 shRNA lentiviral particle treatment prevented the cannabinoid agonist-induced up-regulation and enhanced 5-HT2A receptor-mediated calcium release. Repeated cannabinoid exposure was also associated with enhanced phosphorylation of CB2 receptors and increased interaction between β-arrestin 2 and ERK1/2. These latter phenomena were also significantly inhibited by GRK5 shRNA lentiviral treatment. Our results suggest that sustained activation of CB2 receptors, which up-regulates 5-HT2A receptor signaling, enhances GRK5 expression; the phosphorylation of CB2 receptors; and the β-arrestin 2/ERK interactions. These data could provide a rationale for some of the adverse effects associated with repeated cannabinoid agonist exposure.
Introduction
We have recently reported that repeated exposure to cannabinoid agonists induces a strong up-regulation and increases the activity of 5-HT2A2 receptors in rat prefrontal cortex (PFCx) and in two neuronal cell models (1–4). This cannabinoid-mediated up-regulation of 5-HT2A receptors was 1) induced by nonselective CB1/CB2 and selective CB2 receptor agonists (2, 4) and 2) inhibited by selective CB2, but not CB1, shRNA lentiviral particles, suggesting that CB2 receptors mediate this phenomenon (3). Moreover, this up-regulation of 5-HT2A receptors was β-arrestin 2- and ERK1/2-dependent because it was inhibited in cells stably transfected with β-arrestin 2 shRNA lentiviral particles (3) and by ERK1/2 inhibitors (2, 4).
The clinical manifestations of this CB2 receptor-induced up-regulation of 5-HT2A receptors are currently under discussion. It is noteworthy that recent and independent clinical studies provide evidence indicating that sustained use of nonselective cannabinoid agonists may precipitate the onset of mental disorders associated with dysfunction of 5-HT2A receptor neurotransmission in PFCx, such as anxiety, schizophrenia, and psychosis (5–9). Accordingly, recent preclinical studies have indicated that chronic, but not acute, exposure to non-selective (10, 11) or selective CB2 receptor agonists induced anxiety-like behaviors in rodents (12).
CB2 receptors have been identified in postsynaptic neurons in several brain areas of the limbic brain, including brain areas such as the PFCx, hippocampus, and amygdala (13–16). The CB2 receptor is a prototypical G-protein-coupled receptor (GPCR) that couples to the Gi/o class of G-proteins and can activate ERK1/2 signaling in either a G-protein- or β-arrestin-dependent pathway (17, 18). The different signaling and trafficking profiles of this receptor would depend on the nature of post-translational modifications, such as phosphorylation by G-protein receptor kinases (GRKs) that modify the interaction between this receptor and associated signaling proteins (such as β-arrestins and G-proteins) (17) and desensitization of this receptor (19).
Here we study the role of GRKs in the cannabinoid-induced up-regulation of 5-HT2A receptors. GRKs, such as GRK2, exert important roles in the desensitization and inhibition of β-arrestin 2 (βArr2) signaling of GPCRs (20, 21). Of note, recent results demonstrate that some GRKs, such as GRK5 and/or GRK6, also regulate βArr2 signaling-mediated ERK1/2 activation (20). Here we report that agonists of cannabinoid receptors differentially regulate the expression of GRK proteins, which would contribute to regulation of 5-HT2A receptors in neuronal cells. We hypothesize that the data presented here could provide, at least in part, a molecular mechanism by which repeated exposure to cannabinoids might be relevant to the pathophysiology of some cognitive and mood disorders by up-regulating and enhancing the activity of 5-HT2A receptors.
EXPERIMENTAL PROCEDURES
Drugs
CP55940, GP1a, ACEA, PD198306, MDL11,939, and SB242084 were purchased from Tocris (Ellisville, MO). Serotonin creatine sulfate complex was purchased from Sigma-Aldrich.
Animal Experimental Protocol
Male Sprague-Dawley rats (225–275 g; Harlan Laboratories, Indianapolis, IN) were housed two per cage in a temperature-, humidity-, and light-controlled room (12-h light/dark cycle, lights on 7:00 a.m. to 7:00 p.m.). Food and water were available ad libitum. All procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals as approved by the University of Kansas Institutional Animal Care and Use Committee.
After arrival, the rats were allowed to acclimate to their environment for at least 4 days prior to the start of the treatment period. Eight rats were randomly assigned to each group; cage mates were assigned to the same treatment group. The body weight of each rat was recorded every other day. All solutions were made fresh before administration, and rats were injected with either vehicle (Tween 80/ethanol/saline (1:1:18); 1 ml/kg, intraperitoneally) or CP55940 (0.05 mg/kg, intraperitoneally) once per day for 7 days. Rats were sacrificed by decapitation 48 h after the last CP55940 injection. The brains were immediately removed, and the PFCx was dissected and frozen in dry ice.
Phosphoprotein Purification
Phosphorylated proteins were separated by an affinity chromatography procedure using a phosphoprotein purification kit from Qiagen (Valencia, CA) as described in detail previously (22). Immunodetection by phospho-specific antibodies has shown that the kit yields a complete separation of non-phosphorylated (flow-through) and phosphorylated proteins (elution fraction) (23). Briefly, tissue or cells were homogenized in 200 μl of phosphoprotein lysis buffer containing 0.25% (w/v) CHAPS solution, protease inhibitor mixture, and Benzonase. These homogenates were incubated for 30 min at 4 °C and then centrifuged for 30 min at 10,000 × g and 4 °C. Thermo Scientific Pierce BCA assay reagents were utilized to determine the protein concentrations of the supernatants, and then 3.5 mg of total protein, adjusted to 0.1 mg/ml with phosphoprotein lysis buffer containing 0.25% CHAPS, was run through the phosphoprotein purification columns. The non-phosphorylated proteins were washed out of the columns with 35 ml of phosphoprotein lysis buffer, and the bound phosphorylated proteins were eluted with phosphoprotein elution buffer. Eluted fractions containing phosphorylated proteins were collected. The isolated phosphoprotein fractions were concentrated using Nanostep ultrafiltration columns with a molecular mass cut-off of 10 kDa. Thermo Scientific Pierce BCA assay reagents were utilized to determine the protein concentrations, and samples were analyzed by Western blot.
Western Blot
Membrane or cytosol fractions were isolated using the ProteoExtractTM native membrane protein extraction kit (Calbiochem). Expression of GRK5, GRK2, GRK6, or CB2 was determined by Western blot as described previously (3, 4). GRK5, GRK2, GRK6, and CB2 antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Antibodies were used at the following dilutions: GRK5 (1:1,000), GRK2 (1:1,000), GRK6 (1:1,000), and CB2 (1:1,000). The specificity of these antibodies has been verified in the literature (24–26). Films were analyzed densitometrically as described (3, 4). All samples were standardized to controls and normalized to their respective actin levels.
Quantitative Real-time PCR
These reactions were prepared using the QuantiFast SYBR Green PCR kit (Qiagen, Valencia, CA) and the ABI 7500 fast real-time PCR system (Applied Biosystems, Foster City, CA), and then data were analyzed using the comparative cycle threshold (Ct) method as described previously (3, 4). The primers used were as follows: 5-HT2A (forward, 5′-AACGGTCCATCCACAGAG-3′; reverse, 5′-AACAGGAAGAACACGATGC-3′), GRK5 (forward, 5′-GAACCACCAAAGAAAGGGCTG-3′; reverse, 5′-CTAGCTGCTTCCAGTGGAG-3′), GRK2 (forward, 5′-GATGAGGAGACACAAAAGGAATC-3′; reverse, 5′-TCAGAGGCCGTTGGCACTGCCACGCTG-3′), GRK6 (forward, 5′-TTTGGGCTGGATGGGTCTGTTC-3′; reverse, 5′-CGCTGCAGTTCCCACAGCAATC-3′), and GAPDH (forward, 5′-TGGAGTCTACTGGCGTCTTCAC-3′; reverse, 5′-GGCATGGACTGTGGTCATGA-3′). These primers have been validated in the literature (27, 28).
Cell Culture Protocol
CLU213 cells, a neuronal cell line that co-expresses 5-HT2A, D2, CB1, and CB2 receptors, were purchased from Cedarlane Laboratories (Burlington, NC). CLU213 cells were grown on 100-mm2 plates treated by vacuum gas plasma (Corning Inc.) and maintained in 5% CO2 at 37 °C, in Dulbecco's modified Eagle's medium (DMEM; Mediatech Inc., Manassas, VA) containing 10% fetal bovine serum (FBS; Thermo Scientific, Logan, UT).
Effect of Non-selective CB1/CB2 Receptor Agonist on GRK5 and GRK2 Expression
CLU213 cells were incubated with either vehicle (ethanol, 0.01% final concentration) or CP55940 (CB1/CB2 agonist, 1 nm) (29) for 72 h. Cells were washed (three times) with PBS every 24 h, and fresh vehicle or 1 nm CP55940 was added. Expression of GRK5, GRK6, or GRK2 was determined by Western blot or qRT-PCR.
Effect of Selective CB1 or CB2 Receptor Agonists on GRK5 mRNA
CLU213 cells were incubated with either vehicle (ethanol, 0.01% final concentration), GP1a 1 nm (selective CB2 agonist, Ki = 0.037 and 353 nm for CB2 and CB1 receptors, respectively) (30), or 15 nm ACEA (selective CB1 agonist, Ki = 1.4 nm and 3.1 μm for CB1 and CB2 receptors, respectively) (31) for 72 h. Cells were washed (three times) with PBS every 24 h, and fresh vehicle, GP1a, or ACEA was added. qRT-PCR for GRK5 was performed.
