

1. Introduction
In the discipline of pediatric cancer biology, neuroblastoma signifies an oncologic conundrum given the clinical range with which it presents. Prognosis correlates with age and the degree of differentiation, and thus, outcomes vary from high rates of survival (with even possible tumor regression) to recurrence and mortality. While the standard of treatment is chemotherapy, radiation, and/or surgical resection, there is growing evidence that aggressive neuroblastomas are resistant to our therapies. To this end, research has been focused on the molecular mechanisms behind differentiation, cell survival and apoptosis, angiogenesis, and metastasis to elucidate where the process goes awry. The basis of this research has led to the development of novel therapies that are directed towards key targets, some of which are quite promising. While discussing clinical background, this article aims to provide a synopsis of the latest, up and coming developments in the field of neuroblastoma.
2. Epidemiology
Neuroblastoma is the most common extra-cranial solid tumor in infants and children, representing 8%-10% of all childhood tumors. It accounts for approximately 15% of all cancer-related deaths in the pediatric population.1 The incidence of neuroblastoma is 10.2 cases per million children under 15 years of age,2 and nearly 500 new cases are reported annually. While 90% of cases are diagnosed before the age of 5, 30% of those are within the first year. The median age of diagnosis is 22 months.3 Rarely does it present in adolescence and adulthood, but outcomes are much poorer in this age group. There does not appear to be an increased prevalence among races, but there is a slight predilection for males (1.2:1).1
With a family history noted in 1-2% of diagnoses, there are reports of autosomal dominant patterns of inheritance.3 In such pedigrees, patients are frequently diagnosed at an earlier age (median age of 9 months) than those with sporadic disease and are more likely to have associated multiple primary cancers. Neuroblastoma has also been diagnosed in conjunction with other congenital conditions such as Hirschsprung's disease, congenital hypoventilation disorder, and neurofibromatosis type 1.1 There was early interest in the co-occurrence of neuroblastoma and neurofibromatosis, as they are both disorders of neural crest cells. However, this may represent coincidence rather than a true association.4
Outcomes in patients with neuroblastoma have improved steadily over the last 30 years with 5-year survival rates rising from 52% to 74%.2 The improvement is likely secondary to improved cure rates in the low-risk group, who have survival rates of up to 92%. It is estimated that 50-60% of patients in the high-risk group experience relapse,2 and as such, they have only seen a modest decrease in mortality. In a study by the International Neuroblastoma Risk Group, the median time to relapse was 13.2 months, and 73% of those who relapsed were 18 months or older. Taken together, their overall survival rates remain quite abysmal (∼20% at 5 years) despite more aggressive therapies.5
3. Clinical Presentation
Multiple factors play into a patient's clinical presentation since it depends largely on tumor location, size, degree of invasion, effects from catecholamine secretion, and symptoms due to paraneoplastic syndromes. Nearly 65% of tumors arise in the abdomen with half of those localized to the medulla of the adrenal gland. However, they can occur in the neck (5%), chest (20%), or pelvis (5%), and 1% of patients have no detectable primary.1, 6 Many patients are asymptomatic, yet some may present with constitutional symptoms (malaise, fevers, and weight loss), an enlarging mass, pain, abdominal distension, lymphadenopathy, or respiratory distress secondary to compression or hepatomegaly. Pelvic masses may cause constipation or difficulty urinating, while thoracic involvement can cause dysphagia, dyspnea, or rarely, thoracic outlet syndrome.7 For cervical tumors, a patient may develop Horner's syndrome, and in up to 15% of patients, epidural extension may result in neurological deficits such as progressive paralysis.1
At the time of diagnosis, 50% of patients present with localized disease while 35% already have regional lymph node spread.1 Metastasis can occur by hematogenous and/or lymphatic route, seeding bone marrow, liver, and bone. Commonly, the orbits are involved, which manifests as periorbital swelling and proptosis (“raccoon eyes”). When dissemination occurs to the skin, patients develop blue subcutaneous nodules known as blueberry muffin syndrome. Surprisingly, this is associated with a favorable prognosis with likely spontaneous tumor regression.
Because of its neuroendocrine properties, neuroblastoma has the potential to secrete catecholamines, which results in early-onset hypertension and tachycardia. Patients may also experience paraneoplastic syndromes. Examples include intractable diarrhea with electrolyte disturbances due to release of vasoactive intestinal peptide (VIP), encephalomyelitis, or sensory neuropathy. There have been reports of the development of opsoclonus-myoclonus syndrome (OMS), which occurs when antibodies cross-react with cerebellar tissue.8 The characteristic symptoms and signs of OMS include rapid, conjugate eye nystagmus with involuntary spasms of the limbs. Interestingly, the patients with intractable diarrhea due to VIP secretion or OMS generally tend to present with less aggressive neuroblastomas. Nonetheless, symptomatic paraneoplastic syndromes are quite rare and are estimated to occur in < 0.01% of all cancers.9
4. Classification and Staging
Neuroblastoma belongs to a group collectively known as peripheral neuroblastic tumors, which also includes intermixed ganglioneuroblastoma, ganglioneuroma, and nodular ganglioneuroblastoma. Neuroblastoma can further be divided based on the degree of neuroblastic differentiation (undifferentiated, poorly differentiated, and differentiating) and the mitosis-karyorrhexis index (MKI) (low, intermediate, or high).10 Histologically, it has limited Schwannian cell production, is stroma-poor, and has abundant neuroblasts.8
The International Neuroblastoma Pathology Classification has been used to predict prognosis based on the histopathology of the tumor and age of the patient. This system takes into account the degree of cell differentiation, MKI, and the presence of Schwann cells. Following these guidelines, the unfavorable group encompasses patients with any tumor over 60 months; undifferentiated tumors with a high MKI at any age; and undifferentiated or poorly differentiated tumors with intermediate or high MKI in children older than 18 months.11
Staging is dictated by the International Neuroblastoma Staging System (INSS) (Table 1). The Children's Oncology Group (COG) currently stratifies patients into low-, intermediate-, or high-risk categories based on the patient's age at diagnosis, INSS stage, tumor histopathology, DNA index, and MYCN amplification status (Table 2).12 Since the INSS is based on tumor resection, a new system, called the International Neuroblastoma Risk Group Staging System (INRGSS), was introduced in hopes of better stratifying pretreatment patient risk based on clinical criteria and image-defined risk factors (Fig. 1). This staging system classifies neuroblastoma into L1 (localized disease that does not involve vital structures and is confined to one body compartment); L2 (localized disease with image-defined risk factors); M (distant metastatic disease); and MS (metastatic disease confined to the skin, liver and/or bone marrow in children < 18 months of age).13 Based on this, patients can be sorted into pretreatment very low-, low-, intermediate-, and high-risk groups.14 This has implications as to what treatment algorithm is recommended.
