

Abstract
JAK2 activity is tightly controlled through a self-inhibitory effect via its JAK homology domain 2 (JH2), which restricts the strength and duration of JAK2/STAT3 signaling under physiological conditions. Although multiple mutations within JAK2, which abrogate the function of JH2 and sustain JAK2 activation, are widely observed in hematological malignancies, comparable mutations have not been detected in solid tumors. How solid tumor cells override the autoinhibitory effect of the JH2 domain to maintain constitutive activation of JAK2/STAT3 signaling remains puzzling. Herein, we demonstrate that AGK directly interacted with the JH2 domain to relieve inhibition of JAK2 and activate JAK2/STAT3 signaling. Overexpression of AGK sustained constitutive JAK2/STAT3 activation, consequently promoting the cancer stem cell population and augmenting the tumorigenicity of esophageal squamous cell carcinoma (ESCC) cells both in vivo and in vitro. Furthermore, AGK levels significantly correlated with increased STAT3 phosphorylation, poorer disease-free survival, and shorter overall survival in primary ESCC. More importantly, AGK expression was significantly correlated with JAK2/STAT3 hyperactivation in ESCC, as well as in lung and breast cancer. These findings uncover a mechanism for constitutive activation of JAK2/STAT3 signaling in solid tumors and may represent a prognostic biomarker and therapeutic target.
Introduction
JAK2, a member of the Janus (JAK) family of nonreceptor protein tyrosine kinases, regulates signaling via multiple cytokine receptors, such as the interleukin-6 (IL-6), erythropoietin, leptin, and interferon-γ receptors (1–3). Functioning as a prototypical kinase to mediate the phosphorylation of STAT3, JAK2 plays a crucial role in regulating the JAK/STAT3 signaling pathway, which is hyperactivated in a wide range of tumor types (2, 3). Recent advances have shown that the JAK2/STAT3 pathway is involved in the maintenance of the cancer stem cell (CSC) population. It has been reported that JAK/STAT3 signaling is required for induction of the pluripotency factor NANOG and the chemoresistant phenotype in liver CSCs (4). Activation of the JAK/STAT3 pathway in glioblastoma is essential for the maintenance of the tumor stem cell–like phenotype, such as sphere formation, expression of pluripotency-associated markers, and tumorigenicity (5). Conversely, blockade of JAK2 activation in breast cancer results in a reduction of the CD44+/CD24– CSC population and a loss of tumorigenicity in vivo (6). Disruption of constitutively activated JAK2/STAT3 signaling has also been found to inhibit tumorigenicity and tumor progression in multiple types of cancer (7–9).
JAK2 kinase is composed of 7 JAK homology (JH) domains, namely JH1-7, from the carboxyl terminal to the amino terminal. The JH1 domain functions as the kinase domain of JAK2, and transphosphorylation of the tyrosine 1007 and 1008 residues in the JH1 domain facilitates activation of JAK2. The JH3-7 region of JAK2 is essential for receptor interactions (1, 10, 11). Interestingly, basal JAK2 activity is shown to be tightly controlled by its JH2 domain, which can physically interact with and inhibit the kinase activity of the JH1 domain (12, 13). Mutation or deletion of the JH2 domain in Drosophila JAK or human JAK2 results in hyperactivation of the kinase (14, 15). Importantly, the discovery of a large number of mutations within the JH2 domain, which result in persistent JAK2 activation in hematological malignancies, strongly supports the notion that overriding JH2-mediated JAK2 inhibition is crucial for JAK2 hyperactivation in cancer. The most common JAK2 mutation that inhibits the function of JH2, JAK2 V617F, is a driver mutation in hematological malignancies, such as polycythemia vera, essential thrombocythemia, and primary myelofibrosis (16–18). However, JAK2 mutations leading to a loss of function in the JH2 domain are rarely reported in solid tumors, despite the fact that persistent JAK2 activity is also widely observed (3, 19). This raises the possibility that a potent, nonmutation-driven mechanism may serve to override JH2-mediated inhibition of JAK2 and thus sustain constitutive activation of JAK2 in solid tumors.
Acylglycerol kinase (AGK), a multisubstrate lipid kinase, catalyzes the production of lysophosphatidic acid and phosphatidic acid from monoacylglycerol and diacylglycerol (20–22). Overexpression of AGK leads to activation of EGFR and promotes the proliferation and migration of prostate cancer cells, suggesting that AGK might act as a potent oncogene (20, 23). However, the clinical significance of AGK and its associated signaling pathways remain unclear. Herein, we report that AGK is markedly overexpressed in esophageal squamous cell carcinoma (ESCC) and correlates with poorer disease-free survival and shorter overall survival in primary ESCC. Furthermore, we found that AGK directly binds to the JH2 domain of JAK2 and blocks JH2-mediated inhibition of JAK2, resulting in constitutive activation of JAK2/STAT3 signaling and propagation of the CSC population in ESCC in vitro and in vivo. More importantly, AGK expression was shown to correlate significantly with STAT3-regulated signatures in ESCC, lung cancer, and breast cancer patient gene expression profiles. These findings uncover a mutation-independent mechanism of JH2 inhibition that sustains activation of JAK2 in solid tumors.
Results
Identification of AGK as a JH2 domain–interacting protein that activates the JAK2/STAT3 pathway.