Lentivirus and Stable Transduction of shRNAs in CLU213 Cells
GRK5 shRNA (rat), β-arrestin 2 shRNA (rat), CB1 shRNA (rat), CB2 shRNA (rat), control shRNA lentiviral particles, Polybrene, and puromyocin were purchased from Santa Cruz Biotechnology, Inc. Optimal transduction conditions were determined, and transfection of cells with lentiviral particles was conducted as described previously (3). Cells were analyzed for GRK5, β-arrestin 2, CB1, or CB2 knockdown 1 week after initiation of puromyocin selection.
Effect of β-Arrestin 2, CB2, or CB1 shRNA Lentivirus Transfection on Cannabinoid-induced Up-regulation of GRK5 mRNA
After confirming that treatment with the β-arrestin 2, CB2, or CB1 shRNA lentivirus significantly reduced the respective protein levels, β-arrestin 2, CB2, or CB1 shRNA-treated cells were treated with either vehicle (ethanol, 0.01% final concentration) or GP1a (1 nm) for 72 h. Cells were washed (three times) with PBS every 24 h, and fresh vehicle or 1 nm GP1a was added.
Effect of a Selective ERK1/2 Inhibitor on GP1a-induced Increases in GRK5 mRNA
CLU213 cells were pretreated with either vehicle (ethanol, 0.01% final concentration) or PD 198306 (200 nm) (32, 33). 20 min later, cells were incubated with either vehicle (ethanol, 0.01% final concentration) or GP1a (1 nm) for 72 h. Cells were washed (three times) with PBS every 24 h, and pretreatment and treatment were repeated.
Effect of GRK5 shRNA Lentivirus Transfection on Cannabinoid-induced Up-regulation of 5-HT2A Receptors
After confirming that treatment with the GRK5 shRNA lentivirus reduced GRK5 mRNA and protein levels ∼70%, control or GRK5 shRNA-treated cells were treated with either vehicle (ethanol, 0.01% final concentration), CP55940 (1 nm), or GP1a (1 nm) for 72 h.
Calcium Assay to measure 5-HT2A Receptor Activity
Optimal conditions were elucidated using different buffers, plating densities, agonists, time courses, etc., and through reference to previously published protocols (34, 35). Cells were plated at 30,000 cells/well in complete medium and grown to 90% confluence on black-sided 96-well plates. 24 h prior to measuring calcium release, medium was changed to serum-free medium. After a 24-h incubation in serum-free medium, cells were washed (twice) with Kreb's medium (135 mm NaCl, 5.9 mm KCl, 1.5 mm CaCl2, 1.2 mm MgCl2, 11.6 mm Hepes, 11.5 mm d-glucose, pH 7.3) and incubated with 4 μm Fluo 3-AM in 200 μl of Kreb's medium for 60 min at 37 °C in the dark. After loading, cells were washed (twice) with Kreb's medium and incubated in 200 μl of Kreb's medium for 30 min to allow for de-esterfication of intracellular AM esters. Finally, cells were stimulated with a single injection of 5-HT, and the response was recorded for 1 min in 6-s intervals. Fluo 3-AM fluorescence using 485-nm excitation and 528-nm emission was measured with a BioTek fluorescence plate reader (34, 35).
Initial experiments were performed after loading cells with Fluo 3-AM and included serotonin (5-HT) dose response (0.1 nm to 10 μm 5-HT) experiments with MDL11,939 or SB242084 (5-HT2A and 5-HT2C receptor antagonists, respectively) (34, 35). The MDL11,939 Kd for 5-HT2A and 5-HT2C receptors is 0.54 and 86 nm, respectively (34, 35). The SB242084 Kd for 5-HT2C and 5-HT2A receptors is 0.48 and 144 nm, respectively (34, 35). Cannabinoid experiments were conducted using naive cells or transfected cells (control, GRK5, or CB2 shRNA lentiviral particles). These cells were pretreated with either vehicle, CP55940 (1 nm), GP1a (1 nm), or ACEA (15 nm) over 72 h. Cells were washed (three times) with PBS every 24 h, and fresh vehicle or cannabinoid agonist were added. After loading cells with Fluo 3-AM, cells were treated with 10 nm SB242084, to inhibit 5-HT2C receptor-mediated Ca2+ release, for 20 min during the 30-min incubation with Kreb's medium and then stimulated with either 0.1 nm, 1 nm, 0.4 nm, 10 nm, 100 nm, 1 μm, or 10 μm 5-HT (34, 35).
Effect of GRK5 shRNA Lentivirus Transfection on Cannabinoid-induced CB2 Receptor Phosphorylation or β-Arrestin 2/ERK Interaction
Cells stably transfected with control or GRK5 shRNA lentivirus particles were treated with vehicle (ethanol, 0.01% final concentration) or GP1a (1 nm) for 72 h. Cells were washed (three times) with Kreb's buffer every 24 h, and fresh vehicle or GP1a was added. Phosphorylated proteins were isolated from cells, and Western blot was used to determine the expression of phosphorylated CB2 receptors as described above. Co-immunoprecipitation of β-arrestin 2/ERK was examined following the protocol listed below. Expression of phosphorylated CB2 receptors was determined by Western blot as described above.
Co-immunoprecipitation
These experiments were conducted with the Thermo Scientific Pierce co-immunoprecipitation kit following the manufacturer's protocol and as described in detail previously (1). The β-arrestin 2 and ERK1/2 antibody was purchased from Santa Cruz Biotechnology, Inc. Samples were analyzed by Western blot using ERK1/2 antibody. The specificity of the β-arrestin 2 or ERK1/2 antibody has been verified (1).
Statistics
All data are expressed as the mean ± S.E., where n indicates the number of rats or cell culture plates per group. Data were analyzed by an unpaired Student's t test or ANOVA.
RESULTS
Chronic exposure to cannabinoid receptor agonists could mediate the cannabinoid-induced up-regulation of 5-HT2A receptors, at least in part, through changes in the phosphorylation status of CB2 receptors by GRK proteins. We initially examined the effect of repeated exposure to CP55940 (CB1/CB2 receptor agonist) on the phosphorylation status of the CB2 receptors in rat PFCx (Fig. 1A). Phosphorylated proteins were separated from the PFCx of vehicle- and CP55940-treated rats, and Western blot was conducted as explained previously. We found a significant (p < 0.01) increase in the phosphorylation of CB2 receptors in CP55940-treated rats compared with vehicle-treated animals (121 ± 12% increase; Fig. 1A). Of note, CP55940 treatment did not significantly (p > 0.05) modify the total CB2 receptor protein expression in PFCx homogenate compared with vehicle-treated controls (Fig. 1B).
FIGURE 1.

CP55940-induced enhanced phosphorylation of CB2 receptors, increased GRK5 expression levels, and reduced GRK2 expression levels in rat PFCx. Rats were injected with CP55940 (0.05 mg/kg, intraperitoneally) once a day for 7 days. After decapitation, the brains were collected, and PFCx was dissected. A, phosphorylated proteins were separated and detected as described under “Experimental Procedures.” 30 μg of isolated phosphorylated protein was used in Western blot detection. B–E, CB2 receptor and GRK protein levels were evaluated by Western blot. Proteins (8 μg) were resolved by SDS-PAGE, and antibodies for CB2 receptor (A and B), GRK5 (C), GRK6 (D), and GRK2 (E) were used to detect the proteins of interest. Representative Western blots are shown, and integrated optical density was calculated as described under “Experimental Procedures.” β-Actin was used as a loading control. F, GRK5, GRK6, and GRK2 mRNA levels were evaluated by qRT-PCR as described under “Experimental Procedures.” **, p < 0.01; *, p < 0.05, significant effect of CP55940 treatment compared with vehicle-treated controls. The data represent mean ± S.E. (error bars) (n = 6–8).
Because CP55940-enhanced expression of selective GRKs could underlie the increased phosphorylation of CB2 receptor, we studied the effect of CP55940 exposure on the protein levels of GRK5, GRK6, and GRK2 proteins in rat PFCx. Chronic administration of CP55940 produced significant (p < 0.01) increases in GRK5 protein levels (58 ± 6% increase compared with controls; Fig. 1C). However, we did not detect any significant (p > 0.05) changes in GRK6 protein levels in PFCx (Fig. 1D). GRK2 protein levels were significantly (p < 0.01) reduced after repeated CP55940 exposure in rat PFCx (24 ± 2% decrease; Fig. 1E). We also examined what effect CP55940 treatment had on GRK5, GRK6, and GRK2 mRNA levels (Fig. 1F). GRK5 mRNA levels were significantly (p < 0.01) increased in PFCx of CP55940-treated rats (62 ± 0.4% increase compared with controls). GRK6 mRNA levels did not significantly (p > 0.05) change, whereas GRK2 mRNA levels were significantly (p < 0.05) reduced in PFCx of CP55940-treated rats (22 ± 0.1% reduction compared with vehicle-treated controls; Fig. 1F).
We then used a neuronal cell line, CLU213 cells, to determine if CP55940 treatment shifts GRK expression similar to shifts found in rat PFCx and to better examine the mechanisms involved in the cannabinoid-induced up-regulation of GRK5 proteins. Here we treated cells with CP55940 for 72 h in order to assess the effect of repeated cannabinoid agonist exposure on the expression of GRKs because our previous findings show that repeated, but not single, exposure to cannabinoid agonists up-regulates 5-HT2A receptor protein expression (1–4). CP55940 treatment in these cells significantly (p < 0.01) increased GRK5 protein levels (67 ± 3% increase compared with vehicle-treated controls) without significant (p > 0.05) changes in the protein levels of GRK6 (Fig. 2, A and B, respectively). On the other hand, GRK2 protein levels were significantly (p < 0.01) reduced compared with controls (24 ± 2% decrease; Fig. 2C). CP55940 treatment also significantly (p < 0.05) increased GRK5 mRNA levels and significantly (p < 0.05) reduced GRK2 mRNA levels compared with vehicle-treated cells (69 ± 0.2% increase and 24 ± 0.05% decrease, respectively). There was no significant (p > 0.05) change in GRK6 mRNA levels in CP55940-treated cells compared with vehicle-treated controls (Fig. 2D).
