

Background: Cdc37 is a kinase-specific co-chaperone inhibiting the ATPase activity of Hsp90.
Results: Caenorhabditis elegans Cdc37 binds to the M-domain of Hsp90, exposing a novel functional interaction site.
Conclusion: Inhibition of Hsp90 by Cdc37 is caused by conformational restriction of the N-M domain motility.
Significance: Understanding the Cdc37-Hsp90 interaction improves also the knowledge on kinase activation, the main reason for the focus on Hsp90 in cancer research.
Keywords: Analytical Ultracentrifugation, Molecular Chaperone, Nucleotide, Protein Conformation, Protein-Protein Interactions, Co-chaperone
Abstract
The ATPase-driven dimeric molecular Hsp90 (heat shock protein 90) and its cofactor Cdc37 (cell division cycle 37 protein) are crucial to prevent the cellular depletion of many protein kinases. In complex with Hsp90, Cdc37 is thought to bind an important lid structure in the ATPase domain of Hsp90 and inhibit ATP turnover by Hsp90. As different interaction modes have been reported, we were interested in the interaction mechanism of Hsp90 and Cdc37. We find that Cdc37 can bind to one subunit of the Hsp90 dimer. The inhibition of the ATPase activity is caused by a reduction in the closing rate of Hsp90 without obviously bridging the two subunits or affecting nucleotide accessibility to the binding site. Although human Cdc37 binds to the N-terminal domain of Hsp90, nematodal Cdc37 preferentially interacts with the middle domain of CeHsp90 and hHsp90, exposing two Cdc37 interaction sites. A previously unreported site in CeCdc37 is utilized for the middle domain interaction. Dephosphorylation of CeCdc37 by the Hsp90-associated phosphatase PPH-5, a step required during the kinase activation process, proceeds normally, even if only the new interaction site is used. This shows that the second interaction site is also functionally relevant and highlights that Cdc37, similar to the Hsp90 cofactors Sti1 and Aha1, may utilize two different attachment sites to restrict the conformational freedom and the ATP turnover of Hsp90.
Introduction
Molecular chaperones, as the highly conserved Hsp90 (heat shock protein 90), are responsible for protein folding, cell signaling, and the assembly of multiprotein complexes (1, 2). More than one hundred cellular proteins were identified as clients of Hsp90 ((3) and the laboratory of Didier Picard). Hsp90 clients originate from distinct protein classes: transcription factors such as p53 or steroid hormone receptors and kinases such as Src, Raf, and many others (4–7). The activation and stabilization of these client proteins makes Hsp90 itself an interesting drug target for the treatment of cancer and other diseases, in which Hsp90 clients are involved.
In the cytoplasm of vertebrates, two distinct isoforms of Hsp90 exist: an inducible form (Hsp90α) and a constitutively expressed form (Hsp90β) (8, 9). The same is true for Saccharomyces cerevisiae (S. cerevisiae) encoding the isoforms Hsc82 and Hsp82. Caenorhabditis elegans (C. elegans) instead expresses only one gene for cytosolic Hsp90 (DAF-21) (10). All Hsp90 proteins, from bacteria to mammals, share a common domain organization and are able to hydrolyze ATP after distinct conformational changes (9, 11). Each subunit of the Hsp90 dimer consists of an N-domain, an M-domain, and a C-terminal domain. The N-domain contains the nucleotide binding site and is connected by a long linker sequence to the M-domain. The C terminus is the dimerization site of Hsp90. The chaperone itself is a very slow ATPase, with yeast Hsp90 hydrolyzing one unit of ATP/min (12) and human Hsp90 hydrolyzing only 0.05 units of ATP/min (13). Nevertheless, the ATPase cycle is conserved from HtpG of Escherichia coli to human Hsp90 (9, 14). ATP binding leads to rearrangements of the three domains from an open V-shaped conformation to a closed conformation. At the beginning of this process, a short segment of the N-domain, the ATP lid, flaps over the ATP binding pocket to initiate the assembly of the catalytic center (15). The ATPase activity of Hsp90 and client binding is affected by co-chaperones. These cofactors can be grouped in tetratricopeptide repeat domain containing co-chaperones such as Sti1 and protein phosphatase 5 (PPH-5)2 and non-tetratricopeptide repeat containing co-chaperones such as p23/Sba1, Aha1, and Cdc37.
Cdc37 is a kinase-specific co-chaperone, which is required to assemble stable Hsp90-kinase complexes. Cdc37 itself becomes phosphorylated at Ser-14 during kinase turnover by casein kinase 2α (16–18). Additionally, Cdc37 is inhibiting the ATPase activity of Hsp90 (19). It is known that the N-terminal part of Cdc37 is responsible for kinase binding and the middle domain for Hsp90 interaction, whereas the C-terminal part is of unknown function (20). A binding site between yeast Hsp90 and human Cdc37 could be identified based on a crystal structure of a truncation mutant of human Cdc37 (hCdc37, amino acids 138–378) and the N-terminal domain of yeast Hsp90 (21). Further contacts between other domains of both proteins remain elusive. Surprisingly, however, yeast Cdc37 does not bind yeast Hsp90 in the normal concentration range (19, 21), implying that certain features of the interaction are only weakly conserved.
Currently, different mechanisms for the inhibitory function of Cdc37 toward Hsp90 are discussed. On the one hand, it is assumed that Cdc37 positions itself in between the two Hsp90 subunits and prevents the closing of the N-terminal domains, leading to disruption of ATP turnover (21, 22). On the other hand, there are hints that Cdc37 is located at the outer surface of Hsp90 (23). To get a more detailed view of the interaction site of Cdc37 and the conformational rearrangements upon Hsp90 binding, we investigated these questions for Cdc37 proteins from nematodes and humans.
EXPERIMENTAL PROCEDURES
Cloning, Protein Expression, and Purification
The C. elegans homolog proteins of Cdc37 (CDC-37, W08F4.8), Hsp90 (DAF-21, C47E8.5), and all of their truncation mutants, as well as Aha1 (C01G10.8) and PPH-5 (Y39B6A.2) were obtained as described previously (24, 25). The same is true for human Hsp90 and its variants (9), Aha1, Cdc37, and its variants and for yeast Hsp90 (26). All proteins were cloned in an E. coli expression vector and purified with a His6 tag. Transformed BL21-CodonPlus (DE3)RIL cells were grown to an A600 of 0.8, and expression was induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside. Thereafter, the cells were harvested and lysed using the TS 0.75 cell disruption instrument (Constant Systems, Ltd., Northants, UK). The His6-tagged proteins were then purified with three columns. Initially proteins were applied onto a HisTrap 5-ml column (GE Healthcare), and eluted with 300 mm imidazole. ResourceQ ion exchange chromatography was used as a second purification step and size exclusion chromatography was used as a third step on either a Superdex 75 or 200 HiLoad column (both GE Healthcare) were performed. The proteins were then dialyzed against their storage buffer (40 mm HEPES/KOH, pH 7.5, 20 mm KCl, 1 mm DTT). Concentrations of frozen protein aliquots were in between 50 and 300 μm.