To explore the mechanism by which solid tumor cells override the autoinhibitory effect of JH2 to maintain activation of JAK2/STAT3 signaling, affinity purification and mass spectrometry (MS) were used to identify JH2-interacting proteins in ECa109 ESCC cells. As shown in Figure 1, A and B, and Supplemental Figure 1A (supplemental material available online with this article; doi: 10.1172/JCI68143DS1), AGK and 7 other proteins were identified as potent JH2-interacting proteins. Importantly, reciprocal coimmunoprecipitation and Western blot assays further demonstrated that AGK could form a complex with JAK2 and STAT3, suggesting that AGK might be involved in the regulation of JAK2/STAT3 signaling (Figure 1C). Indeed, we found that among these JH2-interacting partners, overexpression of AGK dramatically increased, whereas silencing of AGK decreased, STAT3 luciferase reporter activity and the expression levels of phosphorylated JAK2 (p-JAK2, Tyr1007-1008) and phosphorylated STAT3 (p-STAT3, Tyr705) (Figure 1, D and E and Supplemental Figure 1B). Furthermore, through analysis of AGK expression and STAT3-regulated gene signatures via gene set enrichment analysis (GSEA) (24, 25) in published ESCC patient expression profiles, we found that AGK levels between normal and tumor tissues and within tumors were positively correlated with the STAT3-activated gene signatures and inversely correlated with the STAT3-suppressed gene signatures (Figure 1F and Supplemental Figure 2). Taken together, these results suggest that AGK contributes to the activation of JAK2/STAT3 signaling in ESCC.
Figure 1. Identification of AGK as a JH2 domain–interacting protein that activates the JAK2/STAT3 pathway.

(A) Lysates from ECa109 cells transfected with HA-JH2 were immunoprecipitated using anti-HA affinity agarose, followed by MS peptide sequencing. AGK was identified in the precipitate together with the 7 other indicated proteins. (B) Representative MS plots and sequences of peptides from AGK. (C) Immunoprecipitation assay revealing that AGK interacted with JAK2 and STAT3. (D) STAT3 luciferase reporter activity was analyzed in AGK-transduced, AGK-silenced, and control cells. Error bars represent the means ± SD of 3 independent experiments. *P < 0.05. (E) Western blot analysis of p-STAT3 (Tyr705), total STAT3, p-JAK2 (Tyr1007-1008), and total JAK2 expression in the indicated cells. α-Tubulin was used as a loading control. (F) GSEA plot showing that AGK expression positively correlated with STAT3-activated gene signatures (DAUER_STAT3_TARGETS_UP, V$STAT3_01/02) and inversely correlated with STAT3-suppressed gene signatures (DAUER_STAT3_TARGETS_Dn) in published ESCC patient gene expression profiles (NCBI/GEO/GSE20347 and GSE29001, n = 79). IP, immunoprecipitation.
AGK interacts with JAK2 via binding directly to its JH2 domain.
To further investigate the physical association between AGK and the JAK2/STAT3 complex, the effect of AGK knockdown on the interaction between endogenous JAK2 and STAT3 was examined. As shown in Figure 2A (left panel), AGK silencing did not reduce the binding affinity of JAK2 for STAT3, indicating that AGK does not contribute to JAK2/STAT3 interaction. Though knockdown of STAT3 did not lead to an obvious effect on the interaction between JAK2 and AGK, silencing JAK2 dramatically decreased the interaction between AGK and STAT3 (Figure 2A, middle and right panels). These observations suggest that the AGK-STAT3 interaction occurs in an indirect manner and that AGK-mediated activation of JAK2/STAT3 signaling might be dependent on JAK2.
Figure 2. AGK enhances JAK2 activity by blocking JH2-mediated autoinhibition of JAK2.

(A) Depletion of AGK did not affect the interaction between JAK2 and STAT3 (left panel). Depletion of STAT3 did not alter the interaction between JAK2 and AGK (middle panel). Depletion of JAK2 impaired the interaction between AGK and STAT3 (right panel). (B) Schematic illustration of wild-type JAK2 and the truncated JAK2. (C) Coimmunoprecipitation assay showing that AGK specifically interacted with the JH2 domain of JAK2. (D) Immunoprecipitated HA-tagged JAK2 and the JH2 domain were gel purified, transferred to a membrane, and incubated with His-tagged recombinant AGK, then detected using an antibody specific to 6 × His or the HA tag. Recombinant His-tagged AGK was used as a control. (E) Expression of p-JAK2 (Tyr1007-1008) in whole-cell lysates (WCL) from HEK293T cells transfected with vector or AGK (left panel). Total JAK2 was used as a control. Tyrosine phosphorylation status of the immunoprecipitated JH2 domain from HEK293T cells cotransfected with JH2 and vector or AGK (right panel). (F) In vitro kinase assay of immunoprecipitated JAK2 incubated with or without recombinant AGK. Recombinant STAT3 was used as the substrate. (G) The expression of p-STAT3 (Tyr705) and p-JAK2 (Tyr1007-1008) in the indicated cells infected with wild-type AGK or a kinase-dead AGK mutant (AGK G126E). (H) Immunoprecipitation assay indicating that the mutated AGK (AGK G126E) interacted with JAK2 in the indicated cells.
Next, we examined whether AGK specifically interacts with the JH2 domain of JAK2. We constructed 3 truncated JAK2 fragments: JH1, JH2, and JH3-7, the 3 major functional regions of JAK2 (Figure 2B). We performed an immunoprecipitation assay which demonstrated that AGK only interacted with the JH2 fragment of JAK2 (Figure 2C). Importantly, far-Western blot analysis revealed that both immunoprecipitated full-length JAK2 and the JH2 fragment interacted with recombinant His-tagged AGK (Figure 2D), indicating that AGK interacted with JAK2 by directly binding to its JH2 domain.
AGK sustains JAK2 activation via blockage of JH2-mediated autoinhibition of JAK2.
