

Background: A controlled approach as opposed to conventional toxic drugs to activate p53 is applicable for tumors and metabolic and inflammatory diseases.
Results: A 0.1-ms pulse width mild electrical stimulation (MES) activated p53 function in epithelial cell lines.
Conclusion: MES induced p53 phosphorylation via p38 MAPK signaling and G2 cell cycle arrest without cell death.
Significance: MES works as a non-cytotoxic and controllable p53 activator.
Keywords: Electrophysiology, p38, p53, Signaling, Stress, G2 Arrest, Mild Electrical Stimulation, p53 Activation
Abstract
Exogenous low-intensity electrical stimulation has been used for treatment of various intractable diseases despite the dearth of information on the molecular underpinnings of its effects. Our work and that of others have demonstrated that applied electrical stimulation at physiological strength or mild electrical stimulation (MES) activates the PI3K-Akt pathway, but whether MES activates other molecules remains unknown. Considering that MES is a form of physiological stress, we hypothesized that it can activate the tumor suppressor p53, which is a key modulator of the cell cycle and apoptosis in response to cell stresses. The potential response of p53 to an applied electrical current of low intensity has not been investigated. Here, we show that p53 was transiently phosphorylated at Ser-15 in epithelial cells treated with an imperceptible voltage (1 V/cm) and a 0.1-ms pulse width. MES-induced p53 phosphorylation was inhibited by pretreatment with a p38 MAPK inhibitor and transfection of dominant-negative mutants of p38, MKK3b, and MKK6b, implying the involvement of the p38 MAPK signaling pathway. Furthermore, MES treatment enhanced p53 transcriptional function and increased the expression of p53 target genes p21, BAX, PUMA, NOXA, and IRF9. Importantly, MES treatment triggered G2 cell cycle arrest, but not cell apoptosis. MES treatment had no effect on the cell cycle in HCT116 p53−/− cells, suggesting a dependence on p53. These findings identify some molecular targets of electrical stimulation and incorporate the p38-p53 signaling pathway among the transduction pathways that MES affects.
Introduction
Endogenous and exogenous electrical currents exert some influence over how cells behave and interact with one another at the cellular and organismal levels. The endogenous biological electrical system is driven by exchange of extracellular and intracellular ions and is an integral part in maintaining basic physiological functions (1). In recent years, exogenous electrical stimulation has been used for the treatment of cranial nerve dysfunction, inflammation, bone injury, pain, and cancer cell penetration for drug delivery (2–6). Given the mounting evidence on the positive effects of exogenous electrical current, it is not surprising that it has been employed in a clinical setting (2, 7, 8), notwithstanding that the molecular mechanisms of its action are not well understood. A possible mechanism of the effects of applied electrical stimulation could be the activation of signal transduction pathways, as proposed by Seegers et al. (9). Indeed, studies by our group and others have demonstrated that electrical stimulation activates the PI3K-Akt pathway, resulting in the process of wound healing (10) and amelioration of hyperglycemia (11, 12). We have also shown that, together with heat shock, which elevates the level of the heat shock protein Hsp72, mild electrical stimulation (MES)2 attenuates hepatic ischemia/reperfusion injury in mice (13–15), ameliorates the diabetic phenotype and protects pancreatic β-cells in a diabetes mouse model (11, 16), reduces inflammatory markers in healthy male subjects (17), and decreases proteinuria and renal inflammation in an Alport syndrome mouse model (18). In the latter disease model, MES was shown to activate not only the PI3K-Akt pathway but also the p38 MAPK signaling pathway (18). It is not surprising that, like other forms of physiological mechanical stresses such as shear stress, MES can activate the signal transduction pathways described above (9, 19). Because we found previously that MES exerts protective effects (11, 14, 18) and other reports have shown that electrical current impedes tumor cell proliferation (20, 21), we asked whether MES affects the expression of p53, a tumor suppressor known for its cell-protective functions via a network of signaling pathways.
p53 is activated by diverse cell stresses such as DNA damage, cell starvation, oncogene activation, telomere elongation, cell adhesion, and heat and mechanical stresses (22–28). It is predicted that there are still other biological stresses that can activate p53. Of note, there have been no reports on the effects of low-intensity electrical current on the p53 protein. p53 is rapidly degraded by the ubiquitin-proteasome system in normal cellular states, but factors that lead to p53 activation can phosphorylate and stabilize p53 (29). Activated p53 is translocated to the nucleus to act as a transcription factor for its numerous target genes. These target genes are effectors of cell cycle arrest, DNA repair, and apoptosis (30). In addition to the antiproliferative genes up-regulated by p53, some molecules involved in metabolism and in response to inflammation have been reported to be targets of p53 (31, 32). Many studies have also shown that p53 is crucial in anti-inflammatory responses (33–37) and in the transcription of the innate immune receptor TLR3 (Toll-like receptor 3) (38). Hence, functional activation of p53 could be beneficial not only for therapeutic application in cancer but also for treatment of inflammatory diseases. In this study, we investigated whether MES, an applied electrical current of physiological strength, can activate p53 in epithelial cells. Our results are the first to reveal that MES can activate p53 and its target genes, leading to G2 phase arrest. These findings add to our growing knowledge of the underlying mechanisms of the effects of electrical stimulation.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
5-Fluorouracil (5-FU) was purchased from Wako (catalog no. 068-01401). SB203580 was from Calbiochem (catalog no. 559389). The following antibodies were obtained from Santa Cruz Biotechnology: anti-p53 (DO-1; sc-126), anti-p21 (C-19; sc-397), anti-MDM2 (SMP14; sc-965), normal mouse IgG (sc-2025), and anti-actin (I-19; sc-1616). Anti-phospho-p53 (Ser-15; catalog no. 9284), anti-p38 (catalog no. 9212), anti-phospho-p38 (Thr-180/Tyr-182; catalog no. 9211), and anti-phospho-ATF2 (Thr-71; catalog no. 9221) antibodies were from Cell Signaling Technology. HRP-conjugated secondary antibodies were from Jackson ImmunoResearch Laboratories.
Cell Culture, Treatment, and Transfection
Human colorectal cancer HCT116 p53+/+ and HCT116 p53−/− cells were kind gifts from Dr. Bert Vogelstein (The Johns Hopkins University, Baltimore, MD). Cells were maintained in DMEM/Ham's F-12 medium supplemented with 10% (v/v) FBS, 100 IU/ml penicillin, and 100 μg/ml streptomycin. Human lung adenocarcinoma cells (A549), human hepatoma cells (HepG2), and human embryonic kidney cells (HEK293) were obtained from American Type Culture Collection. These cell lines were maintained in DMEM containing 10% FBS and antibiotics. All cell lines were cultured at 37 °C in a humidified atmosphere of 5% CO2 as described previously (39). HCT116 cells were treated with 10 μm SB203580 or dimethyl sulfoxide (control) for 1 h before treatment with MES. Transient transfection of plasmids was performed using HilyMax transfection reagent (catalog no. H357, Dojindo Molecular Technologies) following the recommended protocol. HilyMax diluted with Opti-MEM I (catalog no. 31985, Invitrogen) was mixed with total DNA at a DNA/HilyMax ratio of 1:4, and the complex was added to subconfluent cells. The dominant-negative (DN) constructs transfected into cells, p38-DN, MKK3b-DN, and MKK6b-DN, were described previously (40).
