

Background: Information was not available about prototoxin LYNX1 amino acid residues involved in binding to muscle and neuronal nicotinic receptors.
Results: A series of water-soluble LYNX1 (ws-LYNX1) mutants was obtained and their interaction with nicotinic receptors was analyzed.
Conclusion: There are both common and selective ws-LYNX1 residues recognizing distinct receptor types.
Significance: For the first time, several functionally important residues in ws-LYNX1 are identified.
Keywords: Computer Modeling, Electrophysiology, Mutagenesis, Neurotoxin, Nicotinic Acetylcholine Receptors, LYNX1, Acetylcholine-binding Proteins, Ionic Currents, Radioligand Assay
Abstract
Human LYNX1, belonging to the Ly6/neurotoxin family of three-finger proteins, is membrane-tethered with a glycosylphosphatidylinositol anchor and modulates the activity of nicotinic acetylcholine receptors (nAChR). Recent preparation of LYNX1 as an individual protein in the form of water-soluble domain lacking glycosylphosphatidylinositol anchor (ws-LYNX1; Lyukmanova, E. N., Shenkarev, Z. O., Shulepko, M. A., Mineev, K. S., D'Hoedt, D., Kasheverov, I. E., Filkin, S. Y., Krivolapova, A. P., Janickova, H., Dolezal, V., Dolgikh, D. A., Arseniev, A. S., Bertrand, D., Tsetlin, V. I., and Kirpichnikov, M. P. (2011) NMR structure and action on nicotinic acetylcholine receptors of water-soluble domain of human LYNX1. J. Biol. Chem. 286, 10618–10627) revealed the attachment at the agonist-binding site in the acetylcholine-binding protein (AChBP) and muscle nAChR but outside it, in the neuronal nAChRs. Here, we obtained a series of ws-LYNX1 mutants (T35A, P36A, T37A, R38A, K40A, Y54A, Y57A, K59A) and examined by radioligand analysis or patch clamp technique their interaction with the AChBP, Torpedo californica nAChR and chimeric receptor composed of the α7 nAChR extracellular ligand-binding domain and the transmembrane domain of α1 glycine receptor (α7-GlyR). Against AChBP, there was either no change in activity (T35A, T37A), slight decrease (K40A, K59A), and even enhancement for the rest mutants (most pronounced for P36A and R38A). With both receptors, many mutants lost inhibitory activity, but the increased inhibition was observed for P36A at α7-GlyR. Thus, there are subtype-specific and common ws-LYNX1 residues recognizing distinct targets. Because ws-LYNX1 was inactive against glycine receptor, its “non-classical” binding sites on α7 nAChR should be within the extracellular domain. Micromolar affinities and fast washout rates measured for ws-LYNX1 and its mutants are in contrast to nanomolar affinities and irreversibility of binding for α-bungarotoxin and similar snake α-neurotoxins also targeting α7 nAChR. This distinction may underlie their different actions, i.e. nAChRs modulation versus irreversible inhibition, for these two types of three-finger proteins.
Introduction
The gene LYNX1 was the first found to encode a protein of the well known Ly6 family but was but detected in the mammalian brain rather than in the immune system (1). Miwa et al. (2) named the putative protein LYNX1, where “Ly” is borrowed from Ly6 and “nx” from neurotoxins. Similar to other members of this family, the protein LYNX1 has the same arrangement of disulfide bridges as three-finger snake venom neurotoxins and shares with them a similar three-dimensional structure (3). The principal difference of LYNX1 from snake neurotoxins is the presence at its C terminus of a glycosylphosphatidylinositol anchor by which it is attached to the membrane in the vicinity of neuronal nicotinic acetylcholine receptors (nAChRs),3 thus modulating their functioning (4). In the past decade, the involvement of LYNX1, LYNX2, and other relevant Ly6 members was demonstrated in regulation of behavior (5, 6), retinal plasticity (7), and some other processes, including lung cancer cell growth (8–10).
To elucidate the mechanism of LYNX1 action on nAChRs, it would be of interest to compare its functional properties with those of snake venom neurotoxins because for the latter, comprehensive information is compiled from affinity labeling, mutagenesis, and electrophysiology (see Refs. 11–13). Moreover, there are x-ray structures of snake venom α-neurotoxins in complexes with the acetylcholine-binding protein (AChBP, a model for the ligand-binding domains of nAChRs and other Cys-loop receptors) and with the ligand-binding domain of human α1 nAChR subunit (14, 15). However, current ideas about the functions of LYNX1 and its congeners are based only on co-immunoprecipitation experiments and overexpression or knock-out of the respective genes, because as an individual protein LYNX1 was obtained only recently, in the form of its water-soluble domain lacking the glycosylphosphatidylinositol anchor (ws-LYNX1) (3). The protein competed with radioactive α-bungarotoxin (125I-αBgt) for binding to AChBP and Torpedo californica nAChR, evidently targeting the classical binding sites for agonists and competitive antagonists. However, there was no competition at neuronal α7 nAChR, and the observed effects on the current amplitudes at heterologously expressed α7 nAChR were apparently due to binding outside of the classical site (3).
In this study, we first map the binding surfaces of ws-LYNX1 essential for recognition of different targets. From the computer model of the ws-LYNX1 complex with AChBP (3), in loops II and III of ws-LYNX1, several mutations were chosen (see Fig. 1) that were expected to affect binding to AChBP and/or to muscle-type nAChRs. We also planned to check whether the mutated residues might be important for binding to α7 nAChR. Because this receptor subtype exhibits very rapid desensitization, patch clamp analysis of the ws-LYNX1 mutants was performed on the nondesensitizing chimera α7-GlyR, which consists of the α7 extracellular ligand-binding domain and the transmembrane domain of α1 glycine receptor (16).
FIGURE 1.

Mutants of ws-LYNX1. Disposition of the chosen mutations as side chains of the mutated residues is shown on the polypeptide backbone of ws-LYNX1 taken from PDB code 2L03 (3). I, II, and III mark the N-terminal, central, and C-terminal loops of the ws-LYNX1 in the three-finger spatial structure. In the right table, the sequences of oligonucleotide primers used for mutagenesis of ws-LYNX1 are presented with bold underlined positions of mutations. The measured by MALDI mass spectrometry molecular masses of the recombinant proteins which correspond within experimental error to the calculated masses of ws-LYNX1 mutants (amino acid residues 1–73) with an additional Met residue at the N terminus and five closed disulfide bridges are collected in the table on the left.
EXPERIMENTAL PROCEDURES
Cloning, Bacterial Expression, and Structural Analysis of ws-LYNX1 Mutants
The ws-LYNX1 mutant genes were engineered using site-directed mutagenesis. Plasmid pET-22b(+)/ws-LYNX1 obtained previously (17) was used as a template for PCR reactions. Oligonucleotides used for site-directed mutagenesis are given in Fig. 1. The ws-LYNX1 mutant genes were cloned into the expression vector pET-22b(+) (Novagen) at the NdeI and BamHI restriction sites. Escherichia coli expression, purification, and refolding of ws-LYNX1 mutants were done as described in Ref. 17 with minor changes. Briefly, ws-LYNX1 mutants were extracted from inclusion bodies after incubation with 50 mm NaPi, 8 m urea, 1 mm Tris-(2-carboxyethyl)phosphine, 5 mm DTT, pH 7.4. Then, reduced ws-LYNX1 mutants were purified on a SP-Sepharose resin (GE Healthcare) equilibrated in 50 mm NaPi, 8 m urea, 5 mm DTT, pH 5.0. The proteins were eluted by a gradient of NaCl. Fractions containing reduced ws-LYNX1 mutants were concentrated using membranes with 1-kDa cut-off (Millipore). Refolding of ws-LYNX1 mutants was induced by dissolving of reduced proteins in a 60-fold excess of a renaturation buffer (50 mm Tris/HCl, 1.5 m urea, 0.5 m l-Arg, 0.3 mm GSH, and 3 mm GSSG, pH 9.5) to the final protein concentration 0.1 mg/ml. Renaturation was performed during 3 days at 4 °C. The refolded ws-LYNX1 mutants were analyzed and purified on a reverse-phase C4 HPLC column (4.6 × 250 mm, A300, Jupiter, Phenomenex).