Table 1. The International Neuroblastoma Staging System.
| Stage 1 | Localized tumor with complete gross excision with or without microscopic residual disease; ipsilateral and contralateral LN negative for tumor microscopically |
| Stage 2A | Unilateral tumor with incomplete gross resection; ipsilateral and contralateral LN negative for tumor microscopically |
| Stage 2B | Unilateral tumor with or without complete gross excision with ipsilateral LN positive for tumor; contralateral LN negative microscopically |
| Stage 3 | Tumor infiltrating across the midline with or without regional LN involvement, localized unilateral tumor with contralateral regional LN involvement, or midline tumor with bilateral extension by infiltration (unresectable) or by LN involvement |
| Stage 4 | Any primary tumor with dissemination to distant LN, bone, bone marrow, liver, skin, or other organs (except as defined for stage 4S) |
| Stage 4S | Localized primary tumor (as defined for stage 1 or 2) with dissemination limited to skin, liver, or bone marrow (limited to infants <1 yr of age) |
LN: lymph nodes
Table 2. Children's Oncology Group Risk Stratification.
| INSS | Age | MYCN Status | Shimada | DNA Index | Risk Group |
|---|---|---|---|---|---|
| 1 | 0-21 yrs | Any | Any | Any | Low |
| 2A/2B | < 1 yr | Any | Any | Any | Low |
| ≥ 1 - 21 yrs | Normal | Any | – | Low | |
| ≥ 1 - 21 yrs | Amplified | Favorable | – | Low | |
| ≥ 1 - 21 yrs | Amplified | Unfavorable | – | High | |
| 3 | < 1 yr | Normal | Any | Any | Intermediate |
| < 1 yr | Amplified | Any | Any | High | |
| ≥ 1 - 21 yrs | Normal | Favorable | – | Intermediate | |
| ≥ 1 - 21 yrs | Normal | Unfavorable | – | High | |
| ≥ 1 - 21 yrs | Amplified | Any | – | High | |
| 4 | < 1 yr | Normal | Any | Any | Intermediate |
| < 1 yr | Amplified | Any | Any | High | |
| ≥ 1 - 21 yrs | Any | Any | – | High | |
| 4S | < 1 yr | Normal | Favorable | >1 | Low |
| < 1 yr | Normal | Any | =1 | Intermediate | |
| < 1 yr | Normal | Unfavorable | Any | Intermediate | |
| < 1 yr | Amplified | Any | Any | High |
INSS: International Neuroblastoma Staging System
Figure 1.
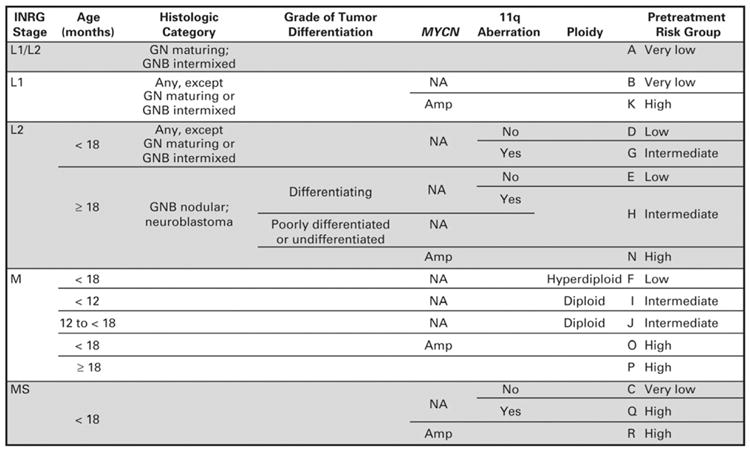
International Neuroblastoma Risk Group (INRG) Classification. L1, localized tumor confined to one body compartment and with absence of image-defined risk factors (IDRFs); L2, locoregional tumor with presence of one or more IDRFs; M, distant metastatic disease (except stage MS); MS, metastatic disease confined to skin, liver and/or bone marrow in children < 18 months of age; EFS, event-free survival (From Cohn et al, The International Neuroblastoma Risk Group (INRG) Classification System: An INRG Task Force Report.J Clin Onc; 2009; Fig. 2, p.295; with permission).