MES Treatment
For MES treatment, cells were plated on 6-cm culture dishes and treated at 80% confluency. MES treatment of cells has been described previously (11). Briefly, the culture plate cover had slits at two sides designed to accommodate insulated wires bearing a pair of flat rubber electrodes, which were fitted at the walls of the culture plate and in contact with the culture medium. The electrodes were connected to a Model WF1973 multifunction generator (NF Corp.). Electrical stimulation of cells was delivered using 6 V (∼1 V/cm) and 55 pulses/s (pps) with the indicated pulse widths of 0.01, 0.1, 1, and 10 ms (12) or without pulse (∞ ms). After the indicated treatment times, the culture media were replaced with fresh media, and cells were re-incubated until the assays were performed.
Immunoblotting and Immunoprecipitation
For the detection of p53, phospho-p53 (Ser-15), p21, and actin, cells were lysed on ice for 30 min with immunoprecipitation lysis buffer (41) containing 1% protease inhibitor mixture (Sigma). Samples were diluted with dilution buffer (41) and mildly sonicated. Samples were centrifuged, and lysates were collected. For analysis of p38, phospho-p38, phospho-ATF2, and cleaved caspase-3 expression, cells were snap-frozen in liquid nitrogen and then scraped with lysis buffer (11) containing 1% protease inhibitor mixture. Samples were centrifuged, and supernatants were collected. To recover nuclear extracts, the cell pellet was resuspended in 400 μl of cold buffer containing 10 mm HEPES (pH 7.9), 10 mm KCl, 0.1 mm EDTA, 0.1 mm EGTA, 1 mm dithiothreitol, and 0.5 mm phenylmethylsulfonyl fluoride by gentle pipetting. The cells were allowed to swell on ice for 15 min, after which 25 μl of a 10% solution of Nonidet P-40 (Nacalai Tesque) was added, and the tube was vigorously vortexed for 10 s. The homogenate was centrifuged for 30 s in a microcentrifuge. The nuclear pellet was resuspended in 50 μl of ice-cold buffer containing 20 mm HEPES (pH 7.9), 0.4 m NaCl, 1 mm EDTA, 1 mm EGTA, 1 mm dithiothreitol, and 1 mm phenylmethylsulfonyl fluoride, and the tube was vigorously rocked at 4 °C for 15 min on a shaking platform. The nuclear extract was centrifuged for 5 min in a microcentrifuge at 4 °C, and the supernatant (∼55 μl) was frozen in aliquots at −80 °C. Protein lysates were subjected to SDS-PAGE and Western blot analysis. To analyze the interaction between p53 and MDM2 or phospho-p38, MES-treated HCT116 cells were first cross-linked with 1 mm dithiobis(succinimidyl propionate) (catalog no. 22586, Thermo Scientific) for 30 min at room temperature. The cells were then lysed with immunoprecipitation lysis buffer or radioimmune precipitation assay buffer. Cell lysates were incubated for 12 h at 4 °C with 2 μg of anti-p53 monoclonal antibody or mouse control IgG, and reacted proteins were immobilized in protein G-Sepharose beads (catalog no. 17-0618-01, GE Healthcare) for p53/MDM2 interaction. Cell lysates were incubated with 2 μg of anti-p53 monoclonal antibody or mouse control IgG conjugated to protein G magnetic beads (catalog no. DB10003, VERITAS) for 1 h at room temperature for p53/phospho-p38 interaction. Immunoprecipitates were washed, eluted, and subjected to immunoblotting. Blots of protein lysates, immunoprecipitated samples, or input fraction were probed with the indicated antibodies and visualized using SuperSignal (catalog no. 34077, Pierce).
Quantitative RT-PCR
Total RNA was isolated using RNAiso reagent (catalog no. 9109, Takara Bio Inc.) following the manufacturer's instructions. Real-time quantitative RT-PCR analyses were carried out with SYBR Green Master Mix (catalog no. 4309155, Applied Biosystems) as described previously (38). The threshold cycle values for each gene amplification were normalized by subtracting the threshold cycle value calculated for GAPDH (internal control). The normalized gene expression values are expressed as the relative quantity of gene-specific mRNA. The sequences of primers used for quantitative PCR are listed in Table 1.
TABLE 1.
Primers used for quantitative PCR
| Primer | Forward | Reverse |
|---|---|---|
| Human p21 | 5′-CTGGAGACTCTCAGGGTCGAAA-3′ | 5′-GATTAGGGCTTCCTCTTGGAGAA-3′ |
| Human BAX | 5′-TTTGCTTCAGGGTTTCATCC-3′ | 5′-CAGTTGAAGTTGCCGTCAGA-3′ |
| Human NOXA | 5′-GCTGGAAGTCGAGTGTGCTA-3′ | 5′-CCTGAGCAGAAGAGTTTGGA-3′ |
| Human PUMA | 5′-GACGACCTCAACGCACAGTA-3′ | 5′-AGGAGTCCCATGATGAGATTGT-3′ |
| Human IRF9 | 5′-CAAGTGGAGAGTGGGCAGTT-3′ | 5′-ATGGCATCCTCTTCCTCCTT-3′ |
| Human GAPDH | 5′-TCCACTGGCGTCTTCACC-3′ | 5′-GGCAGAGATGATGACCCTTTT-3′ |
ChIP Assay
ChIP assay was performed as described previously (38). Cross-linking of HCT116 cells was performed using formaldehyde (1% final concentration). After adding the stop reaction buffer (0.125 m glycine), cells were washed with PBS, lysed on ice using cell lysis buffer (38), and then homogenized in a Dounce homogenizer. The crude nuclei were resuspended in nuclear lysis buffer and incubated on ice for 10 min. The samples were sonicated on ice and microcentrifuged. The chromatin solution was precleared and immunoprecipitated with 2 μg of anti-p53 mouse monoclonal antibody or mouse IgG. Washing, cross-link reversal, and DNA extraction were performed as described previously (38). The primers used recognize a fragment of the human p21 or GAPDH promoter (42).
Flow Cytometry
Cultured cells were trypsinized and collected 6 h after MES treatment. Cells were fixed and permeabilized with 70% ethanol in PBS at 4 °C. Cells were incubated on ice for 1 h with 10 μg/ml propidium iodide (catalog no. P-4170, Sigma) and 0.2 mg/ml RNase in PBS, protected from light. Propidium iodide-stained cells were analyzed using a FACSCanto flow cytometer (BD Biosciences). 3 × 104 cells from each sample were counted, and the cell cycle phase ratio was assessed.