The homogeneity of the refolded ws-LYNX1 mutants was confirmed by SDS-PAGE, analytical HPLC, and mass spectrometry. The measured molecular masses of the mutants (see Fig. 1) within experimental error coincide with the calculated masses of the water-soluble domain of human LYNX1 (amino acid residues 1–73), with an additional Met residue at the N terminus, five closed disulfide bridges, and the introduced mutations. Formation of disulfide bonds for the refolded ws-LYNX1 mutants was additionally confirmed using Ellman's reagent. CD spectroscopy (data not shown) of the refolded ws-LYNX1 mutants revealed a secondary structure within experimental error coinciding with the secondary structure of ws-LYNX1 (α-helix, 6%; β-sheet, 39%; β-turn, 22%; irregular, 33%). The one-dimensional 1H NMR spectra (data not shown) also confirmed proper folding for the all ws-LYNX1 mutants.
Binding of ws-LYNX1 and Its Mutants to nAChR and AChBPs
The binding of ws-LYNX1 and its mutants to nAChR-enriched T. californica membranes, α7-GlyR chimera transfected in HEK cells and Lymnaea stagnalis AChBP (Ls-AChBP) (see Fig. 2) was carried out in competitive experiments with 125I-αBgt as described previously for ws-LYNX1 itself (3). Shortly, the compounds (in concentrations up to 30 μm) were preincubated 3 h with the nAChR-enriched Torpedo membranes (final concentration of toxin-binding sites, 1.25 nm), α7-GlyR transfected HEK cells (final concentration of toxin-binding sites, 12.7 pm) or the Ls-AChBP (final concentration of toxin-binding sites, 2.4 nm). Bindings with Torpedo membranes were carried out in 50 μl of 20 mm Tris/HCl buffer, containing 1 mg/ml of BSA, pH 8.0 (for HEK cells, this buffer contained additionally protease inhibitor mixture), and with the Ls-AChBP, in 50 μl of binding buffer (PBS, containing 0.7 mg/ml of BSA and 0.05% Tween 20, pH 7.5) at 25 °C. After that, 125I-αBgt (∼2000 Ci/mmol) was added to final concentration of 0.15–0.58 nm for 5 min followed by filtration of reaction mixture on GF/C filters (Whatman, Maidstone, UK) presoaked in 0.25% polyethylenimine (in the case of membranes or HEK cells) or on double DE-81 filters (Whatman) presoaked in binding buffer (for Ls-AChBP). Unbound radioactivity was removed from the filters by washes (3 × 3 ml) with the respective incubation buffers. Nonspecific binding was determined in the presence of 10 μm α-cobratoxin (3 h of preincubation). Competition data analyses were fit using ORIGIN (version 7.5; OriginLab Corp.) to a one-site dose-response curve by Equation 1,
where IC50 is the concentration at which 50% of the sites are inhibited and nH is the Hill coefficient.
FIGURE 2.

Binding of ws-LYNX1 and its mutants to different targets in radioligand assays with 125I-αBgt. The compounds were tested in their binding potency on AChBP from L. stagnalis (A and B), nAChR-enriched membranes from T. californica (C), and α7-GlyR chimera transfected HEK cells (D). Inhibition effects in A and C were evaluated at the concentrations of 20 and 30 μm, respectively. Each column and point is mean ± S.E. of three independent experiments. In B, the Hill equation (y = 100/(1 + ([toxin]/IC50)nH)) was fitted to normalized data (% of control binding). The calculated parameters IC50 and nH were 17 ± 2 μm and 1.4 ± 0.2 for ws-LYNX1 (stars, solid line), 8.2 ± 0.6 μm and 1.1 ± 0.1 for P36A mutant (filled circles, solid line), 6.0 ± 0.2 μm and 1.2 ± 0.1 for R38A mutant (crosses, dotted line), and 8.2 ± 0.3 μm and 0.8 ± 0.1 for Y57A mutant (circles, dashed line). Saturation binding of 125I-αBgt to α7-GlyR chimera-transfected HEK cells (D) gave KD 0.39 ± 0.02 nm from the equation, −y = Bmax/(1 + KD/x). No inhibition of 125I-αBgt binding to chimera with ws-LYNX1 in the concentration range from 1 to 30 μm was detected (D, top inset). Equipotency of α-cobratoxin (αCbt) and αBgt at concentration of 10 μm (used both in radioligand and electrophysiological experiments to completely bind/block AChBPs, Torpedo, and α7 nAChRs) is demonstrated in competition with 125I-αBgt for binding to α7-GlyR chimera as compared with total binding (100%) in the absence of α-cobratoxin/αBgt (D, bottom inset).
Saturation binding with α7-GlyR chimera-transfected HEK cells was determined using 125I-αBgt concentrations in the range from 0.05 to 0.6 nm. Nonspecific 125I-αBgt binding was determined in this case in the presence of 10 μm αBgt. Equilibrium binding data were fit using ORIGIN (version 7.5) to a one-site model according to Equation 2,
where B(x) is the radioligand specifically bound at a free concentration x (determined by subtraction of the amount of bound and adsorbed radioligand from the total amount added to incubation mixture), Bmax is the maximal specific bound radioligand, and KD is the dissociation constant.
Cell Culture Preparation and Electrophysiological Recording
Chinese hamster ovary (CHO-K1) cells were obtained from the American Type Tissue Culture Collection (ATCC, Molsheim, France) and maintained as described previously (18). One day before the transfection, cells were plated on the coverslips (12–14 mm in diameter), which were placed inside 35-mm cell culture dishes with 2 ml of medium. CHO-K1 cells were transfected with ∼3 μg/1 μl cDNA of α7-GlyR chimera (16), kindly provided by Dr. L. Prado de Carvalho, using the Lipofectamine 2000 transfection protocol (Life Technology). To facilitate identification of α7-GlyR-expressing cells, GFP (0.5 μg/μl) was added to transfection medium. Three hours after the initial exposure of the cells to the cDNAs, a fresh cDNA-containing solution replaced the old one. Electrophysiological recordings were performed on fluorescent cells 48–72 h after transfection. Whole-cell recordings were conducted on CHO-K1 cells at room temperature (20–25 °C) using an EPC-9 amplifier (HEKA Elektronik). Cells were continuously superfused with external solution containing the following: 140 mm NaCl, 2 mm CaCl2, 2.8 mm KCl, 4 mm MgCl2, 20 mm HEPES, 10 mm glucose; pH 7.4; 320–330 milliosmoles. The intracellular patch pipette solution contained the following: 140 mm CsCl, 6 mm CaCl2, 2 mm MgCl2, 2 mm MgATP, 0.4 mm NaGTP, 10 mm HEPES/CsOH, 20 mm BAPTA/KOH; pH 7.3; 290 milliosmoles. Pipettes were pulled from borosilicate glass capillaries (Harvard Apparatus, Ltd.) and had resistances of 5–8 megaohms.
