

Background: The mechanism by which KCa3.1 regulates cell proliferation remains elusive.
Results: KCa3.1 regulates the expression of transcription factors and cyclins by controlling intracellular calcium levels in activated vascular smooth muscle cells (VSMCs).
Conclusion: KCa3.1 is an important regulator of the calcium-dependent proliferation machinery in VSMCs.
Significance: KCa3.1 modulation constitutes a therapeutic target for cell proliferative diseases such as atherosclerosis.
Keywords: Atherosclerosis, Calcium, Calcium Signaling, Cell Proliferation, Potassium Channels, Vascular Biology, Vascular Smooth Muscle Cells
Abstract
The intermediate conductance calcium-activated potassium channel KCa3.1 contributes to a variety of cell activation processes in pathologies such as inflammation, carcinogenesis, and vascular remodeling. We examined the electrophysiological and transcriptional mechanisms by which KCa3.1 regulates vascular smooth muscle cell (VSMC) proliferation. Platelet-derived growth factor-BB (PDGF)-induced proliferation of human coronary artery VSMCs was attenuated by lowering intracellular Ca2+ concentration ([Ca2+]i) and was enhanced by elevating [Ca2+]i. KCa3.1 blockade or knockdown inhibited proliferation by suppressing the rise in [Ca2+]i and attenuating the expression of phosphorylated cAMP-response element-binding protein (CREB), c-Fos, and neuron-derived orphan receptor-1 (NOR-1). This antiproliferative effect was abolished by elevating [Ca2+]i. KCa3.1 overexpression induced VSMC proliferation, and potentiated PDGF-induced proliferation, by inducing CREB phosphorylation, c-Fos, and NOR-1. Pharmacological stimulation of KCa3.1 unexpectedly suppressed proliferation by abolishing the expression and activity of KCa3.1 and PDGF β-receptors and inhibiting the rise in [Ca2+]i. The stimulation also attenuated the levels of phosphorylated CREB, c-Fos, and cyclin expression. After KCa3.1 blockade, the characteristic round shape of VSMCs expressing high l-caldesmon and low calponin-1 (dedifferentiation state) was maintained, whereas KCa3.1 stimulation induced a spindle-shaped cellular appearance, with low l-caldesmon and high calponin-1. In conclusion, KCa3.1 plays an important role in VSMC proliferation via controlling Ca2+-dependent signaling pathways, and its modulation may therefore constitute a new therapeutic target for cell proliferative diseases such as atherosclerosis.
Introduction
The intermediate conductance calcium-activated potassium channel KCa3.1 (also known as KCNN4 and IKCa), a member of the calcium-activated potassium channel (KCa)4 family, tightly binds the Ca2+ sensor calmodulin near its C-terminal domain. KCa3.1 is opened by a small rise in free cytosolic Ca2+ ([Ca2+]i) due to Ca2+-calmodulin-mediated cross-linking in the subunits of the channel tetramer (1). Channel activation induces membrane hyperpolarization, which promotes Ca2+ influx. KCa3.1 is highly expressed in a variety of nonexcitable and proliferating cells (2), and an increase in KCa3.1 expression has been associated with cancer development, immune disorders, and vascular inflammation. In particular, this channel plays a critical role in the proliferation of smooth muscle cells (3, 4), endothelial cells (5), lymphocytes including B- and T-cells (6, 7), fibroblasts (8), stem cells (9), and several cancer cells (10).
Vascular smooth muscle cell (VSMC) proliferation is a crucial event in the development of vascular diseases such as atherosclerosis and restenosis (11, 12). VSMCs proliferate, migrate, and invade the intima in response to growth factors and vascular injury, resulting in atherosclerotic fibrous cap formation and intimal hyperplasia following angioplasty and stent placement. Electrophysiological properties of VSMCs dramatically change as they proliferate (13). In the contractile form of VSMCs, Ca2+ influx through voltage-dependent Ca2+ channels causes VSMC contraction and co-activation of large conductance KCa channels (BK), which in turn induces Ca2+ channel closure through repolarization. In contrast, in proliferating VSMCs, this mechanism is down-regulated, and Ca2+ influx through Ca2+ release-activated Ca2+ channels is maintained by membrane hyperpolarization induced by KCa3.1 activation. We recently found that vascular remodeling following myocardial infarction (14) and chronic inhibition of nitric oxide synthesis (15) is associated with increased KCa3.1 expression in rat hearts, suggesting a pathophysiological role for this channel. Consistently, KCa3.1 expression and activity are increased in VSMCs activated by mitogens, in intimal VSMCs of restenotic lesions following vascular injury, and in atherosclerotic plaques in humans, swine, rats, and mice (3, 11, 12, 16). Importantly, pharmacological blockade, siRNA knockdown, or genetic deficiency of KCa3.1 suppresses VSMC activation (including proliferation, migration, and excessive oxidant production) and attenuates the development of restenosis and atherosclerosis in swine, rats, and mice (3, 12, 17, 18). Activation of the transcription factor AP-1 is associated with KCa3.1 induction in T cells (7). In VSMCs, an elevation of AP-1 activity is linked with proliferation (19, 20), implying that this transcription factor may play a role in KCa3.1 induction in VSMCs. In addition, the repressor element 1-silencing transcription factor (REST) represses KCa3.1 gene expression (21), suggesting that AP-1 and REST may operate in a coordinated manner for KCa3.1 induction in VSMCs. Although a rise in [Ca2+]i plays a crucial role in the regulation of cell proliferation via controlling intracellular signaling pathways (13, 22, 23), the mechanism by which KCa3.1 regulates cell proliferative processes remains unknown.
We hypothesized that KCa3.1 blockade or knockdown would suppress VSMC proliferation by inhibiting the mitogen-induced rise in [Ca2+]i and subsequent mitogenic signaling pathways, whereas KCa3.1 stimulation or overexpression would confer the opposite effects. To test this hypothesis, we examined the effect of pharmacological KCa3.1 blockade or activation, gene silencing, or overexpression on platelet-derived growth factor-BB (PDGF)-induced proliferation of VSMCs, focusing on changes in [Ca2+]i, activation of mitogenic signaling pathways, cell cycle progression, morphology, and phenotypic characteristics in human coronary artery VSMCs (HCSMCs).
EXPERIMENTAL PROCEDURES
Cell Culture
HCSMCs (Cell Applications) were grown as reported previously (3). All experiments were performed between passages 5 and 7. Cells were seeded and cultured up to 70% confluence in smooth muscle growth medium (Cell Applications). Before each assay, cells were serum-starved and synchronized for 48 h in smooth muscle basal medium. HCSMCs in a quiescent state were stimulated for 1–48 h with PDGF (20 ng/ml; R&D Systems). None of the treatments performed in this study altered the viability of VSMCs, as judged by trypan blue exclusion (data not shown).
Small Interfering RNA Transfection
siRNA transfection was performed as reported previously (3). Optimization of siRNA concentration (12.5 nm; Ambion), transfection time, and cell density were determined by quantifying KCa3.1 mRNA and protein expression (3).