FIGURE 2.

CP55940-induced increased GRK5 and reduced GRK2 expression levels in CLU213 cells. Cells were incubated with either vehicle (ethanol, 0.01% final concentration) or CP55940 (1 nm) for 72 h. Cells were washed (three times) with PBS every 24 h, and fresh vehicle or CP55940 (1 nm) was added. A and B, Western blots to show alteration or no alteration in GRK5 (A), GRK6 (B), or GRK2 (C) protein levels after repeated CP55940 treatment. Proteins (8 μg) were resolved by SDS-PAGE as described under “Experimental Procedures.” D, qRT-PCR to assess GRK5, GRK6, and GRK2 mRNA levels. **, p < 0.01; *, p < 0.05, significant effect of CP55940 treatment compared with vehicle-treated controls. The data represent mean ± S.E. (error bars) (n = 3).
We then aimed to identify the cannabinoid receptor involved in the up-regulation of GRK5 because this could mediate the enhanced phosphorylation of CB2 receptors detected in rat PFCx. Cells were treated with either vehicle, GP1a (1 nm) (selective CB2 agonist), or ACEA (15 nm) (selective CB1 agonist) over 72 h. We found that GP1a treatment significantly (p < 0.01) increased (69 ± 0.05% increase), whereas ACEA did not significantly (p > 0.5) modify GRK5 mRNA levels compared with controls (Fig. 3A). This evidence suggested that CB2 receptors could mediate the up-regulation of GRK5. We then tested the effect of GP1a on GRK5 mRNA levels in either control, CB1 shRNA, or CB2 shRNA stably transfected cells over 72 h. We have previously shown that treatment with the CB1 or CB2 shRNA lentiviral particles significantly reduces CB1 or CB2 receptor expression, respectively (3). We found that treatment with GP1a significantly (p < 0.01) increased GRK5 mRNA levels in control and CB1 shRNA-treated cells (73 ± 0.03 and 73 ± 0.04% increase compared with controls, respectively). It is noteworthy that CB2 shRNA treatment prevented (p < 0.01) the GP1a-induced increases in GRK5 mRNA levels (Fig. 3B). Neither control, CB1, nor CB2 shRNA lentivirus treatment significantly (p > 0.01) modified basal GRK5 mRNA levels. The two-way ANOVA for GRK5 mRNA showed significant main effects of transfection (F2,17 = 20.4, p < 0.0001) and cannabinoid agonist treatment (F1,17 = 187, p < 0.0001). There was a significant interaction between transfection and cannabinoid agonist treatment (F2,17 = 22.8, p < 0.0001) on GRK5 mRNA levels.
FIGURE 3.

GP1a, a selective CB2 receptor agonist, up-regulated GRK5 via CB2 receptors, β-arrestin 2, and ERK1/2 signaling in CLU213 cells. A, cells were incubated with either vehicle (ethanol 0.01% final concentration), 1 nm GP1a, or 15 nm ACEA for 72 h. qRT-PCR was used to show the effect of selective CB1 or CB2 receptor agonist treatment on GRK5 mRNA levels. **, p < 0.01 significant effect of GP1a treatment compared with vehicle-treated controls. B, cells stably transfected with control, CB1, or CB2 shRNA lentiviral particles were treated with vehicle or 1 nm GP1a for 72 h. qRT-PCR was used to examine the effect of CB1 or CB2 receptor knockdown on GP1a-induced increases in GRK5 mRNA levels. **, p < 0.01, significant effect of GP1a treatment on GRK5 mRNA levels in control or CB1 shRNA lentivirus-treated cells compared with vehicle-treated controls. ##, p < 0.01, significant effect of CB2 shRNA lentivirus transfection on the GP1a-induced up-regulation of GRK5. *, p < 0.05, significant effect of GP1a treatment in CB2 shRNA-transfected cells compared with vehicle-treated/CB2 shRNA-transfected cells. C, cells were pretreated with vehicle or 200 nm PD198306, potent ERK1/2 inhibitor, and then treated with vehicle or 1 nm GP1a for 72 h. qRT-PCR was used to show the effect of PD198306 pretreatment on GP1a-induced increases in GRK5 mRNA levels. **, p < 0.01, significant effect of GP1a treatment compared with vehicle-treated controls. ##, p < 0.01, significant effect of PD198306 pretreatment on GP1a-induced increases in GRK5 mRNA levels compared with vehicle-treated controls. D, cells were stably transfected with control or β-arrestin 2 shRNA lentivirus particles and then treated with vehicle or 1 nm GP1a for 72 h. qRT-PCR was used to examine the effect of β-arrestin 2 knockdown on GP1a-induced increases in GRK5 mRNA levels. **, p < 0.01, significant effect of GP1a treatment on GRK5 mRNA levels in control shRNA lentivirus-treated cells compared with vehicle-treated controls. ##, p < 0.01, significant effect of β-arrestin 2 shRNA lentivirus transfection on the GP1a-induced up-regulation of GRK5. *, p < 0.05, significant effect of GP1a treatment in β-arrestin 2 shRNA-transfected cells compared with vehicle-treated β-arrestin 2 shRNA-transfected cells. The data represent mean ± S.E. (error bars) (n = 3).
Next, we investigated whether the ERK1/2 signaling pathway may be involved in the cannabinoid-induced up-regulation of GRK5. CB2 receptors are positively coupled to the ERK1/2 signaling pathway, and cannabinoid agonists, such as Δ9-THC, can regulate the expression of some GRKs through the ERK1/2 signaling pathway (36). We used PD198306, a selective ERK1/2 inhibitor (37), to study the effect of GP1a-induced ERK1/2 activation on GRK5 up-regulation. GP1a treatment significantly (p < 0.01) increased GRK5 mRNA levels compared with vehicle-treated cells (77 ± 2% increase; Fig. 3D). This up-regulation of GRK5 was prevented (p < 0.01) by pretreatment with PD198306. This ERK1/2 inhibitor pretreatment did not significantly (p > 0.05) modify basal levels of GRK5 mRNA. The two-way ANOVA for GRK5 mRNA showed significant main effects of PD198306 pretreatment (F1,11 = 22.4, p < 0.0015) and GP1a treatment (F1,11 = 43.3, p < 0.0002). There was a significant interaction between PD198306 and GP1a treatment (F1,11 = 35.2, p < 0.0003).
We also used cells stably transfected with either β-arrestin 2 or control shRNA lentiviral particles to study the contribution of β-arrestin 2 in the cannabinoid-induced up-regulation of GRK5. We have previously shown that treatment with β-arrestin 2 shRNA lentiviral particles significantly reduces β-arrestin 2 protein expression by ∼80% compared with control shRNA-treated cells (1, 3). Here, β-arrestin 2- or control shRNA-treated cells were incubated with vehicle or GP1a for 72 h. GP1a significantly (p < 0.01) up-regulated GRK5 mRNA in control shRNA-treated cells by 73 ± 6%. The cannabinoid agonist-induced up-regulation of GRK5 mRNA levels was significantly (p < 0.01) reduced in cells stably transfected with β-arrestin 2 shRNA lentiviral particles (29 ± 5% increase compared with controls; Fig. 3E). The two-way ANOVA showed main effects of transfection (F1,11 = 8.72, p < 0.0183), CB2 agonist treatment (F1,11 = 52.3, p < 0.0001), and a main interaction between these two factors (F1,11 = 14.3, p < 0.0054) on GRK5 mRNA levels.
In order to determine whether GRK5 is involved in the cannabinoid-induced up-regulation of 5-HT2A receptors, we used cells stably transfected with GRK5 or control shRNA lentiviral particles. GRK5 shRNA lentiviral particle treatment significantly (p < 0.01) reduced both GRK5 mRNA (Fig. 4A) and protein levels (Fig. 4B) by ∼80% compared with control-treated cells. As expected, CP55940 up-regulated 5-HT2A mRNA in control shRNA-treated cells (93 ± 2% increase; Fig. 4C). Of note, CP55940-induced increases in 5-HT2A mRNA were significantly (p < 0.01) reduced in cells stably transfected with GRK5 shRNA (Fig. 4C). In contrast, CP55940 treatment in GRK5 shRNA-treated cells significantly (p < 0.01) increased 5-HT2A mRNA levels by only 29 ± 0.03%. The two-way ANOVA showed main effects of transfection (F1,11 = 65.3, p < 0.0001), CP55940 (F1,11 = 231, p < 0.0001) on 5-HT2A mRNA levels and a main interaction between these two factors (F1,11 = 58.1, p < 0.0001). Similarly, GP1a up-regulated 5-HT2A mRNA in control shRNA cells (97 ± 0.02% increase; Fig. 4D), and GP1a-induced increases in 5-HT2A mRNA were significantly (p < 0.01) reduced in cells stably transfected with GRK5 shRNA (70% reduction compared with GP1a effect on control cells; Fig. 4D). The two-way ANOVA showed main effects of transfection (F1,11 = 40.7, p < 0.0002), GP1a (F1,11 = 124, p < 0.0001), and a main interaction between these two factors (F1,11 = 42.2, p < 0.0002) on 5-HT2A mRNA levels.
FIGURE 4.