5. Genomics
From a genomic standpoint, neuroblastoma occurs in the setting of chromosomal losses and gains. DNA mutations can fall into two prognostic categories: whole-chromosome gains and segmental chromosomal aberrations. The former results in hyperdiploidy that is associated with a favorable prognosis, while the latter is characterized by MYCN amplification and regional gains or losses that tend to be associated with worse outcomes.
It has been shown that deletion of the chromosome 1p36 region occurs in 70% of tumors and may correlate with an increased risk of relapse in patients with localized tumors.15, 16 Interestingly, a promising candidate suppressor gene in the 1p36 region is CHD5, which is expressed in the nervous system and controls cellular proliferation, senescence, and apoptosis. Studies have confirmed that CHD5 expression is low in neuroblastoma tumors that have a chromosome 1p deletion, and when these cells were transfected with CHD5, clonogenicity and tumor growth were decreased.17 Whether this has any prognostic significance is unknown, but it does appear to play a role in neuroblastoma tumorigenesis.
Gain of chromosome 17q occurs in approximately 80% of neuroblastomas, making it the most common genetic aberration. Whereas whole-chromosome 17 gain is associated with a good prognosis,16 unbalanced translocations of chromosome 17q with either 1p or 11q have been reported to be an independent poor prognostic factor. Likewise, deletions of chromosome 11q have been identified in 15–22% of neuroblastomas and are also associated with unfavorable patient outcomes and a lessened time of progression-free survival.8 Interestingly, chromosome 11q and 3p deletions are often found together and occur in high-stage tumors without MYCN amplification that is typical of aggressive neuroblastoma.16 With 3p deletions frequently noted in patients who are older at the time of diagnosis (median age of 60.5 months), some speculate that 3p loss is an event that occurs later in oncogenesis.18
In instances of familial neuroblastoma, recent evidence points to mutations of the anaplastic lymphoma kinase (ALK) oncogene as the putative cause. ALK is a tyrosine kinase receptor whose expression is important in neural differentiation, proliferation, and survival. Its malignant transformation is thought to arise from missense mutations that are linked to chromosomal region 2p23-24 and result in constitutive kinase activity. However, ALK can also be mutated at specific hotspots, such as R1275Q and F1174L, in up to 12% of sporadic cases, with the latter mutation being the most common.19 Currently, efforts are being made to determine the pathways by which ALK exerts its effects and whether inhibitors of ALK would be feasible therapeutically.
Lastly, the MYCN oncogene, which is amplified at chromosome 2p24 in 25% of cases, is used as a biomarker for disease stratification. MYCN amplification is defined as ≥10 gene copies per nuclei since tumors with fewer than this do not behave as aggressively.20 MYCN amplification is found in 30–40% of stage 3 and 4 neuroblastomas, but only in 5% of localized or stage 4S neuroblastomas. It encodes for multiple transcription factors that, when overexpressed, lead to deregulated growth and proliferation.4 Several MYCN downstream targets have been identified, including p53, Aurora A kinase, and MDM2.21 However, the exact mechanisms remain unknown.
6. Diagnosis
Based on clinical presentation, the index of suspicion must be high. Initial diagnostic testing should include basic blood work (complete blood count, serum electrolytes, liver function tests) and a chest radiograph, which may reveal calcifications or a posterior mediastinal mass. Additional diagnostic findings will include increased levels of urine or serum catecholamines or catecholamine metabolites (dopamine, vanillylmandelic acid, and homovanillic acid). Patients may have elevated levels of nonspecific biomarkers such as lactate dehydrogenase (>1500 U/ml), ferritin (>142 ng/ml), and neuron-specific enolase (>100 ng/ml) that may be associated with advanced stage and/or relapse.15
For diagnostic imaging, a computed tomography scan of the neck, chest, and/or abdomen is the gold standard, as it can simultaneously localize the tumor and determine the degree of involvement (Fig. 1). Ultrasound may be used initially to characterize the mass. A magnetic resonance imaging may be useful if there is concern for spinal extension, and imaging of the brain is only necessary in the setting of neurological symptoms. While not routinely used, a 123/131iodine-meta-iodobenzylguanidine (MIBG) scan is valuable in both the detection of primary tumor and metastases since metaiodobenzylguanidine, a norepinephrine analog, is selectively concentrated in sympathetic tissue. It has also proven to be practical in surveillance of treatment response and recurrence. The role of fluorodeoxyglucose positive emission tomography (FDG-PET) remains controversial, but a small retrospective study has suggested that while MIBG is generally more sensitive for the detection of lesions, FDG-PET may be better at localizing soft tissue metastases.22
Despite advances in diagnostic medicine, the diagnosis of neuroblastoma can only be confirmed pathologically with tissue obtained from tumor or bone marrow. Specimens can be obtained either during resection of the primary tumor or as an open biopsy for unresectable disease. Bilateral posterior iliac crest marrow aspirates are required to exclude metastatic disease. Molecular studies, such as fluorescent in situ hybridization (FISH), can be performed on tissue samples to note ploidy, MYCN amplification, and the presence of other chromosomal aberrations.