Lactate Dehydrogenase Assay
Cells were assayed for lactate dehydrogenase (LDH) release according to the protocol described previously (43). Briefly, media were collected and centrifuged at 12,000 rpm for 15 min. Supernatants were transferred to fresh tubes, and cell pellets were lysed by the addition of 1% Triton X-100 solutions for 30 min at 37 °C. The remaining attached cells in plates were also lysed with 1% Triton X-100 solution. Culture media and lysates were subjected to LDH assay using a cytotoxicity detection kit (catalog no. 11644793, Roche Applied Science) according to the manufacturer's instructions. LDH release is expressed as the percentage of LDH in the medium over the total LDH (medium and lysate). Values are means ± S.D. of triplicate testing for a representative experiment. At least two independent experiments were performed.
Statistical Analysis
For statistical analysis, the data were analyzed by Student's t test or by one-way analysis of variance with Dunnett's test or the Tukey-Kramer multiple comparison test (JMP software, SAS Institute) as indicated in the figure legends. A p value of <0.05 was considered statistically significant.
RESULTS
MES at a Pulse Width of 0.1 ms Induces Phosphorylation of p53 in HCT116 Cells
It has been suggested that pulse width is an important factor that decides the effect of electrical stimulation on human cancer cells (44), but whether electrical stimulation impacts on the tumor suppressor protein p53 is unclear. To test whether electrical stimulation can activate p53, we treated HCT116 cells with electrical current at different pulse widths. Utilizing the MES conditions we employed previously (12), cells plated in 6-cm culture dishes were stimulated for 10 min with low-intensity current maintained at 6 V or ∼1 V/cm and 55 pps at various pulse widths (0.01, 0.1, 1, or 10 ms) or in the absence of pulse (∞ ms) (Fig. 1A and supplemental Fig. S1A). Protein lysates, extracted from cells 1 h after MES treatment, were analyzed by immunoblotting. Intriguingly, we detected an increase in phosphorylation of p53 at Ser-15 at 0.01-, 0.1-, and 1-ms pulse widths (Fig. 1B, p-p53). Quantification analysis showed that MES at a 0.1-ms pulse width significantly induced p53 phosphorylation, whereas 0.01- and 1-ms pulse widths tended to increase phosphorylated p53 expression (Fig. 1C). A 10-ms millisecond pulse width and ∞ ms did not alter the level of phosphorylated p53 compared with the control. These data suggest that electrical stimulation at a 0.1-ms pulse width can induce p53 phosphorylation. To investigate whether phosphorylation of p53 by MES is dependent primarily on pulse width or whether it is also affected by electrical input energy, we assessed the effect of MES at a lower energy, which was achieved by changing the reference voltage (V0) point from −3 V, the V0 used in the above experiments, to 0 V while maintaining the amplitude of the waveform at 6 V. The electrical input energy (P) at 0–6 V (P0V∼6V) is lower than that of −3 to +3 V (P−3V∼+3V) as shown in supplemental Fig. S1 (A–C). Comparison of the effect of MES with different electrical energies revealed that p53 was phosphorylated at relatively similar levels (Fig. 1D), implying that phosphorylation of p53 by MES depends mainly on pulse width rather than electrical input energy. In the subsequent experiments, we used P−3V∼+3V, consistent with the conditions in the previous studies we undertook (11, 12, 18).
FIGURE 1.

MES at a pulse width of 0.1 ms induces phosphorylation of p53. A, pulse shape with the indicated pulse width in milliseconds of the electrical stimulation used for treatment. DC, direct current. B, HCT116 cells were treated for 10 min with MES (1 V/cm, 55 pps) at various pulse widths (0.01, 0.1, 1, and 10 ms) or in the absence of pulse (∞ ms). Cell lysates were extracted 1 h after MES treatment. Phospho-p53 (Ser-15; p-p53) and p53 were detected by Western blot analysis. Actin served as a loading control. con, control. C, blots of phospho-p53 were quantified by densitometry using Image Gauge software (version 4.23, Fujifilm). Quantified blots were normalized to actin and are expressed as -fold increase relative to the control. Error bars indicate the mean ± S.D. (n = 3). *, p < 0.05 (assessed by analysis of variance with Dunnett's multiple comparison test). D, cells were treated for 30 min with MES (0.1 ms) at an amplitude of 6 V with a reference voltage (V0) at −3 or 0 V, and lysates were recovered 1 h after treatment. Cell lysates were analyzed by immunoblotting with the indicated antibodies. Actin served as an internal control.
MES Phosphorylates p53 Transiently and Cumulatively in Several Epithelial Cell Lines
To further characterize the effect of MES on p53 phosphorylation, we assessed the sustainability of p53 activation by MES at 1, 3, and 6 h after treatment. The data revealed that MES-induced p53 phosphorylation was highest at 1 h after treatment and then dipped and returned to the basal level at 3 and 6 h post-treatment, respectively (Fig. 2A), indicating a transient effect of MES on p53 activation. We also determined whether longer MES treatment times could increase the level of phosphorylated p53. A time-dependent increase in phospho-p53 expression was observed. MES treatment for 30 min resulted in a higher expression level of phosphorylated p53 compared treatment for 10 or 20 min (Fig. 2B). These results suggest that phospho-p53 accumulates upon MES treatment. The activation of p53 by MES was observed not only in HCT116 cells but also in other human epithelial cell lines such as lung adenocarcinoma A549 cells, hepatoma HepG2 cells, and embryonic kidney HEK293 cells (Fig. 2C). Images of the cells revealed no differences in cellular morphology after MES treatment compared with control cells (supplemental Fig. S2), suggesting that MES does not induce cellular damage.
FIGURE 2.

MES phosphorylates p53 transiently and cumulatively in several epithelial cell lines. A, HCT116 cells were treated with MES (0.1 ms) for 10 min, and protein lysates were isolated at 0, 1, 3, or 6 h after treatment. p-p53, phospho-p53. B, cells were treated with MES (0.1 ms) for 0, 10, 20, or 30 min, and protein lysates were recovered 1 h after treatment. C, HCT116, A549, HEK293, and HepG2 cells were treated with MES (0.1 ms) for 10 min, and lysates were extracted 1 h after treatment. con, control. For A–C, cell lysates were analyzed by immunoblotting with the indicated antibodies. Actin served as an internal control.
MES Induces p53 Phosphorylation at Ser-15, but Not at Ser-20 or Ser-46, and p53 Translocation to the Nucleus
Previous studies revealed that a variety of p53 functions are selectively regulated by post-transcriptional modification of p53. Ser-15, Ser-20, and Ser-46 of p53 are known to be key regulatory phosphorylation sites of p53 function. The level of p53 activation by MES treatment (0.1 ms, 1 V/cm, 55 pps) for 30 min was somewhat similar to that induced by a 24-h treatment with 1 μm 5-FU, a DNA-damaging reagent that activates p53 (Fig. 3A). These data imply that, under the conditions we employed, MES could activate p53 to a relatively mild degree and at a faster time compared with 5-FU. MES treatment for 30 min specifically phosphorylated Ser-15 of p53, but not Ser-20 or Ser-46 (Fig. 3A). In contrast, treatment with 5-FU at low (5 μm) and high (10 and 50 μm) doses increased the phosphorylation of Ser-15, Ser-20, and Ser-46, respectively (Fig. 3A). p53 expression was negatively regulated by the E3 ubiquitin ligase MDM2. MES inhibited p53/MDM2 interaction 1 h after treatment (Fig. 3B). MES treatment under this condition had no effect on the cytosolic expression level of non-phosphorylated p53. However, nuclear expression of non-phosphorylated p53 was increased by MES (Fig. 3C). These data suggest that MES-induced p53 Ser-15 phosphorylation inhibits p53/MDM2 interaction and enhances basal p53 nuclear translocation.