ATPase Activity Assay of Hsp90
The ATPase activity of Hsp90 in absence and presence of co-chaperones was measured with an ATP regenerating system described previously (12). The ATPase premix contains phosphoenolpyruvate, NADH, l-lactate dehydrogenase, and pyruvate kinase (Roche Applied Science). Measurements were carried out in standard buffer (40 mm HEPES/KOH, pH 7.5, 20 mm KCl) and 5 mm MgCl2 at 30 °C. The Hsp90 concentration was 3 μm in the reaction mixture, whereas the concentration of the added co-chaperones was 15 μm. To detect background activities, the specific Hsp90 inhibitor radicicol (Sigma-Aldrich) was added toward the end of the measurements, and the remaining background was subtracted. The specific ATPase activity of Hsp90 was calculated as described previously using the Origin software (OriginLab) and Equation 1.
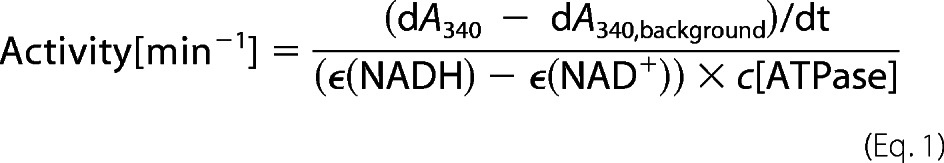 |
Analytical Ultracentrifugation with Labeled Cdc37
Human and nematodal Cdc37 were labeled with Alexa Fluor 488 C5-maleimide (Invitrogen). Label was added in 3-fold molar excess to 1.0 mg/ml protein in a buffer containing 40 mm HEPES/KOH, pH 7.5, and 20 mm KCl. After an incubation time of 1 h at 20 °C, DTT was added to a final concentration of 20 mm to stop the reaction. Free label was separated from the labeled protein using a Superdex 75HR column (GE Healthcare). The labeling efficiency and concentration of the labeled protein were determined with the following equation. A280 is the absorbance of the protein at 280 nm, Amax is the absorbance of the dye at its absorption maximum, CF280 (the correction factor) is 0.11, and ϵdye is 71000 m−1 cm−1 according to the manufacturer.
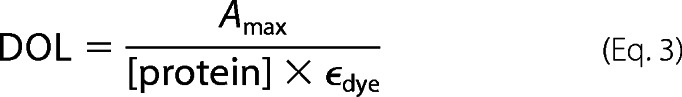 |
Measurements were performed in a Beckman ProteomeLab XL-A with a fluorescence detection system (Aviv Biomedical, Lakewood, NY) and a Ti-50 rotor (Beckman-Coulter) at 20 °C and 42,000 rpm. Labeled Cdc37 was applied at a concentration of 500 nm in combination with unlabeled putative binding partners (3 to 10 μm) and if applicable with nucleotide (4 mm). Evaluations were done using dc/dt analysis as described (27). To obtain the s20,w values, the plots were fitted with Gaussian or bi-Gaussian functions.
Fluorescence Resonance Energy Transfer Measurements
FRET measurements were performed as described elsewhere (26). We used N-terminal labeled yeast Hsp90 in which an Asp at position 61 was mutated to Cys. As an acceptor, the fluorescent dye ATTO-550-maleimide (ATTO-Tec GmbH, Siegen, Germany) and as a donor ATTO-488-maleimide was coupled to yHsp90. Analyses were carried out in a 40 mm HEPES/KOH, pH 7.5, 20 mm KCl, and 5 mm MgCl2 buffer at 30 °C in a Fluoromax 3 fluorescence spectrophotometer (Horiba JobinYvon, Kyoto, Japan). Fluorescently labeled proteins were applied at 200 nm each, and kinetics were measured in absence and presence of hCdc37. In case of the subunit exchange the hCdc37 concentration was 10 μm. Subunit exchange was initiated by adding a 10-fold excess of unlabeled yeast Hsp90. The N-terminal closing reaction of Hsp90 was initiated with 2 mm ATPγS and the closing rate was measured in the presence of 3, 6, and 10 μm of the co-chaperone.
Kinetics of Nucleotide Binding
Stopped flow measurements were performed with a Biologic SFM-400 instrument (BioLogic Science Instruments, Claix, France) and the fluorescent nucleotide analog adenosinetriphospho-γ-(N′-methylanthraniloylaminobutyl)-phosphoramidate (PγMABA-ATP) (28). The binding of nucleotide to Hsp90s was measured with variable PγMABA-ATP concentrations at an excitation wavelength of 362 nm and detection of emission with a Photomultiplier tube and long-pass filter with 400 nm cut off (400FG03-25, LOT Oriel Group). The sample reservoirs and detection cell were thermostatted to an average temperature of 21.8 ± 0.3 °C. The protein concentrations in the reservoir were 6 μm Hsp90 and 21 μm Cdc37. These solutions were diluted 3-fold upon mixing with various concentrations of PγMABA-ATP. The resulting time traces were analyzed with a single exponential equation with the Program Grafit (version 7.0; Erathicus, Ltd.), and the resulting observed rate constants (λ) were plotted versus the nucleotide concentration. The resulting straight line indicates the rate constant for association (kon) from the slope and rate constant for dissociation (koff) from the intercept with the y axis (28). The resulting dissociation constant KD,calc could be calculated from koff/kon.
Radioactivity Dephosphorylation Assay
Radioactive phosphorylated proteins were obtained as described previously (15). CeCdc37 was phosphorylated with [γ-32P]ATP using 0.1 milliunits of casein kinase 2α at 30 °C for 180 min. Remaining ATP was hydrolyzed by addition of apyrase for 30 min at 30 °C. Phosphorylated CeCdc37 was then incubated with 3 μm of either CeHsp90, CeHsp90-MC, CeHsp90-C or CeHsp90-ΔMEEVD and 2 μm PPH-5 at 20 °C. After 10 and 60 min, samples were taken, and dephosphorylation was stopped by addition of Laemmli buffer (29) and boiling at 95 °C for 5 min. The relative degree of phosphorylation of CeCdc37 was analyzed by SDS-PAGE followed by phosphoimaging on a Typhoon 9200 Phosphoimager (Amersham Biosciences, Freiburg, Germany).
RESULTS
Hsp90-hCdc37 Complexes Show Conserved ATPase Inhibition
We had previously investigated the interaction between C. elegans Hsp90 (CeHsp90) and its endogenous co-chaperone CeCdc37 (25). There, we had observed an interaction of CeCdc37 with the open and closed conformation of Hsp90, which deviates from interaction modes observed for other eukaryotic Hsp90 systems. We thus aimed at gaining more information on the interaction sites between Hsp90 and Cdc37 also in the human Hsp90 system (hHsp90 and hCdc37, respectively).