RNA Extraction, Reverse Transcription, and Quantitative PCR
Real-time PCR (iCycler, Bio-Rad or 7900 HT real-time PCR system, Applied Biosystems) was performed using iQ SYBR Green supermix to quantify transcript levels for KCa3.1 and GAPDH as described (3). Primers were designed using Beacon Designer software 3.0 (PREMIER Biosoft International).
Western Blotting
Total cell lysates or membrane fractions (10–50 μg) were analyzed by Western blotting as reported previously (3). Briefly, harvested cells or membrane fractions were lysed in a buffer containing CHAPS (1% w/v), protease inhibitor mixture (Sigma), 1 mm sodium orthovanadate, and 10 mm sodium fluoride. The protein content of each sample was determined using an RC DC protein assay (Bio-Rad). Proteins were resolved on a 10% SDS-polyacrylamide gel and subsequently transferred to a PVDF membrane (Bio-Rad). The following primary antibodies were used: a polyclonal antibody against human and mouse KCa3.1 (46 kDa) obtained from sera of rabbits immunized with the following oligopeptide: H-LNASYRSIGALNQVRC-NH2 (S4-S5 of human and mouse KCa3.1) (3), anti-l-caldesmon, anti-calponin-1, anti-PDGF β-receptors, anti-cyclin A, anti-cyclin B1, anti-cyclin D1, anti-cyclin E, anti-β-actin (Santa Cruz Biotechnology), anti-cAMP-response element-binding protein (CREB), anti-phospho-CREB (Ser 133), anti-c-Fos (Cell Signaling), and anti-neuron-derived orphan receptor-1 (NOR-1) (R&D Systems). Secondary antibodies were from Santa Cruz Biotechnology. COLO 320DM cell lysate was used as a positive control for KCa3.1, and HeLa nuclear extract was used for c-Fos and NOR-1 (Santa Cruz Biotechnology). Representative immunoblots from three or more experiments are shown.
Cell Proliferation and Migration Assays
Cell proliferation (ELISA kit, Roche Applied Science) and migration were examined as we reported previously (3). VSMC migration was stimulated with PDGF for 8 h in Transwell plates (Corning).
Cell Cycle Analysis
As reported previously (3), HCSMCs were stimulated with 10% FBS for 24 h. After fixation, cells were treated with ribonuclease A (250 μg/106 cells) for 30 min at 37 °C and stained with propidium iodide (50 μg/ml) for 30 min at 4 °C. The ratio of cells in each cell cycle phase was determined by flow cytometry (FACSCalibur: BD Biosciences) using CellQuest software (BD Biosciences).
Intracellular Ca2+ Measurement
To measure [Ca2+]i in VSMCs after 48 h of treatment (24), cells were incubated with Fluo-4 AM (10 μm; Invitrogen) for 25 min at room temperature. Fluorescence images were captured and analyzed using an inverted epifluorescence microscope (Nikon TE200) with a 40× Plan Fluor objective, a high speed wavelength switcher (Lambda DG-4 from Sutter Instrument), a PC-controlled digital CCD camera (Hamamatsu C4742-95), and MetaMorph software (Universal Imaging). Fluorescence was measured at 488 nm, and recording emission was measured at 523 nm. Images were analyzed with MetaMorph software.
Patch Clamping
Serum-starved VSMCs or cells treated for 48 h with PDGF or with PDGF in the presence of TRAM-34 (a specific blocker of KCa3.1 (3, 25)), EBIO (an activator of KCa3.1 and small conductance KCa2 channels (26)), or SKA-31 or NS309 (more potent and specific KCa3.1 activators (27, 28)) were patch-clamped in the whole-cell mode of the patch clamp technique using an EPC-10 amplifier. KCa3.1 currents were elicited by dialysis with an aspartate-based pipette solution containing 3 μm free Ca2+ and voltage ramps from −120 to 40 mV of 200-ms duration applied every 10 s. Whole-cell KCa3.1 conductances were calculated from the slope at −80 mV where the KCa3.1 currents are not “contaminated” by inwardly rectifying potassium channel, voltage-gated potassium channel (Kv), or BK currents. The KCa3.1 whole-cell conductance was then divided by the KCa3.1 single channel conductance (11 picosiemens) to determine the KCa3.1 channel number per cell.
Determination of Cell Morphology
Cell morphology was analyzed as reported previously (29). After 48 h of treatment, phase-contrast images of ∼20 randomly chosen fields per condition and per experiment were taken. The following morphological parameters were calculated from the cell boundary. 1) Cell length was determined along the principal axis of traction, which is a unique axis and presumably coincides with the principal actin bundles. 2) Cell width was measured in the direction perpendicular to the principal axis of traction. The maximum cell width was taken, ignoring thin cell protrusions. 3) The shape index was defined as the ratio of the cell length and width.
KCa3.1 Overexpression
For pharmacological induction, HCSMCs were treated with a combination of phorbol-12-myristate-13-acetate (PMA, 40 nm, a specific activator of protein kinase C; Calbiochem) and cyclosporin A (CsA, 100 nm, an inhibitor of calcineurin; Sigma-Aldrich) in smooth muscle basal medium for 48 h (7). For viral induction, replication-defective lentiviral vectors pseudotyped with vesicular stomatitis virus G-protein were produced as described previously (30). Positive colonies expressing eGFP were identified by fluorescent microscopy 3 days after the final transduction step. Using this approach, 50–75% of HCSMCs were eGFP-positive.
Statistics
All data are expressed as mean ± S.E. Student's t test and analysis of variance (for one-way and nonparametric tests) were performed using SigmaStat version 3 (SPSS Inc.) (3). Computations were followed by a Bonferroni's corrected t test when significant differences were noted. Statistical significance was defined as a value of p < 0.05.
RESULTS
KCa3.1 Regulates VSMC Proliferation via Controlling [Ca2+]i
We first examined whether KCa3.1 regulates VSMC proliferation via controlling [Ca2+]i. As we reported previously (3), a 48-h stimulation with PDGF caused a significant increase in de novo DNA synthesis in HCSMCs as demonstrated by a BrdU incorporation assay (Fig. 1A). Lowering [Ca2+]i with BAPTA (30 μm, an intracellular Ca2+ chelator (31)) suppressed the PDGF-induced increase in DNA synthesis, whereas BAPTA alone had no effect (Fig. 1A). On the other hand, a forced rise in [Ca2+]i with A23187 (0.01–1 nm, a Ca2+ ionophore (31)) increased DNA synthesis in the absence of PDGF and enhanced PDGF-induced responses in a dose-dependent manner (Fig. 1B). Fluorescence microscopy using Fluo-4 showed that a 48-h stimulation with PDGF increased fluorescence intensity in HCSMCs (Fig. 1C), which was inhibited by co-treatment with a specific KCa3.1 blocker, TRAM-34 (100 nm (3, 25)), or by silencing KCa3.1 expression (Fig. 1C). NOR-1 and c-fos are immediate early genes that are activated in a [Ca2+]i-sensitive manner in proliferating VSMCs (32–34). As shown in Fig. 1D, PDGF-induced expression of c-Fos and NOR-1 was inhibited by TRAM-34 but not by TRAM-7 (100 nm, an inactive analog of TRAM-34 (3, 25)). In addition, KCa3.1 knockdown also reduced the expression of both pro-oncogenes. Inhibition of PDGF-induced VSMC proliferation by KCa3.1 blockade with TRAM-34 (100 nm) was suppressed by A23187 in a dose-dependent manner (Fig. 2A). Similarly, the inhibitory effect of KCa3.1 knockdown was also suppressed by co-treatment with A23187 (Fig. 2B). These data indicate that KCa3.1 regulates PDGF-induced VSMC proliferation via controlling the rise in [Ca2+]i and subsequent signaling pathways.