It has been demonstrated that JH2 domain–mediated autophosphorylation is responsible for JH2-mediated JAK2 inhibition (26, 27). Therefore, we tested whether AGK-JH2 interaction can affect the phosphorylation status of JH2. Since there is currently no commercially available JH2 phosphorylation–specific antibody, we immunoprecipitated the ectopically expressed JH2 domain and then examined its phosphorylation status using a phosphotyrosine-specific antibody. As shown in Figure 2E, overexpression of AGK drastically reduced the phosphorylation level of JH2 but elevated the expression of p-JAK2 (Tyr1007-1008), suggesting that AGK induced JAK2 kinase activity via inhibition of JH2 autophosphorylation. Furthermore, an in vitro kinase assay showed that incubation of recombinant STAT3 with AGK alone did not result in phosphorylation of STAT3 (Tyr705). However, AGK could dramatically increase the phosphorylation level of STAT3 (Tyr705) mediated by JAK2 (Figure 2F). Interestingly, the duration of STAT3 activation induced by IL-6 stimulation was dramatically prolonged in AGK-transduced cells and reduced in AGK-silenced cells (Supplemental Figure 3), indicating that overexpression of AGK sustained JAK2/STAT3 signaling. Moreover, we found that the kinase-dead AGK mutant, AGK G126E (20), could still form a complex with JAK2, and overexpression of AGK G126E also increased the phosphorylation level of STAT3 (Figure 2, G and H). Taken together, these results further support the notion that AGK-mediated activation of JAK2/STAT3 signaling occurs through the induction of JAK2 activity via the suppression of JH2 autophosphorylation.
AGK promotes ESCC tumorigenesis in vivo.
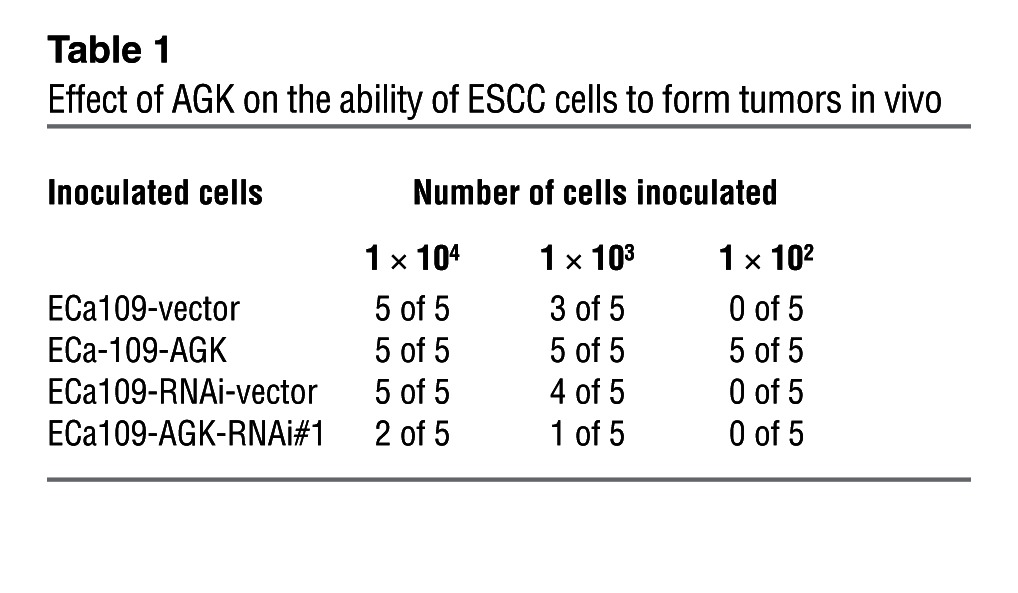
In an effort to understand the effect of AGK on activation of JAK2/STAT3 signaling, we subcutaneously inoculated different numbers of cells mixed with Matrigel into the inguinal folds of NOD/SCID mice. As shown in Figure 3, A and B, the tumors formed by AGK-transduced ESCC cells were dramatically larger than the vector control tumors when 1 × 104 or 1 × 103 cells were implanted. Conversely, AGK-silenced cells formed much smaller tumors and presented lower rates of tumorigenesis (Figure 3A and Table 1). Importantly, only AGK-overexpressing cells formed tumors when 1 × 102 cells were implanted (Figure 3A and Table 1). Furthermore, immunohistochemistry (IHC) revealed that AGK overexpression increased, whereas AGK silencing decreased, the phosphorylation levels of both JAK2 and STAT3 in tumor xenografts (Figure 3C). These results indicate that AGK activates the JAK2/STAT3 pathway and strongly promotes ESCC tumorigenesis in vivo.
Figure 3. AGK enhances the tumorigenicity of ESCC cells in vivo.

(A) The tumors formed by AGK-transduced ESCC cells were larger than the vector control tumors. Conversely, the tumors formed by AGK-silenced cells were smaller than the tumors formed by the RNAi-vector cells. 1 × 104 and 1 × 103 of the indicated cells were implanted into NOD/SCID mice (upper and middle panels). 1 × 102 of the indicated cells were implanted into NOD/SCID mice (lower panels); only the AGK-transduced cells formed tumors. Representative images of the tumors are shown. (B) Growth curves for tumor formation after implantation of different numbers of the indicated cells. Mean tumor volumes are plotted. (C) Representative images of tumor tissue formed by the indicated cells stained with H&E or antibodies against AGK, p-STAT3 (Tyr705), or p-JAK2 (Tyr1007-1008). Scale bars: 1 cm (A); 100 μm (C).
Table 1.
Effect of AGK on the ability of ESCC cells to form tumors in vivo
AGK promotes the stem cell population and stem cell–like phenotype in ESCC.
