

Abstract
It has been shown that eNOS uncoupling occurs in hypertension and atherosclerosis. However its causal role in vascular pathogenesis has not been previously characterized. Here, we challenged eNOS pre-uncoupled hph-1 mice (deficient in eNOS cofactor tetrahydrobiopterin biosynthetic enzyme GTPCHI) with Ang II (0.7 mg/kg/day, 14 days). Both wild-type (WT) and hph-1 groups developed hypertension similarly up to day 6 to 7. Thereafter approximately 14% of Ang II-infused (0.7 mg/kg/day) hph-1 mice (n=72) started to die suddenly of ruptured AAA. Among the survivors, 65% developed AAA, resulting in a total morbidity rate of 79%. In contrast, none of the Ang II-infused WT mice died or developed AAA. Ang II progressively deteriorated eNOS uncoupling in hph-1 mice, while augmenting H4B and nitric oxide (NO•) deficiencies. The abundance of the H4B salvage enzyme dihydrofolate reductase (DHFR) in the endothelium was decreased in hph-1 mice and further diminished by Ang II infusion. Intriguingly, restoration of DHFR expression by oral administration of folic acid (FA), or overexpression of DHFR, completely prevented AAA formation in Ang II-infused hph-1 mice while attenuating progressive uncoupling of eNOS. Folic acid also attenuated vascular remodelling and inflammation characterized by medial elastin break down, augmented MMP2 activity and activation of MMP9, as well as macrophage infiltration. In conclusion, these data innovatively suggest a causal role of eNOS uncoupling/H4B deficiency in AAA formation. Therefore oral FA administration, endothelium targeted DHFR gene therapy, and perhaps other countermeasures directed against eNOS uncoupling, could be used as new therapeutics for AAA.
Keywords: abdominal aortic aneurysm (AAA), eNOS uncoupling, hph-1 mice, tetrahydrobiopterin (H4B), angiotensin II, folic acid (FA), dihydrofolate reductase (DHFR)
INTRODUCTION
By producing nitric oxide (NO•) to rapidly inactivate superoxide (O2•−) and other reactive oxygen species (ROS), endothelial nitric oxide synthase (eNOS) protects vascular cells from oxidative damage. Accumulating evidence has demonstrated that when eNOS cofactor tetrahydrobiopterin (H4B) is deficient, eNOS becomes dysfunctional to produce O2•− rather than NO• 1-13. This uncoupling of eNOS could deteriorate endothelial dysfunction, making it extremely difficult to correct. One important pathological agonist capable of transforming eNOS into the uncoupled state is angiotenisn II (Ang II) 1,2,5. We have previously shown that Ang II uncouples eNOS via transient activation of NADPH oxidase (NOX) and consequent hydrogen peroxide-dependent, endothelium-specific deficiency in H4B salvage enzyme dihydrofolate reductase (DHFR) 1,5.
Ang II plays an important role in the pathogenesis of vascular diseases such as hypertension and atherosclerosis, acting via well-characterized mechanisms such as vasoconstriction, activation of vascular NOX and ROS-dependent inflammatory and hypertrophic signaling 14-18. Uncoupling of eNOS however, represents a novel mechanism whereby Ang II causes prolonged oxidative stress 1,2,5. Nevertheless, it has remained unclear whether uncoupled eNOS is directly involved in the pathogenesis of vascular disease. Tetrahydrobiopterin (H4B) deficiency subsequent to a mutation in GTP cyclohydrolase 1 (GTPGH1) induces hyperphenylalaninemia in mice 19,20. At baseline hph-1 mice have reduced NO• bioavailability but preserved vasorelaxation due to hydrogen peroxide-dependent compensation 19. After crossing with C57BL6 mice for over 10 generations we have genotyped and characterized the hph-1 mice using electron spin resonance for detection of superoxide (O2•−) production. What we found was that while in the wild-type (WT) animals L-NAME increased O2•− production due to the loss of NO•, L-NAME attenuated O2•− production in hph-1 mice, implicating uncoupling of eNOS. Therefore hph-1 mice can serve as an excellent model system to study contribution to vascular pathogenesis of uncoupled eNOS/H4B deficiency.
To examine whether uncoupled eNOS exaggerate pathological effects of Ang II to amplify hypertension or augment vascular remodeling, we infused WT and hph-1 mice with Ang II for 14 days. Mean blood pressure (MBP) monitored by an intra-carotid telemetry method was found increased in both groups up to day 6-7. While MBP in WT mice continued to rise, it started to decline in hph-1 mice, which was associated with sudden death (13.9%, n=72). Immediate post-mortem inspection revealed rupturing abdominal aortic aneurysm (AAA). Approximately 65% of the surviving hph-1s developed AAA, resulting in a total morbidity rate of 79% (n=72). We further characterized AAA and revealed that progressive DHFR deficiency and uncoupling of eNOS in Ang II-infused hph-1 underlie extensive vascular remodeling, inflammation and AAA formation. Intriguingly, restoration of DHFR expression by oral administration of folic acid (FA), or overexpression of DHFR, completely prevented AAA formation in Ang II-infused hph-1 mice. These treatments also blunted progressive uncoupling of eNOS, as well as vascular remodeling and inflammation characterized by MMP activation, elastin breakdown, collagen remodeling and macrophage infiltration. Therefore these innovative findings represent first evidence that eNOS uncoupling/H4B deficiency plays a causal role in AAA formation, and that oral FA administration, DHFR-targeted therapy and perhaps other countermeasures directed against eNOS uncoupling, could serve as novel and powerful therapeutic regimes for AAA, the severe and prevalent human disease for which no pharmacological treatment is currently available.