FIGURE 3.

MES enhances phosphorylation of p53 at Ser-15 and p53 recruitment to the nucleus. A, HCT116 cells were treated with MES (0.1 ms) for 30 min, and lysates were recovered 1 h after MES treatment; or cells were treated with 5-FU at the indicated concentrations for 24 h. Protein lysates were analyzed by Western blotting using the indicated antibodies. p-p53, phospho-p53; con, control. B, HCT116 cells were cross-linked 1 h after MES treatment. Protein lysates were immunoprecipitated (IP) with anti-p53 antibody (DO-1) or mouse IgG. Immunoprecipitated lysates and input samples were immunoblotted (IB) and analyzed using the indicated antibodies. C, cytosolic proteins and nuclear extract of HCT116 cells were recovered 1 h after MES treatment. Protein lysates were analyzed by Western blotting using the indicated antibodies. γ-Tubulin and HDAC2 serve as internal controls of the cytosol and nuclear fractions, respectively.
MES Induces p53 Phosphorylation via p38 MAPK Signaling
The results described above prompted us to ask how MES increases p53 phosphorylation. A well known mediator of p53 phosphorylation, p38, is activated by DNA damage, osmotic shock, and many other biological stresses and directly interacts with p53 and other substrates to phosphorylate them (45–47). To investigate the role of p38 in MES-induced p53 phosphorylation, we used the p38-specific inhibitor SB203580. HCT116 cells were pretreated with dimethyl sulfoxide (control) or SB203580 (10 μm) for 1 h before treatment with MES (0.1 ms, 1 V/cm, 55 pps) for 30 min. Interestingly, SB203580 inhibited the MES-induced phosphorylation of p53 (Fig. 4, A and B). MES also increased the levels of phosphorylated p38 and phosphorylated ATF2, which is a p38 downstream molecule (Fig. 4A). The phosphorylation of ATF2 by MES treatment was suppressed by pretreatment with SB203580. In contrast, inhibition by SP600125 and KU55933, antagonists of the JNK1/2 and ATM (ataxia telangiectasia mutated) pathways, respectively, which are also known to affect p53 activation (48, 49), did not block the MES-induced phosphorylation of p53 (supplemental Fig. S3). The DN mutant of p38 and those of MKK3b and MKK6b, which are p38 signal mediators, also inhibited MES-induced p53 phosphorylation (Fig. 4C). Next, we checked the interaction of p53 and p38 proteins using the immunoprecipitation method after MES treatment. The p53/phospho-p38 interaction was detected in MES-treated cells (supplemental Fig. S4). Collectively, these data suggest that MES induces activation of p38 MAPK signaling and association of p53 and phospho-p38 proteins. The interaction between p53 and phospho-p38 leads to p53 phosphorylation.
FIGURE 4.

MES induces p53 activation via the MKK3b-MKK6b-p38 pathway. A, HCT116 cells were cultured for 1 h in the presence of dimethyl sulfoxide (control (con)) or 10 μm SB203580 and then treated with MES (0.1 ms, 1 V/cm, 55 pps) for 30 min. Protein lysates were extracted 1 h after MES treatment and analyzed by Western blotting using the indicated antibodies. The experiments were performed in triplicate. p-p53, phospho-p53. B, densitometric analysis of phosphorylated p53 expression was performed using Image Gauge software. The quantified blots were normalized to actin. Error bars indicate the mean ± S.D. (n = 3). *, p < 0.05 versus the control (analyzed by Student's t test); n.s., not significant. DMSO, dimethyl sulfoxide. C, HCT116 cells were transfected with the pcDNA3.1 (control vector), p38-DN, MKK3b-DN, or MKK6b-DN plasmid. 36 h after transfection, cells were treated with MES (0.1 ms, 1 V/cm, 55 pps) for 30 min. Protein lysates were extracted 1 h after MES treatment, and Western blotting was performed.
MES Can Enhance p53 Transcriptional Activity
We next determined whether MES-induced p53 activation has the potential to promote the transcriptional function of p53. HCT116 wild-type (p53+/+) and p53 knock-out (p53−/−) cells were treated with MES (0.1 ms, 1 V/cm, 55 pps) for 30 min. The mRNA expression of known p53 target genes such as p21 (50), BAX (51), NOXA (52), PUMA (53) and IRF9 (54) was analyzed by real-time quantitative PCR. The mRNA levels of these p53 target genes were up-regulated 4 h after MES treatment in HCT116 p53+/+ cells, but not in HCT116 p53−/− cells (Fig. 5, A–E). Consistent with these data, p21 protein expression was increased after MES treatment in HCT116 wild-type cells (Fig. 2A). Next, to determine whether MES enhances the binding of p53 to its target gene promoter, we analyzed the recruitment of p53 to the p21 promoter region by ChIP assay of nuclear lysates from HCT116 p53+/+ cells 3 h after MES treatment and 12 h after 5-FU treatment at 50 μm. Although the effect of MES was weaker than that of 5-FU, immunoprecipitation with anti-p53 antibody, but not with control IgG, and subsequent PCRs revealed that MES increased the recruitment of p53 to the p21 promoter region (Fig. 5F). These results show that MES is capable of stimulating the transcriptional activity of p53.
FIGURE 5.

p53 target genes are up-regulated by MES in HCT116 wild-type (p53+/+) cells. A–E, HCT116 p53+/+ and HCT116 p53−/− cells were treated with MES (0.1 ms, 1 V/cm, 55 pps) for 30 min, and total RNA was extracted 4 h after treatment. The expression levels of p53 target genes (p21, BAX, NOXA, PUMA, and IRF9) were analyzed by quantitative RT-PCR. GAPDH was used as an internal control. The experiments were performed in triplicate. Error bars indicate the mean ± S.E. *, p < 0.05 versus the control (assessed by Student's t test); **, p < 0.01 versus the control; n.s., not significant. con, control. F, nuclear extracts were obtained from HCT116 p53+/+ cells 3 h after treatment with MES (0.1 ms, 1 V/cm, 55 pps, 30 min) or 12 h after treatment with 5-FU (50 μm). p53 binding to the p21 promoter was assessed by conventional PCR using primers that recognize the p53 consensus site in the p21 promoter. The GAPDH promoter was used as negative control. IP, immunoprecipitate.