Considering the ability of AGK to induce tumorigenesis in a very small number of cells, we suspected that AGK might be involved in the promotion of the CSC population in ESCC. We therefore conducted a tumor sphere formation assay to examine the effect of AGK on self-renewal of spherogenic ESCC cells. Notably, AGK-transduced cells formed approximately 2-fold more spheres with an approximately 2- to 10-fold higher cell content compared with the spheres formed by vector control cells. Conversely, AGK-silenced cells formed approximately 4-fold fewer spheres with an approximately 3- to 7-fold lower cell content compared with vector control cells (Figure 4, A–C, and Supplemental Figure 4).
Figure 4. AGK promotes the ESCC CSC population in vitro.

(A) Representative images of spheres formed by the indicated cells. Scale bar: 100 μm. (B and C) Histograms showing the mean number of spheres (B) and the fold change in the number of cells per sphere (C) in spheres formed by the indicated cells. (D) Hoechst 33342 dye exclusion assay showing that overexpressing AGK promoted, whereas silencing AGK attenuated, the SP cells in the indicated cells. (E) Real-time PCR analysis of the expression of pluripotency-associated markers, including ABCG2, SOX2, OCT4, NANOG, and BMI1, in the indicated cells. Error bars represent the means ± SD of 3 independent experiments. *P < 0.05.
It has been reported that the side population (SP) is a subpopulation of cells that may exhibit stem cell–like characteristics and that CD44 expression correlates with the tumorigenicity of ESCC cells (28–30). Consistent with previous reports, our analysis also shows that SP+ cells sorted from ESCC cells had a higher proportion of CD44+ cells compared with SP– cells, and SP+ cells and CD44+ cells sorted from ESCC cells exhibited a higher clonogenic ability and higher expression of pluripotency-associated markers, including ABCG2, SOX2, OCT4, NANOG, and BMI1 (Supplemental Figure 5, A–C). We then further examined the effect of AGK on the regulation of the proportion of SP+ cells and CD44+ cells. As shown in Figure 4D, AGK overexpression increased the proportion of SP+ cells from 0.66% to 8.12% in ECa109 cells, and from 0.22% to 3.81% in KYSE510 cells. Conversely, silencing AGK decreased the proportion of SP+ cells from 0.64% to 0.14% in ECa109 cells, and from 0.22% to 0.09% in KYSE510 cells (Figure 4D). Similarly, the CD44+ population and the expression of multiple pluripotency-associated factors dramatically increased in AGK-transduced cells but decreased in AGK-silenced or JAK2-silenced cells (Supplemental Figure 6, A and B, and Figure 4E). Collectively, our results indicate that overexpression of AGK promotes the stem cell population and stem cell–like phenotype in ESCC.
JAK2/STAT3 signaling is required for the cancer stem cell–promoting effect of AGK.
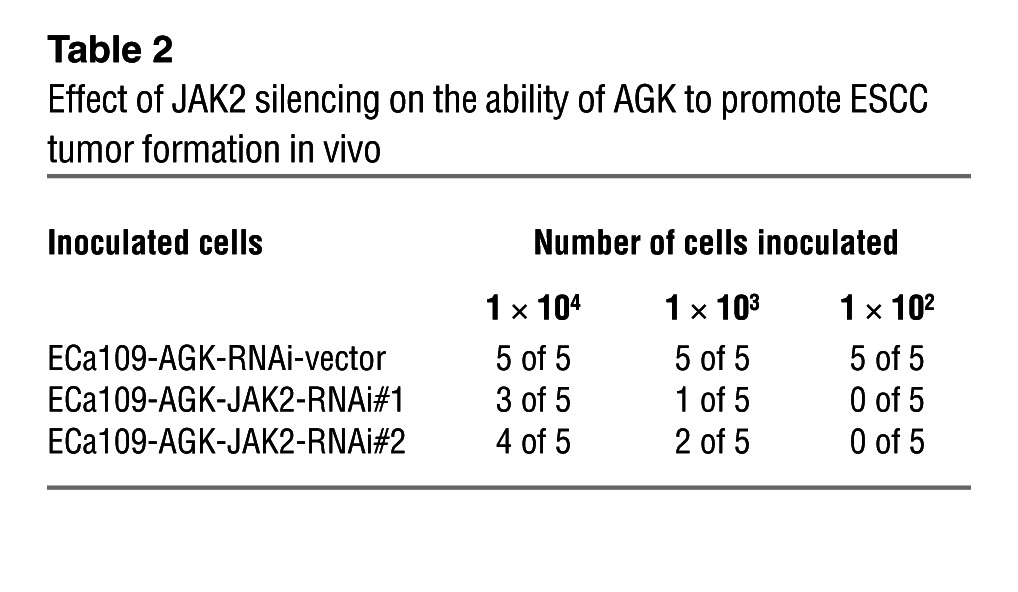
Next, we investigated whether JAK2/STAT3 activation is responsible for the promoting effect of AGK on the CSC population in ESCC. As shown in Figure 5, A–C, Supplemental Figure 7A, and Table 2, silencing JAK2 in AGK-transduced cells resulted in a drastic reduction of sphere-forming ability and a reversal of AGK-induced tumorigenicity in vivo. The increased proportion of SP+ and CD44+ cells, elevated expression of pluripotency-associated markers, and increased p-STAT3 expression induced by AGK overexpression could also be dramatically abrogated by JAK2 knockdown or by treatment with JAK2 inhibitors (Figure 5, D and E, Supplemental Figure 7B, and Supplemental Figure 8). Conversely, depletion of STAT3 also abolished the ability of AGK to promote sphere formation (Figure 5, F–H, and Supplemental Figure 7C). Together, these results suggest that the JAK2/STAT3 pathway is required for the promoting effect of AGK on cancer stem cell–associated phenotypes in ESCC.