Two independent application systems were used in this study. First, for rapid replacement of solutions, a system of two parallel rectangular tubes, 100 μm in diameter, located at a distance of 40–50 μm from the tested cell, was used. The movement of tubes was controlled by a computer-driven fast exchange system (SF 77A Perfusion Fast-Step, Warner). Second, for efficient delivery of small amount of mutants, we used a pressure application system. ws-LYNX1 and its mutants were applied under pressure to the surface of studied cells via a glass pipette (tip diameter, ∼3–5 μm) using a picospritzer (TooheySpritzer, Toohey Company, Fairfield, NJ). The application pipette filled with ws-LYNX1 or mutant samples dissolved in external solution at final concentration 10 μm, was positioned at a distance of 20–50 μm from the recording cell. The duration of application varied from 90 to 240 s. Cells with a low input resistance (<150 megaohms) and a rapid run down (>30% with repetitive application) were excluded from analysis. The results obtained are presented in Figs. 3–5.
FIGURE 3.

Patch clamp characterization of the chimeric α7-GlyR (A–D) versus GlyR (E and F) with two representatives of the Ly6/neurotoxin family of three-finger proteins, α-cobratoxin (A) and ws-LYNX1 (B–F). A, mean amplitudes from nine cells in control (before addition of tested compound) (left), after 90 s of α-cobratoxin (αCbt) application (middle) and after 5 min of washout (right). An asterisk represents statistically significant difference p ≤ 0.05 (analysis of variance test). Lines show comparison of each test column with control one. B, mean amplitudes from 16 cells in control (left), after 90 s of ws-LYNX1 (10 μm) application (middle) and after 5 min of washout (right). Note the irreversible inhibition by α-cobratoxin and partially reversible by ws-LYNX1. An asterisk means statistically significant difference p ≤ 0.05 (analysis of variance test). C shows detailed presentation of the current amplitude (in %) changes for each of 22 cells (see B) relatively to the amplitude of the control (when no ws-LYNX1 was added). D, superimposed traces of whole-cell currents induced by rapid application of nicotine (Nic; 30 μm) in control (black traces), after application of ws-LYNX1, 10 μm (gray traces), and after a 5-min washout (dark gray traces). In a, b, and c are shown examples of different exposure times to ws-LYNX1: 90 s, 180 s and 240 s, respectively. E, traces of whole-cell currents induced by rapid application of glycine (Gly, 30 μm) before treatment with ws-LYNX1 (control, left), after application of ws-LYNX1 (10 μm) (middle) and after 5 min of washout of ws-LYNX1 (right) at whole-cell recordings from CHO-K1 cells expressing human α1 glycine receptor subunits. Note absence of ws-LYNX1-induced inhibition. F, mean amplitudes of glycine-induced currents from 16 cells in control (left), after 90 s of ws-LYNX1 application (middle), and after 5 min of washout (right).
FIGURE 4.

Patch clamp analysis of the interactions between the chimeric α7-GlyR and ws-LYNX1 mutants. A, the study of T35A mutant is represented as mean nicotine-induced current amplitudes from seven cells in control (left; a), after 90 s of mutant (10 μm) application (middle) and after 5 min of washout (right), and amplitudes of nicotine-induced currents in control (black; b), after application of 10 μm mutant (white), and after 5 min of washout (shaded) for each of seven recorded cells. Note the absence of inhibition. B, the study of P36A mutant is represented as superimposed traces of whole-cell currents induced by rapid application of nicotine (Nic; 30 μm (a)) in control, after 90 s of application of mutant (10 μm) and after 5 min of washout and mean nicotine-induced current amplitudes from five cells in control (left; b), after 90 s of mutant (10 μm) application (middle) and after 5 min of washout (right). An asterisk means statistically significant difference p ≤ 0.05 (analysis of variance test). Effects of the other ws-LYNX1 mutants on the amplitudes of nicotine-induced currents at whole-cell recordings from CHO-K1 cells expressing the α7-GlyR chimera are represented in C–H as mean amplitudes from 6–16 cells before treatment with compounds (control, left), after 90-s ws-LYNX1 mutant (10 μm) application (middle), and after 5 min of washout (right). Names of mutants are shown below the middle columns.
FIGURE 5.

Summary of the effects of ws-LYNX1 and its mutants on the modulation of nicotine-induced whole-cell currents in CHO-K1 cells expressing α7-GlyR. The amplitude of the current of the control (no ws-LYNX1 is added) is taken as zero, its decrease (in %) from the starting amplitude exerted by ws-LYNX1 or its mutants (10 μm) indicated below the horizontal axis (disposition close to this axis or on it means a decrease or complete lack of the inhibitory activity). On the contrary, a rectangle above this axis shows that the respective mutant slightly increased the current amplitude.
Human embryonic kidney cells (HEK 293) were cultured in DMEM (Paneko) supplemented with 10% fetal bovine serum (HyClone), 5 mm glutamine, gentamicin, and amphotericin B. The cells were grown in a humidified atmosphere of 5% CO2 at 37 °C. One day before the transfection, cells were plated onto 25-ml culture flasks. Transfection of HEK 293 cells for transient expression of α7-GlyR chimera was carried out by Lipofectamine 2000 transfection protocol, as described for CHO-K1 cells. To assess α7-GlyR expression levels at 72 h following transfection, cells were removed from flasks by Versen solution and used for radioligand binding assays.
Mutagenesis of α7-GlyR Chimera
The Y168A mutation was obtained by PCR amplification of α7-GlyR chimera in pMT3 vector with the following primers: 5′-caaatgcaagaagcagatatatccggcgctatttcaaatg-3′ and 5′-gccggatatatctgcttcttgcatttg-3′ by using Phusion® High-Fidelity DNA polymerase. Amplification product was analyzed after 20 cycles of the following thermal steps: 10 s of initial denaturation at 98 °C, 10 s of denaturation at 98 °C, 30 s of annealing at 60 °C, and 4 min of extension at 72 °C. The product obtained was restricted with DpnI and then transformed into XL-Lada supercompetent cells. The mutation was confirmed by DNA sequencing.
Two-electrode Voltage Clamp Analysis of Y168A Mutant of α7-GlyR Chimera Expressed in Xenopus Oocytes
Oocytes were obtained from benzocaine-anesthetized Xenopus by dissecting abdomen and removing necessary amount of ovarium. Then, part of ovarium was treated by collagenase during 2–3 h. Afterward, single oocytes were transferred to ND96 electrophysiology buffer (5 mm HEPES, 95 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 2 mm MgCl2, pH 7.5). To achieve nAChR expression, each oocyte was injected with 2 ng of plasmid DNA containing wild-type or Y168A substituted α7-GlyR chimera cDNA insert. After 36–48 h of incubation at 18 °C in ND96 solution, current recordings were performed.
All recordings were made using Turbo TEC-03X amplifier (npi electronic GmbH, Tamm, Germany) and 3 m KCl-containing electrodes with resistance ∼0.1 megaohms. Membrane potential was clamped at −60 mV. During the experiment, each oocyte was perfused by ND96 solution until the leakage current reached a steady state. Then, acetylcholine was applied to cell. If cell responded to acetylcholine application and amplitude of evoked current did not change after 5 min, the oocyte was considered suitable for further experiments. Applications of agonist with or without tested compound (α-cobratoxin, αBgt, or ws-LYNX1) were performed for 5 min each. Before application of the mixture of acetylcholine and tested compound, the preliminary 5-min application of the tested compound alone was carried out. The current recorded in response to application of agonist in the presence of tested compound was compared with previous acetylcholine-induced current.
All gathered data were plotted using Origin (version 7.5) and QtiPlot software by Equation 3,
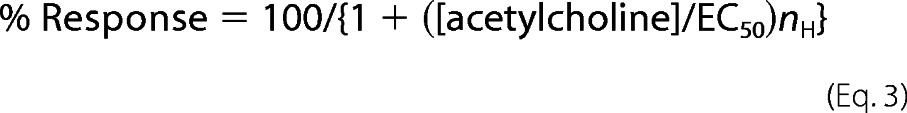 |
where EC50 is a half-maximal effective concentration and nH is the Hill coefficient.