FIGURE 1.

KCa3.1 regulates PDGF-induced rise in [Ca2+]i and signaling pathway activation. A and B, lowering [Ca2+]i with BAPTA suppressed (A) and a forced rise in [Ca2+]i with A23187 enhanced (B) PDGF-induced proliferation of HCSMCs. C and D, both blockade with TRAM-34 and siRNA knockdown of KCa3.1 reduced PDGF-induced rise in [Ca2+]i (C) and inhibited PDGF-induced expression of c-Fos and NOR-1 (D). *, p < 0.05 versus control, and †, p < 0.05 versus PDGF alone. neg. siRNA, negative control siRNA.
FIGURE 2.
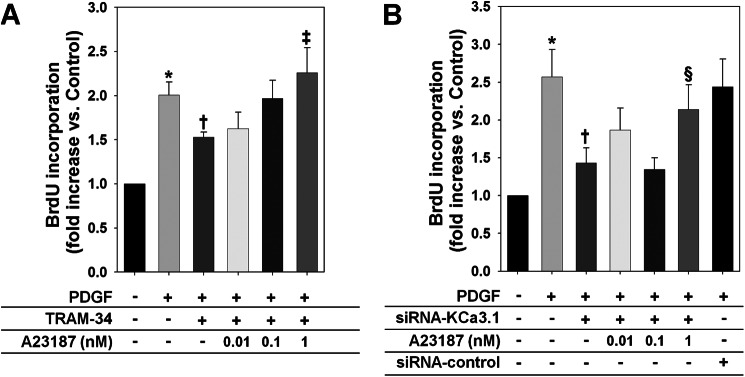
A forced rise in [Ca2+]i suppresses the inhibitory effects of KCa3.1 blockade or knockdown on PDGF-induced VSMC proliferation. A and B, A23187 blocked the inhibition of PDGF-induced HCSMC proliferation by TRAM-34 (A) and KCa3.1 knockdown (B). *, p < 0.05 versus control, †, p < 0.05 versus PDGF alone, ‡, p < 0.05 versus PDGF+TRAM-34, and §, p < 0.05 versus PDGF+siRNA-KCa3.1.
Paradoxical Inhibition of PDGF-induced HCSMC Proliferation by KCa3.1 Stimulation
To test the hypothesis that full activation of KCa3.1 with pharmacological activators would enhance VSMC proliferation during PDGF exposure by augmenting the rise in [Ca2+]i, EBIO (an activator of KCa3.1 and small conductance KCa2 channels (26)) was applied at 100 or 300 μm during PDGF-induced proliferation of HCSMCs. These cells predominantly express KCa3.1 but not small conductance KCa2 channels (3). PDGF-induced increase in DNA synthesis was unexpectedly inhibited by co-treatment with EBIO in a dose-dependent manner, whereas EBIO alone had no significant effect (Fig. 3A). The antiproliferative effect of KCa3.1 stimulation with EBIO was also confirmed using a cell count assay (data not shown). PDGF-induced chemotaxis of HCSMCs was also inhibited by 300 μm EBIO (fold increase: PDGF+EBIO 7.5 ± 3.0, p < 0.05 versus PDGF 17.4 ± 4.0, n = 5), and EBIO alone had no effect (EBIO 1.2 ± 0.1). Fluorescence microscopy using Fluo-4 revealed that co-treatment with EBIO (300 μm) reduced the PDGF-induced rise in [Ca2+]i in HCSMCs (Fig. 3B). To confirm the antiproliferative effect of KCa3.1 stimulation, two more potent and specific KCa3.1 activators, SKA-31 (27) and NS309 (28), were also tested. SKA-31 inhibited PDGF-induced proliferation at 0.5 μm, whereas SKA-16, an inactive derivative of SKA-31, had no effect (Fig. 3C). NS309 also exhibited an antiproliferative effect at 10 nm. Both activators had no effect in the absence of PDGF (data not shown).
FIGURE 3.
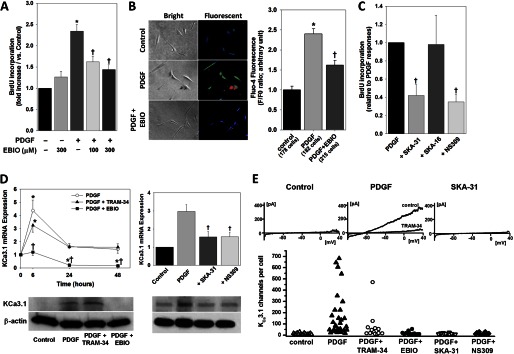
Paradoxical inhibition of VSMC activation by KCa3.1 stimulation. A, PDGF-induced increase in BrdU incorporation was markedly inhibited by EBIO in a dose-dependent manner. B, PDGF-induced rise in [Ca2+]i was reduced by EBIO (300 μm). C, two other chemically distinct KCa3.1 activators, SKA-31 (0.5 μm) and NS309 (10 nm), also inhibited BrdU incorporation, whereas SKA-16 (0.5 μm), an inactive derivative of SKA-31, had no effect. D, KCa3.1 activation attenuated PDGF-induced increases in KCa3.1 mRNA and membranous protein expression in HCSMCs. E, functional KCa3.1 expression in HCSMCs determined by whole-cell patch clamp. Although serum-starved cells expressed on average 12.2 ± 7 KCa3.1 channels per cell (n = 16), PDGF stimulation increased channel numbers to 163 ± 194 (n = 32, p = 0.003 versus control). TRAM-34 treatment did not significantly reduce channel numbers (66 ± 116, n = 15), whereas the activators EBIO (16.7 ± 13.9 channels, n = 14, p = 0.007 versus PDGF), SKA-31 (12.2 ± 4.8, n = 15, p = 0.004 versus PDGF), and NS309 (16.1 ± 17.9, n = 12, p = 0.005 versus PDGF) drastically reduced functional KCa3.1 channel numbers. *, p < 0.05 versus control, and †, p < 0.05 versus PDGF alone.