MES Induces p53-dependent G2 Arrest with No Cytotoxicity
Because activation of p53 impacts the cell cycle and apoptosis, we assessed the effect of MES on these p53 functions. We performed propidium iodide staining on MES-treated HCT116 p53+/+ and HCT116 p53−/− cells and analyzed the cell cycle progression by flow cytometry. Interestingly, MES treatment (0.1 ms, 1 V/cm, 55 pps) for 30 min statistically increased the G2/M population in HCT116 p53+/+ cells. In contrast, MES increased the S phase population but had no effect on the G2/M population in HCT116 p53−/− cells. The G1 population was decreased in both cell lines by MES treatment (Fig. 6, A and B). These data indicate that MES can induce p53-dependent G2 arrest in HCT116 cells. Consistent with the role of p38 MAPK in the activation of p53 by MES, pretreatment with SB203580 inhibited the effect of MES on cell cycle distribution (Fig. 6C). MES at ∞ ms did not affect cell cycle distribution (Fig. 6C), in line with the data showing that MES at ∞ ms did not increase phosphorylated p53 (Fig. 1, B and C). p53 also mediates cell death by regulating apoptosis-related genes (29). To evaluate the potential apoptotic effect of MES, we analyzed cell death by LDH assay. No change in LDH release was observed between the control and MES-treated groups (Fig. 6D). Furthermore, cleaved caspase-3, a marker of cell apoptosis, was not detected by immunoblotting in MES-treated cells (Fig. 6E). These data show that MES-induced p53 activation results in G2 arrest, but not cell apoptosis.
FIGURE 6.
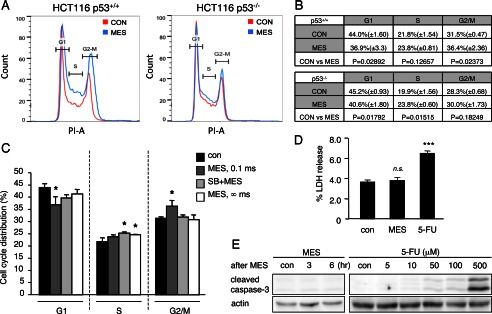
MES induces p53-dependent cell cycle arrest. A and B, HCT116 p53+/+ and HCT116 p53−/− cells were treated with MES (0.1 ms, 1 V/cm, 55 pps) for 30 min. 6 h after MES treatment, cells were washed, fixed, and stained with propidium iodide (PI). Propidium iodide-stained cells were analyzed by flow cytometry to determine cell cycle distribution. The experiments were performed in triplicate, and representative graphs are shown in A. The mean ± S.D. and p values were calculated and are shown in B. CON, control. C, HCT116 cells were pretreated with the p38 pathway inhibitor SB203580 (SB) for 1 h and then treated with MES (0.1 ms or ∞ ms) for 30 min. Cells were stained with propidium iodide 6 h after MES treatment, and the cell population was analyzed by flow cytometry. Data are presented as the percent distribution of cells in the indicated cell cycle. Error bars are the mean ± S.D. (n = 3). *, p < 0.05 (assessed by analysis of variance with the Tukey-Kramer test). D, HCT116 p53+/+ cells were treated with MES (0.1 ms) for 30 min or 5-FU (400 μm) for 24 h. LDH release was assessed 6 h after MES treatment. The experiments were performed in triplicate. Error bars indicate the mean ± S.D. ***, p < 0.001 versus the control )assessed by analysis of variance with the Tukey-Kramer test); n.s., not significant. E, HCT116 p53+/+ cells were treated with MES (0.1 ms) for 30 min, and protein lysates were isolated 3 or 6 h after MES treatment. Cells were treated with the indicated concentrations of 5-FU for 24 h, and protein lysates were recovered for analysis. Cleaved caspase-3 was detected by immunoblotting. Actin served as a loading control.
DISCUSSION
Applied low-intensity electrical current closely resembles the currents produced by the human body and is the most common type of current used in research (55). A number of studies using low-intensity current have shown that it impacts on cell proliferation and signal transduction pathways (9, 56). Because electrical stimulation can disrupt cancer cell replication (20) and can be considered a form of physical stressor, we analyzed its effect on p53, a protein that is responsive to cell stresses and an important modulator of the cell cycle. In this study, we found that application of imperceptible voltage (1 V/cm) and short pulse width (0.1 ms) electrical stimulation led to the activation of p53, which subsequently resulted in G2 arrest. Although MES-induced p53 activation up-regulated apoptosis-related genes such as BAX, NOXA, and PUMA (Fig. 5, B–D), it did not lead to induction of cell apoptosis. This is thought to be due to the transient and mild effect of p53 activation by MES (Figs. 2A and 3A). In addition, DNA damage-independent and reversible p53 activation has a tendency to induce G2 arrest rather than G1 arrest (57). G2 arrest in the absence of DNA damage is known to prevent apoptosis caused by metabolic stress in HCT116 cells (58, 59). For these reasons, MES treatment induced p53-dependent G2 phase cell cycle arrest without instigating apoptosis (Fig. 6). Intriguingly, MES treatment slightly induced S phase arrest under conditions in which the p38-p53 signaling pathway was disrupted in HCT116 cells, such as in the absence of p53 (Fig. 6, A and B) or inhibition of the p38 pathway (Fig. 6C). We previously showed that MES also activates the MAPKs JNK1 and JNK2 (18), which were reported to promote S phase arrest (59). It is possible that, in the absence of a functional p38-p53 signaling pathway, MES activates JNK1/2, leading to S phase arrest.
MES transiently phosphorylated p53 at Ser-15 and increased the p53 expression level in the nucleus (Figs. 2A and 3C). Phosphorylation of p53 at Ser-15 and Ser-20 inhibits interaction between MDM2 and p53 (29). It has been reported that biological stress induces p53 Ser-46 phosphorylation and leads to apoptosis when thresholds are exceeded (60). This step is important for stabilizing the p53 protein (29). Thus, we investigated MES-induced p53 phosphorylation at Ser-15, Ser-20, and Ser-46 and found that MES can induce p53 phosphorylation at Ser-15, but not at Ser-20 or Ser-46 (Fig. 3A). This site-specific phosphorylation is apparently sufficient for the observed increase in p53-mediated G2 arrest. As to why MES phosphorylates Ser-15 and not Ser-20 and Ser-46 is unknown. It could be an as yet unexplored characteristic of MES and a combination of factors such as the conditions we employed here that caused the Ser-15 phosphorylation. It is notable that the 0.1-ms pulse width, but not the 10-ms pulse width, induced Ser-15 phosphorylation. Moreover, the absence of pulse (∞ ms) did not affect the level of phosphorylated p53 (Fig. 1, B and C), suggesting that a specific pulse width, i.e. 0.1 ms, is important for MES-induced Ser-15 phosphorylation of p53.