Computer Modeling
For the computer modeling of ws-LYNX1 and αBgt complexes with the AChBP, Torpedo nAChR, and α7 nAChR, we used the docking program ZDOCK (version 3.0.2) (19). The AChBP structure was taken from PDB code 1YI5 (14), and the structure of the Torpedo nAChR ligand-binding domain was taken from PDB code 2BG9 (20). The structure of AChBP from Aplysia californica mutated to human α7 nAChR ligand binding domain (PDB code 3T4M) (21) and the MODELLER program (version 9.10) (22) were used to create a model for the α7 nAChR extracellular domain. The final model of a homopentamer was obtained by sequential fitting of the α7 subunit extracellular domain structure to the coordinates of the template AChBP subunit structure (PDB code 3T4M) with the PyMOL program (version 1.4.1) (23). The structure of ws-LYNX1 was taken from the PDB code 2L03 (3) and that of α-bungarotoxin was taken from PDB code 2QC1 (15).
RESULTS
Structural Characterization of ws-LYNX1 Mutants
Mutations (Fig. 1) were chosen based on the computer model of ws-LYNX1 complex with L. stagnalis AChBP, which indicated a possible role for the fragment 35–40 of the ws-LYNX1 central loop II and for the C-terminal loop III region 54–59. For expression and purification of mutants, we used generally the same procedures as those applied for ws-LYNX1 itself (3, 17). The correctness of the primary structures of the mutants was confirmed by DNA sequencing of the plasmids containing mutant genes and by MALDI mass spectrometry of recombinant proteins showing a close match of the calculated and determined molecular masses (Fig. 1). None of the mutations considerably disturbed a three-dimensional structure of ws-LYNX1 as was evidenced by CD and NMR spectroscopy (data not shown).
Binding of ws-LYNX1 and Its Mutants to AChBP and Torpedo nAChR
With Ls-AChBP (Fig. 2A), two mutations (T35A and T37A) did not have any effect on the binding capacity, whereas removal of the positive charges (K40A and K59A) somewhat decreased the affinity. On the contrary, several mutations (P36A, R38A, Y54A, and Y57A) enhanced the affinity as is clearly seen both in Fig. 2A and from the respective displacement curves (Fig. 2B). However, in neither case, there was a loss of activity suggesting that none of the mutated residues in the loops II or III was strictly essential for binding to Ls-AChBP.
The ws-LYNX1 mutants were also tested in competition with the 125I-αBgt for binding to membrane preparations of T. californica nAChR (Fig. 2C). The affinity was preserved and even slightly increased in the case of T35A mutation, it dropped slightly for P36A and T37A, whereas for other mutations, it was totally or almost totally (Y57A) lost. Thus, according to radioligand analysis, the Torpedo nAChR binding site has more strict requirements to ws-LYNX1 as compared with those of Ls-AChBP.
Patch Clamp Analysis of the Interaction of ws-LYNX1 and Its Mutants with the a7-GlyR
We used the α7-GlyR chimera (16) as a model of α7 nAChR. It consists of two distinct parts: extracellular domain of chick α7 nAChR and the rest (transmembrane and cytoplasmic domains) of α1 human GlyR. This α7-GlyR chimera was selected for two main reasons: (i) at transient transfection in cell lines, it effectively forms recombinant pentameric channels producing whole-cell inward currents up to 2 nA on application of acetylcholine or nicotine; (ii) in contrast to native α7 nAChR (24, 25) and the chimera composed of the α7 nAChR and subtype 3 of 5-hydroxytryptamine receptor (26, 27), the α7-GlyR does not show desensitization (16). Moreover, activation kinetics of this chimeric receptor is very slow, which allows recording of responses reliably and independently of small variations in the speed of perfusion. Previously, it was shown that acetylcholine competed with 125I-αBgt for binding to α7-GlyR (16). Here, we provided additional evidence showing that the α7-GlyR chimera is indeed an adequate mimic of α7 nAChR to analyze the action of ws-LYNX1 and its mutants. Fig. 2D shows the saturation curve characterizing the affinity of 125I-αBgt to the α7-GlyR chimera-transfected HEK cells. The measured KD of 0.39 ± 0.02 nm is very close to that characterizing the toxin binding to α7 nAChR in the GH4C1 cell line (28). Previously, it was shown (3) that ws-LYNX1 did not displace 125I-αBgt from the α7 nAChR, which is also true for the α7-GlyR chimera (Fig. 2D, top inset). This chimeric receptor also bound with equal potency both α-cobratoxin and α-bungarotoxin (Fig. 2D, bottom inset). In patch clamp studies, application of 20 nm of α-cobratoxin for 90 s to the α7-GlyR chimera caused inhibition of nicotine-induced currents (Fig. 3A), similarly to what takes place upon the action of α-bungarotoxin or α-cobratoxin on the wild-type α7 nAChR or chimera composed of the α7 nAChR and subtype 3 of 5-hydroxytryptamine receptor (11, 26, 29).
The effect of ws-LYNX1 was tested on 22 CHO-K1 cells. Its application did not change the input resistance of cells estimated from leakage currents (29 ± 8 pA and 26 ± 7 pA, respectively, before and after 90–240-s applications of 10 μm ws-LYNX1). On 16 cells, application of ws-LYNX1 during 90 s caused a partially reversible decrease in the amplitude of the whole-cell currents induced by application of 30 μm nicotine. Mean amplitudes before addition of ws-LYNX1 (control) and after 90 or 180 s of treatment with 10 μm ws-LYNX1 were 674 ± 149 pA and 478 ± 102 pA (p < 0.05), respectively, i.e. mean inhibition (n = 16) was 23.9 ± 3.2% (Fig. 3B) with variations of inhibition from 10 to 50%. In three cells, the effect of ws-LYNX1 was not observed, whereas in the other four cells, responses to nicotine were non-reversibly increased from 5 to 22% (Fig. 3C). This effect might probably result from detachment of recorded cells from the surface of cover glass and increasing the surface area available for nicotine. Prolongation of 10 μm ws-LYNX1 application resulted in stronger inhibition of nicotine-induced currents (Fig. 3D). In general, the major effect of treatment with 10 μm ws-LYNX1 is the reversible inhibition of α7-GlyR, which is consistent with the earlier reported inhibition of human α7 nAChR by 10 μm ws-LYNX1 (3).
Before testing the ws-LYNX1 mutants on α7-GlyR, we examined the interaction of ws-LYNX1 with the “wild-type” α1-GlyR. ws-LYNX1 had no effect on the currents induced in this receptor (Fig. 3, E and F), indicating that in the α7-GlyR chimera, the ws-LYNX1 binding site is confined within the limits of the attached α7 extracellular domain. Therefore, although the ws-LYNX1 binding site in α7 nAChR does not coincide with the classical binding site for agonists and competitive antagonists (3), it should also be within the ligand-binding domain.
The results obtained for ws-LYNX1 mutants are presented in Figs. 4 and 5. Most mutations resulted in a strong decrease of inhibitory effect on nicotine-induced currents. For instance, the T35A mutation completely abolished the inhibitory activity (Fig. 4A, panel a) as clearly seen for all seven analyzed cells (Fig. 4A, panel b). Mutations of the neighbor residues T37A and R38A in the loop II also abrogated the inhibition (Fig. 4, C and D). Although a slight increase of the current amplitudes was detected for the R40A mutant, it was irreversible with further potentiation at washout (Fig. 4E). This effect might result from an irreversible increase of responses, as shown for some cells in the Fig. 3C (see also Fig. 3F). We suggest that the R40A mutation abolished the inhibitory activity of the ws-LYNX1. On the contrary, the P36A mutant had even higher inhibitory activity than ws-LYNX1 with mean inhibition of 32% at application for 90 s (Fig. 4B). A tendency to potentiate the nicotine-induced current was brought about by Y57A mutation (Fig. 4G) in loop III and a tendency to a weak inhibition was preserved by mutations of Tyr-54 and Lys-59 residues (Fig. 4, F and H). The summary of electrophysiology data is schematically presented in Fig. 5.