To clarify the mechanisms responsible for this paradoxical inhibition, we first analyzed the expression level of KCa3.1 in HCSMCs treated with PDGF in the presence of TRAM-34 or the KCa3.1 activators. As we reported previously (3), PDGF induced a remarkable increase in KCa3.1 mRNA expression in HCSMCs (Fig. 3D). TRAM-34 (100 nm) had little effect on PDGF-induced increase in KCa3.1 expression. Contrarily, EBIO (300 μm) markedly attenuated PDGF-induced KCa3.1 up-regulation. Western blotting corroborated the mRNA analysis, showing that EBIO abolished PDGF-induced membranous KCa3.1 expression in HCSMCs, whereas TRAM-34 modestly increased KCa3.1 expression. SKA-31 (0.5 μm) and NS309 (10 nm) also suppressed the PDGF-induced increase in KCa3.1 transcripts at 6 h of PDGF treatment, consistent with the inhibition of protein expression at 48 h of PDGF treatment. Whole-cell patch clamp experiments (Fig. 3E) further confirmed the mRNA and Western blotting observations. Although PDGF stimulation alone induced robust KCa3.1 currents, cells stimulated with PDGF in the presence of EBIO, SKA-31, and NS309 exhibited only barely detectable KCa3.1 currents, similar to serum-starved cells. TRAM-34-treated cells showed a trend toward reduced KCa3.1 expression, but did not differ significantly from PDGF-treated cells. These findings indicate the existence of a negative feedback mechanism whereby chronic pharmacological stimulation of KCa3.1 paradoxically inhibits the rise in [Ca2+]i by attenuating KCa3.1 expression and activity, leading to suppression of PDGF-induced VSMC proliferation.
KCa3.1 Regulates Cell Cycle Progression
We next studied the effect of TRAM-34 or EBIO treatment on the activation of CREB and on the expression of c-Fos, two factors playing an important role in PDGF-induced VSMC proliferation (32, 35). Western blot experiments showed that PDGF induced CREB phosphorylation at Ser-133 and c-Fos up-regulation in HCSMCs (Fig. 4A), which were inhibited by both TRAM-34 and EBIO (100 nm and 300 μm, respectively). Consistent with the fact that the CREB/c-Fos pathway is associated with cell cycle progression via regulating cyclin expression (35), PDGF treatment increased the expression of cyclins A, B1, D1, and E in HCSMCs (Fig. 4B). TRAM-34 reduced the expression of cyclins A and B1, and EBIO suppressed all tested cyclins. Flow cytometry analysis of the cellular DNA content showed that 10% FBS evoked cell cycle progression with fewer G0/G1 and more S and G2/M cells (Fig. 4C), which was reduced by TRAM-34 (100 nm) at the transitions from G0/G1 to S and from S to G2/M phases, whereas EBIO (300 μm) more remarkably inhibited cell cycle progression, especially at the transition from G0/G1 to S phase. siRNA knockdown of KCa3.1 also inhibited cell cycle progression in a manner similar to EBIO (data not shown). These data indicate that KCa3.1 plays a crucial role in the regulation of cell cycle signaling pathways during VSMC proliferation.
FIGURE 4.

KCa3.1 blockade and stimulation inhibit cell cycle progression. A, Western blotting showed that PDGF treatment increased the levels of c-Fos and phosphorylated CREB at Ser-133 (p-CREB Ser133) with no change in total CREB content. This effect was inhibited after treatment with either TRAM-34 or EBIO. B, PDGF-induced expression of cyclins A, B1, D1, and E was suppressed by EBIO, whereas TRAM-34 decreased cyclins A and B1, but not D1 and E. C, flow cytometry showed that both TRAM-34 and EBIO inhibited 10% FBS-induced cell cycle progression at the transitions from G0/G1 to S and S to G2/M phases.
VSMC Morphological and Phenotypical Changes after KCa3.1 Blockade or Stimulation
PDGF treatment changed the cell morphology, conferring a longer and thinner cell shape in HCSMCs (Fig. 5A). TRAM-34 (100 nm) reduced PDGF-induced cell elongation and thinning. In contrast, EBIO treatment (300 μm) remarkably enhanced PDGF-induced cell elongation and thinning, and this enhancement was inhibited by co-treatment with TRAM-34. HCSMCs treated with SKA-31 or NS309 also displayed similar morphological characteristics (ratio of cell length/width; PDGF+SKA-31 25 ± 2 and PDGF+NS309 23 ± 2, p < 0.05 versus PDGF alone 15 ± 2, n = 83 cells). Treatment of HCSMCs with PDGF increased the expression of l-caldesmon, a dedifferentiation marker, and decreased the expression of calponin-1, a differentiation marker, accompanied by a decreased expression of membranous PDGF β-receptors, a known negative feedback response (36) (Fig. 5B). TRAM-34 treatment did not affect these changes; however, the KCa3.1 activators abolished the membranous expression of PDGF β-receptors, another dedifferentiation marker (37), and prevented PDGF-induced up-regulation of l-caldesmon and down-regulation of calponin-1. Furthermore, A23187 failed to suppress the inhibitory effect of EBIO (300 μm) on PDGF-induced DNA synthesis (Fig. 5C). Therefore, these data indicate that KCa3.1 stimulation converted the VSMC phenotype to one that is resistant to PDGF-induced proliferation by disrupting the axis of PDGF β-receptors and its Ca2+ regulatory mechanism.
FIGURE 5.

Effect of KCa3.1 blockade or stimulation on PDGF-induced changes in VSMC morphology and phenotypic state. A, the left panel shows representative images of cell morphology of HCSMCs before and after PDGF treatment in the presence or absence of TRAM-34 or EBIO. Summarized data of cell length, width, and the ratio of length/width in HCSMCs following each treatment are presented in the right panel. PDGF treatment confers longer and thinner cell morphology. TRAM-34 inhibited and EBIO enhanced cell elongation and thinning, whereas TRAM-34 blocked the effect of EBIO. B, in Western blotting, PDGF up-regulated l-caldesmon and down-regulated calponin-1, accompanied by a decrease in PDGF β-receptors on the membrane. Although TRAM34 did not change the expression pattern of these markers, KCa3.1 activators abolished the expression of PDGF β-receptors concomitantly with the suppression of PDGF-induced alteration in the expression of VSMC phenotypic markers. C, A23187 had no effect on EBIO-induced inhibition of VSMC proliferation. *, p < 0.05 versus control, †, p < 0.05 versus PDGF alone, and ‡, p < 0.05 versus PDGF+EBIO.
KCa3.1 Overexpression Initiates and Enhances VSMC Proliferation
Two methods of pharmacological and genetic manipulations were used to increase KCa3.1 expression in quiescent VSMCs. As reported in T cells (7), treatment with PMA and CsA for 48 h increased KCa3.1 mRNA and protein expression with little de novo DNA synthesis (PMA+CsA treatment 1.0 ± 0.2-fold of control, p = not significant versus control, n = 3) in HCSMCs, which can be compared with KCa3.1 expression 48 h after exposure to PDGF (Fig. 6A). Following removal of PMA+CsA, these cells displayed a modest but statistically significant proliferative activity in the absence of any mitogen as compared with quiescent cells (Fig. 6B). PDGF-induced proliferation was also enhanced in PMA+CsA-pretreated, KCa3.1-overexpressing cells 24 h after exposure to PDGF as compared with control cells.