METHODS
Animals, Ang II infusion and blood pressure measurements
The hph-1 mice (originally in CBA background) 19 were backcrossed with C57BL6 mice for more than 10 generations and genotyped based on a protocol by Koo et al. 21. Only homozygote hph-1 mice were used for experiments. Wild-type (WT) and hph-1 male mice at 24 weeks of age were infused with Ang II (0.7 mg/kg/day) using subcutaneously implanted osmotic pumps (Durect Corp.). During the 14-day infusion, blood pressure was monitored by telemetry method. Wireless blood pressure probes were implanted into the animals 10 days prior to the implantation of the osmotic pumps. The catheter of the blood pressure probe was inserted into the left carotid artery, while the body of the probe was inserted into the right flank. Animals were given 1 week to recover from the surgery. After this period, blood pressure was measured for 3 days to obtain a baseline. The osmotic pumps were then implanted on the 10th day after surgery. Measurements were made daily from 9am to 4pm at 250 Hz sampling rate. Average blood pressure was calculated daily as the average of the entire recording period. The use of animals and experimental procedures were approved by the Institutional Animal Care and Usage Committee at the University of California Los Angeles (UCLA).
Electron spin resonance determination of aortic nitric oxide and superoxide production, HPLC determination of aortic H4B content, and Western Blot determination of endothelial DHFR expression were performed as previously published 1,2.
Endothelial cell isolation from mouse aortas
Upon harvest, whole aorta was cut open longitudinally, and then digested with collagenase (0.6 mg/ml) at 37°C for 20 min prior to gentle removal with a cotton applicator. The supernatant containing endothelial cells were centrifuged and lysed for Western blot analysis.
Oral administration of folic acid and DHFR overexpression
WT and hph-1 mice were started on continuous oral administration of folic acid (FA, 15 mg/kg/day), or tail vein transfection of DHFR expression vector (pcDNA3.1-DHFR) 5 in lipid-based reagent from Altogen Biosystems every other day for 14 days, starting 2 days prior to Ang II infusion. The mice were monitored twice per day during the infusion period of 2 weeks, and aortas were harvested for assessment of AAA formation, endothelial DHFR expression, and eNOS uncoupling activity by day 14. Animals that died suddenly during the infusion period of 14 days were inspected for aneurysm immediately and aortas were freshly harvested for experiments.
Ultrasound detection of abdominal aorta size
Animals were anesthetized with isoflurane and placed on a temperature-controlled table, which also measures ECG for heart rate. Isoflurane levels were adjusted throughout the experiment to maintain heart rate between 400-500 bpm while keeping the animal sufficiently anesthetized. Hair was removed from the abdomen using a hair removal cream, and pre-heated ultrasound transmission gel was applied onto the abdomen area. An ultrasound probe (Velvo 770, Visualsonics) was placed on the gel to visualize aorta transversely. The aorta was identified using Doppler measurement for the presence of pulsatile flow. Consistent localization of image acquisition was insured by visualizing the aorta immediately superior to the branch of the left renal artery in all animals. Images were recorded and saved onto a PC computer for offline area analysis.
H&E and VVG stainings were conducted following standard histological protocols.
Macrophage stainings
Formalin fixed and paraffin embedded tissue were sectioned at 5 μm. Paraffin was removed by washing with xylene, then rehydrated with descending concentrations of ethanol. Antigen retrieval was performed by immersing the sections in an antigen retrieval buffer (10uM citric acid, 0.01% Tween 20) at 98.5°C for 20 minutes. Sections were washed in PBS plus 0.1% triton (PBS-T), then blocked with 2% normal goat serum in PBS-T at room temperature for 3 hours. Sections were then incubated with primary antibody (Mac-3, BD Pharmingen, 2%, in PBS-T) overnight at 4°C. After washing with PBS-T for 1 hour, sections were incubated with secondary antibody (Alexa fluor 488, 2% in PBS-T) for 2 hours at room temperature in the dark. After washing with PBS-T for 1 hour, sections were dehydrated with ascending grades of ethanol, then in xylene. Sections were mounted with Permount medium, and pictures taken with a confocal microscope (Leica, SP1 inverted).
MMP activity assays were performed as previously published 22.
Statistical Analysis
Comparisons among different treatment groups were performed by ANOVA. When differences were indicated, the Dunnet’s post hoc test was employed. Statistical significance was set for p<0.05. All grouped data shown in the figures were presented as mean±SEM.