Figure 5. Silencing JAK2 reverses the CSC-promoting effect of AGK.

(A) Representative images of spheres formed by the indicated cells. (B and C) Histograms showing the mean number of spheres (B) and the fold change in the number of cells per sphere (C) in spheres formed by the indicated cells. (D) Hoechst 33342 dye exclusion assay showing that JAK2 silencing reversed the ability of AGK to promote the SP cells in the indicated cells. (E) Real-time PCR analysis of the expression of pluripotency-associated markers in the indicated cells. (F) Representative images of spheres formed by the indicated cells. (G and H) Histograms showing the mean number of tumor spheres (G) and the fold change in the number of cells per sphere (H) in spheres formed by the indicated ESCC cells. Error bars represent the means ± SD of 3 independent experiments. *P < 0.05. Scale bars: 100 μm (A and F).
Table 2.
Effect of JAK2 silencing on the ability of AGK to promote ESCC tumor formation in vivo
AGK overexpression correlates with progression and poor prognosis in human ESCC.
To investigate whether AGK contributes to the pathogenesis of ESCC, we examined the expression of AGK in ESCC cell lines and human ESCC tissues. As shown in Figure 6, A and B, and Supplemental Figure 9, A and B, AGK was differentially upregulated at both the protein and mRNA levels in all 11 ESCC cell lines analyzed compared with 2 primary normal esophageal epithelial cells (NEECs), and in all 8 ESCC patient samples compared with the paired adjacent non-tumor tissues, indicating that AGK is overexpressed in ESCC. We also consistently observed that the levels of AGK were associated with p-JAK2 (Tyr1007-1008; P = 0.032, r = 0.596) and p-STAT3 (Tyr705; P = 0.007, r = 0.703) expression in ESCC cell lines (Figure 6A).
Figure 6. Overexpression of AGK correlates with poor prognosis in human ESCC.

(A) Western blot (left panel) and correlation analyses (right panel) of AGK, p-STAT3 (Tyr705), total STAT3, p-JAK2 (Tyr1007-1008), and total JAK2 expression in 2 primary cultured human NEECs and 11 ESCC cell lines. α-Tubulin was used as a loading control. (B) Western blot (left panel) and correlation analyses (right panel) of AGK expression in 8 paired primary ESCC tissues (T) and the matched adjacent non-tumor tissues (ANT) from the same patient. α-Tubulin was used as a loading control. (C) IHC staining indicating that AGK expression is upregulated in human ESCC compared with normal esophageal tissues. Normal esophageal tissue (upper panel); ESCC tissue (lower panel). Scale bar: 100 μm. (D and E) Kaplan-Meier analysis of 5-year overall survival (D) and 5-year disease-free survival (E) for ESCC patients stratified by low versus high expression of AGK (log-rank test; P < 0.001, P < 0.001, respectively; n = 247). Quantification of IHC using a staining index (SI; see Supplemental Methods). Samples with an SI greater than or equal to 8 were considered to have high expression and samples with an SI less than 8 were considered to have low expression.
To further investigate the clinical significance of AGK in ESCC, AGK expression was examined in 247 cases of ESCC using IHC (Supplemental Table 1). As illustrated in Figure 6C, AGK was markedly upregulated in ESCC, but was only marginally detectable in normal esophageal tissues. Statistical analyses revealed that AGK expression correlated with clinical stage (P < 0.001), TNM classification (T: P = 0.008, N: P < 0.001, M: P < 0.001, respectively), tumor grade (P = 0.002), and recurrence or uncontrolled progression (P = 0.012) in ESCC (Supplemental Table 2). Importantly, high AGK expression was associated with poorer prognosis and poorer disease-free survival in ESCC patients (P < 0.001; P < 0.001; Figure 6, D and E). Moreover, univariate and multivariate survival analyses indicated that AGK expression was recognized as an independent prognostic factor for both overall and disease-free survival in ESCC patients (Supplemental Tables 3 and 4). Taken together, our results suggest a possible link between overexpression of AGK and ESCC progression.
AGK expression correlates with STAT3 activation in ESCC.
Finally, we examined whether the AGK/JAK2/STAT3 axis identified in ESCC cells is clinically relevant. As shown in Figure 7, A and B, correlation studies showed that AGK expression positively correlated with the phosphorylation levels of JAK2 and STAT3 in ESCC specimens (P < 0.001, P < 0.001, respectively). These results were further confirmed in 8 freshly collected ESCC specimens, in which AGK expression positively correlated with the expression of p-STAT3 (Tyr705; P = 0.031, r = 0.753) and p-JAK2 (Tyr1007-1008; P = 0.018, r = 0.797), and STAT3 transcriptional activity (P = 0.039, r = 0.732; Figure 7, C and D). We also found that AGK levels positively correlated with the expression of pluripotency markers in the same 8 ESCC specimens and ESCC datasets (Supplemental Figure 10, A–D). Importantly, AGK expression also correlated with the expression of STAT3-regulated gene signatures in both lung cancer and breast cancer datasets (Figure 8A and Supplemental Figure 11, A and B). Consistently, depletion of AGK in both lung cancer and breast cancer cell lines resulted in decreased expression of p-JAK2 (Tyr1007-1008) and p-STAT3 (Tyr705) and reduced STAT3 transcriptional activity (Figure 8, B and C). These observations further support the notion that AGK contributes to JAK2/STAT3 activation in solid tumors, which results in tumor aggressiveness and poorer clinical outcome (Figure 8D).