FIGURE 6.

Pharmacological overexpression of KCa3.1 initiates VSMC proliferation and enhances PDGF-induced proliferative activity. A, exposure to PMA and CsA increased KCa3.1 mRNA and protein expression in HCSMCs, comparably to PDGF stimulation. B, BrdU incorporation assay showed that de novo DNA synthesis was significantly increased in PMA+CsA-pretreated, KCa3.1-overexpressing cells either with or without PDGF. *, p < 0.05 versus control, and †, p < 0.05 versus PDGF.
A lentiviral vector system was subsequently utilized to genetically modify HCSMCs with either eGFP-tagged hKCa3.1 (KCa3.1-tg cells) or eGFP-tagged luciferase (Luc-tg cells) as a control. As confirmed by the expression of eGFP, KCa3.1-tg cells showed a high level of KCa3.1 mRNA and protein expression as compared with Luc-tg cells (Fig. 7A). BrdU incorporation assay performed in the absence of PDGF revealed that the activity of de novo DNA synthesis in KCa3.1-tg cells was higher than those in nontransduced and Luc-tg cells (Fig. 7B). PDGF-induced proliferation was also enhanced in KCa3.1-tg cells (Fig. 7B). The levels of phosphorylated CREB, c-Fos, and NOR-1 were higher in KCa3.1-tg cells than in Luc-tg cells before, and 1 h after, exposure to PDGF (Fig. 7C). These data confirm that KCa3.1 regulates PDGF-dependent signaling pathways of VSMC proliferation.
FIGURE 7.

KCa3.1 overexpression initiates and potentiates VSMC proliferation by activating PDGF-dependent transcription factors. A, representative images of eGFP-tagged KCa3.1- (KCa3.1-tg) or luciferase- (Luc-tg) overexpressing HCSMCs are shown in the left panel. eGFP expression was detected in both transduced cell types. Increased expression of KCa3.1 transcript and protein in KCa3.1-tg cells is shown in the right panel. Non-tg, control cells. B, cell proliferative activity was increased in KCa3.1-tg cells as compared with Luc-tg cells in the absence or presence of PDGF. C, Western blotting showed that the levels of phosphorylated CREB (p-CREB), c-Fos, and NOR-1 were increased in KCa3.1-tg HCSMCs as compared with Luc-tg cells before and after exposure to PDGF. *, p < 0.05 versus Luc-tg cells.
DISCUSSION
The major novel findings of this study are three-fold. First, both pharmacological blockade and gene knockdown of KCa3.1 inhibit HCSMC proliferation, whereas KCa3.1 overexpression has the opposite effect. Second, pharmacological KCa3.1 stimulation unexpectedly but markedly attenuates HCSMC proliferation by diminishing the expression of KCa3.1 and PDGF β-receptors and by inhibiting further dedifferentiation and cell cycle progression. Third, PDGF-induced HCSMC proliferation is associated with KCa3.1-dependent regulation of the rise in [Ca2+]i and the subsequent expression of transcription factors and cyclins that orchestrate cell cycle progression. Taken together, these findings suggest that KCa3.1 plays a key role in the regulation of VSMC proliferation.
KCa3.1 regulates the proliferation of a variety of cell types, including VSMCs. Previous studies have demonstrated that KCa3.1 regulates cell membrane potential and Ca2+ influx, and its blockade or knockdown inhibits the proliferation of B- and T-cells (6, 7), airway smooth muscle cells (4), fibroblasts (8), cancer cells (10), and VSMCs (3, 12, 13, 18, 21). Moreover, in stem cells, KCa3.1 regulates the cell cycle progression via cyclins D1 and E (9). However, the mechanism by which KCa3.1 regulates cell proliferation in many cell types is still not well understood.
Ca2+ is a major second messenger for a variety of cell activation processes. PDGF binds to and activates its specific tyrosine kinase receptors, leading to a sustained increase in [Ca2+]i in VSMCs (31). Here, PDGF-induced HCSMC proliferation was suppressed by chelating [Ca2+]i with BAPTA and augmented by a forced rise in [Ca2+]i with A23187. These results are consistent with the effects of BAPTA and A23187 on PDGF-induced VSMC chemotaxis (31), indicating an essential role for [Ca2+]i in the regulation of PDGF-induced VSMC activation. It is widely accepted that in a variety of nonexcitable cells, KCa3.1 is activated by a small rise in [Ca2+]i (∼100 nm) following Ca2+ release from intracellular stores and subsequent Ca2+ influx. The resultant K+ efflux causes membrane hyperpolarization that maintains Ca2+ entry by increasing an electrical gradient, thereby playing a role in the activation of Ca2+-dependent signal pathways (38). In VSMCs, an increase in [Ca2+]i activates specific signaling pathways. CREB, a mitogen-induced transcriptional factor, is phosphorylated in response to a rise in [Ca2+]i and subsequently induces mitogenic immediate early genes such as c-fos and NOR-1 (32–34). In the present study, blockade or siRNA-mediated targeting of KCa3.1 suppressed PDGF-induced HCSMC proliferation, concomitantly with an inhibition of the rise in [Ca2+]i, CREB phosphorylation, and immediate early gene expression, and these effects were abolished by a forced rise in [Ca2+]i with A23187. In contrast, KCa3.1 overexpression induced and enhanced VSMC proliferation by activating these signaling pathways. KCa3.1 is associated with VSMC proliferation to a variety of mitogens in different species (3, 12, 39). Taken together, these data indicate that VSMC proliferation is associated with KCa3.1-dependent regulation of Ca2+-dependent signaling pathways.