RESULTS
Ang II infusion of hph-1 mice induces AAA formation
Twenty-four week old male wild-type (WT, n=72) and hph-1 mice (n=72) were infused with Ang II (0.7 mg/kg/day) for 14 days and twice per day. Approximately 14% of Ang II-infused hph-1 mice died suddenly of ruptured AAA within 14 days (Fig. 1B). Among the survivors, 65% developed AAA, resulting in a total morbidity rate of 79% (Fig. 1A-1B). In contrast, none of the Ang II-infused WT mice or untreated hph-1 mice died or developed AAA (Fig. 1A). The mean blood pressure (MBP) was monitored by an intra-carotid telemetry method and was found modestly higher in hph-1 mice as compared with WT mice at baseline (i.e., day 0 Ang II infusion, Fig. 1C). During Ang II infusion, MBP was increased in both genotypes up to day 6 (WT: 101±3 to 140±2 mmHg; hph-1: 112±3 to 140±2 mmHg, Fig. 1C). Thereafter, MBP declined in hph-1 mice, whereas it continued to rise in WT mice (Fig. 1C).
Figure 1. Ang II induces AAA formation in hph-1 mice.

Wild-type and hph-1 mice were infused with Ang II (0.7 mg/kg/day) for 14 days. (A) Representative appearance of abdominal aortas in the different experimental groups of WT, WT/Ang II, hph-1 and hph-1/Ang II by day 14. Only Ang II-infused hph-1 mice developed AAA, with visible evidence of hemorrhage. H&E staining (arrows) of the AAA segment revealed thrombus formation. (B) AAA morbidity and mortality rates (no-AAA vs. non-lethal AAA vs. lethal AAA) in Ang II-infused hph-1 mice at 21%, 65% and 14% respectively. (C) Changes in mean blood pressure (MBP) in Ang II-infused hph-1 mice. The MBP was monitored by an intra-carotid telemetry method (Data Sciences International) continuously for 14 days.
Ang II infusion augments deficiency of H4B and NO•, and aggravates eNOS uncoupling, in hph-1 mice
Freshly isolated aortas from Ang II-infused WT and hph-1 mice were subjected to HPLC determination of H4B content, and electron spin resonance (ESR) determination of NO• and superoxide (O2•−) production 1,2. As compared to WT mice, the hph-1 mice exhibited reduced aortic H4B bioavailability (Fig. 2A), which was further diminished by Ang II infusion (1.9±0.2 to 1.0±0.1 pmol/mg protein, Fig. 2A). Ang II infusion also significantly reduced aortic H4B bioavailability in WT mice (5.1±0.2 to 3.8±0.4 pmol/mg protein). Of note, aortic NO• production mirrored these changes in Ang II-infused WT and hph-1 mice (Fig. 2B). Aortic O2•− production was determined in the presence or absence of L-NAME. Basal O2•− production was twice as high in hph-1 mice as compared with WT mice (Fig. 2C, compare sham hph-1 versus sham WT). Whereas L-NAME increased O2•− production as a result of inhibiting coupled eNOS in untreated WT mice (Fig. 2C), L-NAME decreased O2•− production in Ang II-infused WT mice, consistent with uncoupling of eNOS in response to Ang II, as we reported previously 1,5 (Fig. 2C). In hph-1 mice, L-NAME decreased O2•− production in untreated mice, consistent with uncoupled eNOS at baseline, in the absence of Ang II infusion (Fig. 2C). Importantly, Ang II infusion further uncoupled eNOS in hph-1 mice, as demonstrated by the substantially higher production of O2•− that was completely inhibited by L-NAME (Fig. 2C).
Figure 2. Ang II infusion augments deficiency of H4B and NO•, and aggravates eNOS uncoupling, in hph-1 mice.
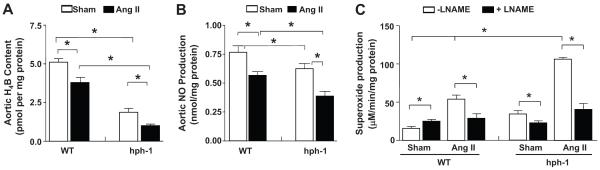
Wild-type and hph-1 mice were infused with Ang II (0.7 mg/kg/day) for 14 days, after which aortas were harvested for: (A) aortic H4B content; (B) aortic NO• production; and (C) aortic O2•− production in the presence or absence of L-NAME. *p<0.05
Folic acid restores endothelial DHFR expression, H4B bioavailability and prevents eNOS uncoupling in Ang II-infused hph-1 mice
Wild-type and hph-1 mice were started on oral folic acid (FA, 15 mg/kg/day) 2 days prior to Ang II infusion (0.7 mg/kg/day), and treated throughout the study period of 14 days. Both endothelial cells (ECs digested off aortas, see Methods section) and EC-denuded aortas were analyzed for DHFR expression. As shown, hph-1 mice exhibited reduced endothelial DHFR expression, which was further reduced by Ang II infusion. FA treatment restored endothelial DHFR expression in hph-1 mice to near WT levels (Fig. 3A). The H4B deficiencies induced by Ang II in both WT and hph-1 mice were likewise prevented by FA treatment, resulting in H4B levels that were even higher than baseline in both groups (Fig. 3B). Moreover, FA prevented eNOS uncoupling in both Ang II-infused WT and hph-1 mice, as evidenced by complete attenuation of L-NAME-sensitive O2•− production (Fig. 3C).
Figure 3. Folic acid prevents eNOS uncoupling in Ang II-infused hph-1 mice via restoration of endothelial DHFR expression.