Comparison of the Experimental Results on Interactions of ws-LYNX1 Mutants with Ls-AChBP, Torpedo nAChR, and α7-GlyR
We first compared the effects of mutations in loop II (Fig. 1). The T35A mutant is close in its activity to ws-LYNX1 at Ls-AChBP and has a slightly higher inhibitory activity against Torpedo nAChR (Fig. 2, A and C). On the contrary, the T35A mutation completely abolished the inhibitory activity against α7-GlyR chimera (Fig. 4A). Thus, the Thr-35 residue is not strictly important for attaching to the classical binding sites at Ls-AChBP and Torpedo nAChR, but it is required to decrease the current amplitude in the α7-GlyR chimera.
The P36A mutation increased the affinity for Ls-AChBP (Fig. 2, A and B) but slightly decreased it toward Torpedo nAChR (Fig. 2C). Interestingly, this mutant diminished the current amplitude in the α7-GlyR chimera more strongly than ws-LYNX1 (Figs. 4B and 5). This finding reflects certain differences between the classical binding sites in Torpedo nAChR and Ls-AChBP and also shows that Pro-36 of ws-LYNX1 is involved in the interactions, both with the classical binding site (as most clearly seen for Ls-AChBP) and with a binding region in the α7-GlyR chimera. The T37A mutation does not affect the ws-LYNX1 affinity for Ls-AChBP, slightly decreases the binding activity to Torpedo nAChR (Fig. 2), and abolishes the ws-LYNX1 inhibitory activity against the α7-Gly chimera (Fig. 4C).
The removal of the positive charge in the tip of the central loop II (R38A mutation) increased the affinity for Ls-AChBP (Fig. 2, A and B), almost abolished the affinity toward Torpedo nAChR (Fig. 2C), and completely destroyed the activity against the α7-GlyR chimera (Fig. 4D). The deletion of another positive charge (K40A mutation) retained some activity against Ls-AChBP (Fig. 2A) but completely abolished both the binding to Torpedo nAChR (Fig. 2C) and inhibition of the current amplitude in the α7-GlyR chimera (Fig. 4E).
Mutations in loop III did not strongly change ws-LYNX1 binding to Ls-AChBP, but destroyed the binding activity toward Torpedo nAChR, with the exception of Y57A mutant, which preserved some activity (Fig. 2). The Y54A and K59A mutants had a very weak inhibitory activity against the α7-GlyR chimera (Fig. 4, F and H). Thus, none of the substituted residues in the loop III appears to be strictly essential for binding to the classical binding site in Ls-AChBP, but they are required for recognition both of the binding sites for agonists and competitive antagonists in the Torpedo nAChR, as well as for the still enigmatic binding center in the α7-GlyR chimera.
Comparison of Putative Binding Sites for Three-finger Proteins, ws-LYNX1, and α-Bungarotoxin, by Computer Modeling
For ws-LYNX1, together with the structure predicted previously (3), where the main contribution to binding at the classical binding site for agonists and competitive antagonists is mainly from the loop II, we found two other orientations: one with the similar role of loops II and III and another one with the predominant input from loop III. In addition, attachment of ws-LYNX at the upper part of the AChBP inner cavity appeared possible. However, docking of ws-LYNX1 to Torpedo nAChR revealed one major form with contributions from loops II and III. A minor binding mode involved the entrance to the lumen of the extracellular domain. This minor form was also found for α-bungarotoxin bound to the Torpedo nAChR. However, the dominant form of αBgt binding mode was at the classical binding site of this receptor revealed in the crystalline complexes. The results of docking ws-LYNX1 and α-bungarotoxin to α7 nAChR were in general similar to those for Torpedo nAChR with one exception: computation predicted also a binding mode where ws-LYNX1 is attached to the outer side of the extracellular domain, with the C-terminal tail (where the glycosylphosphatidylinositol anchor is attached in LYNX1) pointing to the membrane (Fig. 6). Thus, computer modeling indicates a larger variety of binding models for ws-LYNX1 than for α-bungarotoxin.
FIGURE 6.

Putative model of the complex between ws-LYNX1 and α7 nAChR obtained from computer modeling. Two adjacent receptor subunits (yellow and orange) are shown for clarity. Interactions of ws-LYNX1 (green) with the Cys- and C-loops of α7 nAChR are not shown. Legends for α7 nAChR amino acid residues are given in italic type. The C terminus of ws-LYNX1 is marked with an asterisk. The key residues involved in interactions are shown in a ball-and-stick model. The interactions geometry is not optimized because the model was obtained as a result of rigid-body docking procedure.
Analysis of ws-LYNX1 Interaction with the Y168A Mutant of α7-GlyR Chimera
Identification of functionally important residues, which interact with ws-LYNX1, is a different challenging task, especially when it concerns the still enigmatic area in the α7 nAChR. However, because for the latter, the computations gave several candidates, we decided to check one residue - Tyr-168. The mutant Y168A was tested by expressing the mutated gene in Xenopus laevis oocytes (Fig. 7). The mutation did not disrupt channel opening in response to acetylcholine application (Fig. 7A), with the EC50 value for acetylcholine even lower for the mutated receptor (40 ± 7 μm versus 170 ± 20 μm for the wild-type α7-GlyR chimera). The mutated receptor also retained the sensitivity to the inhibiting action both of α-cobratoxin and α-bungarotoxin (Fig. 7B). The IC50 values for α-cobratoxin were 58 ± 7 nm versus 12 ± 2 nm for the Y168A mutant and wild-type chimera, respectively (data not shown), which corresponds to inhibition constants 8 ± 1 nm for mutated and 5 ± 1 nm for the wild-type α7-GlyR chimera. However, for the Y168A mutant, virtually no inhibition of currents induced either by nicotine or acetylcholine was observed upon addition of ws-LYNX1 (Fig. 7C). This result indicates an important role of Tyr-168 and supports the binding mode shown in Fig. 6.
FIGURE 7.

Analysis of ws-LYNX1 interaction with the Y168A mutant of the α7-GlyR chimera expressed in Xenopus oocytes. Oocytes were injected with 2 ng of wild-type (WT) or Y168A mutant α7-GlyR chimera cDNA. After 36–48 h of incubation at 18 °C, two-electrode voltage clamp measurements were performed. In A, the dose-response curves for acetylcholine (ACh) action on wild-type (circles) and Y168A mutant (squares) α7-GlyR chimera are presented. The calculated EC50 values were 170 ± 20 μm and 40 ± 7 μm, respectively. B, superimposed traces of currents induced by rapid application of acetylcholine (500 μm) in control (before addition of αBgt or ws-LYNX1), after application of compounds (20 nm αBgt or 20 μm ws-LYNX1) and after a 5-min washout. The currents measured in oocytes expressing wild-type α7-GlyR chimera are placed on the left; expressing Y168A mutated receptor are shown on the right. C, column representation of ws-LYNX1 action on WT and Y168A mutant α7-GlyR chimera (n = 4, p = 0.05). The current amplitudes registered before application of ws-LYNX1 was taken as 100% of normalized current (control). For wild-type α7-GlyR chimera expressed in Xenopus oocytes, the mean current amplitude in the presence of ws-LYNX1 was 85 ± 8% in contrast to that of for Y168A-mutated form (106 ± 3%).