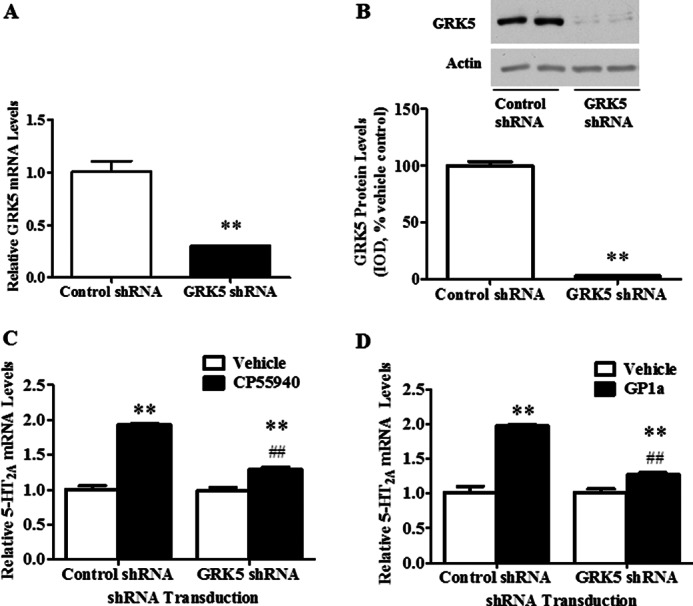
GRK5 is necessary for the CP55940 and GP1a-induced up-regulation of 5-HT2A receptors in CLU213 cells. Cells were transfected with control or GRK5 shRNA lentiviral particles as described under “Experimental Procedures.” A, qRT-PCR to show reduced GRK5 mRNA levels in cells treated with GRK5 shRNA lentiviral particles. B, Western blot to show reduced GRK5 protein levels in cells treated with GRK5 shRNA lentiviral particles. A and B, **, p < 0.01, significant effect of GRK5 shRNA compared with control shRNA-treated cells. C and D, cells stably transfected with control or GRK5 shRNA lentiviral particles were treated with either vehicle, 1 nm CP55940, or 1 nm GP1a for 72 h. C, qRT-PCR to show the effect of GRK5 knockdown on CP55940-induced increases on 5-HT2A receptor mRNA levels. **, p < 0.01, significant effect of CP55940 treatment on 5-HT2A receptor mRNA levels in control shRNA lentivirus-transfected cells compared with vehicle-treated controls. ##, p < 0.01, significant effect of GRK5 shRNA lentivirus transfection on the CP55940-induced up-regulation of 5-HT2A receptors. **, p < 0.01, significant effect of CP55940 treatment in GRK5 shRNA-transfected cells compared with vehicle-treated GRK5 shRNA-transfected cells. D, qRT-PCR to show the effect of GRK5 knockdown on GP1a-induced increases in 5-HT2A receptor mRNA levels. **, p < 0.01, significant effect of GP1a treatment on 5-HT2A receptor mRNA levels in control shRNA lentivirus-transfected cells compared with vehicle-treated controls. ##, p < 0.01, significant effect of GRK5 shRNA lentivirus transfection on the GP1a-induced up-regulation of 5-HT2A receptors. **, p < 0.01, significant effect of GP1a treatment in GRK5 shRNA-transfected cells compared with vehicle-treated GRK5 shRNA-transfected cells. The data represent mean ± S.E. (error bars) (n = 3).
In the next studies, we examined the role of GRK5 in the cannabinoid-induced increases in 5-HT2A receptor activity. We have previously reported that repeated CP55940 treatment in this neuronal cell culture model significantly enhances 5-HT2A receptor-mediated phosphoinositol hydrolysis (2). Here, we studied the effects of GRK5 shRNA treatment on the 5-HT2A receptor-mediated calcium (Ca2+) release. We began conducting dose response experiments with 5-HT as described under “Experimental Procedures.” We used 5-HT in these experiments because previous studies have shown that the maximal response to (−)-1–2,5-dmiethoxy-4-iodoamphetamine HCl (a 5HT2A/2C receptor agonist) is lower than the maximal response to 5-HT in two different cell lines (35). A dose-response experiment in CLU213 cells showed that 5-HT stimulated Ca2+ release in a dose-dependent way (Fig. 5A) with an EC50 of 0.11 ± 0.02 nm. To confirm that this response was the result of stimulation of 5-HT2A but not 5-HT2C receptors, we measured the effect of MDL11,939 or SB242084 (5-HT2A and 5-HT2C receptor antagonists, respectively) (34, 35) in the 5-HT-induced Ca2+ release in a neuronal cell model. MDL11,939 and SB242084 inhibited the 5-HT-mediated Ca2+ release with different affinities. Whereas the MDL11,939 IC50 was ∼1 nm, the SB242084 IC50 was 0.1 μm, suggesting that the 5-HT-mediated Ca2+ release in CLU213 cells is mainly mediated by 5-HT2A receptors at the concentration of 5-HT (0.1 nm) used in this assay (Fig. 5B). 10 nm SB242084 was added to the preincubation medium in the cannabinoid assays to prevent the activation of 5-HT2C receptors. Based on the Kd provided under “Experimental Procedures,” the fractional occupancy of 5-HT2C and 5-HT2A receptors at this dose of SB242084 is 95 and 7%, respectively.
FIGURE 5.

Repeated CP55940 or GP1a pretreatment enhances 5-HT2A receptor-mediated (Ca2+) calcium release in CLU213 cells. Calcium assays were optimized and conducted as described under “Experimental Procedures.” A, cells were stimulated with increasing doses of 5-HT. B, cells were preincubated with increasing doses of MDL11,939 (selective 5-HT2A receptor antagonist, IC50 = 1 nm) or SB242084 (selective 5-HT2C receptor antagonist, IC50 = 0.1 μm) for 20 min and then stimulated with 1 μm 5-HT. C, cells were pretreated with vehicle or 1 nm CP55940 for 72 h. Cells were then preincubated with 10 nm SB242084 for 20 min to inhibit 5-HT2C receptor-mediated Ca2+ release and then stimulated with either 0.1 or 0.4 nm 5-HT. **, p < 0.01, significant effect of CP55940 pretreatment/5-HT stimulation or 5-HT stimulation on Ca2+ release in cells. ##, p < 0.01, significant effect of CP55940 pretreatment/5-HT stimulation compared with 5-HT stimulation in cells. D, cells were treated with vehicle or 1 nm GP1a for 72 h. Cells were then preincubated with 10 nm SB242084 for 20 min, to inhibit 5-HT2C receptor-mediated Ca2+ release and then stimulated with either 0.1 nm, 1 nm, 0.4 nm, 10 nm, 100 nm, 1 μm, or 10 μm 5-HT. **, p < 0.01, significant effect of 0.1 nm to 10 μm of 5-HT stimulation compared with vehicle-treated controls. ##, p < 0.01, significant effect of GP1a pretreatment/5-HT stimulation compared with 5-HT-stimulated cells. The data represent mean ± S.E. (error bars) (n = 3).
We then studied the effect of repeated exposure (72 h) to either CP55940 or GP1a on the 5-HT-mediated Ca2+ release in neuronal cells (Fig. 5, C and D). Whereas in vehicle-treated cells, 0.1 and 0.4 nm 5-HT produced significant (p < 0.01) increases (100 and 122%, respectively), in Ca2+ release in neuronal cells, these increases were significantly (p < 0.01) higher in cells treated with CP55940 (220 and 253% at 0.1 nm and 0.4 nm 5-HT, respectively). The two-way ANOVA showed main effects of CP55940 treatment (F1,17 = 70,421, p < 0.0001), 5-HT treatment (F2,17 = 123,689, p < 0.0001), and a main interaction between them (F2,17 = 17,910, p < 0.0001). Repeated GP1a exposure (72 h) also enhanced the 5-HT-mediated Ca2+ release in CLU213 cells (Fig. 5D). Indeed, we observed a 2-fold increase (p < 0.01) in the maximal 5-HT-mediated responses in GP1a-treated cells compared with vehicle controls. However, no changes (p > 0.05) were detected in the EC50 values between control- and GP1a-treated cells (0.098 ± 0.004 and 0.095 ± 0.009 nm, respectively).
Our previous results suggest that chronic exposure to agonists of CB2 receptors enhance the 5-HT2A receptor-mediated Ca2+ release receptors in our neuronal cell model. Our next experiment aimed to confirm the role of CB2 receptors in this phenomenon (Fig. 6A). Here, control or stably transfected cells with CB2 shRNA were incubated with either vehicle, CP55940 (1 nm), ACEA (20 nm), or GP1a (1 nm) for 72 h. Both CP55940 and GP1a induced a significant (p < 0.01) and approximately 2-fold increase in the 5-HT-mediated Ca2+ release in control cells (220.6 ± 1.6 and 215.4 ± 3.0% increase over maximal response for CP55940- and GP1a-treated cells, respectively). ACEA, a selective CB1 receptor agonist, did not significantly (p > 0.05) modify the 5-HT-mediated Ca2+ responses in control cells. On the other hand, neither CP55940 (1 nm), GP1a (1 nm), nor ACEA (15 nm) significantly (p > 0.05) modified the 5-HT-mediated Ca2+ responses in cells stably transfected with CB2 shRNA. The two-way ANOVA showed a significant main effect of transfection (F1,23 = 2235.8, p < 0.0001), cannabinoid treatment (F3,23 = 3279.8, p < 0.0001), and a main interaction between them (F3,23 = 766.5, p < 0.0001).
FIGURE 6.