EBIO at concentrations relatively specific to KCa3.1 (26) in HCSMCs (which predominantly express KCa3.1 but not small conductance KCa2 channels (3)) also suppressed PDGF-induced rise in [Ca2+]i, proliferation, and migration, accompanied by lower expression and activity of KCa3.1 and PDGF β-receptors. SKA-31 and NS309, two chemically distinct and much more potent KCa3.1 activators, had similar effects. The following mechanisms may explain the inhibitory effects of KCa3.1 activators. 1) Because membrane potential is crucial for cell proliferation (40, 41), excessive membrane hyperpolarization due to full activation of KCa3.1 (3) will stop proliferation in a nonspecific manner. 2) Upon PDGF stimulation, KCa3.1 activators stimulate a small number of KCa3.1 channels remaining on the cell membrane (3). The resultant over-hyperpolarization causes an excess of Ca2+ entry that favors cell differentiation rather than proliferation (43). 3) Consistent with a study in HaCaT keratinocyte and C6 glioma cell lines where EBIO also inhibited proliferation (42), an incisive general mechanism of negative feedback abolishes KCa3.1 expression, leading to lowered [Ca2+]i and thereby attenuated proliferation during the prolonged stimulation. It is unlikely that these activators nonspecifically evoke these effects, independently of KCa3.1 stimulation, because the morphological changes induced by EBIO were suppressed by TRAM-34; SKA-16, an inactive analog of SKA-31, had no effects; and KCa3.1-deficient VSMCs exhibit an almost complete loss of proliferative response to PDGF (3). As compared with TRAM-34, EBIO treatment resulted in a stronger inhibition of PDGF-induced signaling pathways and cell cycle progression, with a lesser induction of cyclins. This effect could be explained by the fact that KCa3.1 stimulation abolished the expression of PDGF β-receptors, whereas TRAM-34 had no additive effect to the PDGF-induced down-regulation of this receptor (36). Interestingly, the KCa3.1 activators were also more effective at reducing PDGF-induced increases in KCa3.1 expression. In addition, a forced rise in [Ca2+]i with A23187 restored the proliferative response in KCa3.1 blocker-treated VSMCs (where [Ca2+]i levels were lowered but the expression of PDGF β-receptors was unaltered) but not in KCa3.1 activator-treated cells (where both [Ca2+]i levels and PDGF β-receptor expression were reduced). Therefore, the abolishment of PDGF β-receptor expression is an additional mechanism for diminished responsiveness to PDGF in KCa3.1 activator-treated VSMCs. Indeed, we show here that treatment with KCa3.1 activators prevents PDGF-induced up-regulation of l-caldesmon and down-regulation of calponin-1. Because the mechanisms of PDGF-induced KCa3.1 gene activation in VSMCs remain to be determined, the precise mechanism by which KCa3.1 activators abolish KCa3.1 gene expression needs further investigation.
Cyclins bound to specific cyclin-dependent kinases control cell cycle progression; in particular, cyclin D1 mainly controls G1 phase, cyclins A and E control S phase, whereas cyclins A and B1 control mitotic phase (44). In addition, a rise in [Ca2+]i, phosphorylated CREB, and immediate early genes play important roles in the expression and activation of cyclins, in the induction of resting cell (G0) reentry into the cell cycle, in DNA synthesis at G1/S transition, and in mitosis at G2/M transition in a cooperative fashion (45–47). In this study, TRAM-34 inhibited PDGF-induced expression of cyclins A and B1, but not D1 and E, along with a reduction in the rise in [Ca2+]i, CREB phosphorylation, and c-Fos and NOR-1 expression. This effect is consistent with the finding that TRAM-34 inhibited FBS-induced cell cycle progression at transitions from G1 to S and G2 to M phases. In contrast, EBIO more effectively inhibited FBS-induced cell cycle progression at the transition from G1 to S phase, concomitantly with a stronger inhibition in PDGF-induced expression of c-Fos and cyclins A, B1, D1, and E, indicating that KCa3.1 stimulation could prevent the reentry of resting cells (G0) into the cell cycle.
PDGF induces VSMC elongation and thinning through the migratory machinery. Our previous observation that blockade of KCa3.1 inhibits PDGF-induced VSMC migration (3) is consistent with the present study showing that TRAM-34 inhibits PDGF-induced cell elongation and thinning, whereas KCa3.1 stimulation enhances these changes in a TRAM-34-sensitive manner. Thus, KCa3.1 may play a crucial role in the regulation of protrusive activity. Other possible mechanisms include the contribution of KCa3.1 to cell volume regulation that modulates morphological changes and the proliferative response (48, 49). Furthermore, in contrast to the effects of TRAM-34, a forced rise in [Ca2+]i with A23187 failed to suppress the inhibitory effect of EBIO on proliferation. KCa3.1 stimulation decreased l-caldesmon and PDGF β-receptor expression, VSMC dedifferentiation markers, and increased calponin-1 expression, a differentiation marker. These effects were accompanied by a strong suppression of KCa3.1 expression, the appearance of a spindle-like shape, and the suppression of signaling pathways and cyclin expression. Thus, the effects of KCa3.1 stimulation cannot be explained only by their direct effects on channel function, implying a possible link between KCa3.1 and VSMC differentiation/dedifferentiation genes.
KCa3.1 overexpression induced VSMC proliferation in the absence of PDGF via signaling pathways similar to those activated by this growth factor. Moreover, KCa3.1 overexpression also enhanced the response to PDGF. Similar enhanced cell proliferation has been reported for the ether-a-go-go K+ channel (Kv10.1), which also regulates Ca2+ influx and is expressed in up to 70% of human cancers (50). Possible mechanisms include: 1) increased channel expression may cause membrane hyperpolarization and Ca2+ influx through nonspecific cation channels, leading to CREB-dependent activation of mitogenic genes (33), and 2) KCa3.1 may be increased in intracellular organelles such as mitochondria, and may maintain the driving force for the store Ca2+ efflux with counter-influx of K+ (51).
In summary, KCa3.1 blockade or knockdown reduces PDGF-induced VSMC proliferation via inhibiting Ca2+-dependent signaling pathways and cell cycle progression, whereas KCa3.1 overexpression has the opposite effects. Pharmacological KCa3.1 stimulation suppresses these responses more markedly via abolishing KCa3.1 and PDGF β-receptor expression. Therefore, targeting KCa3.1 with specific activators, in addition to blockers and gene therapy, may constitute a new therapeutic approach for the prevention of diseases with increased cell proliferative activity such as atherosclerosis.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 HL080173, HL080173-02S1, P20 RR018751, and P20 GM103513 (to H. M.); RO1 GM076063 (to H. W.); P01 HL68769 (to D. D. G.); and RO1 DK090123 (to F. P.). This work was also supported by an Advancing a Healthier Wisconsin grant from the Medical College of Wisconsin.
- KCa
- calcium-activated potassium channel
- CREB
- cAMP-response element-binding protein
- CsA
- cyclosporin A
- EBIO
- 1-ethyl-2-benzimidazolinone
- HCSMC
- human coronary artery smooth muscle cell
- NOR-1
- neuron-derived orphan receptor-1
- NS309
- 6,7-dichloro-1H-indole-2,3-dione 3-oxime
- PMA
- phorbol-12-myristate-13-acetat
- SKA-16
- 2-amino-6-[methylsulfonyl]benzothiazole
- SKA-31
- naphtho[1,2-d]thiazol-2-ylamine
- TRAM-34
- 1-[(2-chlorophenyl)diphenylmethyl]-1H-pyrazole
- VSMC
- vascular smooth muscle cell
- BAPTA
- 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- eGFP
- enhanced green fluorescent protein
- Luc
- luciferase
- tg
- eGFP-tagged.