DISCUSSION
In this study, we identified several amino acid residues of ws-LYNX1 implicated in its interactions with the L. stagnalis AChBP, T. californica nAChR, or α7 nAChR. With the first two targets, the activity of mutants was assessed by radioligand analysis via competition with 125I-αBgt. For electrophysiology experiments aimed at α7 nAChR, we have chosen the α7-GlyR chimera as a model (16). Its suitability follows both from the literature data and our results, showing the sensitivity to long chain α-neurotoxins (Figs. 3A and 7B), whereas the advantage is a lack of desensitization, even at very long applications of agonist, and a slow rate of activation ensuring reliable monitoring of ionic current amplitudes. ws-LYNX1 at 10 μm diminished the current amplitude in the α7-GlyR chimera (Fig. 3, B–D) as efficiently as it did at the human α7 nAChR (3). Thus, the α7-GlyR chimera is indeed an appropriate model for analyzing the action of ws-LYNX1 mutants on α7 nAChR.
As shown recently, ws-LYNX1 competed with 125I-αBgt for binding to AChBPs and Torpedo nAChR, thus pointing to its interaction with the classical binding sites for agonists and competitive antagonists (3). It was found that while attacking these two targets, some mutants retained ws-LYNX1 competing capacity, whereas other mutants almost lost competing capacity. ws-LYNX1 did not compete with 125I-αBgt at human α7 nAChR, attaching outside this classical site (3). In the present work, we also observed no competition between ws-LYNX1 and 125I-αBgt for binding to the α7-GlyR chimera (Fig. 2D). We demonstrated that ws-LYNX1 inhibited currents in the α7-GlyR chimera but had no effect on the currents induced in the wild-type human GlyR (Fig. 3, E and F). This indicates that the extracellular loop between transmembrane fragments M2 and M3 or the transmembrane domain of the α7-GlyR chimera is not essential for binding. Thus, a putative ws-LYNX1 binding site in this chimera should lie within the grafted extracellular domain of α7 nAChR. This finding may simplify future mapping of the ws-LYNX1 binding sites in the α7 nAChR by limiting the area to search for the extracellular domain.
The adequacy of the prepared mutants for probing binding interfaces in ws-LYNX1 follows from their mass spectrometry, CD, and 1H-NMR spectra (data not shown). Other data support the fact that against one target, an individual mutant may show full activity of the starting ws-LYNX1, whereas against another target, the activity can be lost completely. Under “Results,” we compared the effects exerted by the same mutation on such three targets as Ls-AChBP, Torpedo nAChR and α7-GlyR chimera.
In our previous publication (3), we presented the arguments justifying the use of ws-LYNX1 as a model to get first information about the LYNX1 mechanism of action. When this manuscript was accepted, Miwa and Walz (30) demonstrated considerable differences in the effects of LYNX1 and water-soluble LYNX1 in mouse models but also emphasized an interest in the activity of the latter and its possible biomedical application.
Several amino acid residues of ws-LYNX1 important for its interaction with the above-mentioned targets were identified in the present work. The modes of binding to the muscle-type Torpedo nAChR and to neuronal α7 nAChR appear to have considerable differences, as some mutations exert opposite effects on the interaction with these receptors. However, certain loci of the ws-LYNX1 molecule can recognize both classical and non-classical binding sites, as seen for the P36A mutation, increasing the affinity of binding to Ls-AChBP and inducing even a greater decrease in the current amplitude at α7-GlyR chimera than measured with ws-LYNX1. In addition to shedding light on the ws-LYNX1 binding surfaces, mutations might find practical applications in future demonstrating that the inhibitory activity of ws-LYNX1 can be either increased or transformed into current-enhancing effects, as well as giving rise to analogues with a higher selectivity to a particular nAChR subtype.
It is of interest to compare our results on ws-LYNX1 with the literature data on snake toxins in an attempt to understand why toxins block the activity of nAChRs, whereas LYNX1 and other endogenous prototoxins such as SLURP-1 (31) with three-dimensional structures similar to that of toxins, exert diverse modulatory effects. Long chain α-neurotoxins such as α-bungarotoxin or α-cobratoxin (containing an additional disulfide in the loop II and a long C-terminal tail) block muscle-type and α7 nAChRs with nanomolar Ki values; short chain α-neurotoxins lack that additional disulfide and block with similar affinity only the muscle-type nAChRs (see Refs. 11 and 13). For both of these groups of toxins, the major role in binding belongs to the loop II (especially to the positively charged and aromatic residues at its tip), whereas loop III appears to participate in binding only in the case of short α-neurotoxins (11, 13–15, 29, 32–34). Thus, the first major distinction in the type of action between α-neurotoxins and ws-LYNX1 is the following: neurotoxins bind with nanomolar affinity and almost irreversibly (could not be washed out completely within an hour), whereas binding of ws-LYNX1 and its mutants is characterized by micromolar affinity and reversibility (can be washed out in minutes). In addition, for ws-LYNX1 interaction with the Torpedo and α7 nAChRs, both loops II and III are important and, in this respect, ws-LYNX1 is closer to short chain neurotoxins.
ws-LYNX1 binding to different targets can be also compared with the interaction of nAChRs with the so-called “non-conventional toxins.” A common structural feature of this group and ws-LYNX1 (see Fig. 8) is the disposition of the additional fifth disulfide not in loop II but in the N-terminal loop I (see Refs. 11 and 33). One such toxin, candoxin from Bungarus candidus, blocks both the muscle and α7 nAChRs with nanomolar affinity (35), whereas another non-conventional toxin, weak toxin from the cobra Naja kaouthia, acts on these receptors only with micromolar affinity (36). Thus, ws-LYNX1 in terms of affinities for nAChRs is close to weak toxin, but this toxin still shows virtual irreversibility of action (36). Interestingly, a common feature of ws-LYNX1 and weak toxin is their action on muscarinic acetylcholine receptors (3, 37).
FIGURE 8.
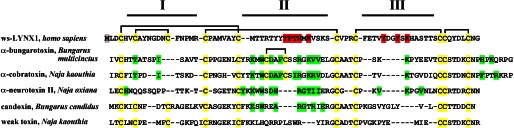
Alignment of the amino acid sequences of ws-LYNX1, long chain α-neurotoxins (α-bungarotoxin and α-cobratoxin), short chain α-neurotoxin (neurotoxin II), and non-conventional neurotoxins (candoxin and weak toxin). The rectangles show the disposition of the disulfide bridges, and the Cys residues are colored in yellow; I–III designate the respective disulfide loops in the three-finger structure of these proteins. The residues mutated in ws-LYNX1 are highlighted in red; the artificially introduced Met1 residue is colored in gray. Green marks the residues of the proteins that participate in interaction with nAChRs according to literature data.
Possible distinctions in the binding modes between ws-LYNX1 and snake venom α-neurotoxins were revealed by computer modeling of the respective complexes (see Fig. 6). We used docking program ZDOCK (version 3.0.2) (19), which utilizes a rigid body-docking algorithm and shows good results in the protein-protein complex predictions (38, 39). For αBgt complexes with Ls-AChBP, Torpedo, and α7 nAChRs, it predicted one major binding mode at the classical binding site for agonists and competitive antagonists, with dominant contributions coming from αBgt central loop II, in accordance with relevant x-ray structures (14, 15). In contrast, for the ws-LYNX1-AChBP complex, three orientations at the classical binding site appear possible, with one of them with the dominant role of loop III. In binding to the Torpedo nAChR classical site, both loops II and III of ws-LYNX1 appear to be important. Surprisingly, computer modeling with the above two targets and with α7 nAChR also predicts for ws-LYNX1 such modes where it touches the upper part of the inner cavity in AChBP and a similar region of the nAChR extracellular domains, whereas for αBgt, in all three cases, the probability of such modes is very low. The most interesting, that only in the case of α7 nAChR, the computations generated a structure where ws-LYNX1 is in contact with the extracellular domain outer surface as well as with both C-loop and Cys-loop (Fig. 6). Such an interaction mode leaves the classical binding site for agonists and competitive antagonists of α7 nAChR unoccupied. It seems that among α7 nAChR, amino acid residues interacting with ws-LYNX1 are Tyr-168 and Arg-205. The first residue forms stacking interactions with Tyr-34 from ws-LYNX1, which are stabilized by ion-paired Asp-175 (α7 nAChR) and Lys-40 (ws-LYNX1). Thr-35 and Thr-37 of ws-LYNX1 form a bidentate ion pair with Arg-205. The ws-LYNX1 C terminus is close to phospholipid bilayer, which would have been present in the whole-size receptor. From available x-ray structures, it is known that α-neurotoxins are attached approximately in the middle of the extracellular domains (14, 15), nevertheless, proposed ws-LYNX1 disposition does not seem unrealistic. αBgt was quite close to a bilayer in the low-resolution structure of its complex with Torpedo nAChR (40) and the conclusion about α-neurotoxin II binding to phospholipid bilayer through its disulfide-containing core was made in (34, 41).