Wild-type and hph-1 mice were started on oral administration of folic acid (FA, 15 mg/kg/day) 2 days prior to Ang II infusion (0.7 mg/kg/day), and treated throughout the study period of 14 days, after which aortas were harvested for: (A) endothelial and non-endothelial DHFR expression in aortic preparations; (B) aortic H4B content; and (C) aortic eNOS uncoupling activity (indicated by L-NAME-sensitive O2•− production). *p<0.01
Folic acid treatment prevents AAA formation and normalizes blood pressure in Ang II-infused hph-1 mice
For this study, a total of 21 FA treated Ang II-infused hph-1 mice was used, and none of these animals developed AAA. Statistical analysis using a 2 × 2 contingency table shows this reduction in AAA development was significant (p<0.0001). On day 0 and 14, abdominal aorta (AA) dimensions were monitored using ultrasound (Velvo 770 high-resolution echo system equipped with a 45 MHz transducer, Visualsonics). Ang II infusion induced dramatic expansion of AA in hph-1 mice (0.37 to 1.96 mm2), which was prevented by FA treatment (0.39 to 0.52 mm2, Fig. 4A-4D). By contrast, in WT mice, Ang II induced a minimal increase in AA size (0.53 to 0.66 mm2). In addition, FA was highly effective in attenuating Ang II-induced hypertension in WT mice (Fig. 4E), and it also prevented the decline in MBP in Ang II-infused hph-1 mice (Figure 4F). These data suggest that in WT mice infused with Ang II, uncoupling of eNOS leads to elevated MBP, most likely consequent to reduced NO• bioavailability, while in hph-1 mice, which exhibit a more profound degree of eNOS uncoupling, the excessive production of ROS paradoxically lowers MBP, or that the reduction in MBP is mediated by ROS-independent mechanisms.
Figure 4. Folic acid prevents AAA formation and normalizes blood pressure in Ang II-infused hph-1 mice.

Wild-type and hph-1 mice were started on oral administration of folic acid (FA, 15 mg/kg/day) 2 days prior to Ang II infusion (0.7 mg/kg/day), and treated throughout the study period of 14 days. At day 0 and day 14, abdominal ultrasound (Velvo 770 high-resolution echo system, Visualsonics) was performed to assess abdominal aorta (AA) dimensions (A-D) in Ang II-infused WT and hph-1 mice treated with or without FA. Aortic cross-sectional areas are depicted by blue circles, and calculated areas are listed below each image. MBP was assessed by telemetry during the course of the study (E-F), as described in Figure 1.
Folic acid prevents progressive uncoupling of eNOS and vascular remodeling in Ang II-infused hph-1 mice
To examine the effects of FA on eNOS uncoupling at different time points during AAA development, we followed O2•− production on day 4 and day 8 day after initiation of Ang II infusion. Aortas were harvested and subjected to ESR detection of O2•− in the presence or absence of L-NAME. In WT mice, eNOS uncoupling activity remained steady in response to Ang II for both time points examined. As is obvious in Fig. 5A, L-NAME-sensitive O2•− production that is reflective of eNOS uncoupling activity, was similar on day 4 and day 8. In contrast, Ang II induced progressive uncoupling of eNOS in hph-1 mice. As demonstrated in Fig. 5B, the L-NAME inhibitable fraction of O2•− production was augmented on day 8 comparing to what was observed on day 4. Importantly, FA consistently suppressed eNOS uncoupling activity in hph-1 mice at both time points.
Figure 5. Folic acid prevents progressive uncoupling of eNOS in Ang II-infused hph-1 mice.
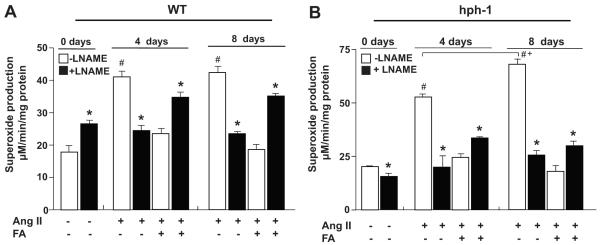
Wild-type (WT) and hph-1 mice were started on oral administration of folic acid (FA, 15 mg/kg/day) 2 days prior to Ang II infusion (0.7 mg/kg/day), and treated throughout the study period of 14 days. Aortas were harvested on day 0, 4 or 8 days for analysis of O2•− production in the presence or absence of L-NAME in (A) WT, and (B) hph-1, mice. *p<0.05 vs L-NAME, #p<0.05 vs WT sham, +p<0.05 vs 4 days
In additional experiments, we found that FA also prevented Ang II-induced vascular remodeling in hph-1 mice. As indicated by H&E staining, FA abrogated medial degradation and adventitial inflammatory cell recruitment in Ang II-infused hph-1 mice (Fig. 6A). More specifically, FA attenuated medial elastin flattening, and rarefaction of elastin fibers, as shown by VVG staining (Figure 6B). Infiltrating macrophages, one of the major sources of matrix degradation enzymes including MMP9, were also dramatically upregulated with Ang II-infused mice, which was completely attenuated with FA treatment (Fig. 6C). Furthermore, FA attenuated Ang II-induced augmentation of MMP2 activation and MMP9 activation in hph-1 mice, while it also inhibited Ang II-induced MMP2 activation in WT mice (Fig. 7)
Figure 6. Folic acid prevents vascular remodeling in Ang II-infused hph-1 mice.