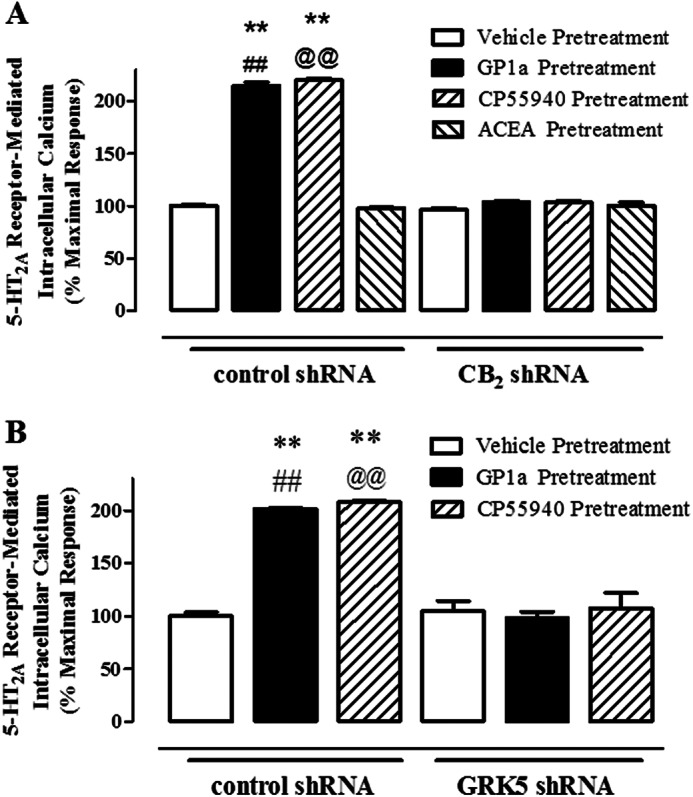
CB2 and GRK5 are necessary for the CP55940 and GP1a-induced increases in 5-HT2A receptor-mediated Ca2+ release in CLU213 cells. Cells were stably transfected with either control, CB2, or GRK5 shRNA lentiviral particles as described under “Experimental Procedures.” A, cells stably transfected with control or CB2 shRNA lentiviral particles were pretreated with either vehicle, 1 nm CP55940, 1 nm GP1a, or 15 nm ACEA for 72 h. Cells were then preincubated with 10 nm SB242084 for 20 min and then stimulated with 0.4 nm 5-HT. B, cells stably transfected with control or GRK5 shRNA lentiviral particles were pretreated with either vehicle, 1 nm CP55940, or 15 nm GP1a. Cells were then preincubated with 10 nm SB242084 for 20 min and then stimulated with 0.4 nm 5-HT. **, p < 0.01, significant effect of CP55940 pretreatment/5-HT stimulation or GP1a pretreatment/5-HT stimulation in control shRNA-transfected cells compared with vehicle-treated controls. ##, p < 0.01, significant effect of GP1a pretreatment/5-HT stimulation in control shRNA-transfected cells compared with GP1a pretreatment/5-HT stimulation in CB2 or GRK5 shRNA-transfected cells. @@, p < 0.01, significant effect of CP55940 pretreatment/5-HT stimulation in control shRNA-transfected cells compared with CP55940 pretreatment/5-HT stimulation in CB2 or GRK5 shRNA-transfected cells. The data represent mean ± S.E. (error bars) (n = 3).
Our next aim was to determine whether GRK5 plays a significant role in the 5-HT-mediated Ca2+ release in a neuronal cell model. Control cells or cells stably transfected with GRK5 shRNA were incubated with either vehicle, CP55940 (1 nm) or GP1a (1 nm), for 72 h. In control cells, 5-HT (0.1 nm)-mediated Ca2+ release was significantly increased by both CP55940 and GP1a treatment (205 ± 3% and 201 ± 4% increase over control for CP55940 or GP1a, respectively). It is noteworthy that in cells stably transfected with GRK5 shRNA, neither CP55940 nor GP1a induced significant increases in the 5-HT-mediated Ca2+ release. The two-way ANOVA showed a significant main effect of transfection (F1,17 = 215.2, p < 0.0001) and cannabinoid treatment (F2,17 = 55.97, p < 0.0001) and a main interaction between them (F2,17 = 46.58, p < 0.0001). In summary, these results suggest that GRK5 plays a pivotal role in the CB2 receptor-induced up-regulation of 5-HT2A receptors in our neuronal cell model.
We then examined the role of GRK5 in the cannabinoid agonist-induced phosphorylation of CB2 receptors. Cells stably transfected with either GRK5 or control shRNA lentiviral particles were treated with vehicle or GP1a (1 nm) for 72 h, and phosphorylated proteins were isolated as described under “Experimental Procedures.” We found that GP1a treatment significantly (p < 0.01) enhanced phosphorylation of CB2 receptors in control shRNA-treated cells by 36 ± 7% (Fig. 7A). Of note, this GP1a-induced enhanced phosphorylation of CB2 receptors was prevented (p < 0.01) in cells stably transfected with GRK5 shRNA lentiviral particles. No significant differences in the CB2 receptor phosphorylation levels were detected between vehicle and GP1a in cells stably transfected with GRK5 shRNA lentiviral particles. The two-way ANOVA for phosphorylated CB2 showed main effects of transfection (F1,43 = 21, p < 0.0001) and GP1a (F1,43 = 4.8, p < 0.0333). There was a significant interaction between transfection and GP1a treatment (F1,43 = 8.8, p < 0.005) on phosphorylated CB2 receptors. We then examined the effects of GRK5 shRNA lentivirus treatment and GP1a treatment on CB2 receptor protein levels in whole cell lysates. Repeated GP1a treatment did not significantly (p > 0.05) modify CB2 receptor protein levels in whole cell lysates (Fig. 7B). Furthermore, GRK5 shRNA lentivirus particle transfection did not significantly (p > 0.05) alter the basal levels of CB2 receptors (Fig. 7B).
FIGURE 7.
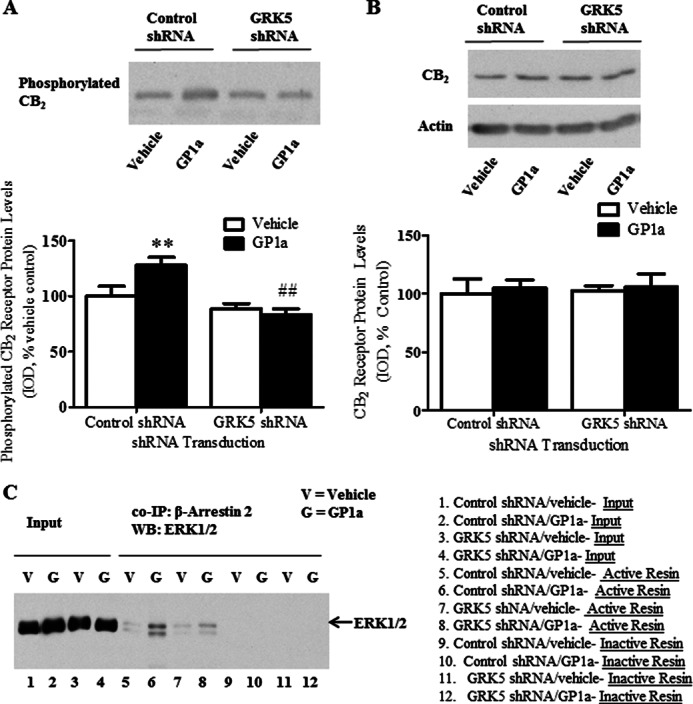
GRK5 mediates GP1a-induced increases in CB2 phosphorylation and enhanced β-arrestin 2/ERK interaction in CLU213 cells. To examine the role of GRK5 in the GP1a-induced increases in CB2 receptor phosphorylation and β-arrestin 2/ERK interaction, cells stably transfected with control of GRK5 shRNA lentiviral particles were treated with either vehicle or 1 nm GP1a for 72 h. A, phosphorylated proteins were separated and detected as described under “Experimental Procedures.” 30 μg of isolated phosphorylated protein was used in Western blot detection. **, p < 0.01, significant effect of GP1a treatment on CB2 phosphorylation levels in control shRNA lentivirus-transfected cells compared with vehicle-treated controls. ##, p < 0.01, significant effect of GRK5 shRNA lentivirus transfection on the GP1a-induced increases in CB2 phosphorylation. The data represent mean ± S.E. (n = 3). B, CB2 receptor protein levels in whole cell lysate were evaluated by Western blot as described under “Experimental Procedures.” Representative Western blots are shown, and β-actin was used as a loading control. The data represent mean ± S.E. (n = 3). C, co-immunoprecipitation was used to examine the role of GRK5 in GP1a-induced increases in β-arrestin 2/ERK interaction. Co-immunoprecipitation was conducted as described under “Experimental Procedures.” Negative controls (lanes 9–12) received the same concentration of β-arrestin 2 antibody except that the coupling resin was replaced with control agarose resin that is not amine-reactive. All columns were incubated with whole cell lysate (300 μg) from vehicle-treated (lanes 1 and 3) or GP1a-treated (lanes 4 and 6) cells. Cell lysate (15 μg of protein) was used as an input control (lanes 1–4). The data represent mean ± S.E. (error bars) (n = 4).
We recently reported chronic CP55940 treatment can enhance β-arrestin 2 and ERK1/2 interaction in rat PFCx (1). This cannabinoid agonist-induced enhanced interaction could be mediated by GRK5. Here, we used co-immunoprecipitation protocols to study the effect of GP1a treatment on the physical interaction between β-arrestin 2 and ERK1/2 in control- and GRK5 shRNA lentivirus-transfected cells (Fig. 7C). We used β-arrestin 2 antibody as bait and ERK1/2 antibody as prey. Inactive columns, which are unable to bind β-arrestin 2 antibody, were used as a control as described under “Experimental Procedures.” We found that ERK1/2 co-precipitates with β-arrestin 2 when we used β-arrestin 2 as bait (Fig. 7C, lanes 5–8). Interestingly, we detected a significant 4-fold increase in the interaction between β-arrestin 2 and ERK1/2 in GP1a/control shRNA-treated cells compared with vehicle/control shRNA-treated cells (Fig. 7C, lanes 5 and 6, vehicle- and GP1a-treated, respectively). In GRK5 shRNA-treated cells, this GP1a-induced enhanced interaction between β-arrestin 2 and ERK1/2 was significantly reduced (∼1.5-fold) compared with GP1a/control shRNA-treated cells (lanes 6 and 8, GP1a/control shRNA and GP1a/GRK5 shRNA, respectively). No co-precipitation of β-arrestin 2 and ERK1/2 was detected using the inactive columns (Fig. 7C, lanes 9–12). The two-way ANOVA for β-arrestin 2/ERK co-immunoprecipitation showed main effects of transfection (F1,15 = 718.4, p < 0.0001) and GP1a (F1,15 = 1252, p < 0.0001). There was a significant interaction between transfection and GP1a treatment (F1,15 = 649.6, p < 0.0001) on β-arrestin 2/ERK co-immunoprecipitation.