7. Treatment
The mainstay of treatment consists of chemotherapy, surgical resection, and/or radiotherapy. However, many aggressive neuroblastomas have developed resistance to chemotherapeutic agents, making the likelihood of relapse quite high. Treatment can be hindered by aggressive neuroblastomas that exhibit drug resistance, and this may be related to the selection of clones that express MRP1 (multidrug resistant-associated protein).23 The treatment algorithm is dependent on the patient's stage and risk stratification. The goal of induction chemotherapy is to achieve remission by reducing the tumor burden, which then allows for a more complete resection when indicated. Induction chemotherapy consists of some combination of cyclophosphamide, doxorubicin, cisplatin, melphalan, carboplatin, etoposide, topotecan, ifosfamide, and vincristine. After induction, treatment is consolidated with one or more courses of high-dose chemotherapy to induce bone marrow ablation, which necessitates autologous hematopoietic stem cell support. Rescue is not without complication as it can lead to growth failure, endocrinopathy, and the occurrence of secondary malignancies.24
For high-risk patients, radiotherapy is needed for local and metastatic control. It is indicated when there is minimal residual disease post induction chemotherapy and resection. Current radiation protocols utilize a dose of 2100 cGy, and based on the practices at the Memorial Sloan-Kettering Cancer Center, the combination of chemotherapy, surgery, and radiation therapy has resulted in a local relapse rate of <10%.20 Radiation is contraindicated for intraspinal tumors because it can lead to vertebral damage, growth arrest, and scoliosis. Yet, it may be necessary for palliation in the setting of pain or acute neurological symptoms due to cord compression.8
Surgery is recommended based on the ability to obtain complete resection, which is itself dependent on location and invasiveness of the tumor. In preoperative planning, important considerations include tumor size, extent of adherence and/or extension into adjacent structures, and the likelihood of surgical cure. Resection is correlated with a reduced risk of local recurrence, especially in combination with induction chemotherapy and local radiotherapy. Thus, surgical resection should be pursued only after adjuvant therapy because it both increases the degree of complete excision and decreases morbidity. For stage 1-2B tumors, resection is recommended. For more advanced-stage neuroblastoma (stages 3 and 4), surgical intervention is limited to an open biopsy. For those infants who are stage 4S, surgical resection is not recommended since these tumors tend to spontaneously differentiate and regress.
Presently, patients receive six courses of 13-cis-retinoic acid (CRA) to eradicate residual disease that may still be present despite meeting imaging criteria for complete remission. This treatment is based on the finding that high-dose therapy with 13-cis-retinoic acid given after chemoradiation significantly improved event-free survival in high-risk neuroblastoma. Side effects, such as skin dryness and cheilitis, are the dose-limiting factor, and consequently, CRA therapy consists of 2-week courses alternating with 2 weeks for mucocutaneous recovery.25
Trials involving myeloablative chemotherapy and 131I-MIBG have been underway in an effort to minimize adverse side effects by making therapies more targeted. Previous studies have shown that 131I-MIBG exhibits activity against refractory neuroblastoma with response rates ranging from 10-50%.26 In a phase I trial of 131I-MIBG therapy for relapsed neuroblastoma, myelosuppression was the most significant toxicity at doses >15 mCi/kg, as nearly half of the patients enrolled required hematopoietic cell transfusion. Despite this, the response rate (36%), event-free survival (18% at 1 year), and overall survival (49% at 1 year; 29% at 2 years) were found to be significantly higher in patients older than 12 years and who had fewer than three prior treatment regimens.27 Subsequently, a phase I dose escalation study of 131I-MIBG with myeloablative chemotherapy and stem cell rescue showed a significant response rate of 25% in patients with primary refractory disease. Given these findings, 131I-MIBG may prove to be useful in conjunction with other treatment modalities.
The total length of therapy averages nearly one year, and most treatment failures are due to minimal residual disease that was not eradicated following high-dose chemotherapy. While the aim of further treatment is remission, prolonged disease stabilization is usually the reality seeing as how most patients who relapse eventually die from disease progression. Even patients who achieve a cure with initial therapy remain at risk for developing long-term complications related to treatment, including hearing loss, infertility, and second malignancies.28
8. Current Research Milestones and Proposed Novel Therapies
A. Retinoic Acid
Retinoids are vitamin A derivatives that can cause arrest of cell growth and induce differentiation of human neuroblastoma cells. While 13-cis-retinoic acid is being used clinically for this purpose, fenretinide (4-HPR) is another retinoid derivative currently in phase I trials and has been reported to inhibit growth in vitro in a dose-dependent manner. At the higher end of the dose spectrum, it also seems to be active against retinoic acid-resistant neuroblastoma cell lines that harbor mutations in the retinoic acid receptor (RAR). Unlike all-trans and 13-cis retinoic acids, 4-HPR does not induce differentiation, but is actually cytotoxic and pro-apoptotic and may do so in a receptor-independent manner.25, 29 So far, it appears that 4-HPR is minimally toxic with no evidence of myelosuppression; the most common side effect reported is decreased night vision.
B. mTOR inhibitors
One promising target is the mammalian target of rapamycin (mTOR), which is a serine/threonine protein kinase that regulates cell growth, proliferation, motility, metabolism, and survival. It can exert its effects through several different signaling pathways, notably the phosphatidylinositol-3 kinase (PI3K)-AKT pathway. Rapamycin, a bacterial byproduct, is the classically cited mTOR inhibitor, and it does so by complexing with the intracellular receptor FKBP12 to bind directly to the FKBP12-rapamycin binding (FRB) domain on mTOR.30 This leads to activation of downstream effectors, such as cyclin D1, p21, and HIF1a/b. Treatment with mTOR inhibitors leads to downregulation of MYCN and vascular endothelial growth factor (VEGF). Temsirolimus, a rapamycin analogue, is being investigated in phase II trials in patients with relapsed neuroblastoma. One study showed that treatment with an insulin-like growth factor (IGF)-1 receptor inhibitor and temsirolimus in vitro was more effective at inducing cell death than either agent alone.31 Known side effects include skin rashes, stomatitis, and hyperglycemia.