Wild-type and hph-1 mice were treated with our without folic acid (FA, 15 mg/kg/day) beginning 2 days prior to Ang II (0.7 mg/kg/day) or vehicle infusion, and treated throughout the study period of 14 days, after which aortas were harvested for (A) H&E staining (black arrows and red arrows indicating FA-induced changes in media and adventitia respectively); (B) VVG staining (black arrows showing elastin changes); (C) Macrophage staining indicating macrophage infiltraiton
Figure 7. Folic acid prevents Ang II-induced MMP2 and MMP9 activation in hph-1 mice.

Wild-type and hph-1 mice were treated with our without folic acid (FA, 15 mg/kg/day) beginning 2 days prior to Ang II (0.7 mg/kg/day) or vehicle (sham) infusion. After the 14 day study period, aortas were harvested to assess MMP activity. (A) Representative zymogram showing MMP2 and MMP9 activities. (B) Quantitative data of MMP2 activity. (C) Quantitative data of MMP9 activity. *p<0.05 vs sham.
DHFR gene therapy recouples eNOS and prevents AAA formation in Ang II-infused hph-1 mice
We have previously shown that DHFR gene therapy is effective in recoupling eNOS in Ang II-infused WT mice 1. To test whether DHFR gene therapy is enough to overcome its deficiency in Ang II-infused hph-1 mice that seems crucial for AAA formation, WT or hph-1 mice were transfected with DHFR prior to initiation of Ang II infusion and throughout the entire infusion period. Endothelial DHFR expression was markedly enhanced after successful in vivo transfection of a DHFR-containing expression vector (Fig. 8A) 5, and this was effective in completely attenuating augmented eNOS uncoupling in Ang II-infused hph-1 mice (L-NAME instead increased O2•− production as in WT controls, Fig. 8B). None of the Ang II-infused hph-1 mice in which DHFR was overexpressed developed AAA.
Fig. 8. DHFR overexpression recouples eNOS in Ang II-infused hph-1 mice.

Wild-type and hph-1 mice were subjected to tail vein transfection of DHFR expression vector (pcDNA3.1-DHFR) 5 in lipid-based reagent from Altogen Biosystems every other day for 14 days, starting 2 days prior to Ang II infusion. At the end of the 14 day infusion, aortas were harvested for: (A) endothelial DHFR expression in aortic preparations by western blotting; (B) aortic superoxide production in the presence or absence of L-NAME. *p<0.05 vs. WT sham, #p<0.05 vs. L-NAME, +p<0.05 vs. hph-1 sham
DISCUSSION
The most significant finding of the present study is the first demonstration of a causal role of eNOS uncoupling/H4B deficiency, and the therapeutic potential of eNOS recoupling, in AAA formation. In hph-1 mice where eNOS is uncoupled at baseline, we found that Ang II infusion induces AAA formation in conjunction with further uncoupling of eNOS. Treatment with oral FA effectively prevented both eNOS uncoupling and AAA formation in hph-1 mice. Moreover, FA attenuated Ang II-induced vascular remodelling in both hph-1 and WT mice, and modulated blood pressure responses to Ang II differentially in both groups of animals. These findings suggest that eNOS uncoupling predisposes to AAA formation, and that strategies directed at eNOS recoupling could be of benefit in treating this vascular disorder.
Elevated parameters of oxidative stress have been detected both systemically and locally in human AAA 23,24. Moreover, increased aortic oxidative stress has been reported in conjunction with AAA induced experimentally in animal models, and countermeasures against oxidative stress have proven effective in preventing AAA formation in Ang II-infused mice, though not in humans 25-27. Despite a previously established role of vascular smooth muscle, whether or not other cellular or enzymatic sources of oxidative stress are involved in the pathogenesis of AAA however, remain to be fully understood. Here, we report for the first time that uncoupled eNOS can contribute to oxidative stress leading to severe vascular remodeling and AAA formation in a murine model. The infrarenal pattern of the AAA is very similar to what is found in humans. In addition, the pathological features we observed in mice with uncoupled eNOS resemble those observed in human AAA, including adventitial inflammation, activation of MMPs and matrix degradation (Fig. 6 and 7). Moreover, the AAA was prone to rupture leading to sudden death, as demonstrated by a mortality rate of 14% in these Ang II-infused hph-1 mice (Fig. 1B). Of note, the uncoupling process makes eNOS a peroxynitrite generator, implicating that peroxynitrite, rather than other ROS, might serve as an important redox-signaling mediator for AAA formation. Deletion of eNOS in high fat fed apolipoprotein E (apoE) null mice resulted in spontaneous AAA formation, although the incidence rate was much lower at 25% 28,29. Notably, eNOS uncoupling has also been observed in apoE deficient mice at baseline 7,30. Mice deficient in eNOS exhibit increased oxidative stress consequent to a loss in NO• production. However, these mice do not develop eNOS uncoupling, since they lack functional eNOS protein. Taken together, these data suggest that an eNOS uncoupling-dependent NO•/ROS imbalance in the vasculature, rather than loss of NO• production per se, is more profoundly inductive of AAA formation. Our data also suggest a novel role for endothelial cells in oxidant generation that is involved in promoting AAA formation.