Initially, we checked whether there is a cross-species reactivity between human, C. elegans, and yeast proteins, which can be expected based on the high homology of Hsp90s. Therefore, we labeled hCdc37 with Alexa Fluor 488 (*hCdc37) and subjected it to analytical ultracentrifugation (aUC) in the absence and presence of Hsp90 of these three organisms. Indeed we could observe a similar interaction in all three cases (Fig. 1A), implying that the homology between eukaryotic Hsp90 is sufficiently high to interact in a conserved manner.
FIGURE 1.

hCdc37 binds to Hsp90 from different species and inhibits their ATPase activity. A, individual scans of a sedimentation velocity experiment of 500 nm *hCdc37 alone (black circle) and in the presence of 3 μm of Hsp90 from human (red triangle), yeast (blue square), and C. elegans (green rhombus) in 40 mm HEPES/KOH, pH 7.5, 20 mm KCl (standard buffer) were monitored by fluorescence detection at 42,000 rpm. B, ATPase activity assays of 3 μm yeast and C. elegans Hsp90 were measured alone and in combination with 10 μm hCdc37 in standard buffer plus 5 mm MgCl2 at 30 °C.
We further tested whether the ATPase activity of the non-human Hsp90s is inhibited by hCdc37. To this end, the Hsp90 proteins from C. elegans and yeast were used, and changes of their activity were recorded in the presence of hCdc37 (Fig. 1B). As reported previously (19, 21), hCdc37 suppressed the ATPase activity of yHsp90, but additionally, it could also suppress the ATPase activity of CeHsp90. These findings point out that Hsp90 proteins are sufficiently conserved to interact with Cdc37 from different species.
hCdc37 Influences Conformational Changes of Hsp90
Having shown that ATP turnover is strongly affected in the presence of hCdc37, we aimed at further defining the mode of ATPase inhibition in Hsp90-hCdc37 complexes. Thus, we applied a previously established FRET system, in which yHsp90 is labeled at its N-terminal domain with an acceptor and donor dye (26, 30). Using this FRET system, we initially tested whether hCdc37 has an influence on the subunit exchange of yHsp90. Therefore, we mixed ATTO-488 and ATTO-550 labeled yHsp90 and incubated until the fluorescence signal reached equilibrium. Then, unlabeled Hsp90 was added to disrupt the preformed FRET complexes in presence and absence of saturating amounts of hCdc37. The kinetics of the increase of the donor fluorescence were recorded (Fig. 2A). The rate constants of both kinetics are very similar, indicating that the subunit exchange is not altered by the presence of hCdc37. Thus, in contrast to the cofactors Aha1, p23, and Sti1 (30, 31), hCdc37 does not utilize both sides of the Hsp90 dimer but binds to each subunit independently. Next, we wanted to know whether the rate of the N-terminal closing reaction induced by ATPγS is affected (Fig. 2B). Indeed, the presence of 3, 6, or 10 μm hCdc37 leads to an observable reduction in the closing reaction implying that the co-chaperone hinders the two N-terminal domains to dimerize in response to ATPγS.
FIGURE 2.

Mechanism of interaction of hCdc37 with Hsp90. A and B show the influence of hCdc37 on an established yHsp90 FRET system (26). In A, kinetics of the subunit exchange after adding 4 μm unlabelded yHsp90 are presented. The donor channel fluorescence in the absence of hCdc37 is shown in black (traces and fit) and in its presence is shown in red. B represents the influence of hCdc37 on the closing rate of yHsp90 upon addition of 2 mm ATPγS. Acceptor channel fluorescence without the co-chaperone is shown in black and in combination with 3 μm (red), 6 μm (blue), and 10 μm hCdc37 (magenta).
Cdc37s Do Not Influence the Nucleotide Accessibility
One explanation for the reduced closing rate could be that the bound hCdc37 restricts access of nucleotide to the binding pocket of Hsp90. We thus used stopped-flow experiments to determine the binding parameters for Hsp90s and preformed Cdc37-Hsp90 complexes. Using MABA-labeled ATP (Fig. 3A), the kon and koff rates of Hsp90 were obtained in the presence and absence of Cdc37. In all tested combinations of Cdc37 and Hsp90 (hCdc37 + hHsp90, hCdc37 + yHsp90, CeCdc37 + CeHsp90), binding of MABA-ATP to Hsp90 is fast, regardless of the presence of the co-chaperone (Table 1). This shows that the nucleotide binding pocket is still fully accessible in the Cdc37-Hsp90 complex.
FIGURE 3.
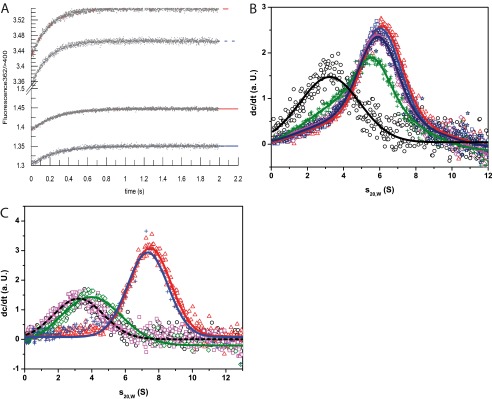
Nucleotide binding to Hsp90 in presence of Cdc37. A shows the recorded kinetics of 2 μm CeHsp90 in absence (blue) and presence of 7 μm CeCdc37 (red) after addition of 7.5 μm MABA-ATP (solid lines) or 15 μm MABA-ATP (dashed lines). B, sedimentation velocity experiment in standard assay buffer and 5 mm MgCl2 with 500 nm *hCdc37 (black circle) and in complex with 3 μm yHsp90 alone (red triangle) and rising concentrations of ATPγS (blue square, 0.5 mm; magenta rhombus, 1 mm; green cross, 4.0 mm) and 4 mm ADP (navy blue star) to define the binding ability were measured. C, the binding ability of hHsp90 fragments toward hCdc37 was tested using sedimentation velocity experiments. 500 nm *hCdc37 either alone (black circle) or after addition of 3 μm of hHsp90 (red triangle), hHsp90-lidless (magenta square), or hHsp90-N (green rhombus) were monitored with fluorescence detection in standard assay buffer at 42,000 rpm. Additionally, a competition experiment was performed in which 12 μm hHsp90-N was added to a preformed complex out of 500 nm *hCdc37 and 3 μm hHsp90 (blue cross).
TABLE 1.