REFERENCES
- 1. Berkefeld H., Fakler B., Schulte U. (2010) Ca2+-activated K+ channels: from protein complexes to function. Physiol. Rev. 90, 1437–1459 [DOI] [PubMed] [Google Scholar]
- 2. Wulff H., Castle N. A. (2010) Therapeutic potential of KCa3.1 blockers: recent advances and promising trends. Expert Rev. Clin. Pharmacol. 3, 385–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Toyama K., Wulff H., Chandy K. G., Azam P., Raman G., Saito T., Fujiwara Y., Mattson D. L., Das S., Melvin J. E., Pratt P. F., Hatoum O. A., Gutterman D. D., Harder D. R., Miura H. (2008) The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J. Clin. Invest. 118, 3025–3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shepherd M. C., Duffy S. M., Harris T., Cruse G., Schuliga M., Brightling C. E., Neylon C. B., Bradding P., Stewart A. G. (2007) KCa3.1 Ca2+ activated K+ channels regulate human airway smooth muscle proliferation. Am. J. Respir. Cell Mol. Biol. 37, 525–531 [DOI] [PubMed] [Google Scholar]
- 5. Grgic I., Eichler I., Heinau P., Si H., Brakemeier S., Hoyer J., Köhler R. (2005) Selective blockade of the intermediate-conductance Ca2+-activated K+ channel suppresses proliferation of microvascular and macrovascular endothelial cells and angiogenesis in vivo. Arterioscler. Thromb. Vasc. Biol. 25, 704–709 [DOI] [PubMed] [Google Scholar]
- 6. Wulff H., Knaus H. G., Pennington M., Chandy K. G. (2004) K+ channel expression during B cell differentiation: implications for immunomodulation and autoimmunity. J. Immunol. 173, 776–786 [DOI] [PubMed] [Google Scholar]
- 7. Ghanshani S., Wulff H., Miller M. J., Rohm H., Neben A., Gutman G. A., Cahalan M. D., Chandy K. G. (2000) Up-regulation of the IKCa1 potassium channel during T-cell activation: molecular mechanism and functional consequences. J. Biol. Chem. 275, 37137–37149 [DOI] [PubMed] [Google Scholar]
- 8. Peña T. L., Rane S. G. (1999) The fibroblast intermediate conductance KCa channel, FIK, as a prototype for the cell growth regulatory function of the IK channel family. J. Membr. Biol. 172, 249–257 [DOI] [PubMed] [Google Scholar]
- 9. Tao R., Lau C. P., Tse H. F., Li G. R. (2008) Regulation of cell proliferation by intermediate-conductance Ca2+-activated potassium and volume-sensitive chloride channels in mouse mesenchymal stem cells. Am. J. Physiol. Cell Physiol 295, C1409–C1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chou C. C., Lunn C. A., Murgolo N. J. (2008) KCa3.1: target and marker for cancer, autoimmune disorder and vascular inflammation? Expert. Rev. Mol Diagn. 8, 179–187 [DOI] [PubMed] [Google Scholar]
- 11. Neylon C. B., Lang R. J., Fu Y., Bobik A., Reinhart P. H. (1999) Molecular cloning and characterization of the intermediate-conductance Ca2+-activated K+ channel in vascular smooth muscle: relationship between KCa channel diversity and smooth muscle cell function. Circ. Res. 85, e33-e43 [DOI] [PubMed] [Google Scholar]
- 12. Köhler R., Wulff H., Eichler I., Kneifel M., Neumann D., Knorr A., Grgic I., Kämpfe D., Si H., Wibawa J., Real R., Borner K., Brakemeier S., Orzechowski H. D., Reusch H. P., Paul M., Chandy K. G., Hoyer J. (2003) Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circulation 108, 1119–1125 [DOI] [PubMed] [Google Scholar]
- 13. Neylon C. B. (2002) Potassium channels and vascular proliferation. Vascul. Pharmacol. 38, 35–41 [DOI] [PubMed] [Google Scholar]
- 14. Saito T., Fujiwara Y., Fujiwara R., Hasegawa H., Kibira S., Miura H., Miura M. (2002) Role of augmented expression of intermediate-conductance Ca2+-activated K+ channels in postischaemic heart. Clin. Exp. Pharmacol. Physiol. 29, 324–329 [DOI] [PubMed] [Google Scholar]
- 15. Terata Y., Saito T., Fujiwara Y., Hasegawa H., Miura H., Watanabe H., Chiba Y., Kibira S., Miura M. (2003) Pitavastatin inhibits upregulation of intermediate conductance calcium-activated potassium channels and coronary arteriolar remodeling induced by long-term blockade of nitric oxide synthesis. Pharmacology 68, 169–176 [DOI] [PubMed] [Google Scholar]
- 16. Tharp D. L., Wamhoff B. R., Turk J. R., Bowles D. K. (2006) Upregulation of intermediate-conductance Ca2+-activated K+ channel (IKCa1) mediates phenotypic modulation of coronary smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 291, H2493–H2503 [DOI] [PubMed] [Google Scholar]
- 17. Tharp D. L., Wamhoff B. R., Wulff H., Raman G., Cheong A., Bowles D. K. (2008) Local delivery of the KCa3.1 blocker, TRAM-34, prevents acute angioplasty-induced coronary smooth muscle phenotypic modulation and limits stenosis. Arterioscler. Thromb. Vasc. Biol. 28, 1084–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Si H., Grgic I., Heyken W. T., Maier T., Hoyer J., Reusch H. P., Köhler R. (2006) Mitogenic modulation of Ca2+-activated K+ channels in proliferating A7r5 vascular smooth muscle cells. Br. J. Pharmacol. 148, 909–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miano J. M., Vlasic N., Tota R. R., Stemerman M. B. (1993) Localization of Fos and Jun proteins in rat aortic smooth muscle cells after vascular injury. Am. J Pathol. 142, 715–724 [PMC free article] [PubMed] [Google Scholar]
- 20. Sylvester A. M., Chen D., Krasinski K., Andrés V. (1998) Role of c-Fos and E2F in the induction of cyclin A transcription and vascular smooth muscle cell proliferation. J. Clin. Invest. 101, 940–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheong A., Bingham A. J., Li J., Kumar B., Sukumar P., Munsch C., Buckley N. J., Neylon C. B., Porter K. E., Beech D. J., Wood I. C. (2005) Downregulated REST transcription factor is a switch enabling critical potassium channel expression and cell proliferation. Mol. Cell 20, 45–52 [DOI] [PubMed] [Google Scholar]
- 22. Barlow C. A., Rose P., Pulver-Kaste R. A., Lounsbury K. M. (2006) Excitation-transcription coupling in smooth muscle. J. Physiol. 570, 59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dolmetsch R. E., Lewis R. S., Goodnow C. C., Healy J. I. (1997) Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 386, 855–858 [DOI] [PubMed] [Google Scholar]