DISCUSSION
Cannabinoid agonists produce their physiological effects through the activation of two G-protein-coupled cannabinoid receptors in the brain, the CB1 and CB2 receptors (18, 38). CB1 and CB2 receptors bind endocannabinoids, synthetic cannabinoids, and cannabinoids found in nature (such as in Cannabis sativa) with high affinity (18, 38). Although only CB1 receptors were initially identified in the brain (39), later studies have also identified CB2 receptors in several brain areas, including PFCx, hippocampus, amygdala, substantia nigra, and cerebellum (13, 14), triggering a reevaluation of the possible roles that CB2 receptors might play in the brain.
We have previously reported that repeated exposure to either nonselective cannabinoid agonists or selective CB2 receptor agonists up-regulates and enhances the activity of 5-HT2A receptors in rat PFCx and neuronal cell models (1–4). CB2 receptors can couple to the Gi/o class of G-proteins to regulate transient ERK1/2 signaling, whereas β-arrestin 2 may be involved in the long term regulation of ERK1/2 signaling (1, 17, 40, 41). Recent evidence has highlighted that neuronal CB1 receptors can modulate ERK1/2 signaling through Gi/o and multiple tyrosine kinase receptors (41). Although G-protein mediated activation of ERK1/2 is transient and peaks within 2–5 min (42, 43), β-arrestins can form a scaffolding complex with ERK1/2 to regulate long term ERK1/2 activity (42–44). Although the mechanisms of the cannabinoid-induced up-regulation of 5-HT2A receptors have not been completely identified, our results suggest that activation of the β-arrestin 2 and ERK1/2 signaling pathway mediates this phenomenon that is dependent on CB2, but not CB1, receptors (2, 4). The key role of β-arrestin 2 in this up-regulation seems to involve an enhanced cannabinoid-induced interaction between β-arrestin 2 and ERK1/2 in rat PFCx (1).
Recent reports suggest that certain GRK proteins could trigger the activation of the β-arrestin 2 and ERK1/2 signaling pathway (45). The classical role described for GRK proteins is to trigger the desensitization of GPCRs (42, 46). Indeed, GRK2 and GRK3 proteins would phosphorylate the serine and threonine residues within the intracellular loops and carboxyl-terminal tail domains of GPCRs to uncoupled them from their G-proteins and, hence, trigger the desensitization of their corresponding signaling pathway (42, 46). GRK2 and GRK3 proteins would also inhibit β-Arrestin signaling in a Gβγ-dependent pathway (45, 47). On the other hand, GRK5 and GRK6 proteins would have new roles in the signaling of GPCR that would relate to their ability to trigger the activation of the β-arrestin 2/ERK1/2 signaling pathway in a G-protein-independent way (21, 45, 47). Indeed, overexpression of GRK5 and/or GRK6 has been found to enhance β-arrestin 2-mediated ERK1/2 activation, whereas overexpression of GRK2 and/or GRK3 abolishes β-arrestin 2-mediated ERK1/2 activation (45).
Here we report that repeated CP55940 treatment increases CB2 receptor phosphorylation and selectively increases GRK5 mRNA and protein expression in rat PFCx and a neuronal cell model without changes in the mRNA or protein levels of GRK6. This was also associated with reduced levels of GRK2 mRNA and protein levels in this area of the limbic brain and in cultured cells.
A limited number of reports have studied the effect of cannabinoids on the regulation of GRK protein expression. For instance, multiple tetrahydrocannabinoil (CB1/CB2 receptor agonist) treatments, but not a single tetrahydrocannabinoil treatment, up-regulates GRK2 and GRK4 in the striatum, GRK4 in the cerebellum, and GRK2 in the PFCx and hippocampus (36). To the best of our knowledge, there are currently no other reports detailing the effects of repeated cannabinoid agonist exposure on the expression of GRK proteins. This limited evidence would suggest that chronic exposure to different classes of cannabinoids may have differential effects on expression of GRKs and subsequent regulation of signaling cascades throughout the brain. Furthermore, our previous evidence suggests that repeated CP55940 or GP1a treatment enhances β-arrestin 2-mediated ERK1/2 signaling because we have previously reported that repeated cannabinoid treatment enhances pERK levels over a single cannabinoid exposure (1, 2). We have previously found that the β-arrestin 2 shRNA lentivirus transfection significantly reduces cannabinoid-induced increases in pERK levels (2). The modulation of GRK protein expression by cannabinoids could be contributing to an intensification of this signaling cascade. Interestingly, different agonists and drugs of abuse have been shown to modulate changes in expression of GRKs, and changes in GRK expression have been described in different pathophysiological conditions (48).
Our evidence indicates that the cannabinoid-induced changes in GRK5 expression could be mediated by changes in transcription because we report here that repeated CP55940 treatment increases GRK5 mRNA and protein in rat PFCx and in our neuronal cell culture model. In neuronal cells, we found that a selective CB2 receptor agonist (GP1a), but not a selective CB1 receptor agonist (ACEA), significantly increased GRK5 mRNA levels compared with vehicle-treated controls, suggesting that CB2 receptors mediate the cannabinoid-induced up-regulation of GRK5. Confirmatory evidence of the role of CB2 receptors in the GRK5 up-regulation was provided by studies with either CB1 or CB2 shRNA lentiviral particles (Fig. 3), where the cannabinoid-induced up-regulation of GRK5 was prevented only in CB2 shRNA lentivirus-treated cells. Although the detailed mechanism of cannabinoid-induced up-regulation of GRK5 was not identified in this paper, we speculate that a transcription factor such as nuclear factor immunoglobulin κ chain enhancer-B cell (NF-κB) could mediate this GRK5 up-regulation. The CB2 receptor is positively coupled to the ERK1/2 signaling pathway, which regulates NF-κB (18, 49). Rat, human, and mouse GRK5 promoter contains a consensus sequence for NF-κB, and activation of NF-κB increases GRK5 expression (50).
Here we also investigated the role of GRK5 in the cannabinoid-induced up-regulation of 5-HT2A receptors. Through the use of GRK5 shRNA lentiviral particles, we identified that GRK5 is involved in the cannabinoid-induced up-regulation and enhanced activity of 5-HT2A receptors. Indeed, treatment with GRK5 lentiviral particles significantly reduced the CP55940- and GP1a-induced up-regulation of 5-HT2A receptors without significantly altering basal levels of 5-HT2A receptor mRNA. However, treatment with CP55940 or GP1a significantly increased 5-HT2A mRNA levels in GRK5 shRNA lentivirus-treated cells compared with vehicle-treated controls. This evidence suggests that the CB2 receptor can mediate 5-HT2A receptor up-regulation, at least in part, through GRK5. Here the CP55940- and GP1a-induced increases in 5-HT2A mRNA levels could be attributed to new rates in synthesis and degradation of GRK5 protein due to the GRK5 shRNA lentivirus particle transduction.
Additionally, we found that repeated CP55940 and GP1a treatment significantly increased serotonin-stimulated 5-HT2A receptor-mediated Ca2+ release (Fig. 5, D and E). We have previously reported that repeated CP55940 treatment significantly increases 5-HT2A receptor-mediated phospholipase Cβ activity in rat PFCx- and 5-HT2A receptor-mediated phosphoinositol hydrolysis in a neuronal cell culture model (2). Interestingly, here we provide more evidence that the enhanced 5-HT2A receptor activity would involve the cannabinoid-induced up-regulation of 5-HT2A receptors. As shown in Fig. 5E, repeated GP1a treatment significantly increased the Emax (2-fold increase) without significantly affecting the EC50, which could be explained, at least in part, by the enhanced cannabinoid-induced overexpression (2-fold) of 5-HT2A receptors in neuronal cells and in rat PFCx. The role of CB2 receptors in mediating this phenomenon was identified by either CB2 or GRK5 shRNA treatment. Indeed, CB2 or GRK5 shRNA lentiviral particle treatment prevented CP55940- and GP1a-induced increases in serotonin-stimulated 5-HT2A receptor-mediated Ca2+ release (Fig. 6, A and B).
We also examined the role of GRK5 in the cannabinoid-induced phosphorylation of the CB2 receptor and enhanced β-arrestin 2/ERK interaction (Fig. 7). We found that GRK5 shRNA lentiviral particle treatment reduced the cannabinoid-induced enhanced phosphorylation of the CB2 receptor and the enhanced β-arrestin 2/ERK interaction in a neuronal cell culture model. Here the GP1a-induced increases in CB2 receptor phosphorylation and β-arrestin/ERK interaction in GRK5 shRNA lentivirus-treated cells could be attributed to new rates in synthesis and degradation of GRK5 after GRK5 shRNA lentivirus treatment and/or shifts in GRK6 activity. This evidence indicates that GRK5 is necessary for the cannabinoid-induced up-regulation of 5-HT2A receptors. Although further evidence is required, it is possible to speculate that GRK5-induced phosphorylation of the CB2 receptor and subsequent formation of the β-arrestin 2/ERK scaffolding complex could be an initiating mechanism contributing to the up-regulation and enhanced activity of 5-HT2A receptors. Further experiments are needed to demonstrate this hypothesis.