C. Aurora A Kinase
Aurora A kinase is a serine/threonine kinase important in regulating progression through the cell cycle, particularly during the G2 to M phase transition. Aurora A kinase has an important role in centrosome maturation, spindle assembly, meiotic maturation, and spindle orientation. Selective inhibition of Aurora A kinase results in inhibition of autophosphorylation and p53 phosphorylation, monopolar spindles, and G2-M arrest.32 MLN8237, a reversible Aurora A kinase inhibitor, is being investigated in phase I clinical trials by the COG for patients who have experienced relapse. In vitro and in vivo studies with MLN8237 have shown that in addition to inducing apoptosis, it upregulates p53 and the tumor suppressor genes p21 and p27.33 Dose-escalation and combination therapy studies are currently underway.
D. TrkB Inhibitors
The Trk family is a group of tropomyosin kinases that are known to modulate neuronal differentiation and survival. There are 3 isoforms (TrkA, TrkB, and TrkC) that each has an affinity for a specific ligand (NGF, BDNF, and NT-3 respectively). Ligand binding causes receptor homodimerization, which leads to autophosphorylation, docking of downstream effectors, and activation of PI3K/AKT, phospholipid C (PLC) γ1, and Ras/mitogen-activated protein kinase (MAPK) signaling.34 Their ligands can similarly bind p75, a nonselective and low-affinity death receptor, to induce apoptosis via the c-Jun amino terminal kinase (JNK) pathway.35 While TrkA expression is associated with favorable prognosis, TrkB has been found to correlate with MYCN amplification and clinically aggressive neuroblastoma. In studies, neuroblastoma tumor cells treated with brain-derived nerve factor (BDNF) appear to be less sensitive to cytotoxic drugs, survive in a less than optimal microenvironment, are more invasive, and produce increased levels of VEGF. Furthermore, inhibitors were found to slow growth and induce apoptosis. Thus, the Trk inhibitor CEP-701 is presently being used as an oral phase I agent with the premise that it will be most effective in conjunction with conventional chemotherapeutic agents.
E. Immunotherapy
The ganglioside GD2 is expressed in the surface of tumors of neuroectodermal origin, including neuroblastoma. Anti-GD2 monoclonal antibodies have been developed because they are specific, high affinity, and fairly nontoxic. They kill tumor cells through both complement and cell-mediated lysis. 3F8 is a murine IgG3 monoclonal antibody that has been shown to activate complement on malignant neutralists with ensuing lysis. Moreover, it is thought that granulocyte-macrophage colony-stimulating factor (GM-CSF) can potentially amplify 3F8 antitumor activity by increasing granulocytes.36 However, its clinical use has been complicated by the formation of human anti-mouse antibodies. To circumvent this, chimeric mouse-human antibodies, such as Ch14.18 (a monoclonal antibody against the tumor-associated GD2), have been generated. While they were thought to be less immunogenic, 28% of patients reportedly still developed antibodies in a phase I trial.37 In other studies, beta-glucan has been found to enhance anti-GD2 antibody anti-tumor effects through increasing iC3b-mediated cytotoxicity. An ongoing phase I trial is comparing the efficacy of 3F8 when given with oral beta-glucan. Other trials are focused on using Ch14.18 in conjunction with other agents, such as interleukin-2 and GM-CSF, after high-dose chemotherapy with stem-cell rescue. A recent study demonstrated that immunotherapy with Ch14.18, GM-CSF, and interleukin-2 was found to be associated with significantly improved overall outcome when compared to standard therapy in high-risk group neuroblastoma patients.38
F. Angiogenesis inhibitors
VEGF has been established as an important growth factor that promotes tumor angiogenesis by inducing endothelial cell proliferation. It has been found to play a significant role in tumorigenesis because in hypoxic environments, such as those in rapidly growing tumors, hypoxia-inducing factor (HIF) stimulates VEGF production. This results in downstream activation of several signaling pathways, including PI3K/AKT (increased vascular permeability) and MAPK/ERK (endothelial cell proliferation). In neuroblastoma murine models, VEGF inhibitors were found to decrease cell proliferation.39 Bevacizumab, a human-derived VEGF monoclonal antibody, is FDA-approved for the treatment of colorectal, breast, and lung cancer, and it is now being used in phase II studies in neuroblastoma, as it has not previously been used in children.
G. MDM2 and ODC1 as a MYCN targets
p53 mutations are rare in neuroblastoma, and several studies have shown that it appears to be functional since it is capable of activating downstream apoptotic effectors in response to DNA damage.40 MDM2 is a negative regulator of p53, being a key player in its ubiquitination. It has been suggested that deregulated MDM2 expression may explain aberrant p53 function. Notably, MDM2 is Tran activated by MYCN, and downregulation of MYCN leads to decreased MDM2 expression, p53 stabilization, and cell death. Therefore, dysregulation of MDM2 is thought to decrease p53 activity, and in vitro studies with the MDM2 inhibitor nutlin showed that it could stabilize p53 and induce G1 cell cycle arrest and apoptosis.41, 42 Further investigations are needed, but this class of agents may represent an untapped resource.