Nucleotide binding ability of Hsp90 in presence of Cdc37
Based on stopped flow measurements, in which PγMABA-ATP was added to Hsp90 alone or in complex with Cdc37, the kon, koff, and KD values of the binding reaction were determined.
| kon | koff | KpD,calc | |
|---|---|---|---|
| s−1 μm−1 | s−1 | μm | |
| yHsp90 | 0.07 ± 0.03 | 2.2 ± 0.3 | 31 |
| yHsp90-hCdc37 | 0.10 ± 0.03 | 1.8 ± 0.3 | 18 |
| hHsp90 | 0.17 ± 0.04 | 2.2 ± 0.3 | 13 |
| hHsp90-hCdc37 | 0.22 ± 0.04 | 0.8 ± 0.3 | 4 |
| CeHsp90 | 0.21 ± 0.04 | 2.2 ± 0.3 | 10 |
| CeHsp90-CeCdc37 | 0.18 ± 0.04 | 2.4 ± 0.3 | 13 |
A reason why hCdc37 inhibits the formation of the closed Hsp90 conformation could be that the Hsp90-hCdc37 complex is generally weakened in the presence of ATPγS. To address this, we analyzed the influence of nucleotides on the complex forming ability of *hCdc37 with yHsp90 in the aUC. We could observe that the complex formation is indeed slightly reduced upon addition of 4 mm ATPγS, whereas it showed no alterations in the presence of ADP (Fig. 3B), implying that although nucleotide binding in general is possible to the Hsp90-hCdc37 complex, the closing reaction appears to be not fully compatible with the binding of human Cdc37. All in all, the results indicate that hCdc37 may work to inhibit the formation of N-terminal closed complexes within one subunit of the dimeric Hsp90 protein.
This inhibitory mechanism could be the result of binding to the flexible ATP lid of Hsp90 as suggested previously (21, 32). We thus aimed to test this interaction site by using fragments and deletions of hHsp90 in our aUC setup with *hCdc37 (Fig. 3C). As expected, we could observe a complex formation with hHsp90 and hHsp90-N and no interaction with a mutant lacking the ATP lid (hHsp90-lidless). We further tested whether the N-domain alone can compete with full-length Hsp90 for hCdc37. To this end, a higher amount of hHsp90-N should displace the full-length protein from the *hCdc37-hHsp90 complex. Surprisingly, it was not possible to displace hHsp90 with a 4-fold excess of hHsp90-N. This might indicate that other parts within this subunit could also play a role during the interaction with hCdc37.
Aha1 Binds without Disrupting the hCdc37-Hsp90 Complexes
Having seen that ATPγS can disfavor yHsp90-hCdc37 complexes, we wondered whether nucleotides may influence the Cdc37-Hsp90 complexes of nematode and human systems in a different manner. Therefore, we added ATPγS to the C. elegans system in the aUC (Fig. 4A). The data confirm that CeCdc37 is able to bind to CeHsp90 even in the closed conformation. This indeed appears to be in contrast to the human system (Fig. 4B). hCdc37 seems unable to interact with the closed conformation of Hsp90 as strongly as it does with the open conformation. Based on these results, it is likely that CeCdc37 and hCdc37 have differences regarding the interaction with Hsp90s. This is further evident from our inability to form a protein complex between *CeCdc37 and yeast Hsp90 (data not shown).
FIGURE 4.

Influence of Aha1 and nucleotides on the binding of Cdc37 to Hsp90. In A–D, analytical ultracentrifugation runs were done with either 500 nm *hCdc37 or *CeCdc37. The co-chaperone alone is always shown in black circles. The influence of nucleotides on the Cdc37-Hsp90 complexes is shown in A and B. A, the addition of 3 μm CeHsp90 (red triangle) and further addition of 4 mm ATPγS (blue square) to *CeCdc37 is depicted. B, this graph presents the addition of 3 μm hHsp90 (red triangle) and 4 mm ATPγS (blue square) to *hCdc37. Experiments were performed in 40 mm HEPES, pH 7.5, 80 mm KCl, and 5 mm MgCl2. In C and D, 3 μm Hsp90 were added to the *hCdc37 (red triangle) and, additionally, 10 μm Aha1 (blue square). C shows the complex formation with hHsp90 and hAha1, and D shows the complex formation of *hCdc37 with CeHsp90 and hAha1. Measurements were done in standard assay buffer.
Due to the different binding capability, one might assume that also the Aha1 interaction may deviate between human and nematode systems (33). Aha1 is an ATPase activating cofactor known to bind to the N- and M-domain of Hsp90 (30, 34–36). In the nematode system, it partially can replace Cdc37 during the kinase chaperone cycle (25). To investigate the binding behavior in the human system, we used our common aUC set up and added hAha1 to a complex formed out of hCdc37* and full-length hHsp90. We could observe a peak shift to a higher s20,w value in presence of hAha1 compared with binding of hHsp90 alone (Fig. 4C). We did not see any indications of weaker complex formation as we had seen in similar experiments using the C. elegans system (25). We aimed at understanding, whether this is the consequence of using the human Hsp90 protein. We thus performed an identical experiment using CeHsp90 instead (Fig. 4D). The result was similar. The shift to higher s20,w values upon hAha1 addition was weaker though, implying that we did not fully saturate the binding due to a potentially weaker affinity of hAha1 to CeHsp90. Nevertheless, these data suggest that any differences observed in terms of the partial Aha1-Cdc37 competition may originate from differences between the two Cdc37 proteins.
CeCdc37 and hCdc37 Utilize Different Primary Interaction Sites
We aimed at understanding the differences between the human and nematodal system observed so far. To address this issue, we tested whether differences exist in respect to the binding sites and generated fragments of Hsp90, which omit the reported interaction site at the N terminus (hHsp90-MC and CeHsp90-MC). We then tested the binding of the corresponding Cdc37 to these deletion fragments in our established aUC assay. As expected, *hCdc37 could not interact with hHsp90-MC (Fig. 5A). Surprisingly, binding of CeHsp90-MC to *CeCdc37 was strong instead (Fig. 5A). It resulted in exhaustive binding of *CeCdc37, whereas for full-length CeHsp90 at the identical concentration, unbound CeCdc37* is still present. Thus, CeCdc37 strongly binds to the MC part of Hsp90. This interaction seems to become weaker in presence of the nucleotide binding domain of Hsp90, implying that a partial interference between the N-terminal domain and bound CeCdc37 may occur.
FIGURE 5.

Differential binding of hCdc37 and CeCdc37 to Hsp90-MC. A–C, sedimentation velocity experiment were performed in standard assay buffer at 42,000 rpm. A shows *hCdc37 alone (blue square) and in combination with 3 μm hHsp90-MC (magenta rhombus) and *CeCdc37 alone (black circle) and in the presence of 3 μm CeHsp90-MC (red triangle). B, the same setup as before was carried out except that the experiment was performed in a cross-species approach. *hCdc37 together with CeHsp90-MC is shown in magenta (rhombus) and *CeCdc37 with hHsp90-MC in red (triangle). C, salt dependence of the Cdc37-Hsp90 and Hsp90-MC complex, respectively. Experiments were run for *hCdc37-hHsp90 (blue square), *CeCdc37-CeHsp90 (black circle), and *CeCdc37-CeHsp90-MC (red triangle) in standard assay buffer containing 50, 100, 150, and 250 mm KCl. For analyses, the amplitude of free *Cdc37 was set into relation with complexed *Cdc37 and showed in logarithmic scale as depending on the salt concentration.