In conclusion, this study provides new insight into the cannabinoid agonist regulation of GRK proteins in rat PFCx and neuronal cell culture. Furthermore, this study is the first to show that GRK5 is involved in the cannabinoid-induced up-regulation and enhanced activity of 5-HT2A receptors in neuronal cells. We also identified mechanisms contributing to the up-regulation of GRK5 in a neuronal cell model. Recent and independent clinical studies have provided evidence indicating that sustained use of nonselective cannabinoid agonists may precipitate the onset of mental disorders associated with dysfunction of 5-HT2A receptor neurotransmission in PFCx, such as anxiety, schizophrenia, and psychosis (5–7, 51–54). However, a definitive mechanism by which repeated cannabinoid agonist exposure may be precipitating neuropsychiatric disorders has not been identified. The results presented here and our previous studies (1–4) suggest that GRK5-mediated enhanced phosphorylation of the CB2 receptor and enhanced β-arrestin 2/ERK interaction would drive the up-regulation of 5-HT2A receptors and GRK5. Interestingly, a recent report has linked enhanced function and expression of 5-HT2A receptors in PFCx to enhanced anxiety-like behaviors in rodents (55). Furthermore, the therapeutic benefits of atypical antipsychotics are proposed to be mediated by desensitization of 5-HT2A receptor signaling in PFCx, particularly pyramidal neurons, which are enriched in 5-HT2A receptors (28, 56). Therefore, this study may facilitate a better understanding of mechanisms underlying the etiology of some neuropsychiatric disorders and adverse effects of chronic exposure to cannabinoids. Understanding the mechanisms underlying the adverse effects of repeated cannabinoid exposure is especially critical because accumulating evidence is showing that selective CB2 receptor agonists have wide therapeutic application in the treatment of a variety of different conditions (57–60). This evidence could provide insight into mechanisms that can be targeted to prevent the potential adverse effect while deriving the therapeutic benefits of cannabinoids.
This work was supported, in whole or in part, by National Institutes of Health, NIDA, Grants DA024329 and DA034315. This work was also supported by University of Kansas General Research Fund 2301421 and New Faculty General Research Fund 2302213 awards and University of Kansas startup funds.
- 5-HT2A
- serotonin 2A
- 5-HT
- serotonin
- GRK
- G-protein receptor kinase
- GPCR
- G-protein coupled receptor
- PFCx
- prefrontal cortex
- qRT-PCR
- quantitative RT-PCR
- ANOVA
- analysis of variance
- ACEA
- arachidonyl-2′-chloroethylamide.
REFERENCES
- 1. Franklin J. M., Vasiljevik T., Prisinzano T. E., Carrasco G. A. (2013) Cannabinoid agonists increase the interaction between β-arrestin 2 and ERK1/2 and upregulate β-arrestin 2 and 5-HT2A receptors. Pharmacol. Res. 68, 46–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Franklin J. M., Carrasco G. A. (2013) Cannabinoid receptor agonists upregulate and enhance serotonin 2A (5-HT2A) receptor activity via ERK1/2 signaling. Synapse 67, 145–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Franklin J. M., Vasiljevik T., Prisinzano T. E., Carrasco G. A. (2012) Cannabinoid 2 receptor- and β-Arrestin 2-dependent upregulation of serotonin 2A receptors. Eur. Neuropsychopharmacol., in press: 10.1016/j.euroneuro.2012.06.012 [DOI] [PMC free article] [PubMed]
- 4. Franklin J. M., Carrasco G. A. (2012) Cannabinoid-induced enhanced interaction and protein levels of serotonin 5-HT2A and dopamine D2 receptors in rat prefrontal cortex. J. Psychopharmacol. 26, 1333–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Henquet C., Murray R., Linszen D., van Os J. (2005) The environment and schizophrenia: the role of cannabis use. Schizophr. Bull. 31, 608–612 [DOI] [PubMed] [Google Scholar]
- 6. Kuepper R., van Os J., Lieb R., Wittchen H. U., Höfler M., Henquet C. (2011) Continued cannabis use and risk of incidence and persistence of psychotic symptoms. 10 year follow-up cohort study. BMJ 342, d738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Large M., Sharma S., Compton M. T., Slade T., Nielssen O. (2011) Cannabis use and earlier onset of psychosis. A systematic meta-analysis. Arch. Gen. Psychiatry 68, 555–561 [DOI] [PubMed] [Google Scholar]
- 8. Roth B. L. (2011) Irving Page Lecture. 5-HT2A serotonin receptor biology. Interacting proteins, kinases and paradoxical regulation. Neuropharmacology 61, 348–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carrasco G. A., Van de Kar L. D. (2003) Neuroendocrine pharmacology of stress. Eur. J. Pharmacol. 463, 235–272 [DOI] [PubMed] [Google Scholar]
- 10. O'Shea M., Singh M. E., McGregor I. S., Mallet P. E. (2004) Chronic cannabinoid exposure produces lasting memory impairment and increased anxiety in adolescent but not adult rats. J. Psychopharmacol. 18, 502–508 [DOI] [PubMed] [Google Scholar]
- 11. O'Shea M., McGregor I. S., Mallet P. E. (2006) Repeated cannabinoid exposure during perinatal, adolescent or early adult ages produces similar longlasting deficits in object recognition and reduced social interaction in rats. J. Psychopharmacol. 20, 611–621 [DOI] [PubMed] [Google Scholar]
- 12. García-Gutiérrez M. S., García-Bueno B., Zoppi S., Leza J. C., Manzanares J. (2012) Chronic blockade of cannabinoid CB2 receptors induces anxiolytic-like actions associated with alterations in GABAA receptors. Br. J. Pharmacol. 165, 951–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. den Boon F. S., Chameau P., Schaafsma-Zhao Q., van Aken W., Bari M., Oddi S., Kruse C. G., Maccarrone M., Wadman W. J., Werkman T. R. (2012) Excitability of prefrontal cortical pyramidal neurons is modulated by activation of intracellular type-2 cannabinoid receptors. Proc. Natl. Acad. Sci. U.S.A. 109, 3534–3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gong J. P., Onaivi E. S., Ishiguro H., Liu Q. R., Tagliaferro P. A., Brusco A., Uhl G. R. (2006) Cannabinoid CB2 receptors. Immunohistochemical localization in rat brain. Brain Res. 1071, 10–23 [DOI] [PubMed] [Google Scholar]
- 15. Onaivi E. S., Ishiguro H., Gong J. P., Patel S., Meozzi P. A., Myers L., Perchuk A., Mora Z., Tagliaferro P. A., Gardner E., Brusco A., Akinshola B. E., Hope B., Lujilde J., Inada T., Iwasaki S., Macharia D., Teasenfitz L., Arinami T., Uhl G. R. (2008) Brain neuronal CB2 cannabinoid receptors in drug abuse and depression. From mice to human subjects. PLoS One 3, e1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Brusco A., Tagliaferro P. A., Saez T., Onaivi E. S. (2008) Ultrastructural localization of neuronal brain CB2 cannabinoid receptors. Ann. N.Y. Acad. Sci. 1139, 450–457 [DOI] [PubMed] [Google Scholar]
- 17. Bouaboula M., Poinot-Chazel C., Marchand J., Canat X., Bourrié B., Rinaldi-Carmona M., Calandra B., Le Fur G., Casellas P. (1996) Signaling pathway associated with stimulation of CB2 peripheral cannabinoid receptor. Involvement of both mitogen-activated protein kinase and induction of Krox-24 expression. Eur. J. Biochem. 237, 704–711 [DOI] [PubMed] [Google Scholar]
- 18. Felder C. C., Dickason-Chesterfield A. K., Moore S. A. (2006) Cannabinoids biology. The search for new therapeutic targets. Mol. Interv. 6, 149–161 [DOI] [PubMed] [Google Scholar]
- 19. Bouaboula M., Dussossoy D., Casellas P. (1999) Regulation of peripheral cannabinoid receptor CB2 phosphorylation by the inverse agonist SR 144528. Implications for receptor biological responses. J. Biol. Chem. 274, 20397–20405 [DOI] [PubMed] [Google Scholar]
- 20. Reiter E., Ahn S., Shukla A. K., Lefkowitz R. J. (2012) Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52, 179–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shenoy S. K., Lefkowitz R. J. (2011) β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 32, 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Oien D. B., Carrasco G. A., Moskovitz J. (2011) Decreased phosphorylation and increased methionine oxidation of α-synuclein in the methionine sulfoxide reductase A knockout mouse. J. Amino Acids 2011, 721094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fu J., Naren A. P., Gao X., Ahmmed G. U., Malik A. B. (2005) Protease-activated receptor-1 activation of endothelial cells induces protein kinase Cα-dependent phosphorylation of syntaxin 4 and Munc18c. Role in signaling P-selectin expression. J. Biol. Chem. 280, 3178–3184 [DOI] [PubMed] [Google Scholar]
- 24. Yaniv S. P., Lucki A., Klein E., Ben-Shachar D. (2010) Dexamethasone enhances the norepinephrine-induced ERK/MAPK intracellular pathway possibly via dysregulation of the α2-adrenergic receptor. Implications for antidepressant drug mechanism of action. Eur. J. Cell Biol. 89, 712–722 [DOI] [PubMed] [Google Scholar]
- 25. Yiangou Y., Facer P., Durrenberger P., Chessell I. P., Naylor A., Bountra C., Banati R. R., Anand P. (2006) COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pronin A. N., Carman C. V., Benovic J. L. (1998) Structure-function analysis of G protein-coupled receptor kinase-5. Role of the carboxyl terminus in kinase regulation. J. Biol. Chem. 273, 31510–31518 [DOI] [PubMed] [Google Scholar]