Ang II infusion induced a rapid rise in blood pressure in the hph-1 mice, similar to what was observed in the WT mice. However, in hph-1 mice, blood pressure began to progressively fall after day 6 of Ang II infusion, reaching pre-treatment values by day 11. In contrast, in WT mice, blood pressure continued to rise throughout the course of Ang II infusion. Treatment with FA, which has been demonstrated to re-couple eNOS 1,31,32, attenuated both the rise in blood pressure in WT mice and the fall in blood pressure in hph-1 mice resulted from Ang II infusion, suggesting the involvement of eNOS uncoupling in both responses. In the case of WT animals, the eNOS uncoupling induced by Ang II likely contributed to hypertension by reducing NO• bioavailability. Reduced NO• bioavailability likely also contributed to the increased blood pressure at baseline in hph-1 mice and to the initial pressor response to Ang II infusion (Fig. 1C). The subsequent fall in blood pressure in Ang II infused hph-1 mice also appears to result from eNOS uncoupling, although the exact mechanism remains to be determined. It is possible that the growing aneurysm affects hemodynamics hence blood pressure. Modulation of blood pressure by oxidative stress is complex and dependent on the balance between destruction of NO• and production of ROS that can have vasoconstrictor or vasodilator effects. For example, hydrogen peroxide has been shown to mediate compensatory vasodilatation in hypertensive animals 6. Then scavenging of eNOS-derived hydrogen peroxide by recoupling of eNOS may increase vasocontractility and blood pressure. Considering that WT mice develop sustained hypertension but no AAA formation in response to Ang II infusion, while hph-1 mice develop AAA formation but only transient hypertension, our data are consistent with the previous notions that hypertension is not a decisive risk factor for AAA development, although it may facilitate the disease process 28,29,33.
It was also important to note that eNOS was progressively uncoupled in Ang II-infused hph-1 mice but not in WT mice (Fig. 6), which is consistent with progressive DHFR deficiency in hph-1 mice that was fully corrected by FA treatment (Fig. 3A). These data indicate a critical role for endothelial DHFR deficiency in mediating eNOS uncoupling and AAA formation. DHFR is expressed in vascular cells other than endothelial cells; however, only endothelial DHFR abundance correlates with aortic H4B and NO• bioavailability (Fig. 3A, 1,5).
Previous studies have shown that MMPs, specifically MMP-2 and –9, are major players in the development of AAA 34-36. In our study, we observed an increase in the activities of both of these enzymes in the Ang II infused hph-1 animals (Fig 7B&C), which matches well with those earlier observations. Interestingly, MMP-2 activity was also increased in Ang II infused WT animals, which did not develop AAA. This seems to suggest that MMP2 activation alone is not sufficient for AAA development. Of note, MMP-9 activity was found increased only in Ang II-infused hph-1 mice. Further, macrophage staining (Fig. 6C) in these animals was dramatically more abundant than in the Ang II infused WT animals. These findings are in agreement with previous observations that the source of MMP-9 in AAA is generally macrophages 36. Taken together, our data suggests that Ang II infusion causes an increase in MMP-2 production in the aortas of both WT and hph-1 animals, mostly likely from vascular smooth muscle cells 34,36,37 and endothelial cells 38,39. However, an increase in MMP-9 from stimulated vascular cells and infiltrating macrophages is necessary for AAA to occur.
Perspectives
These data innovatively suggest a causal role of eNOS uncoupling/H4B deficiency in AAA formation and raise the possibility that oral FA administration, DHFR gene therapy, and perhaps other countermeasures directed against eNOS uncoupling, could be of benefit in treating AAA.
Acknowledgments
SOURCES OF FUNDING This work was supported by National Heart, Lung and Blood Institute (NHLBI) Grants HL077440 (HC), HL081571 (HC), HL088975 (HC), HL101228 (PP, JW, HC), HL076684 (NLW), HL62948 (NLW), HL071061 (ZG), American Diabetes Association Award 7-08-RA-23 (HC). The macrophage images were taken using the California NanoSystems Institute Advanced Light Microscopy/Spectroscopy Shared Facility at UCLA, directed by Laurent Bentolila and supported with funding from NIH-NCRR shared resources grant (CJX1-443835-WS-29646) and NSF Major Research Instrumentation grant (CHE-0722519). Kin Siu has been supported by a Vascular Biology Training Grant (NHLBI T32 HL69766, Irela-Arispe) at UCLA (2010-2011).