To check whether the interaction site is determined by the respective Hsp90 protein or by the Cdc37 protein, we analyzed binding of *CeCdc37 to hHsp90-MC and *hCdc37 binding to CeHsp90-MC (Fig. 5B). Here, similar to the experiment described previously, *hCdc37 required the presence of the N-terminal domain, whereas *CeCdc37 bound strongly also to the MC-construct of hHsp90. These results point out that Cdc37 determines the interaction site. Although hCdc37 preferentially binds to the N-terminal domain of both Hsp90s, CeCdc37 apparently strongly favors a binding site in the MC fragment independent of the Hsp90 homolog used.
We further wanted to determine the biophysical properties of the different binding sites. To this end, we investigated the ionic strength dependence of the interaction. To define the hydrophilic nature of the binding site, we measured the formation of the *hCdc37-hHsp90 complex and the *CeCdc37-CeHsp90 complex at increasing KCl concentrations until complex formation was abolished (Fig. 5C). The interaction between CeCdc37 and CeHsp90 was dependent on ionic strength as reported before (25). The interaction of the human proteins also was dependent on ionic strength, but generally, the affinity of complex formation was higher. Additionally, we tested the complex of *CeCdc37 and CeHsp90-MC (Fig. 5C). Here, the hydrophilic properties of the interaction site were much more evident compared with the full-length proteins, suggesting that CeCdc37 might recognize a very hydrophilic site in the Hsp90-MC fragment. This interaction apparently is modified to some extent, when the N-terminal domain is present in the full-length protein.
CeCdc37 Binds to the Middle Domain of Hsp90
Due to the unexpected binding properties of *CeCdc37 for Hsp90s, we wanted to analyze this interaction in a more detailed way. Therefore, we deleted the dimerization site (C-domain) and also generated the individual domain constructs of the nematodal Hsp90 protein (Fig. 6A). We then tested the binding ability of these domains toward *CeCdc37. We detected an interaction with the monomeric NM construct and with the isolated middle domain, whereas no binding was observed for the N-domain and the isolated dimerization site (Fig. 6B). Interestingly, the binding to the isolated M-domain appeared to generate a stronger shift compared with the much larger NM-domain, implying that here as well, the presence of the N-domain reduces the affinity. Thus based on these data, *CeCdc37 appears to have the highest affinity to the M-domain fragment of Hsp90, providing clear evidence for a new interaction site in this protein complex.
FIGURE 6.

Fragmentation of CeHsp90 and binding ability toward CeCdc37. A, schematic representation of the created CeHsp90 fragments with the corresponding hydrophobicity plot. B, sedimentation velocity experiments with 500 nm *CeCdc37 (black circle) and after addition of 3 μm of CeHsp90-NM (red triangle), CeHsp90-N (blue square), CeHsp90-M (magenta rhombus), or CeHsp90-C (green cross) were monitored with fluorescence detection in standard assay buffer at 42,000 rpm. AA, amino acids.
CeCdc37 Binds Hsp90 via Parts of Its N-terminal Domain
Having identified a new interaction site on Hsp90, we wondered whether the established interaction site on hCdc37 is also different in CeCdc37. In the available crystal structure of the yHsp90 N terminus complexed to a hCdc37 fragment, amino acids 138 to 266 of hCdc37 are responsible for Hsp90 binding (21). We initially generated an N-terminal deletion fragment of CeCdc37 (Fig. 7A), where the N-terminal domain up to residue 128 was deleted (Δ128CeCdc37) and another fragment, where the C-terminal domain was omitted from residue 287 (CeCdc37ΔC), generating Cdc37 fragments, which both contain the interaction site reported for hCdc37 (21) We initially tested the interaction with Hsp90 by analyzing the inhibitory effect on the ATPase activity of CeHsp90. As expected the C-terminal truncation did not affect the inhibition and behaved similar to wild-type Cdc37 (Fig. 7B). The deletion of the N-terminal domain in Δ128CeCdc37 instead generated a fragment that was unable to inhibit the ATPase activity of CeHsp90 (Fig. 7B). We thus aimed at analyzing the binding directly. To analyze their binding ability, we used our aUC setup, in which labeled *CeCdc37-CeHsp90 complexes were preformed. Additionally unlabeled CeCdc37 variants were added in large excess to disrupt the complex. The C-terminal deletion fragment was able to disturb the interaction of *CeCdc37-CeHsp90, whereas the fragment without the N-terminal domain was incapable to bind to Hsp90 (Fig. 7C). This observation is surprising given that the deleted amino acids are so far not considered to be part of the binding site. Consequently, we generated more detailed CeCdc37 deletions within the N-terminal domain, truncating either 46 or 97 residues according to linker regions detected in hydrophathy plots (Fig. 7A). Although the truncation of 46 residues could still compete against binding of *CeCdc37 to Hsp90 in the aUC setup, Δ97CeCdc37 was inactive (Fig. 7C). Finally, we analyzed the inhibitory effect and the binding capability of a CeCdc37 fragment containing only amino acids 46 to 287 (Fig. 7A). This fragment could fulfill both tasks: inhibition of Hsp90 (Fig. 7B) and competition with *CeCdc37 for binding to Hsp90 (Fig. 7C). Thus, the fragmentation study of CeCdc37 also supports the presence of a further interaction site with its relevant part between residues 46–97, which apparently in the nematode system contributes most of the binding affinity.
FIGURE 7.

CeCdc37 binds Hsp90 via its N-domain. A, schematic overview of CeCdc37 fragments with corresponding hydrophobicity analysis, in which the hCdc37 is shown in red and CeCdc37 is shown in black. B, ATPase activity of 3 μm CeHsp90 was measured after addition of 10 μm of CeCdc37 fragments in standard assay buffer containing 5 mm MgCl2 at 30 °C. C, competition of 500 nm labeled *CeCdc37 (black circle) from a complex formed with 3 μm CeHsp90 (red triangle) with 10 μm of either CeCdc37 (blue square), Δ46CeCdc37 (magenta rhombus), Δ97CeCdc37 (green cross), Δ128CeCdc37 (navy blue star), CeCdc37ΔC (purple filled circle), or Δ46CeCdc37ΔC (brown filled triangle) were preformed in an aUC setup at 42,000 rpm using fluorescence detection. D, an aUC competition experiment was performed in standard assay buffer with 500 nm of labeled *hCdc37 (black circle) in complex with 3 μm hHsp90 (red triangle) and 10 μm of either hCdc37 (magenta rhombus) or Δ133hCdc37 (blue square) to compete for binding. E shows an aUC competition experiment in standard assay buffer (low salt) and 500 nm of the fluorescence-labeled protein. *CeCdc37 (black circle) is complexed with CeHsp90NM (red triangle), and additionally, 10 μm hCdc37 (blue square) was added to compete for binding.
We performed a similar experiment with human Cdc37. We formed *hCdc37-hHsp90 complexes and disrupted the interaction again with hCdc37 (Fig. 7D). Alternatively, we used the fragment Δ133hCdc37, which lacks the N-terminal domain and closely matches the published interacting fragment of hCdc37. Indeed, we could, in sharp contrast to CeCdc37, observe competition in our assay in a similar manner as for full-length Cdc37, implying that indeed, most of the binding site in human Cdc37 is contributed from this fragment.