Figure 7. Clinical relevance of AGK-induced activation of STAT3 in human ESCC.

(A) The expression levels of p-STAT3 (Tyr705) and p-JAK2 (Tyr1007-1008) were associated with the expression of AGK in 247 primary human ESCC specimens. Two representative cases are shown. Scale bar: 100 μm. (B) Percentage of samples showing low or high p-STAT3 (Tyr705) or p-JAK2 (Tyr1007-1008) expression in 247 primary human ESCC specimens relative to the levels of AGK. Quantification of IHC using an SI (see Supplemental Methods). Samples with an SI greater than or equal to 8 were considered to have high expression, and samples with an SI less than 8 were considered to have low expression. (C and D) Western blot (left panel) and correlation analyses (right panel) of AGK expression with the levels of p-STAT3 (Tyr705), p-JAK2 (Tyr1007-1008), and STAT3 transcriptional activity in 8 freshly collected human ESCC samples. α-Tubulin and OCT-1 DNA-binding complex were used as loading controls, respectively.
Figure 8. AGK expression positively correlated with STAT3 activation signatures in lung cancer and breast cancer.

(A) GSEA plot showing that AGK expression is positively correlated with STAT3-activated gene signatures (DAUER_STAT3_TARGETS_UP, V$STAT3_01/02) and inversely correlated with STAT3-suppressed gene signatures (DAUER_STAT3_TARGETS_Dn) in published lung cancer patient gene expression profiles (NCBI/GEO/GSE19804, n = 120) and breast cancer patient gene expression profiles (NCBI/GEO/GSE3744 and GSE5764, n = 77). (B) Western blot analysis of p-STAT3 (Tyr705), total STAT3, p-JAK2 (Tyr1007-1008), and total JAK2 expression in the indicated cells. α-Tubulin was used as a loading control. (C) STAT3 luciferase reporter activity in the indicated lung cancer cells or breast cancer cells. Error bars represent the means ± SD of 3 independent experiments. *P < 0.05. (D) Hypothetical model illustrating that AGK promotes JAK2 activity via a direct interaction with the JH2 domain, resulting in activation of the JAK2/STAT3 pathway in solid tumors.
Discussion
A novel mechanism regulating JAK2 activity in solid tumors.
For many cytoplasmic tyrosine kinases, intramolecular domain-domain interactions act as another level of negative regulation of their catalytic activity by inhibiting autophosphorylation and preventing aberrant activation of the kinases in response to various activation signals. For instance, the SRC kinases c-SRC and HCK are self-inhibited by association of the intramolecular SRC homology region 2 (SH2) and SH3 domains, which lock the molecule in a conformation that simultaneously disrupts the kinase active site (31, 32). However, mutations abrogating these intramolecular interaction sites result in kinase hyperactivation (33). Interaction of the intramolecular JH1/2 domain of JAK2 has also been found to autoinhibit and terminate basal JAK2 activity, which prevents persistent signal activation and increases inducibility under physiological conditions (12, 13). In agreement with this observation, JAK2 mutations that result in abrogation of JH2 kinase activity have been identified as driver mutations in hematological malignancies (16–18). However, how solid tumors cells, which rarely harbor comparable mutations, override JH2-mediated autoinhibition remains largely unknown. In the current study, we identify AGK as a binding partner of the JH2 domain of JAK2 kinase in ESCC. The interaction between AGK and the JH2 domain blocked the autoinhibitory effect of JH2 on JAK2, thus contributing to elevated basal JAK2 activity and prolonged STAT3 activity. More importantly, AGK expression was also found to correlate with STAT3-regulated signatures in ESCC, lung cancer, and breast cancer patient expression profiles. Thus, our findings uncover a novel mutation-independent mechanism that abrogates the autonegative regulation of JAK2 in solid tumors.
Oncogenic role of AGK in promoting the CSC population in ESCC.
Extensive evidence indicates that CSCs, the subpopulation of tumor cells that are capable of self-renewal and undergo aberrant differentiation processes, are strongly linked to cancer initiation and progression (34). CSCs display greater resistance to radio- and chemotherapy compared with more differentiated tumor cells, which indicates that the CSC subset can escape from conventional cancer therapy to initiate and perpetuate tumorigenesis (35). In multiple independent studies, the CSC population has been associated with poor patient prognosis in ESCC, which is the sixth leading cause of cancer deaths worldwide (28, 36, 37). Huang and colleagues reported that the CSC population in ESCC displays robust resistance to radio- and chemotherapy (38) and correlates with the risk of mortality in this disease. Despite strong evidence for their clinical relevance, the critical factors that regulate the maintenance of the CSC population in ESCC are still poorly explored. In this study, we demonstrate that AGK was markedly upregulated in ESCC, and high AGK expression was associated with poorer prognosis and reduced disease-free survival in ESCC patients. Overexpression of AGK promoted the CSC population and augmented the tumorigenicity of ESCC cells both in vivo and in vitro. Therefore, our findings not only provide a mechanistic insight into the maintenance of CSCs in ESCC, but also represent a target for restraining the CSC population in ESCC.
Biological and clinical lines of evidence have established that NF-κB is constitutively activated in ESCC (39, 40). Interestingly, we found that high levels of NF-κB are recruited to the promoter region of AGK, according to ChIP sequencing tracks in the UCSC genome browser ( http://genome.ucsc.edu/cgi-bin/hgGateway). Meanwhile, the AGK locus is located in the same region as the oncogene BRAF, which has been reported to be amplified in several solid tumor types (41–43), suggesting that overexpression of AGK in ESCC might be associated with genomic amplification. Thus, it would be of great interest to further investigate whether AGK upregulation in ESCC can be attributed to genomic amplification and/or NF-κB–mediated transcriptional upregulation.