Recently, ODC1, an oncogene that encodes for an enzyme needed in polyamine biosynthesis, was tagged as a potential therapeutic candidate. Polyamines enhance transcription, translation, and replication and are, therefore, essential for cell survival as depletion leads to growth arrest and apoptosis. MYCN functions as a transcription factor that dimerizes with another factor known as MAX to form a complex that can then activate ODC1 transcription.43MYCN-amplified tumors have elevated levels of ODC1 and exhibit deregulated polyamine synthesis. In mouse models, studies revealed that treatment with cyclophosphamide and a-difluoromethylornithine (DFMO), an ODC inhibitor, led to an increased overall survival (80% vs. 20% at 3 months) than cyclophosphamide alone.44 It is now being piloted in phase I clinical trials with and without etoposide administration.
H. GRP receptor
Gastrin-releasing peptide (GRP) receptors, a member of G-protein coupled receptor family, are abundantly expressed in undifferentiated neuroblastomas. As in other well-characterized cell surface tyrosine kinase receptor's function in cancers (e.g., epidermal growth factor receptor), GRP receptor may also be an important therapeutic cell membrane target in neuroblastomas. GRP ligand is produced and secreted by neuroblastomas to stimulate its tumor growth, hence it functions as an autocrine growth factor.45 GRP binds to GRP receptors to activate PI3K/AKT pathway in promoting tumorigenesis in neuroblastomas.46 Targeted silencing of GPR-receptors in neuroblastoma cells showed significantly attenuated xenograft growth as well as liver metastases when compared to controls with constitutive expression of GRP receptors.47 Knockdown of GRP receptors demonstrated inhibition of tumor metastasis to liver in vivo (Fig. 2).47
Figure 2.
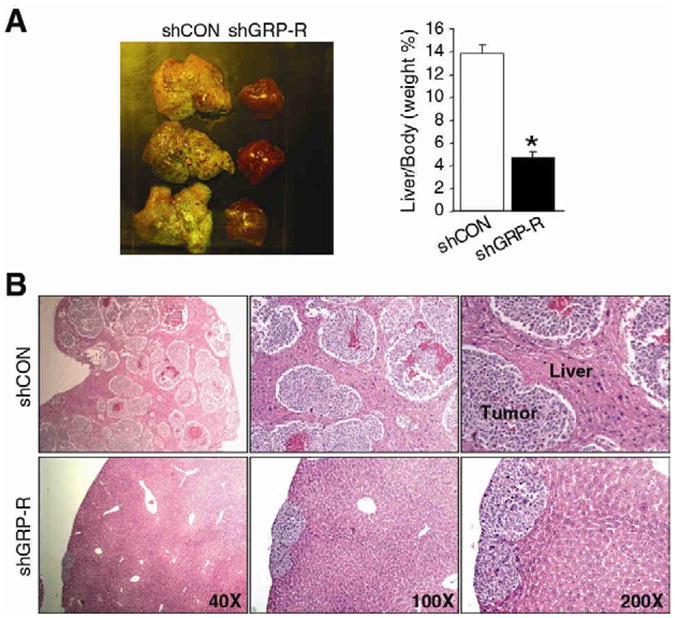
Effects of GRP-receptor knockdown on neuroblastoma metastasis in vivo. Spleen injection liver metastasis murine model was used using human neuroblastoma BE(2)-C cells transfected with pEGFP/shGRP-R (silenced GRP receptors) or pEGFP/shCON (controls). (A) Representative images of livers from mice injected with BE(2)-C/GFP/shGRP-R (short-hairpin silenced GRP-receptors) and control cells (shCON). Silencing of GRP-receptors (shGRP-R; right column) attenuated liver metastasis of BE(2)-C tumors. Quantitative analysis of liver weight relative to body weight (* p<0.05 vs. shCON) shows significantly less weight in GRP-R silenced group (B) Representative H&E-stained sections of livers from mice injected with BE(2)-C/GFP/shCON (controls; upper) and BE(2)-C/GFP/shGRP-R (GRP receptor silenced; lower) cells. Extensive liver metastases are seen in mice from constitutive GRP-receptor expressing BE(2)-C cells (upper). (Adapted from Qiao et al, Proc Natl Acad Sci 2008; Fig. 6, p.12895. Copyright2008 National Academy of Sciences, U.S.A).
9 Conclusions
In spite of the knowledge provided within, neuroblastoma remains a therapeutic enigma. As we are driven to improve outcomes and survival, the ideal therapy also remains elusive. However, there are many fronts on which to attack, and it seems unquestionable that the cure will require a multimodal approach. Part of the solution is to effectively eradicate minimal residual disease, as this appears to put patients at highest risk for relapse and progression. Until then, we remain vulnerable to a cancer that continues to evolve.