We further addressed the issue whether we can also disrupt CeCdc37-containing complexes with hCdc37, as the binding sites may at least partially overlap. To this end, we utilized the NM fragment of CeHsp90 to prevent difficulties originating from the dimeric nature of Hsp90. We could observe two changes of the *CeCdc37-CeHsp90-NM complexes upon addition of hCdc37 (Fig. 7E). First, hCdc37 was able to bind simultaneously with *CeCdc37 to CeHsp90-NM, as observable by a peak shift to higher s20,w values. However, an additional shift to lower s20,w values could be observed. Therefore, hCdc37 might also have a weak affinity toward the M-domain as an additional binding site. These results indicate that the binding interfaces between CeCdc37 and hCdc37 are not entirely independent, but overlap to some extent, implying that besides the primary interface, which is recognized differently by the two Cdc37 proteins, the secondary contact sites may lead to a structurally conserved binding mode for both of them.
The Interaction of N-M Is Functional
Having established an alternative primary interaction site between Hsp90 and CeCdc37, we finally wanted to know whether this interaction site also is functional. In recent studies, it had been observed that Cdc37 can be dephosphorylated as part of the Hsp90 complex by the tetratricopeptide repeat-containing phosphatase protein phosphatase 5 (18). We used the nematodal proteins to assemble this system and studied the dephosphorylation of [32P]CeCdc37 by PPH-5 in presence of either no Hsp90 or different Hsp90 constructs, including Hsp90-MC and a full-length Hsp90 devoid of the tetratricopeptide repeat recognition motif at the C-terminal end (CeHsp90-ΔMEEVD). As expected, full-length Hsp90 resulted in accelerated dephosphorylation of [32P]CeCdc37, but also CeHsp90-MC could exert this effect, whereas the C-domain and CeHsp90-ΔMEEVD were unable to promote the dephosphorylation of [32P]CeCdc37 by PPH-5 (Fig. 8). This result further highlights the importance of the interaction between CeCdc37 and the middle domain of Hsp90 and suggests that this interaction may also contribute functionality to Cdc37 during the chaperoning cycle of kinases.
FIGURE 8.

Functionality of the Hsp90-MC fragment. Radioactive labeled [32P]CeCdc37 was incubated with CeHsp90, CeHsp90-MC, CeHsp90-C, or CeHsp90-ΔMEEVD in presence of PPH-5 to observe its dephosphorylation. Samples were taken at time point 0 (no addition of CeHsp90 and PPH-5) and after 10 and 60 min for the other conditions. Lanes 1 and 12, samples taken at time point 0; lanes 2 and 3, Cdc37 and PPH-5 alone; lanes 4 and 5, samples in the presence of CeHsp90; lanes 6 and 7, in the presence of CeHsp90-MC; lanes 8 and 9, samples in the presence of CeHsp90-C; and lanes 10 and 11, samples in the presence of CeHsp90-ΔMEEVD.
DISCUSSION
The protein Cdc37 plays a major role during the Hsp90 cycle as it is the specific co-chaperone for kinase recruitment. It also inhibits the ATPase activity of Hsp90 (19, 37, 38). Therefore, it is important to gain insight into the formation and the mechanistic features of the ternary Hsp90-Cdc37-kinase complex but also of the binary Hsp90-Cdc37 complex. So far, no full-length structure exists and binding sites primarily were obtained from a crystal structure of the complex between N-terminal yHsp90 and an hCdc37 fragment (amino acids 138–378) (21) and an NMR study of the interacting human fragments (32). Some earlier studies had also proposed binding to the C-terminal domain of Hsp90 (39, 40). In the ternary complex, the kinase is supposed to bind to the first 126 residues of Cdc37 (41).
In this study, we address the binding between Cdc37 and Hsp90 in a more detailed way, and we combined the data into one hypothetical model of Cdc37 function, despite the differences seen between human and nematode Cdc37 proteins (Fig. 9). We, consistent with the previous mechanism, see preferential binding of hCdc37 to the N-terminal domain and a marked slowdown in conformational changes in response to ATPγS treatment. Considering our aUC analysis, the co-chaperone and the nucleotide seem to compete for binding to the N terminus of Hsp90 (Fig. 9, blue interaction site). However, we failed to see the proposed bridging function of hCdc37 in between the two N-terminal subunits of the Hsp90 dimer (21, 23). The co-chaperone appears to trap one Hsp90 monomer in a conformation unfavorable for ATP turnover, independent of the other Hsp90 subunit.
FIGURE 9.

Hypothetical model of Cdc37 binding to Hsp90. The binding behavior of Cdc37 toward Hsp90 is shown in the closed and open conformation. The ATP lid in the N-terminal domain of Hsp90 is depicted in red. The primary interaction site of hCdc37 is highlighted in blue and the one of CeCdc37 is in red. In the open conformation, Hsp90 is bound by both Cdc37s using their primary binding sites and orienting the phosphate to make it accessible to the (not depicted) C-terminal-bound phosphatase. hCdc37 is predominately released in the closed conformation of Hsp90 as the blue interaction site apparently is less accessible in the closed conformation (depicted in the gray box). CeCdc37 instead stays predominantly bound to Hsp90 in presence of nucleotide, as its primary red interaction site stays accessible during the closing reaction. Cdc37 is shown in its phosphorylated form generated by casein kinase II (CKII), but both forms are capable of binding to Hsp90.
Using the nematodal proteins, we identified a different interaction site (Fig. 9, red interaction site). We see preferential binding to the middle domain of Hsp90, and we also observe Cdc37 parts as interaction partners, which previously were not considered to be involved in the interaction with Hsp90. As a weak overlap between the binding sites can be observed, it is likely that Cdc37 utilizes both of them, but with different priorities and also with a certain contribution from the orientation of the N-M domains. Thus, the binding mechanism of Cdc37 shows similarities to that of other Hsp90 co-chaperones such as Aha1 and Sti1, which also interact via two binding sites (30, 31, 42, 43). In this context, it is interesting to note, that the interaction of nematodal Cdc37 with the M-domain of Hsp90 (human or nematode) is weakened by the presence of the N-domain, implying that some parts of the N-M interface may also be part of the interaction site for Cdc37, leading to observable competition between N-domain and Cdc37. It is conclusive that Cdc37 induces a conformation at the N-M interface, arrests the motility, and thus slows down the ability to perform the conformational change required for ATP hydrolysis (Fig. 9).
Based on our results, we assume that hCdc37 and CeCdc37 recognize different primary binding sites with measurable affinity. This interaction in both cases appears to be modified by further contacts outside the primary binding site. For hCdc37, reports exist that also involve the middle domain of Hsp90 in the interaction model (22). For CeCdc37, this interaction interface may be the primary docking site with further secondary interactions in the N-domain, leading to a similar assembly of the proteins in the full-length Hsp90 dimer. This might explain the conserved inhibition of the ATPase activity, which can be seen for both systems.