Contribution of AGK to activation of JAK2/STAT3 signaling.
Recent advances have highlighted the role of JAK2/STAT3 signaling in the maintenance of CSCs, which reinforces the importance of this pathway in tumor recurrence and chemoresistance and indicates the potential curative effects of JAK2/STAT3 pathway inhibition (4–6). Meanwhile, constitutive activation of JAK2/STAT3 signaling is widely observed in ESCC, and disruption of the JAK2/STAT3 pathway can inhibit ESCC tumorigenesis and progression (44, 45), indicating the importance of JAK2/STAT3 signaling during the development and progression of ESCC. Herein, we demonstrated that ectopically expressing AGK significantly increased, whereas silencing AGK decreased, the STAT3 transactivity in ESCC cells. As a major cytokine responsible for activation of JAK2/STAT3 signaling, IL-6, has been demonstrated to play important roles in the promotion of malignant properties in multiple types of cancer. However, through an analysis of previously published gene expression profiles from ESCC tissues, we did not observe a significant correlation between the expression of IL-6 and the STAT3-regulated signatures (Supplemental Figure 12). In contrast, a positive correlation was found between the level of AGK and the STAT3-regulated gene signatures. Therefore, our results demonstrate an important role of AGK in the activation of JAK2/STAT3 signaling, which may occur independently of the effects of IL-6 in ESCC.
Therapeutic potential of AGK in ESCC.
Chemoradiotherapy (CRT) in combination with surgery is emerging as an effective therapeutic strategy in ESCC (46–48). However, the clinical response to CRT varies greatly in ESCC patients. Differences in the overall survival rate of patients receiving CRT followed by surgery, compared with surgery alone, have varied in multiple independent trials (46–50). On the other hand, patients who achieve a pathologic complete response (pathCR) after CRT have an improved survival rate (51, 52). Thus, identification of an effective parameter that can predict the response to CRT may help to determine the optimal therapeutic strategy in ESCC patients. Apart from chemoradioresistance, disease recurrence is another dominant prognostic factor in ESCC and also greatly reduces the effect of treatment (50, 53, 54). It is notable that the CSC population in ESCC has been reported to confer ESCC cells with both chemoradioresistance and the ability to recur (38, 55). Herein, we found that AGK acts as a potent CSC-promoting factor in ESCC, and high AGK expression was associated with poorer overall survival and disease-free survival in ESCC patients. Thus, our results suggest that AGK may be a potent determining factor in patient response to CRT and may have significance for the selection of the optimal therapeutic strategy for ESCC patients.
The discovery of JAK2 V617F as a driver mutation in hematological malignancies has led to the development and clinical trials of JAK2 inhibitors as potent therapeutic agents (56, 57). However, JAK2 inhibitors do not result in a reduced disease burden in most patients, which is thought to be linked to the compensatory effects of other members of the JAK kinase family (56–58). Since the JH2 domain is highly conserved in the JAK family (12), it would be interesting to further investigate whether AGK can also interact with other members of the JAK family and enhance their kinase activities. In this scenario, the inhibition of AGK-JAK interaction using dominant-negative AGK or an AGK-competing peptide may serve as a novel and efficient approach to block constitutive JAK/STAT3 activation in solid tumors.
Methods
Cell lines and primary cell culture.
Primary cultures of NEECs were established from fresh specimens of the adjacent non-tumor esophageal tissue, located more than 5 cm from the cancerous tissue, according to a previous report (59). The esophageal cancer cell lines EC18, ECa109, and HKESC1 were provided by S.W. Tsao and G. Srivastava (University of Hong Kong, Hong Kong, China). ESCC cell lines KYSE30, KYSE140, KYSE180, KYSE410, KYSE510, and KYSE520 were originally obtained from DSMZ, the German Resource Centre for Biological Material (60). The esophageal cancer cell lines were grown in DMEM medium (Invitrogen) supplemented with 10% FBS (HyClone; Thermo Scientific).
Tissue specimens and patient information.
A total of 247 paraffin-embedded, archived ESCCs and specimens were clinically and histopathologically diagnosed at the Sun Yat-sen University Cancer Center from 2000 to 2006. ESCCs and adjacent non-tumor tissues were obtained from resected tumors and adjacent non-tumor esophageal tissues, respectively, and were provided by Sun Yat-sen University Cancer Center and confirmed by pathological review. A total of 10 normal esophageal tissues were obtained by donation from individuals who died in traffic accidents and were confirmed to be free of any prior pathologically detectable conditions. Prior donor consent and approval from the Institutional Research Ethics Committee were obtained.
Plasmids, virus production, and infection of target cells.
The human AGK gene was PCR amplified from cDNA and cloned into the pSin EF2 lentiviral vector. shRNAs targeting AGK, JAK2, and STAT3 were cloned into the pSuper Retro viral vector. Truncated JAK2 fragments were cloned into pCDNA3 vector. 6 × STAT3 binding elements were cloned into pTAL Luc vector to generate STAT3 luciferase reporters. Recombinant His-tagged AGK was expressed using pET-19b vector. All primers and oligonucleotides used in plasmid construction are listed in the Primers and Oligonucleotides table in Supplemental Methods. Transfection of siRNAs or plasmids was performed using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. Stable cell lines expressing AGK and AGK shRNA(s) were generated via retroviral infection using HEK293T cells as previously described (61) and were selected with 0.5 μg/ml puromycin for 10 days.
Western blot analysis.