Footnotes
DISCLOSURES Ling Gao, M.D., Ph.D. None
Kin Lung Siu, Ph.D. None
Karel Chalupsky, Ph.D. None
Andrew Nguyen, B.A. None
Peng Chen, M.D. None
Neal L. Weintraub, M.D. None
Zorina Galis, Ph.D. None
Hua Linda Cai, MD, PhD None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Gao L, Chalupsky K, Stefani E, Cai H. Mechanistic insights into folic acid-dependent vascular protection: dihydrofolate reductase (DHFR)-mediated reduction in oxidant stress in endothelial cells and angiotensin II-infused mice. A Novel HPLC-based Fluorescent Assay for DHFR Activity. J Mol Cell Cardiol. 2009;47:752–760. doi: 10.1016/j.yjmcc.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oak JH, Cai H. Attenuation of angiotensin II signaling recouples eNOS and inhibits nonendothelial NOX activity in diabetic mice. Diabetes. 2007;56:118–126. doi: 10.2337/db06-0288. [DOI] [PubMed] [Google Scholar]
- 3.Cai H. Hydrogen peroxide regulation of endothelial function: origins, mechanisms, and consequences. Cardiovasc Res. 2005;68:26–36. doi: 10.1016/j.cardiores.2005.06.021. [DOI] [PubMed] [Google Scholar]
- 4.Cai H. NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ Res. 2005;96:818–822. doi: 10.1161/01.RES.0000163631.07205.fb. [DOI] [PubMed] [Google Scholar]
- 5.Chalupsky K, Cai H. Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2005;102:9056–9061. doi: 10.1073/pnas.0409594102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laursen JB, Somers M, Kurz S, McCann L, Warnholtz A, Freeman BA, Tarpey M, Fukai T, Harrison DG. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103:1282–1288. doi: 10.1161/01.cir.103.9.1282. [DOI] [PubMed] [Google Scholar]
- 8.Milstien S, Katusic Z. Oxidation of tetrahydrobiopterin by peroxynitrite: implications for vascular endothelial function. Biochem Biophys Res Commun. 1999;263:681–684. doi: 10.1006/bbrc.1999.1422. [DOI] [PubMed] [Google Scholar]
- 9.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA., Jr. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia Y, Tsai AL, Berka V, Zweier JL. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J Biol Chem. 1998;273:25804–25808. doi: 10.1074/jbc.273.40.25804. [DOI] [PubMed] [Google Scholar]
- 11.Wever RM, van Dam T, van Rijn HJ, de Groot F, Rabelink TJ. Tetrahydrobiopterin regulates superoxide and nitric oxide generation by recombinant endothelial nitric oxide synthase. Biochem Biophys Res Commun. 1997;237:340–344. doi: 10.1006/bbrc.1997.7069. [DOI] [PubMed] [Google Scholar]
- 12.Heinzel B, John M, Klatt P, Bohme E, Mayer B. Ca2+/calmodulin-dependent formation of hydrogen peroxide by brain nitric oxide synthase. Biochem J. 1992;281:627–630. doi: 10.1042/bj2810627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pou S, Pou WS, Bredt DS, Snyder SH, Rosen GM. Generation of superoxide by purified brain nitric oxide synthase. J Biol Chem. 1992;267:24173–24176. [PubMed] [Google Scholar]
- 14.Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ Res. 1994;74:1141–1148. doi: 10.1161/01.res.74.6.1141. [DOI] [PubMed] [Google Scholar]
- 15.Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, Harrison DG, Taylor WR, Griendling KK. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension. 1998;32:488–495. doi: 10.1161/01.hyp.32.3.488. [DOI] [PubMed] [Google Scholar]
- 16.Korge P, Honda HM, Weiss JN. Regulation of the mitochondrial permeability transition by matrix Ca(2+) and voltage during anoxia/reoxygenation. Am J Physiol Cell Physiol. 2001;280:C517–526. doi: 10.1152/ajpcell.2001.280.3.C517. [DOI] [PubMed] [Google Scholar]
- 17.Hanna IR, Dikalova A, Hilenski LL, Quinn MT, Griendling KK. Nox1 binds p22phox to form a functional oxidase in vascular smooth muscle cells (VSMCs) CIrculation. 2002;106:164. [Google Scholar]
- 18.Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci. 2003;24:471–478. doi: 10.1016/S0165-6147(03)00233-5. [DOI] [PubMed] [Google Scholar]
- 19.Cosentino F, Barker JE, Brand MP, Heales SJ, Werner ER, Tippins JR, West N, Channon KM, Volpe M, Luscher TF. Reactive oxygen species mediate endothelium-dependent relaxations in tetrahydrobiopterin-deficient mice. Arterioscler Thromb Vasc Biol. 2001;21:496–502. doi: 10.1161/01.atv.21.4.496. [DOI] [PubMed] [Google Scholar]
- 20.Bode VC, McDonald JD, Guenet JL, Simon D. hph-1: a mouse mutant with hereditary hyperphenylalaninemia induced by ethylnitrosourea mutagenesis. Genetics. 1988;118:299–305. doi: 10.1093/genetics/118.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khoo JP, Nicoli T, Alp NJ, Fullerton J, Flint J, Channon KM. Congenic mapping and genotyping of the tetrahydrobiopterin-deficient hph-1 mouse. Mol Genet Metab. 2004;82:251–254. doi: 10.1016/j.ymgme.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 22.Johnson C, Galis ZS. Matrix metalloproteinase-2 and -9 differentially regulate smooth muscle cell migration and cell-mediated collagen organization. Arterioscler Thromb Vasc Biol. 2004;24:54–60. doi: 10.1161/01.ATV.0000100402.69997.C3. [DOI] [PubMed] [Google Scholar]