Acknowledgments
We thank Karen Martin for manuscript preparation. This work was supported by a grant R01 DK61470 from the National Institute of Health and Rally Foundation for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Park JR, Eggert A, Caron H. Neuroblastoma: biology, prognosis, and treatment. Hematol Oncol Clin North Am. 2010;24:65–86. doi: 10.1016/j.hoc.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 2.Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esiashvili N, Anderson C, Katzenstein HM. Neuroblastoma. Curr Probl Cancer. 2009;33:333–360. doi: 10.1016/j.currproblcancer.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 4.Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003;3:203–216. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- 5.London WB, Matthay KK, Ambros PF, et al. Clinical and biological features predictive of survival after relapse of neuroblastoma: A study from the International Neuroblastoma (NB) Risk Group (INRG) Database. J Clin Oncol. 2010;28:9518. [Google Scholar]
- 6.Kushner BH. Neuroblastoma: a disease requiring a multitude of imaging studies. J Nucl Med. 2004;45:1172–1188. [PubMed] [Google Scholar]
- 7.Haase GM, LaQuaglia MP. Neuroblastoma. In: Ziegler MM, Azizkhan RG, Weber TR, editors. Operative Pediatric Surgery. New York: McGraw-Hill; 2003. pp. 1181–1192. [Google Scholar]
- 8.Ishola TA, Chung DH. Neuroblastoma. Surg Oncol. 2007;16:149–156. doi: 10.1016/j.suronc.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 9.Darnell RB, Posner JB. Paraneoplastic syndromes involving the nervous system. N Engl J Med. 2003;349:1543–1554. doi: 10.1056/NEJMra023009. [DOI] [PubMed] [Google Scholar]
- 10.Goto S, Umehara S, Gerbing RB, et al. Histopathology (International Neuroblastoma Pathology Classification) and MYCN status in patients with peripheral neuroblastic tumors: a report from the Children's Cancer Group. Cancer. 2001;92:2699–2708. doi: 10.1002/1097-0142(20011115)92:10<2699::aid-cncr1624>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 11.Janoueix-Lerosey I, Schleiermacher G, Delattre O. Molecular pathogenesis of peripheral neuroblastic tumors. Oncogene. 2010;29:1566–1579. doi: 10.1038/onc.2009.518. [DOI] [PubMed] [Google Scholar]
- 12.Maris JM. The biologic basis for neuroblastoma heterogeneity and risk stratification. Curr Opin Pediatr. 2005;17:7–13. doi: 10.1097/01.mop.0000150631.60571.89. [DOI] [PubMed] [Google Scholar]
- 13.Monclair T, Brodeur GM, Ambros PF, et al. The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol. 2009;27:298–303. doi: 10.1200/JCO.2008.16.6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohn SL, Pearson AD, London WB, et al. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol. 2009;27:289–297. doi: 10.1200/JCO.2008.16.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim S, Chung DH. Pediatric solid malignancies: neuroblastoma and Wilms' tumor. Surg Clin North Am. 2006;86:469–487. xi. doi: 10.1016/j.suc.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 16.Van Roy N, De Preter K, Hoebeeck J, et al. The emerging molecular pathogenesis of neuroblastoma: implications for improved risk assessment and targeted therapy. Genome Med. 2009;1:74. doi: 10.1186/gm74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fujita T, Igarashi J, Okawa ER, et al. CHD5, a tumor suppressor gene deleted from 1p36.31 in neuroblastomas. J Natl Cancer Inst. 2008;100:940–949. doi: 10.1093/jnci/djn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vandesompele J, Baudis M, De Preter K, et al. Unequivocal delineation of clinicogenetic subgroups and development of a new model for improved outcome prediction in neuroblastoma. J Clin Oncol. 2005;23:2280–2299. doi: 10.1200/JCO.2005.06.104. [DOI] [PubMed] [Google Scholar]
- 19.Kelleher FC, McDermott R. The emerging pathogenic and therapeutic importance of the anaplastic lymphoma kinase gene. Eur J Cancer. 2010;46:2357–2368. doi: 10.1016/j.ejca.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 20.Modak S, Cheung NK. Neuroblastoma: Therapeutic strategies for a clinical enigma. Cancer Treat Rev. 2010;36:307–317. doi: 10.1016/j.ctrv.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Bell E, Chen L, Liu T, et al. MYCN oncoprotein targets and their therapeutic potential. Cancer Lett. 2010;293:144–157. doi: 10.1016/j.canlet.2010.01.015. [DOI] [PubMed] [Google Scholar]
- 22.Taggart DR, Han MM, Quach A, et al. Comparison of iodine-123 metaiodobenzylguanidine (MIBG) scan and [18F]fluorodeoxyglucose positron emission tomography to evaluate response after iodine-131 MIBG therapy for relapsed neuroblastoma. J Clin Oncol. 2009;27:5343–5349. doi: 10.1200/JCO.2008.20.5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peaston AE, Gardaneh M, Franco AV, et al. MRP1 gene expression level regulates the death and differentiation response of neuroblastoma cells. Br J Cancer. 2001;85:1564–1571. doi: 10.1054/bjoc.2001.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yalcin B, Kremer LC, Caron HN, et al. High-dose chemotherapy and autologous haematopoietic stem cell rescue for children with high-risk neuroblastoma. Cochrane Database Syst Rev. 2010 doi: 10.1002/14651858.CD006301.pub2. CD006301. [DOI] [PubMed] [Google Scholar]
- 25.Reynolds CP, Matthay KK, Villablanca JG, et al. Retinoid therapy of high-risk neuroblastoma. Cancer Lett. 2003;197:185–192. doi: 10.1016/s0304-3835(03)00108-3. [DOI] [PubMed] [Google Scholar]