It was reported previously that 0.1-ms high-voltage electrical stimulation induces transmembrane potential across the cell plasma membrane (3). Here, we observed that the cellular p38-p53 pathway responded to a 0.1-ms MES pulse width regardless of electrical input energy (Fig. 1D). The mechanism of how cells receive and react to a specific pulse width of MES is unclear, but our results suggest that the MKK3b-MKK6b-p38 MAPK pathway is a key signaling pathway of MES-induced p53 phosphor ylation. p38 activation is mediated by ATM or other unknown pathways in the DNA damage response (61). However, ATM inhibition did not alter MES-induced p53 phosphorylation (supplemental Fig. S3), implying the involvement of an unidentified electrical pulse-sensing molecule in the stimulation of the MKK3b-MKK6b-p38-p53 pathway by MES. Recently, we showed that MES accumulates a cell membrane receptor, specifically the insulin receptor, in lipid rafts and initiates ligand-independent signal transduction (62). However, lipid raft depletion did not abrogate MES-induced p38 and p53 phosphorylation (supplemental Fig. S5). This result suggests that MES activates the MKK3b-MKK6b-p38-p53 pathway through a lipid raft-independent mechanism. It is plausible that non-lipid raft-associated cell membrane proteins could be similarly affected by MES, such as the EGF receptor, TGF-β receptor, and transient receptor potential channel, which are upstream of the MKK3b-MKK6b-p38 MAPK pathway (63, 64).
Methods of p53 activation such as irradiation and hyperthermia and DNA-damaging reagents are clinically applied for cancer treatment because of the tumor-suppressive functions of p53. Moreover, p53 has fundamental roles not only in tumor suppression but also in suppression of the inflammatory cytokine response and metabolism (31, 65, 66). The influence of p53 on these biological events is independent of its apoptotic functions; thus, the approach to induce p53 activity in this context would be different from that of inducing p53 to promote apoptosis. Interestingly, MES treatment exerted a p53-dependent anti-inflammatory effect on the LPS-stimulated inflammatory cytokine response in mouse peritoneal macrophages (supplemental Fig. S6). This finding indicates that MES-induced mild and transient p53 activation has the potential to attenuate infection-mediated inflammation. Various signals that activate p53 may use divergent pathways, and it is becoming clear that this differential activation of p53 results in vastly different effects. In looking beyond the role of p53 in tumor suppression, we foresee a need to explore various ways to modulate the level of p53 activity. For this reason, the ability of MES to induce a transient, relatively low level of p53 activation may be beneficial for p53-based therapeutic applications that do not necessarily warrant cell killing.
Supplementary Material
Acknowledgment
The HCT116 p53+/+ and HCT116 p53−/− cell lines were provided by Dr. Bert Vogelstein.
This work was supported by Grants-in-aid for Science Research 22390015 (to H. K.) and 23590082 (to M. A. S.) from the Ministry of Education, Science, Sports, and Culture (MEXT) of Japan.

This article contains supplemental Figs. S1–S6 and Table S1.
- MES
- mild electrical stimulation
- 5-FU
- 5-fluorouracil
- DN
- dominant-negative
- pps
- pulses/s
- LDH
- lactate dehydrogenase.
REFERENCES
- 1. McCaig C. D., Rajnicek A. M., Song B., Zhao M. (2005) Controlling cell behavior electrically: current views and future potential. Physiol. Rev. 85, 943–978 [DOI] [PubMed] [Google Scholar]
- 2. Griffin M., Bayat A. (2011) Electrical stimulation in bone healing: critical analysis by evaluating levels of evidence. Eplasty 11, e34. [PMC free article] [PubMed] [Google Scholar]
- 3. Sersa G., Miklavcic D., Cemazar M., Rudolf Z., Pucihar G., Snoj M. (2008) Electrochemotherapy in treatment of tumours. Eur. J. Surg. Oncol. 34, 232–240 [DOI] [PubMed] [Google Scholar]
- 4. Gu G., Zhang Z., Wang G., Han F., Han L., Wang K., Liu J., Li W. (2011) Effects of electroacupuncture pretreatment on inflammatory response and acute kidney injury in endotoxaemic rats. J. Int. Med. Res. 39, 1783–1797 [DOI] [PubMed] [Google Scholar]
- 5. Ainsworth L., Budelier K., Clinesmith M., Fiedler A., Landstrom R., Leeper B. J., Moeller L., Mutch S., O'Dell K., Ross J., Radhakrishnan R., Sluka K. A. (2006) Transcutaneous electrical nerve stimulation (TENS) reduces chronic hyperalgesia induced by muscle inflammation. Pain 120, 182–187 [DOI] [PubMed] [Google Scholar]
- 6. Bassett C. A. (1993) Beneficial effects of electromagnetic fields. J. Cell Biochem. 51, 387–393 [DOI] [PubMed] [Google Scholar]
- 7. Utz K. S., Dimova V., Oppenländer K., Kerkhoff G. (2010) Electrified minds: transcranial direct current stimulation (tDCS) and galvanic vestibular stimulation (GVS) as methods of non-invasive brain stimulation in neuropsychology–a review of current data and future implications. Neuropsychologia 48, 2789–2810 [DOI] [PubMed] [Google Scholar]
- 8. Svirskis D., Travas-Sejdic J., Rodgers A., Garg S. (2010) Electrochemically controlled drug delivery based on intrinsically conducting polymers. J. Control. Release 146, 6–15 [DOI] [PubMed] [Google Scholar]
- 9. Seegers J. C., Engelbrecht C. A., van Papendorp D. H. (2001) Activation of signal-transduction mechanisms may underlie the therapeutic effects of an applied electric field. Med. Hypotheses 57, 224–230 [DOI] [PubMed] [Google Scholar]
- 10. Zhao M., Song B., Pu J., Wada T., Reid B., Tai G., Wang F., Guo A., Walczysko P., Gu Y., Sasaki T., Suzuki A., Forrester J. V., Bourne H. R., Devreotes P. N., McCaig C. D., Penninger J. M. (2006) Electrical signals control wound healing through phosphatidylinositol-3-OH kinase-γ and PTEN. Nature 442, 457–460 [DOI] [PubMed] [Google Scholar]
- 11. Morino S., Kondo T., Sasaki K., Adachi H., Suico M. A., Sekimoto E., Matsuda T., Shuto T., Araki E., Kai H. (2008) Mild electrical stimulation with heat shock ameliorates insulin resistance via enhanced insulin signaling. PLoS ONE 3, e4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yano S., Morino-Koga S., Kondo T., Suico M. A., Koga T., Shimauchi Y., Matsuyama S., Shuto T., Sato T., Araki E., Kai H. (2011) Glucose uptake in rat skeletal muscle L6 cells is increased by low-intensity electrical current through the activation of the phosphatidylinositol-3-OH kinase (PI-3K)/Akt pathway. J. Pharmacol. Sci. 115, 94–98 [DOI] [PubMed] [Google Scholar]