- 23.Sakalihasan N, Pincemail J, Defraigne JO, Nusgens B, Lapiere C, Limet R. Decrease of plasma vitamin E (alpha-tocopherol) levels in patients with abdominal aortic aneurysm. Ann N Y Acad Sci. 1996;800:278–282. doi: 10.1111/j.1749-6632.1996.tb33332.x. [DOI] [PubMed] [Google Scholar]
- 24.Miller FJ, Jr., Sharp WJ, Fang X, Oberley LW, Oberley TD, Weintraub NL. Oxidative stress in human abdominal aortic aneurysms: a potential mediator of aneurysmal remodeling. Arterioscler Thromb Vasc Biol. 2002;22:560–565. doi: 10.1161/01.atv.0000013778.72404.30. [DOI] [PubMed] [Google Scholar]
- 25.Yajima N, Masuda M, Miyazaki M, Nakajima N, Chien S, Shyy JY. Oxidative stress is involved in the development of experimental abdominal aortic aneurysm: a study of the transcription profile with complementary DNA microarray. J Vasc Surg. 2002;36:379–385. doi: 10.1067/mva.2002.124366. [DOI] [PubMed] [Google Scholar]
- 26.Nakahashi TK, Hoshina K, Tsao PS, Sho E, Sho M, Karwowski JK, Yeh C, Yang RB, Topper JN, Dalman RL. Flow loading induces macrophage antioxidative gene expression in experimental aneurysms. Arterioscler Thromb Vasc Biol. 2002;22:2017–2022. doi: 10.1161/01.atv.0000042082.38014.ea. [DOI] [PubMed] [Google Scholar]
- 27.Gavrila D, Li WG, McCormick ML, Thomas M, Daugherty A, Cassis LA, Miller FJ, Jr., Oberley LW, Dellsperger KC, Weintraub NL. Vitamin E inhibits abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:1671–1677. doi: 10.1161/01.ATV.0000172631.50972.0f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen J, Kuhlencordt PJ, Astern J, Gyurko R, Huang PL. Hypertension does not account for the accelerated atherosclerosis and development of aneurysms in male apolipoprotein e/endothelial nitric oxide synthase double knockout mice. Circulation. 2001;104:2391–2394. doi: 10.1161/hc4501.099729. [DOI] [PubMed] [Google Scholar]
- 29.Kuhlencordt PJ, Gyurko R, Han F, Scherrer-Crosbie M, Aretz TH, Hajjar R, Picard MH, Huang PL. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation. 2001;104:448–454. doi: 10.1161/hc2901.091399. [DOI] [PubMed] [Google Scholar]
- 30.Alp NJ, McAteer MA, Khoo J, Choudhury RP, Channon KM. Increased endothelial tetrahydrobiopterin synthesis by targeted transgenic GTP-cyclohydrolase I overexpression reduces endothelial dysfunction and atherosclerosis in ApoE-knockout mice. Arterioscler Thromb Vasc Biol. 2004;24:445–450. doi: 10.1161/01.ATV.0000115637.48689.77. [DOI] [PubMed] [Google Scholar]
- 31.Stroes ES, van Faassen EE, Yo M, Martasek P, Boer P, Govers R, Rabelink TJ. Folic acid reverts dysfunction of endothelial nitric oxide synthase. Circ Res. 2000;86:1129–1134. doi: 10.1161/01.res.86.11.1129. [DOI] [PubMed] [Google Scholar]
- 32.Antoniades C, Shirodaria C, Warrick N, Cai S, de Bono J, Lee J, Leeson P, Neubauer S, Ratnatunga C, Pillai R, Refsum H, Channon KM. 5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation. 2006;114:1193–1201. doi: 10.1161/CIRCULATIONAHA.106.612325. [DOI] [PubMed] [Google Scholar]
- 33.Cassis LA, Gupte M, Thayer S, Zhang X, Charnigo R, Howatt DA, Rateri DL, Daugherty A. ANG II infusion promotes abdominal aortic aneurysms independent of increased blood pressure in hypercholesterolemic mice. Am J Physiol Heart Circ Physiol. 2009;296:H1660–1665. doi: 10.1152/ajpheart.00028.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davis V, Persidskaia R, Baca-Regen L, Itoh Y, Nagase H, Persidsky Y, Ghorpade A, Baxter BT. Matrix metalloproteinase-2 production and its binding to the matrix are increased in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 1998;18:1625–1633. doi: 10.1161/01.atv.18.10.1625. [DOI] [PubMed] [Google Scholar]
- 35.Thompson RW, Holmes DR, Mertens RA, Liao S, Botney MD, Mecham RP, Welgus HG, Parks WC. Production and localization of 92-kilodalton gelatinase in abdominal aortic aneurysms. An elastolytic metalloproteinase expressed by aneurysm-infiltrating macrophages. J Clin Invest. 1995;96:318–326. doi: 10.1172/JCI118037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–632. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Galis ZS, Muszynski M, Sukhova GK, Simon-Morrissey E, Unemori EN, Lark MW, Amento E, Libby P. Cytokine-stimulated human vascular smooth muscle cells synthesize a complement of enzymes required for extracellular matrix digestion. Circ Res. 1994;75:181–189. doi: 10.1161/01.res.75.1.181. [DOI] [PubMed] [Google Scholar]
- 38.Herron GS, Banda MJ, Clark EJ, Gavrilovic J, Werb Z. Secretion of metalloproteinases by stimulated capillary endothelial cells. II. Expression of collagenase and stromelysin activities is regulated by endogenous inhibitors. J Biol Chem. 1986;261:2814–2818. [PubMed] [Google Scholar]
- 39.Moscatelli D, Jaffe E, Rifkin DB. Tetradecanoyl phorbol acetate stimulates latent collagenase production by cultured human endothelial cells. Cell. 1980;20:343–351. doi: 10.1016/0092-8674(80)90620-0. [DOI] [PubMed] [Google Scholar]