- 26.Kang TI, Brophy P, Hickeson M, et al. Targeted radiotherapy with submyeloablative doses of 131I-MIBG is effective for disease palliation in highly refractory neuroblastoma. J Pediatr Hematol Oncol. 2003;25:769–773. doi: 10.1097/00043426-200310000-00005. [DOI] [PubMed] [Google Scholar]
- 27.Matthay KK, Yanik G, Messina J, et al. Phase II study on the effect of disease sites, age, and prior therapy on response to iodine-131-metaiodobenzylguanidine therapy in refractory neuroblastoma. J Clin Oncol. 2007;25:1054–1060. doi: 10.1200/JCO.2006.09.3484. [DOI] [PubMed] [Google Scholar]
- 28.Wagner LM, Danks MK. New therapeutic targets for the treatment of high-risk neuroblastoma. J Cell Biochem. 2009;107:46–57. doi: 10.1002/jcb.22094. [DOI] [PubMed] [Google Scholar]
- 29.Wu JM, DiPietrantonio AM, Hsieh TC. Mechanism of fenretinide (4-HPR)-induced cell death. Apoptosis. 2001;6:377–388. doi: 10.1023/a:1011342220621. [DOI] [PubMed] [Google Scholar]
- 30.Yip CK, Murata K, Walz T, et al. Structure of the human mTOR complex I and its implications for rapamycin inhibition. Mol Cell. 2010;38:768–774. doi: 10.1016/j.molcel.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coulter DW, Blatt J, D'Ercole AJ, et al. IGF-I receptor inhibition combined with rapamycin or temsirolimus inhibits neuroblastoma cell growth. Anticancer Res. 2008;28:1509–1516. [PubMed] [Google Scholar]
- 32.Moore AS, Blagg J, Linardopoulos S, et al. Aurora kinase inhibitors: novel small molecules with promising activity in acute myeloid and Philadelphia-positive leukemias. Leukemia. 2010;24:671–678. doi: 10.1038/leu.2010.15. [DOI] [PubMed] [Google Scholar]
- 33.Gorgun G, Calabrese E, Hideshima T, et al. A novel Aurora-A kinase inhibitor MLN8237 induces cytotoxicity and cell-cycle arrest in multiple myeloma. Blood. 2010;115:5202–5213. doi: 10.1182/blood-2009-12-259523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brodeur GM, Minturn JE, Ho R, et al. Trk receptor expression and inhibition in neuroblastomas. Clin Cancer Res. 2009;15:3244–3250. doi: 10.1158/1078-0432.CCR-08-1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thiele CJ, Li Z, McKee AE. On Trk--the TrkB signal transduction pathway is an increasingly important target in cancer biology. Clin Cancer Res. 2009;15:5962–5967. doi: 10.1158/1078-0432.CCR-08-0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kushner BH, Kramer K, Cheung NK. Phase II trial of the anti-G(D2) monoclonal antibody 3F8 and granulocyte-macrophage colony-stimulating factor for neuroblastoma. J Clin Oncol. 2001;19:4189–4194. doi: 10.1200/JCO.2001.19.22.4189. [DOI] [PubMed] [Google Scholar]
- 37.Ozkaynak MF, Sondel PM, Krailo MD, et al. Phase I study of chimeric human/murine anti-ganglioside G(D2) monoclonal antibody (ch14.18) with granulocyte-macrophage colony-stimulating factor in children with neuroblastoma immediately after hematopoietic stem-cell transplantation: a Children's Cancer Group Study. J Clin Oncol. 2000;18:4077–4085. doi: 10.1200/JCO.2000.18.24.4077. [DOI] [PubMed] [Google Scholar]
- 38.Yu AL, Gilman AL, Ozkaynak MF, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. The New England journal of medicine. 2010;363:1324–1334. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shusterman S, Grupp SA, Barr R, et al. The angiogenesis inhibitor tnp-470 effectively inhibits human neuroblastoma xenograft growth, especially in the setting of subclinical disease. Clin Cancer Res. 2001;7:977–984. [PubMed] [Google Scholar]
- 40.Barbieri E, Mehta P, Chen Z, et al. MDM2 inhibition sensitizes neuroblastoma to chemotherapy-induced apoptotic cell death. Mol Cancer Ther. 2006;5:2358–2365. doi: 10.1158/1535-7163.MCT-06-0305. [DOI] [PubMed] [Google Scholar]
- 41.Slack A, Lozano G, Shohet JM. MDM2 as MYCN transcriptional target: implications for neuroblastoma pathogenesis. Cancer Lett. 2005;228:21–27. doi: 10.1016/j.canlet.2005.01.050. [DOI] [PubMed] [Google Scholar]
- 42.Van Maerken T, Speleman F, Vermeulen J, et al. Small-molecule MDM2 antagonists as a new therapy concept for neuroblastoma. Cancer Res. 2006;66:9646–9655. doi: 10.1158/0008-5472.CAN-06-0792. [DOI] [PubMed] [Google Scholar]
- 43.Casero RA, Jr, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov. 2007;6:373–390. doi: 10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 44.Hogarty MD, Norris MD, Davis K, et al. ODC1 is a critical determinant of MYCN oncogenesis and a therapeutic target in neuroblastoma. Cancer Res. 2008;68:9735–9745. doi: 10.1158/0008-5472.CAN-07-6866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim S, Hu W, Kelly DR, et al. Gastrin-releasing peptide is a growth factor for human neuroblastomas. Ann Surg. 2002;235:621–629. doi: 10.1097/00000658-200205000-00003. discussion 629-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kang J, Ishola TA, Baregamian N, et al. Bombesin induces angiogenesis and neuroblastoma growth. Cancer Lett. 2007;253:273–281. doi: 10.1016/j.canlet.2007.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qiao J, Kang J, Ishola TA, et al. Gastrin-releasing peptide receptor silencing suppresses the tumorigenesis and metastatic potential of neuroblastoma. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:12891–12896. doi: 10.1073/pnas.0711861105. [DOI] [PMC free article] [PubMed] [Google Scholar]