- 27. Jones B. W., Song G. J., Greuber E. K., Hinkle P. M. (2007) Phosphorylation of the endogenous thyrotropin-releasing hormone receptor in pituitary GH3 cells and pituitary tissue revealed by phosphosite-specific antibodies. J. Biol. Chem. 282, 12893–12906 [DOI] [PubMed] [Google Scholar]
- 28. Singh R., Jia C., Garcia F., Carrasco G. A., Battaglia G., Muma N. A. (2009) Activation of the JAK-STAT pathway by olanzapine is necessary for desensitization of serotonin2A receptor-stimulated phospholipase C signaling in rat frontal cortex but not serotonin2A receptor-stimulated hormone release. J. Psychopharmacol. 24, 1079–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thomas B. F., Gilliam A. F., Burch D. F., Roche M. J., Seltzman H. H. (1998) Comparative receptor binding analyses of cannabinoid agonists and antagonists. J. Pharmacol. Exp. Ther. 285, 285–292 [PubMed] [Google Scholar]
- 30. Gorantla S., Makarov E., Roy D., Finke-Dwyer J., Murrin L. C., Gendelman H. E., Poluektova L. (2010) Immunoregulation of a CB2 receptor agonist in a murine model of neuroAIDS. J. Neuroimmune Pharmacol. 5, 456–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hillard C. J., Manna S., Greenberg M. J., DiCamelli R., Ross R. A., Stevenson L. A., Murphy V., Pertwee R. G., Campbell W. B. (1999) Synthesis and characterization of potent and selective agonists of the neuronal cannabinoid receptor (CB1). J. Pharmacol. Exp. Ther. 289, 1427–1433 [PubMed] [Google Scholar]
- 32. Pelletier J. P., Fernandes J. C., Brunet J., Moldovan F., Schrier D., Flory C., Martel-Pelletier J. (2003) In vivo selective inhibition of mitogen-activated protein kinase kinase 1/2 in rabbit experimental osteoarthritis is associated with a reduction in the development of structural changes. Arthritis Rheum. 48, 1582–1593 [DOI] [PubMed] [Google Scholar]
- 33. Ciruela A., Dixon A. K., Bramwell S., Gonzalez M. I., Pinnock R. D., Lee K. (2003) Identification of MEK1 as a novel target for the treatment of neuropathic pain. Br. J. Pharmacol. 138, 751–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dai Y., Dudek N. L., Li Q., Muma N. A. (2011) Phospholipase C, Ca2+, and calmodulin signaling are required for 5-HT2A receptor-mediated transamidation of Rac1 by transglutaminase. Psychopharmacology 213, 403–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seitz P. K., Bremer N. M., McGinnis A. G., Cunningham K. A., Watson C. S. (2012) Quantitative changes in intracellular calcium and extracellular-regulated kinase activation measured in parallel in CHO cells stably expressing serotonin (5-HT) 5-HT2A or 5-HT2C receptors. BMC Neurosci. 13, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rubino T., Viganò D., Premoli F., Castiglioni C., Bianchessi S., Zippel R., Parolaro D. (2006) Changes in the expression of G protein-coupled receptor kinases and β-arrestins in mouse brain during cannabinoid tolerance. A role for RAS-ERK cascade. Mol. Neurobiol. 33, 199–213 [DOI] [PubMed] [Google Scholar]
- 37. Aksamitiene E., Kholodenko B. N., Kolch W., Hoek J. B., Kiyatkin A. (2010) PI3K/Akt-sensitive MEK-independent compensatory circuit of ERK activation in ER-positive PI3K-mutant T47D breast cancer cells. Cell. Signal. 22, 1369–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Howlett A. C. (2005) Handb. Exp. Pharmacol. 53–79 [DOI] [PubMed] [Google Scholar]
- 39. Abood M. E., Martin B. R. (1996) Molecular neurobiology of the cannabinoid receptor. Int. Rev. Neurobiol. 39, 197–221 [DOI] [PubMed] [Google Scholar]
- 40. Atwood B. K., Wager-Miller J., Haskins C., Straiker A., Mackie K. (2012) Functional selectivity in CB2 cannabinoid receptor signaling and regulation. Implications for the therapeutic potential of CB2 ligands. Mol. Pharmacol. 81, 250–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dalton G. D., Howlett A. C. (2012) Cannabinoid CB1 receptors transactivate multiple receptor tyrosine kinases and regulate serine/threonine kinases to activate ERK in neuronal cells. Br. J. Pharmacol. 165, 2497–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Reiter E., Lefkowitz R. J. (2006) GRKs and β-arrestins. Roles in receptor silencing, trafficking and signaling. Trends Endocrinol. Metab. 17, 159–165 [DOI] [PubMed] [Google Scholar]
- 43. Shenoy S. K., Drake M. T., Nelson C. D., Houtz D. A., Xiao K., Madabushi S., Reiter E., Premont R. T., Lichtarge O., Lefkowitz R. J. (2006) β-Arrestin-dependent, G protein-independent ERK1/2 activation by the β2 adrenergic receptor. J. Biol. Chem. 281, 1261–1273 [DOI] [PubMed] [Google Scholar]
- 44. Lefkowitz R. J., Shenoy S. K. (2005) Transduction of receptor signals by β-arrestins. Science 308, 512–517 [DOI] [PubMed] [Google Scholar]
- 45. Kim J., Ahn S., Ren X. R., Whalen E. J., Reiter E., Wei H., Lefkowitz R. J. (2005) Functional antagonism of different G protein-coupled receptor kinases for β-arrestin-mediated angiotensin II receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 102, 1442–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang J., Ferguson S. S., Barak L. S., Bodduluri S. R., Laporte S. A., Law P. Y., Caron M. G. (1998) Role for G protein-coupled receptor kinase in agonist-specific regulation of μ-opioid receptor responsiveness. Proc. Natl. Acad. Sci. U.S.A. 95, 7157–7162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ren X. R., Reiter E., Ahn S., Kim J., Chen W., Lefkowitz R. J. (2005) Different G protein-coupled receptor kinases govern G protein and β-arrestin-mediated signaling of V2 vasopressin receptor. Proc. Natl. Acad. Sci. U.S.A. 102, 1448–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Métayé T., Gibelin H., Perdrisot R., Kraimps J. L. (2005) Pathophysiological roles of G-protein-coupled receptor kinases. Cell. Signal. 17, 917–928 [DOI] [PubMed] [Google Scholar]
- 49. Chang F., Steelman L. S., Lee J. T., Shelton J. G., Navolanic P. M., Blalock W. L., Franklin R. A., McCubrey J. A. (2003) Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors. Potential targeting for therapeutic intervention. Leukemia 17, 1263–1293 [DOI] [PubMed] [Google Scholar]
- 50. Islam K. N., Koch W. J. (2012) Involvement of nuclear factor κB (NF-κB) signaling pathway in regulation of cardiac G protein-coupled receptor kinase 5 (GRK5) expression. J. Biol. Chem. 287, 12771–12778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Reilly D., Didcott P., Swift W., Hall W. (1998) Long-term cannabis use. Characteristics of users in an Australian rural area. Addiction 93, 837–846 [DOI] [PubMed] [Google Scholar]
- 52. Clough A. R., d'Abbs P., Cairney S., Gray D., Maruff P., Parker R., O'Reilly B. (2005) Adverse mental health effects of cannabis use in two indigenous communities in Arnhem Land, Northern Territory, Australia. Exploratory study. Aust. N. Z. J. Psychiatry 39, 612–620 [DOI] [PubMed] [Google Scholar]
- 53. Patton G. C., Coffey C., Carlin J. B., Degenhardt L., Lynskey M., Hall W. (2002) Cannabis use and mental health in young people. Cohort study. BMJ 325, 1195–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Troisi A., Pasini A., Saracco M., Spalletta G. (1998) Psychiatric symptoms in male cannabis users not using other illicit drugs. Addiction 93, 487–492 [DOI] [PubMed] [Google Scholar]
- 55. Magalhaes A. C., Holmes K. D., Dale L. B., Comps-Agrar L., Lee D., Yadav P. N., Drysdale L., Poulter M. O., Roth B. L., Pin J. P., Anisman H., Ferguson S. S. (2010) CRF receptor 1 regulates anxiety behavior via sensitization of 5-HT2 receptor signaling. Nat. Neurosci. 13, 622–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ichikawa J., Ishii H., Bonaccorso S., Fowler W. L., O'Laughlin I. A., Meltzer H. Y. (2001) 5-HT2A and D2 receptor blockade increases cortical DA release via 5-HT1A receptor activation. A possible mechanism of atypical antipsychotic-induced cortical dopamine release. J. Neurochem. 76, 1521–1531 [DOI] [PubMed] [Google Scholar]
- 57. Guindon J., Hohmann A. G. (2008) Cannabinoid CB2 receptors. A therapeutic target for the treatment of inflammatory and neuropathic pain. Br. J. Pharmacol. 153, 319–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Morales M., Bonci A. (2012) Getting to the core of addiction. Hooking CB2 receptor into drug abuse? Nat. Med. 18, 504–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sagredo O., González S., Aroyo I., Pazos M. R., Benito C., Lastres-Becker I., Romero J. P., Tolón R. M., Mechoulam R., Brouillet E., Romero J., Fernández-Ruiz J. (2009) Cannabinoid CB2 receptor agonists protect the striatum against malonate toxicity. Relevance for Huntington's disease. Glia 57, 1154–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zarruk J. G., Fernández-López D., García-Yébenes I., Garcia-Gutiérrez M. S., Vivancos J., Nombela F., Torres M., Burguete M. C., Manzanares J., Lizasoain I., Moro M. A. (2012) Cannabinoid type 2 receptor activation downregulates stroke-induced classic and alternative brain macrophage/microglial activation concomitant to neuroprotection. Stroke 43, 211–219 [DOI] [PubMed] [Google Scholar]