The goal of the presented work was to pinpoint the amino acid residues in ws-LYNX1 important for its recognition of different targets. Identification of the active centers in the receptors themselves, especially of the non-classical site in the α7 nAChR, is another important task. Fig. 7 shows that the Y168A mutation, although it does not decrease channel activity, abolishes the ws-LYNX1 capacity to diminish the current amplitude. At present, there are not enough experimental data to give preference to any mode of ws-LYNX1 binding, but this result at least gives support to the model presented in Fig. 6.
In summary, we prepared a series of ws-LYNX1 mutants bearing substitutions in the loops II and III and analyzed their interaction with Ls-AChBP and Torpedo nAChR, where binding occurs at the classical binding sites for agonists/competitive antagonists, as well as with α7-GlyR chimera where ws-LYNX1 is attaching outside of this site. Some of the mutated ws-LYNX1 residues were found to be important to interact with all above-mentioned targets. The other residues are needed for binding the distinct targets. Several mutations completely suppressed the activity, whereas other mutations either increased the inhibitory activity or slightly enhanced the current amplitudes. A comparison with snake toxins indicates that one of the main reasons for different types of activity observed with LYNX1 and α-neurotoxins may be a much tighter binding of the latter resulting in potent inhibitory action. However, our findings demonstrated for ws-LYNX1 much less tight association and more diverse binding modes with different targets. Similar properties of LYNX1 and its congeners may underlie a wider spectrum of their effects, from inhibition of various nAChRs to potentiating their diverse activities.
Acknowledgments
We are grateful to Professors T. Sixma and A. Smit for the samples of acetylcholine binding protein, F. Hucho for membrane-bound nicotinic receptor from T. californica, as well as to Dr. L. Prado de Carvalho for cDNA of α7-GlyR chimera.
This work was supported by the Program “Molecular and Cellular Biology” of the Presidium of Russian Academy of Sciences, Russian Foundation for Basic Research Grants 09-04-01567, 11-04-0111, 11-04-12133, 12-04-01639, and 12-04-01746, Ministry of Education and Science of Russia Project 8268, President of the Russian Federation Grant NSh5597.2012.4, and NeuroCypres EU 7th Framework Programme Grant HEALTH-F4-2008-202088.
- nAChR
- nicotinic acetylcholine receptor
- ws-LYNX1
- water-soluble LYNX1
- AChBP
- acetylcholine-binding protein
- Ls-AChBP
- Lymnaea stagnalis AChBP
- αBgt
- α-bungarotoxin
- α7-GlyR
- chimeric α7 nAChR-glycine receptor
- PDB
- Protein Data Bank.
REFERENCES
- 1. Miwa J. M., Ibanez-Tallon I., Crabtree G. W., Sánchez R., Sali A., Role L. W., Heintz N. (1999) lynx1, an endogenous toxin-like modulator of nicotinic acetylcholine receptors in the mammalian CNS. Neuron 23, 105–114 [DOI] [PubMed] [Google Scholar]
- 2. Miwa J. M., Freedman R., Lester H. A. (2011) Neural systems governed by nicotinic acetylcholine receptors: emerging hypotheses. Neuron 70, 20–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lyukmanova E. N., Shenkarev Z. O., Shulepko M. A., Mineev K. S., D'Hoedt D., Kasheverov I. E., Filkin S. Y., Krivolapova A. P., Janickova H., Dolezal V., Dolgikh D. A., Arseniev A. S., Bertrand D., Tsetlin V. I., Kirpichnikov M. P. (2011) NMR structure and action on nicotinic acetylcholine receptors of water-soluble domain of human LYNX1. J. Biol. Chem. 286, 10618–10627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ibañez-Tallon I., Miwa J. M., Wang H. L., Adams N. C., Crabtree G. W., Sine S. M., Heintz N. (2002) Novel modulation of neuronal nicotinic acetylcholine receptors by association with the endogenous prototoxin lynx1. Neuron 33, 893–903 [DOI] [PubMed] [Google Scholar]
- 5. Tekinay A. B., Nong Y., Miwa J. M., Lieberam I., Ibanez-Tallon I., Greengard P., Heintz N. (2009) A role for LYNX2 in anxiety-related behavior. Proc. Natl. Acad. Sci. U.S.A. 106, 4477–4482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miwa J. M., Stevens T. R., King S. L., Caldarone B. J., Ibanez-Tallon I., Xiao C., Fitzsimonds R. M., Pavlides C., Lester H. A., Picciotto M. R., Heintz N. (2006) The prototoxin lynx1 acts on nicotinic acetylcholine receptors to balance neuronal activity and survival in vivo. Neuron 51, 587–600 [DOI] [PubMed] [Google Scholar]
- 7. Morishita H., Miwa J. M., Heintz N., Hensch T. K. (2010) Lynx1, a cholinergic brake, limits plasticity in adult visual cortex. Science 330, 1238–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Choo Y. M., Lee B. H., Lee K. S., Kim B. Y., Li J., Kim J. G., Lee J. H., Sohn H. D., Nah S. Y., Jin B. R. (2008) Pr-lynx1, a modulator of nicotinic acetylcholine receptors in the insect. Mol. Cell. Neurosci. 38, 224–235 [DOI] [PubMed] [Google Scholar]
- 9. Hruska M., Keefe J., Wert D., Tekinay A. B., Hulce J. J., Ibañez-Tallon I., Nishi R. (2009) Prostate stem cell antigen is an endogenous lynx1-like prototoxin that antagonizes α7-containing nicotinic receptors and prevents programmed cell death of parasympathetic neurons. J. Neurosci. 29, 14847–14854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fu X. W., Rekow S. S., Spindel E. R. (2012) The ly-6 protein, lynx1, is an endogenous inhibitor of nicotinic signaling in airway epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 303, L661–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tsetlin V. I., Hucho F. (2004) Snake and snail toxins acting on nicotinic acetylcholine receptors: fundamental aspects and medical applications. FEBS Lett. 557, 9–13 [DOI] [PubMed] [Google Scholar]
- 12. Karlin A. (2002) Emerging structure of the nicotinic acetylcholine receptors. Nat. Rev. Neurosci. 3, 102–114 [DOI] [PubMed] [Google Scholar]
- 13. Tsetlin V., Utkin Y., Kasheverov I. (2009) Polypeptide and peptide toxins, magnifying lenses for binding sites in nicotinic acetylcholine receptors. Biochem. Pharmacol. 78, 720–731 [DOI] [PubMed] [Google Scholar]
- 14. Bourne Y., Talley T. T., Hansen S. B., Taylor P., Marchot P. (2005) Crystal structure of a Cbtx-AChBP complex reveals essential interactions between snake α-neurotoxins and nicotinic receptors. EMBO J. 24, 1512–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dellisanti C. D., Yao Y., Stroud J. C., Wang Z. Z., Chen L. (2007) Crystal structure of the extracellular domain of nAChR α1 bound to α-bungarotoxin at 1.94 A resolution. Nat. Neurosci. 10, 953–962 [DOI] [PubMed] [Google Scholar]