- 13. Morino S., Suico M. A., Kondo T., Sekimoto E., Yano S., Matsuda T., Matsuno T., Shuto T., Araki E., Kai H. (2008) Mild electrical stimulation increases ubiquitinated proteins and Hsp72 in A549 cells via attenuation of proteasomal degradation. J. Pharmacol. Sci. 108, 222–226 [DOI] [PubMed] [Google Scholar]
- 14. Oba M., Suico M. A., Morino S., Yano S., Matsuno T., Koga T., Sato T., Shuto T., Kai H. (2010) Modified mild heat shock modality attenuates hepatic ischemia/reperfusion injury. J. Surg. Res. 162, 213–220 [DOI] [PubMed] [Google Scholar]
- 15. Kai H., Suico M. A., Morino S., Kondo T., Oba M., Noguchi M., Shuto T., Araki E. (2009) A novel combination of mild electrical stimulation and hyperthermia: general concepts and applications. Int. J. Hyperthermia 25, 655–660 [DOI] [PubMed] [Google Scholar]
- 16. Kondo T., Sasaki K., Matsuyama R., Morino-Koga S., Adachi H., Suico M. A., Kawashima J., Motoshima H., Furukawa N., Kai H., Araki E. (2012) Hyperthermia with mild electrical stimulation protects pancreatic beta-cells from cell stresses and apoptosis. Diabetes 61, 838–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kondo T., Sasaki K., Adachi H., Nakayama Y., Hatemura M., Matsuyama R., Tsuzuroe K., Furukawa N., Motoshima H., Morino-Koga S., Yamashita Y., Miyamura N., Kai H., Araki E. (2010) Heat shock treatment with mild electrical stimulation safely reduced inflammatory markers in healthy male subjects. Obesity Res. Clin. Practice 4, e101–e109 [DOI] [PubMed] [Google Scholar]
- 18. Koga T., Kai Y., Fukuda R., Morino-Koga S., Suico M. A., Koyama K., Sato T., Shuto T., Kai H. (2012) Mild electrical stimulation and heat shock ameliorates progressive proteinuria and renal inflammation in mouse model of Alport syndrome. PLoS ONE 7, e43852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Duan Y., Gotoh N., Yan Q., Du Z., Weinstein A. M., Wang T., Weinbaum S. (2008) Shear-induced reorganization of renal proximal tubule cell actin cytoskeleton and apical junctional complexes. Proc. Natl. Acad. Sci. U.S.A. 105, 11418–11423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kirson E. D., Dbalý V., Tovarys F., Vymazal J., Soustiel J. F., Itzhaki A., Mordechovich D., Steinberg-Shapira S., Gurvich Z., Schneiderman R., Wasserman Y., Salzberg M., Ryffel B., Goldsher D., Dekel E., Palti Y. (2007) Alternating electric fields arrest cell proliferation in animal tumor models and human brain tumors. Proc. Natl. Acad. Sci. U.S.A. 104, 10152–10157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kirson E. D., Gurvich Z., Schneiderman R., Dekel E., Itzhaki A., Wasserman Y., Schatzberger R., Palti Y. (2004) Disruption of cancer cell replication by alternating electric fields. Cancer Res. 64, 3288–3295 [DOI] [PubMed] [Google Scholar]
- 22. Hirao A., Kong Y. Y., Matsuoka S., Wakeham A., Ruland J., Yoshida H., Liu D., Elledge S. J., Mak T. W. (2000) DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 287, 1824–1827 [DOI] [PubMed] [Google Scholar]
- 23. Sherr C. J., Weber J. D. (2000) The ARF/p53 pathway. Curr. Opin. Genet. Dev. 10, 94–99 [DOI] [PubMed] [Google Scholar]
- 24. Lowe S. W., Lin A. W. (2000) Apoptosis in cancer. Carcinogenesis 21, 485–495 [DOI] [PubMed] [Google Scholar]
- 25. Miyakoda M., Suzuki K., Kodama S., Watanabe M. (2002) Activation of ATM and phosphorylation of p53 by heat shock. Oncogene 21, 1090–1096 [DOI] [PubMed] [Google Scholar]
- 26. Saretzki G., Sitte N., Merkel U., Wurm R. E., von Zglinicki T. (1999) Telomere shortening triggers a p53-dependent cell cycle arrest via accumulation of G-rich single-stranded DNA fragments. Oncogene 18, 5148–5158 [DOI] [PubMed] [Google Scholar]
- 27. Nigro J. M., Aldape K. D., Hess S. M., Tlsty T. D. (1997) Cellular adhesion regulates p53 protein levels in primary human keratinocytes. Cancer Res. 57, 3635–3639 [PubMed] [Google Scholar]
- 28. Mayr M., Hu Y., Hainaut H., Xu Q. (2002) Mechanical stress-induced DNA damage and Rac-p38MAPK signal pathways mediate p53-dependent apoptosis in vascular smooth muscle cells. FASEB J. 16, 1423–1425 [DOI] [PubMed] [Google Scholar]
- 29. Kruse J. P., Gu W. (2009) Modes of p53 regulation. Cell 137, 609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vousden K. H., Lu X. (2002) Live or let die: the cell's response to p53. Nat. Rev. Cancer 2, 594–604 [DOI] [PubMed] [Google Scholar]
- 31. Vousden K. H., Ryan K. M. (2009) p53 and metabolism. Nat. Rev. Cancer 9, 691–700 [DOI] [PubMed] [Google Scholar]
- 32. Menendez D., Inga A., Resnick M. A. (2009) The expanding universe of p53 targets. Nat. Rev. Cancer 9, 724–737 [DOI] [PubMed] [Google Scholar]
- 33. Liu G., Park Y. J., Tsuruta Y., Lorne E., Abraham E. (2009) p53 Attenuates lipopolysaccharide-induced NF-κB activation and acute lung injury. J. Immunol. 182, 5063–5071 [DOI] [PubMed] [Google Scholar]
- 34. Müller-Ladner U., Nishioka K. (2000) p53 in rheumatoid arthritis: friend or foe? Arthritis Res. 2, 175–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murphy S. H., Suzuki K., Downes M., Welch G. L., De Jesus P., Miraglia L. J., Orth A. P., Chanda S. K., Evans R. M., Verma I. M. (2011) Tumor suppressor protein (p)53 is a regulator of NF-κB repression by the glucocorticoid receptor. Proc. Natl. Acad. Sci. U.S.A. 108, 17117–17122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Komarova E. A., Krivokrysenko V., Wang K., Neznanov N., Chernov M. V., Komarov P. G., Brennan M. L., Golovkina T. V., Rokhlin O. W., Kuprash D. V., Nedospasov S. A., Hazen S. L., Feinstein E., Gudkov A. V. (2005) p53 is a suppressor of inflammatory response in mice. FASEB J. 19, 1030–1032 [DOI] [PubMed] [Google Scholar]