It is noteworthy that in particular Cdc37 appears to show less conserved biochemical features in eukaryotes, with the dissociation constant of yCdc37 in the range of 100 μm and apparently different primary interaction sites for nematode and human Hsp90. The presented study, similar to other studies determining Cdc37-Hsp90 affinities, is performed in the absence of the client kinase. Although this exposes the interaction sites between Hsp90 and its cofactor, it may not be sufficient to explain affinities in the client-containing complexes. In general, the low affinity of the Cdc37-Hsp90 interaction does not account for the observation that kinase-Cdc37-Hsp90 complexes are stable assemblies that can be purified, immunoprecipitated, and structurally analyzed (23). Therefore, it is likely that the kinase has a sizeable contribution to the overall affinity within the protein complex, suggesting that the assembly of kinase, Cdc37, and Hsp90 might be more stable and structurally defined. Despite this, the identification of a second interaction site between Cdc37 and Hsp90 and the inhibitory function of hCdc37 on the closing rate represent further steps in understanding the chaperoning function of Hsp90 toward its kinase clients and the interaction of this important cofactor with the Hsp90 chaperone machinery.
Acknowledgments
We also gratefully acknowledge the support of the TUM Graduate School's Faculty Graduate Center of Chemistry at the Technische Universität München.
This work was supported by Deutsche Forschungsgemeinschaft Grant RI1873/1-2 (to K. R.) and Fonds der Chemischen Industrie.
- PPH-5
- protein phosphatase 5
- aUC
- analytical ultracentrifugation
- CeCdc37
- C. elegans Cdc37
- hCdc37
- human Cdc37
- CeHsp90
- C. elegans Hsp90
- hHsp90
- human Hsp90
- yHsp90
- yeast Hsp90
- hAha1
- human Aha1
- PγMABA-ATP
- adenosinetriphospho-γ-(N′-methylanthraniloylaminobutyl)-phosphoramidate
- Hsp90-NM
- N-terminal and middle domain of Hsp90
- Hsp90-MC
- middle and C-terminal domain of Hsp90
- N-domain
- N-terminal domain of Hsp90
- M-domain
- middle domain of Hsp90
- C-domain
- C-terminal domain of Hsp90
- a. U.
- artificial units
- PPH-5
- nematode homolog of human PP5.
REFERENCES
- 1. Ashburner M., Bonner J. J. (1979) The induction of gene activity in Drosophilia by heat shock. Cell 17, 241–254 [DOI] [PubMed] [Google Scholar]
- 2. Walsh K. H., Crabb D. W. (1989) The heat-shock response in cultured cells exposed to ethanol and its metabolites. J. Lab. Clin. Med. 114, 563–567 [PubMed] [Google Scholar]
- 3. McClellan A. J., Xia Y., Deutschbauer A. M., Davis R. W., Gerstein M., Frydman J. (2007) Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell 131, 121–135 [DOI] [PubMed] [Google Scholar]
- 4. Galigniana M. D., Harrell J. M., Murphy P. J., Chinkers M., Radanyi C., Renoir J. M., Zhang M., Pratt W. B. (2002) Binding of hsp90-associated immunophilins to cytoplasmic dynein: direct binding and in vivo evidence that the peptidylprolyl isomerase domain is a dynein interaction domain. Biochemistry 41, 13602–13610 [DOI] [PubMed] [Google Scholar]
- 5. Richter K., Buchner J. (2001) Hsp90: chaperoning signal transduction. J. Cell. Physiol. 188, 281–290 [DOI] [PubMed] [Google Scholar]
- 6. Picard D., Khursheed B., Garabedian M. J., Fortin M. G., Lindquist S., Yamamoto K. R. (1990) Reduced levels of hsp90 compromise steroid receptor action in vivo. Nature 348, 166–168 [DOI] [PubMed] [Google Scholar]
- 7. Stancato L. F., Chow Y. H., Hutchison K. A., Perdew G. H., Jove R., Pratt W. B. (1993) Raf exists in a native heterocomplex with hsp90 and p50 that can be reconstituted in a cell-free system. J. Biol. Chem. 268, 21711–21716 [PubMed] [Google Scholar]
- 8. Sreedhar A. S., Kalmár E., Csermely P., Shen Y. F. (2004) Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett. 562, 11–15 [DOI] [PubMed] [Google Scholar]
- 9. Richter K., Soroka J., Skalniak L., Leskovar A., Hessling M., Reinstein J., Buchner J. (2008) Conserved conformational changes in the ATPase cycle of human Hsp90. J. Biol. Chem. 283, 17757–17765 [DOI] [PubMed] [Google Scholar]
- 10. Haslbeck V., Kaiser C. J., Richter K. (2012) Hsp90 in non-mammalian metazoan model systems. Biochim. Biophys. Acta 1823, 712–721 [DOI] [PubMed] [Google Scholar]
- 11. Southworth D. R., Agard D. A. (2008) Species-dependent ensembles of conserved conformational states define the Hsp90 chaperone ATPase cycle. Mol. Cell. 32, 631–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Panaretou B., Prodromou C., Roe S. M., O'Brien R., Ladbury J. E., Piper P. W., Pearl L. H. (1998) ATP binding and hydrolysis are essential to the function of the Hsp90 molecular chaperone in vivo. EMBO J. 17, 4829–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McLaughlin S. H., Smith H. W., Jackson S. E. (2002) Stimulation of the weak ATPase activity of human hsp90 by a client protein. J. Mol. Biol. 315, 787–798 [DOI] [PubMed] [Google Scholar]
- 14. Prodromou C., Panaretou B., Chohan S., Siligardi G., O'Brien R., Ladbury J. E., Roe S. M., Piper P. W., Pearl L. H. (2000) The ATPase cycle of Hsp90 drives a molecular “clamp” via transient dimerization of the N-terminal domains. EMBO J. 19, 4383–4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wandinger S. K., Richter K., Buchner J. (2008) The Hsp90 chaperone machinery. J. Biol. Chem. 283, 18473–18477 [DOI] [PubMed] [Google Scholar]
- 16. Bandhakavi S., McCann R. O., Hanna D. E., Glover C. V. (2003) A positive feedback loop between protein kinase CKII and Cdc37 promotes the activity of multiple protein kinases. J. Biol. Chem. 278, 2829–2836 [DOI] [PubMed] [Google Scholar]
- 17. Shao J., Prince T., Hartson S. D., Matts R. L. (2003) Phosphorylation of serine 13 is required for the proper function of the Hsp90 co-chaperone, Cdc37. J. Biol. Chem. 278, 38117–38120 [DOI] [PubMed] [Google Scholar]