- 24. McAllister J., Ghosh S., Berry D., Park M., Sadeghi S., Wang K. X., Parker W. D., Swerdlow R. H. (2008) Effects of memantine on mitochondrial function. Biochem. Pharmacol. 75, 956–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wulff H., Miller M. J., Hansel W., Grissmer S., Cahalan M. D., Chandy K. G. (2000) Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc. Natl. Acad. Sci. U.S.A. 97, 8151–8156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wulff H., Kolski-Andreaco A., Sankaranarayanan A., Sabatier J. M., Shakkottai V. (2007) Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr. Med. Chem. 14, 1437–1457 [DOI] [PubMed] [Google Scholar]
- 27. Sankaranarayanan A., Raman G., Busch C., Schultz T., Zimin P. I., Hoyer J., Köhler R., Wulff H. (2009) Naphtho[1,2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Mol. Pharmacol. 75, 281–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Strøbaek D., Teuber L., Jørgensen T. D., Ahring P. K., Kjaer K., Hansen R. S., Olesen S. P., Christophersen P., Skaaning-Jensen B. (2004) Activation of human IK and SK Ca2+-activated K+ channels by NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime). Biochim. Biophys. Acta 1665, 1–5 [DOI] [PubMed] [Google Scholar]
- 29. Tolić-Nørrelykke I. M., Wang N. (2005) Traction in smooth muscle cells varies with cell spreading. J. Biomech. 38, 1405–1412 [DOI] [PubMed] [Google Scholar]
- 30. Park F., Ohashi K., Chiu W., Naldini L., Kay M. A. (2000) Efficient lentiviral transduction of liver requires cell cycling in vivo. Nat. Genet. 24, 49–52 [DOI] [PubMed] [Google Scholar]
- 31. Hollenbeck S. T., Nelson P. R., Yamamura S., Faries P. L., Liu B., Kent K. C. (2004) Intracellular calcium transients are necessary for platelet-derived growth factor but not extracellular matrix protein-induced vascular smooth muscle cell migration. J. Vasc. Surg. 40, 351–358 [DOI] [PubMed] [Google Scholar]
- 32. Martínez-González J., Rius J., Castelló A., Cases-Langhoff C., Badimon L. (2003) Neuron-derived orphan receptor-1 (NOR-1) modulates vascular smooth muscle cell proliferation. Circ. Res. 92, 96–103 [DOI] [PubMed] [Google Scholar]
- 33. Pulver R. A., Rose-Curtis P., Roe M. W., Wellman G. C., Lounsbury K. M. (2004) Store-operated Ca2+ entry activates the CREB transcription factor in vascular smooth muscle. Circ. Res. 94, 1351–1358 [DOI] [PubMed] [Google Scholar]
- 34. Rothman A., Wolner B., Button D., Taylor P. (1994) Immediate-early gene expression in response to hypertrophic and proliferative stimuli in pulmonary arterial smooth muscle cells. J. Biol. Chem. 269, 6399–6404 [PubMed] [Google Scholar]
- 35. Mayr B., Montminy M. (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599–609 [DOI] [PubMed] [Google Scholar]
- 36. Kaplan-Albuquerque N., Van Putten V., Weiser-Evans M. C., Nemenoff R. A. (2005) Depletion of serum response factor by RNA interference mimics the mitogenic effects of platelet derived growth factor-BB in vascular smooth muscle cells. Circ. Res. 97, 427–433 [DOI] [PubMed] [Google Scholar]
- 37. Sirois M. G., Simons M., Edelman E. R. (1997) Antisense oligonucleotide inhibition of PDGFR-β receptor subunit expression directs suppression of intimal thickening. Circulation 95, 669–676 [DOI] [PubMed] [Google Scholar]
- 38. Feske S. (2007) Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 7, 690–702 [DOI] [PubMed] [Google Scholar]
- 39. Raicu M., Florea S., Costache G., Popov D., Simionescu M. (2000) Clotrimazole inhibits smooth muscle cell proliferation and has a vasodilator effect on resistance arteries. Fundam. Clin. Pharmacol. 14, 477–485 [DOI] [PubMed] [Google Scholar]
- 40. Sun D., Gong Y., Kojima H., Wang G., Ravinsky E., Zhang M., Minuk G. Y. (2003) Increasing cell membrane potential and GABAergic activity inhibits malignant hepatocyte growth. Am. J. Physiol. Gastrointest. Liver Physiol. 285, G12–G19 [DOI] [PubMed] [Google Scholar]
- 41. Boonstra J., Mummery C. L., Tertoolen L. G., Van Der Saag P. T., De Laat S. W. (1981) Cation transport and growth regulation in neuroblastoma cells. Modulations of K+ transport and electrical membrane properties during the cell cycle. J. Cell Physiol. 107, 75–83 [DOI] [PubMed] [Google Scholar]
- 42. Koegel H., Kaesler S., Burgstahler R., Werner S., Alzheimer C. (2003) Unexpected down-regulation of the hIK1 Ca2+-activated K+ channel by its opener 1-ethyl-2-benzimidazolinone in HaCaT keratinocytes. Inverse effects on cell growth and proliferation. J. Biol. Chem. 278, 3323–3330 [DOI] [PubMed] [Google Scholar]
- 43. Wamhoff B. R., Bowles D. K., McDonald O. G., Sinha S., Somlyo A. P., Somlyo A. V., Owens G. K. (2004) L-type voltage-gated Ca2+ channels modulate expression of smooth muscle differentiation marker genes via a Rho kinase/myocardin/SRF-dependent mechanism. Circ. Res. 95, 406–414 [DOI] [PubMed] [Google Scholar]
- 44. Koledova V. V., Khalil R. A. (2006) Ca2+, calmodulin, and cyclins in vascular smooth muscle cell cycle. Circ. Res. 98, 1240–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Berridge M. J. (1995) Calcium signalling and cell proliferation. Bioessays. 17, 491–500 [DOI] [PubMed] [Google Scholar]
- 46. Choi J., Chiang A., Taulier N., Gros R., Pirani A., Husain M. (2006) A calmodulin-binding site on cyclin E mediates Ca2+-sensitive G1/S transitions in vascular smooth muscle cells. Circ. Res. 98, 1273–1281 [DOI] [PubMed] [Google Scholar]
- 47. Desdouets C., Matesic G., Molina C. A., Foulkes N. S., Sassone-Corsi P., Brechot C., Sobczak-Thepot J. (1995) Cell cycle regulation of cyclin A gene expression by the cyclic AMP-responsive transcription factors CREB and CREM. Mol. Cell. Biol. 15, 3301–3309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brugnara C., Gee B., Armsby C. C., Kurth S., Sakamoto M., Rifai N., Alper S. L., Platt O. S. (1996) Therapy with oral clotrimazole induces inhibition of the Gardos channel and reduction of erythrocyte dehydration in patients with sickle cell disease. J. Clin. Invest. 97, 1227–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rouzaire-Dubois B., Milandri J. B., Bostel S., Dubois J. M. (2000) Control of cell proliferation by cell volume alterations in rat C6 glioma cells. Pflugers Arch. 440, 881–888 [DOI] [PubMed] [Google Scholar]
- 50. Pardo L. A., del Camino D., Sánchez A., Alves F., Brüggemann A., Beckh S., Stühmer W. (1999) Oncogenic potential of EAG K+ channels. EMBO J. 18, 5540–5547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yamashita M., Sugioka M., Ogawa Y. (2006) Voltage- and Ca2+-activated potassium channels in Ca2+ store control Ca2+ release. FEBS J. 273, 3585–3597 [DOI] [PubMed] [Google Scholar]