- 37. Coppé J. P., Patil C. K., Rodier F., Sun Y., Muñoz D. P., Goldstein J., Nelson P. S., Desprez P. Y., Campisi J. (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Taura M., Eguma A., Suico M. A., Shuto T., Koga T., Komatsu K., Komune T., Sato T., Saya H., Li J. D., Kai H. (2008) p53 regulates Toll-like receptor 3 expression and function in human epithelial cell lines. Mol. Cell. Biol. 28, 6557–6567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Taura M., Suico M. A., Fukuda R., Koga T., Shuto T., Sato T., Morino-Koga S., Okada S., Kai H. (2011) MEF/ELF4 transactivation by E2F1 is inhibited by p53. Nucleic Acids Res. 39, 76–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shuto T., Xu H., Wang B., Han J., Kai H., Gu X. X., Murphy T. F., Lim D. J., Li J. D. (2001) Activation of NF-κB by nontypeable Hemophilus influenzae is mediated by Toll-like receptor 2-TAK1-dependent NIK-IKKα/β-IκBα and MKK3/6-p38 MAP kinase signaling pathways in epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 98, 8774–8779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Suico M. A., Yoshida H., Seki Y., Uchikawa T., Lu Z., Shuto T., Matsuzaki K., Nakao M., Li J. D., Kai H. (2004) Myeloid Elf-1-like factor, an ETS transcription factor, up-regulates lysozyme transcription in epithelial cells through interaction with promyelocytic leukemia protein. J. Biol. Chem. 279, 19091–19098 [DOI] [PubMed] [Google Scholar]
- 42. Yugawa T., Handa K., Narisawa-Saito M., Ohno S., Fujita M., Kiyono T. (2007) Regulation of Notch1 gene expression by p53 in epithelial cells. Mol. Cell. Biol. 27, 3732–3742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oba M., Yano S., Shuto T., Suico M. A., Eguma A., Kai H. (2008) IFN-γ down-regulates Hsp27 and enhances hyperthermia-induced tumor cell death in vitro and tumor suppression in vivo. Int. J. Oncol. 32, 1317–1324 [DOI] [PubMed] [Google Scholar]
- 44. Beebe S. J., Fox P. M., Rec L. J., Willis E. L., Schoenbach K. H. (2003) Nanosecond, high-intensity pulsed electric fields induce apoptosis in human cells. FASEB J. 17, 1493–1495 [DOI] [PubMed] [Google Scholar]
- 45. Kishi H., Nakagawa K., Matsumoto M., Suga M., Ando M., Taya Y., Yamaizumi M. (2001) Osmotic shock induces G1 arrest through p53 phosphorylation at Ser33 by activated p38MAPK without phosphorylation at Ser15 and Ser20. J. Biol. Chem. 276, 39115–39122 [DOI] [PubMed] [Google Scholar]
- 46. Freund A., Patil C. K., Campisi J. (2011) p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 30, 1536–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. She Q. B., Chen N., Dong Z. (2000) ERKs and p38 kinase phosphorylate p53 protein at serine 15 in response to UV radiation. J. Biol. Chem. 275, 20444–20449 [DOI] [PubMed] [Google Scholar]
- 48. Fuchs S. Y., Adler V., Pincus M. R., Ronai Z. (1998) MEKK1/JNK signaling stabilizes and activates p53. Proc. Natl. Acad. Sci. U.S.A. 95, 10541–10546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Banin S., Moyal L., Shieh S., Taya Y., Anderson C. W., Chessa L., Smorodinsky N. I., Prives C., Reiss Y., Shiloh Y., Ziv Y. (1998) Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 281, 1674–1677 [DOI] [PubMed] [Google Scholar]
- 50. Bunz F., Dutriaux A., Lengauer C., Waldman T., Zhou S., Brown J. P., Sedivy J. M., Kinzler K. W., Vogelstein B. (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501 [DOI] [PubMed] [Google Scholar]
- 51. Miyashita T., Reed J. C. (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80, 293–299 [DOI] [PubMed] [Google Scholar]
- 52. Oda E., Ohki R., Murasawa H., Nemoto J., Shibue T., Yamashita T., Tokino T., Taniguchi T., Tanaka N. (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288, 1053–1058 [DOI] [PubMed] [Google Scholar]
- 53. Nakano K., Vousden K. H. (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7, 683–694 [DOI] [PubMed] [Google Scholar]
- 54. Muñoz-Fontela C., Macip S., Martínez-Sobrido L., Brown L., Ashour J., García-Sastre A., Lee S. W., Aaronson S. A. (2008) Transcriptional role of p53 in interferon-mediated antiviral immunity. J. Exp. Med. 205, 1929–1938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Balakatounis K. C., Angoules A. G. (2008) Low-intensity electrical stimulation in wound healing: review of the efficacy of externally applied currents resembling the current of injury. Eplasty 8, e28. [PMC free article] [PubMed] [Google Scholar]
- 56. Cucullo L., Dini G., Hallene K. L., Fazio V., Ilkanich E. V., Igboechi C., Kight K. M., Agarwal M. K., Garrity-Moses M., Janigro D. (2005) Very low intensity alternating current decreases cell proliferation. Glia 51, 65–72 [DOI] [PubMed] [Google Scholar]
- 57. Agarwal M. L., Agarwal A., Taylor W. R., Stark G. R. (1995) p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc. Natl. Acad. Sci. U.S.A. 92, 8493–8497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hoeferlin L. A., Oleinik N. V., Krupenko N. I., Krupenko S. A. (2011) Activation of p21-dependent G1/G2 arrest in the absence of DNA damage as an antiapoptotic response to metabolic stress. Genes Cancer 2, 889–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Clotet J., Escoté X., Adrover M. A., Yaakov G., Garí E., Aldea M., de Nadal E., Posas F. (2006) Phosphorylation of Hsl1 by Hog1 leads to a G2 arrest essential for cell survival at high osmolarity. EMBO J. 25, 2338–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Feng L., Hollstein M., Xu Y. (2006) Ser46 phosphorylation regulates p53-dependent apoptosis and replicative senescence. Cell Cycle 5, 2812–2819 [DOI] [PubMed] [Google Scholar]
- 61. Thornton T. M., Rincon M. (2009) Non-classical p38 MAP kinase functions: cell cycle checkpoints and survival. Int. J. Biol. Sci. 5, 44–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Morino-Koga S., Yano S., Kondo T., Shimauchi Y., Matsuyama S., Okamoto Y., Suico M. A., Koga T., Sato T., Shuto T., Arima H., Wada I., Araki E., Kai H. (2013) Insulin receptor activation through its accumulation in lipid rafts by mild electrical stress. J. Cell. Physiol. 228, 439–446 [DOI] [PubMed] [Google Scholar]
- 63. Lambert S., Ameels H., Gniadecki R., Hérin M., Poumay Y. (2008) Internalization of EGF receptor following lipid rafts disruption in keratinocytes is delayed and dependent on p38 MAPK activation. J. Cell. Physiol. 217, 834–845 [DOI] [PubMed] [Google Scholar]
- 64. Kondo T., Sakurai J., Miwa H., Noguchi K. (2013) Activation of p38 MAPK through transient receptor potential A1 in a rat model of gastric distension-induced visceral pain. Neuroreport 24, 68–72 [DOI] [PubMed] [Google Scholar]
- 65. Simelyte E., Rosengren S., Boyle D. L., Corr M., Green D. R., Firestein G. S. (2005) Regulation of arthritis by p53: critical role of adaptive immunity. Arthritis Rheum. 52, 1876–1884 [DOI] [PubMed] [Google Scholar]
- 66. Sen N., Satija Y. K., Das S. (2012) p53 and metabolism: old player in a new game. Transcription 3, 119–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.