- 18. Vaughan C. K., Mollapour M., Smith J. R., Truman A., Hu B., Good V. M., Panaretou B., Neckers L., Clarke P. A., Workman P., Piper P. W., Prodromou C., Pearl L. H. (2008) Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol. Cell. 31, 886–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Siligardi G., Hu B., Panaretou B., Piper P. W., Pearl L. H., Prodromou C. (2004) Co-chaperone regulation of conformational switching in the Hsp90 ATPase cycle. J. Biol. Chem. 279, 51989–51998 [DOI] [PubMed] [Google Scholar]
- 20. MacLean M., Picard D. (2003) Cdc37 goes beyond Hsp90 and kinases. Cell Stress Chaperones 8, 114–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roe S. M., Ali M. M., Meyer P., Vaughan C. K., Panaretou B., Piper P. W., Prodromou C., Pearl L. H. (2004) The Mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 116, 87–98 [DOI] [PubMed] [Google Scholar]
- 22. Zhang W., Hirshberg M., McLaughlin S. H., Lazar G. A., Grossmann J. G., Nielsen P. R., Sobott F., Robinson C. V., Jackson S. E., Laue E. D. (2004) Biochemical and structural studies of the interaction of Cdc37 with Hsp90. J. Mol. Biol. 340, 891–907 [DOI] [PubMed] [Google Scholar]
- 23. Vaughan C. K., Gohlke U., Sobott F., Good V. M., Ali M. M., Prodromou C., Robinson C. V., Saibil H. R., Pearl L. H. (2006) Structure of an Hsp90-Cdc37-Cdk4 complex. Mol. Cell 23, 697–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gaiser A. M., Brandt F., Richter K. (2009) The non-canonical Hop protein from Caenorhabditis elegans exerts essential functions and forms binary complexes with either Hsc70 or Hsp90. J. Mol. Biol. 391, 621–634 [DOI] [PubMed] [Google Scholar]
- 25. Gaiser A. M., Kretzschmar A., Richter K. (2010) Cdc37-Hsp90 complexes are responsive to nucleotide-induced conformational changes and binding of further cofactors. J. Biol. Chem. 285, 40921–40932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hessling M., Richter K., Buchner J. (2009) Dissection of the ATP-induced conformational cycle of the molecular chaperone Hsp90. Nat. Struct. Mol. Biol. 16, 287–293 [DOI] [PubMed] [Google Scholar]
- 27. Stafford W. F., 3rd. (1992) Boundary analysis in sedimentation transport experiments: a procedure for obtaining sedimentation coefficient distributions using the time derivative of the concentration profile. Anal. Biochem. 203, 295–301 [DOI] [PubMed] [Google Scholar]
- 28. Weikl T., Muschler P., Richter K., Veit T., Reinstein J., Buchner J. (2000) C-terminal regions of Hsp90 are important for trapping the nucleotide during the ATPase cycle. J. Mol. Biol. 303, 583–592 [DOI] [PubMed] [Google Scholar]
- 29. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 30. Retzlaff M., Hagn F., Mitschke L., Hessling M., Gugel F., Kessler H., Richter K., Buchner J. (2010) Asymmetric activation of the hsp90 dimer by its cochaperone aha1. Mol. Cell. 37, 344–354 [DOI] [PubMed] [Google Scholar]
- 31. Lee C. T., Graf C., Mayer F. J., Richter S. M., Mayer M. P. (2012) Dynamics of the regulation of Hsp90 by the co-chaperone Sti1. EMBO J. 31, 1518–1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sreeramulu S., Jonker H. R., Langer T., Richter C., Lancaster C. R., Schwalbe H. (2009) The human Cdc37.Hsp90 complex studied by heteronuclear NMR spectroscopy. J. Biol. Chem. 284, 3885–3896 [DOI] [PubMed] [Google Scholar]
- 33. Sun L., Prince T., Manjarrez J. R., Scroggins B. T., Matts R. L. (2012) Characterization of the interaction of Aha1 with components of the Hsp90 chaperone machine and client proteins. Biochim. Biophys. Acta 1823, 1092–1101 [DOI] [PubMed] [Google Scholar]
- 34. Lotz G. P., Lin H., Harst A., Obermann W. M. (2003) Aha1 binds to the middle domain of Hsp90, contributes to client protein activation, and stimulates the ATPase activity of the molecular chaperone. J. Biol. Chem. 278, 17228–17235 [DOI] [PubMed] [Google Scholar]
- 35. Meyer P., Prodromou C., Liao C., Hu B., Mark Roe S., Vaughan C. K., Vlasic I., Panaretou B., Piper P. W., Pearl L. H. (2004) Structural basis for recruitment of the ATPase activator Aha1 to the Hsp90 chaperone machinery. EMBO J. 23, 511–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Koulov A. V., LaPointe P., Lu B., Razvi A., Coppinger J., Dong M. Q., Matteson J., Laister R., Arrowsmith C., Yates J. R., 3rd, Balch W. E. (2010) Biological and structural basis for Aha1 regulation of Hsp90 ATPase activity in maintaining proteostasis in the human disease cystic fibrosis. Mol. Biol. Cell. 21, 871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miyata Y., Nishida E. (2004) CK2 controls multiple protein kinases by phosphorylating a kinase-targeting molecular chaperone, Cdc37. Mol. Cell. Biol. 24, 4065–4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Prodromou C., Pearl L. H. (2003) Structure and functional relationships of Hsp90. Curr. Cancer Drug Targets 3, 301–323 [DOI] [PubMed] [Google Scholar]
- 39. Owens-Grillo J. K., Czar M. J., Hutchison K. A., Hoffmann K., Perdew G. H., Pratt W. B. (1996) A model of protein targeting mediated by immunophilins and other proteins that bind to hsp90 via tetratricopeptide repeat domains. J. Biol. Chem. 271, 13468–13475 [DOI] [PubMed] [Google Scholar]
- 40. Silverstein A. M., Grammatikakis N., Cochran B. H., Chinkers M., Pratt W. B. (1998) p50(cdc37) binds directly to the catalytic domain of Raf as well as to a site on hsp90 that is topologically adjacent to the tetratricopeptide repeat binding site. J. Biol. Chem. 273, 20090–20095 [DOI] [PubMed] [Google Scholar]
- 41. Pearl L. H. (2005) Hsp90 and Cdc37 – a chaperone cancer conspiracy. Curr. Opin. Genet. Dev. 15, 55–61 [DOI] [PubMed] [Google Scholar]
- 42. Richter K., Muschler P., Hainzl O., Reinstein J., Buchner J. (2003) Sti1 is a non-competitive inhibitor of the Hsp90 ATPase. Binding prevents the N-terminal dimerization reaction during the atpase cycle. J. Biol. Chem. 278, 10328–10333 [DOI] [PubMed] [Google Scholar]
- 43. Schmid A. B., Lagleder S., Gräwert M. A., Röhl A., Hagn F., Wandinger S. K., Cox M. B., Demmer O., Richter K., Groll M., Kessler H., Buchner J. (2012) The architecture of functional modules in the Hsp90 co-chaperone Sti1/Hop. EMBO J. 31, 1506–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]