- 16. Grutter T., Prado de Carvalho L., Virginie D., Taly A., Fischer M., Changeux J. P. (2005) A chimera encoding the fusion of an acetylcholine-binding protein to an ion channel is stabilized in a state close to the desensitized form of ligand-gated ion channels. C. R. Biol. 328, 223–234 [DOI] [PubMed] [Google Scholar]
- 17. Shulepko M. A., Lyukmanova E. N., Kasheverov I. E., Dolgikh D. A., Tsetlin V. I., Kirpichnikov M. P. (2011) Bacterial expression of the water-soluble domain of Lynx1, an endogenous neuromodulator of human nicotinic receptors. Russian J. Bioorg. Chem. 37, 543–549 [DOI] [PubMed] [Google Scholar]
- 18. Waseem T., Mukhtarov M., Buldakova S., Medina I., Bregestovski P. (2010) Genetically encoded Cl-Sensor as a tool for monitoring of Cl-dependent processes in small neuronal compartments. J. Neurosci. Methods 193, 14–23 [DOI] [PubMed] [Google Scholar]
- 19. Chen R., Li L., Weng Z. (2003) ZDOCK: an initial-stage protein-docking algorithm. Proteins 52, 80–87 [DOI] [PubMed] [Google Scholar]
- 20. Unwin N. (2005) Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J. Mol. Biol. 346, 967–989 [DOI] [PubMed] [Google Scholar]
- 21. Nemecz A., Taylor P. (2011) Creating an α7 nicotinic acetylcholine recognition domain from the acetylcholine-binding protein: crystallographic and ligand selectivity analyses. J. Biol. Chem. 286, 42555–42565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sali A., Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 23. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.4.1, Schrödinger, LLC, New York [Google Scholar]
- 24. Papke R. L., Porter Papke J. K. (2002) Comparative pharmacology of rat and human α7 nAChR conducted with net charge analysis. Br. J. Pharmacol. 137, 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Friis S., Mathes C., Sunesen M., Bowlby M. R., Dunlop J. (2009) Characterization of compounds on nicotinic acetylcholine receptor α7 channels using higher throughput electrophysiology. J. Neurosci. Methods 177, 142–148 [DOI] [PubMed] [Google Scholar]
- 26. Eiselé J. L., Bertrand S., Galzi J. L., Devillers-Thiéry A., Changeux J. P., Bertrand D. (1993) Chimaeric nicotinic-serotonergic receptor combines distinct ligand binding and channel specificities. Nature 366, 479–483 [DOI] [PubMed] [Google Scholar]
- 27. Rayes D., Spitzmaul G., Sine S. M., Bouzat C. (2005) Single-channel kinetic analysis of chimeric α7–5HT3A receptors. Mol. Pharmacol. 68, 1475–1483 [DOI] [PubMed] [Google Scholar]
- 28. Kasheverov I. E., Zhmak M. N., Fish A., Rucktooa P., Khruschov A. Y., Osipov A. V., Ziganshin R. H., D'hoedt D., Bertrand D., Sixma T. K., Smit A. B., Tsetlin V. I. (2009) Interaction of α-conotoxin ImII and its analogs with nicotinic receptors and acetylcholine-binding proteins: additional binding sites on Torpedo receptor. J. Neurochem. 111, 934–944 [DOI] [PubMed] [Google Scholar]
- 29. Fruchart-Gaillard C., Gilquin B., Antil-Delbeke S., Le Novère N., Tamiya T., Corringer P. J., Changeux J. P., Ménez A., Servent D. (2002) Experimentally based model of a complex between a snake toxin and the α7 nicotinic receptor. Proc. Natl. Acad. Sci. U.S.A. 99, 3216–3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miwa J. M., Walz A. (2012) Enhancement in motor learning through genetic manipulation of the Lynx1 gene. PLoS One 7, e43302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shulepko M. A., Lyukmanova E. N., Paramonov A. S., Lobas A. A., Shenkarev Z. O., Kasheverov I. E., Dolgikh D. A., Tsetlin V. I., Arseniev A. S., Kirpichnikov M. P. (2013) Human Neuromodulator SLURP-1: Bacterial Expression, Binding to Muscle-Type Nicotinic Acetylcholine Receptor, Secondary Structure, and Conformational Heterogeneity in Solution. Biochemistry 78, 204–211 [DOI] [PubMed] [Google Scholar]
- 32. Trémeau O., Lemaire C., Drevet P., Pinkasfeld S., Ducancel F., Boulain J. C., Ménez A. (1995) Genetic engineering of snake toxins. The functional site of Erabutoxin a, as delineated by site-directed mutagenesis, includes variant residues. .J. Biol. Chem. 270, 9362–9369 [DOI] [PubMed] [Google Scholar]
- 33. Kini R. M., Doley R. (2010) Structure, function and evolution of three-finger toxins: mini proteins with multiple targets. Toxicon 56, 855–867 [DOI] [PubMed] [Google Scholar]
- 34. Krabben L., van Rossum B. J., Jehle S., Bocharov E., Lyukmanova E. N., Schulga A. A., Arseniev A., Hucho F., Oschkinat H. (2009) Loop 3 of short neurotoxin II is an additional interaction site with membrane-bound nicotinic acetylcholine receptor as detected by solid-state NMR spectroscopy. J. Mol. Biol. 390, 662–671 [DOI] [PubMed] [Google Scholar]
- 35. Nirthanan S., Charpantier E., Gopalakrishnakone P., Gwee M. C., Khoo H. E., Cheah L. S., Bertrand D., Kini R. M. (2002) Candoxin, a novel toxin from Bungarus candidus, is a reversible antagonist of muscle (αβγδ) but a poorly reversible antagonist of neuronal α7 nicotinic acetylcholine receptors. J. Biol. Chem. 277, 17811–17820 [DOI] [PubMed] [Google Scholar]
- 36. Utkin Y. N., Kukhtina V. V., Kryukova E. V., Chiodini F., Bertrand D., Methfessel C., Tsetlin V. I. (2001) “Weak toxin” from Naja kaouthia is a nontoxic antagonist of α7 and muscle-type nicotinic acetylcholine receptors. J. Biol. Chem. 276, 15810–15815 [DOI] [PubMed] [Google Scholar]
- 37. Mordvintsev D. Y., Polyak Y. L., Rodionov D. I., Jakubik J., Dolezal V., Karlsson E., Tsetlin V. I., Utkin Y. N. (2009) Weak toxin WTX from Naja kaouthia cobra venom interacts with both nicotinic and muscarinic acetylcholine receptors. FEBS J. 276, 5065–5075 [DOI] [PubMed] [Google Scholar]
- 38. Mukherjee S., Zhang Y. (2011) Protein-protein complex structure predictions by multimeric threading and template recombination. Structure 19, 955–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wiehe K., Pierce B., Tong W. W., Hwang H., Mintseris J., Weng Z. (2007) The performance of ZDOCK and ZRANK in rounds 6–11 of CAPRI. Proteins 69, 719–725 [DOI] [PubMed] [Google Scholar]
- 40. Young H. S., Herbette L. G., Skita V. (2003) α-bungarotoxin binding to acetylcholine receptor membranes studied by low angle X-ray diffraction. Biophys. J. 85, 943–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lesovoy D. M., Bocharov E. V., Lyukmanova E. N., Kosinsky Y. A., Shulepko M. A., Dolgikh D. A., Kirpichnikov M. P., Efremov R. G., Arseniev A. S. (2009) Specific membrane binding of neurotoxin II can facilitate its delivery to acetylcholine receptor. Biophys. J. 97, 2089–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]