Western blot analysis was performed using anti-AGK (Epitomics); anti-pSTAT3 (Tyr705), anti-STAT3, anti-pJAK2 (Tyr1007-1008), anti-JAK2, anti-pTyr-100 antibodies (Cell Signaling Technology); and anti-Flag, anti-HA antibodies (Sigma-Aldrich). To control sample loading, the blotting membranes were stripped and re-probed with an anti–α-tubulin antibody or an anti-GAPDH antibody (Sigma-Aldrich).
Immunoprecipitation and MS analysis.
Lysates were prepared from 3 × 107 ECa109 cells transfected with HA-tagged JH2 or vector using lysis buffer (150 mM NaCl, 10 mM HEPES, pH 7.4, 1% NP-40). Lysates were then incubated with HA affinity agarose (Sigma-Aldrich) overnight at 4°C. Beads containing affinity-bound proteins were washed 6 times by immunoprecipitation wash buffer (150 mM NaCl, 10 mM HEPES, pH 7.4, 0.1% NP-40), followed by 2 elutions with 200 μl of 1 M glycine (pH 3.0). The eluates were pooled and concentrated in a 10-kDa MW cut-off filter unit (Millipore) to a volume of 30 μl. After the addition of 10 μl of 4 × sample buffer and denaturation, proteins were separated on SDS polyacrylamide gels stained with Coomassie blue, and JH2-specific bands were subjected to MS analysis.
Chemical reagents.
JAK2 inhibitors II and III (SD-1029) were purchased from Millipore. STAT3 recombinant protein was purchased from Abcam.
Tumor xenografts.
All experimental procedures were approved by the IACUC of Sun Yat-sen University. The NOD/SCID mice were randomly divided into 3 groups (n = 5 per group). Indicated cells of 3 dosages (1 × 104, 1 × 103, 1 × 102) were inoculated s.c. with Matrigel (final concentration of 25%) into the inguinal folds of NOD/SCID mice. Tumor volume was determined using an external caliper and calculated using the equation (L × W2)/2. The mice were scarified 31 days after inoculation and the tumors were excised and subjected to pathologic examination.
Sphere formation assays.
Five hundred cells were seeded in 6-well ultra-low cluster plates and 10 or 20 cells were seeded in 24-well ultra-low cluster plates for 10 days. Spheres were cultured in DMEM/F12 serum-free medium (Invitrogen) supplemented with 2% B27 (Invitrogen), 20 ng/ml EGF, 20 ng/ml bFGF (PeproTech), 0.4% BSA (Sigma-Aldrich), and 5 μg/ml insulin.
Flow cytometric analysis.
Cells were dissociated with trypsin and resuspended at 1 × 106 cells per milliliter in DMEM containing 2% FBS and then preincubated at 37°C for 30 minutes with or without 100 μM verapamil (Sigma-Aldrich) to inhibit ABC transporters. The cells were subsequently incubated for 90 minutes at 37°C with 5 μg/ml Hoechst 33342 (Sigma-Aldrich). Finally, the cells were incubated on ice for 10 minutes and washed with ice-cold PBS before flow cytometric analysis. The data were analyzed by Summit 5.2 software (Beckman Coulter).
Far-Western blot analysis.
For far-Western blot analysis, the indicated proteins were immunoprecipitated by HA-tag affinity gel (Sigma-Aldrich) and resolved by SDS-PAGE, and the proteins were transferred to a PVDF membrane. The membrane was then blocked in 10% skimmed milk for 1 hour at 4°C. As indicated, recombinant His-AGK was added at 5 μg/ml and incubated at 4°C for 18 hours. The blot was then washed 6 times with TBST and subjected to Western blot analysis using anti-His antibody (Sigma-Aldrich).
JAK2 kinase assay.
The immunoprecipitation-purified JAK2 protein (washed in buffer containing 25 mM HEPES, pH 7.4, 250 mM NaCl, 0.5% NP-40, 1 mM EDTA, 0.2% glycerol, and 1 mM PMSF) from HEK293T cells was subsequently washed 3 times with kinase assay buffer (10 mM HEPES, pH 7.4, 50 mM NaCl, 5 mM MgCl2, 5 mM MnCl2, 50 mM NaF, and 0.003 mM Na3VO4). The protein was then incubated with the indicated proteins and 250 μM adenosine triphosphate (ATP). Recombinant STAT3 (500 ng) was used as a substrate for each assay. The reaction system was incubated for 30 minutes at 25°C and then subjected to Western blot analysis.
EMSA.
EMSA was performed using the LightShift Chemiluminescent EMSA kit from Pierce Biotechnology. The following DNA probes containing specific binding sites were used: STAT3: sense, 5′-TCGACATTTCCCGTAAATC-3′, antisense, 5′-GATTTACGGGAAATGTCGA-3′; and OCT-1: sense, 5′-TGTCGAATGCAAATCACTAGAA-3′, antisense, 5′-TTCTAGTGATTTGCATTCGACA-3′.
Microarray data process and visualization.
Microarray data were downloaded from the GEO database ( http://www.ncbi.nlm.nih.gov/geo/) using the accession numbers indicated in Figures 1F and 8A, and in Supplemental Figures 2, 10D, and 12. Microarray data extracts were performed on MeV 4.6 ( http://www.tm4.org/mev/). GSEA was performed using GSEA 2.0.9 ( http://www.broadinstitute.org/gsea/).
Statistics.
All statistical analyses except for microarray data were performed using the SPSS 13.0 (IBM) statistical software package. Pearson’s χ2 test was applied in studying the correlation between AGK expression and clinicopathologic characteristics of ESCC. Survival curves for both AGK-high and AGK-low patients were plotted using the Kaplan-Meier method, and statistical differences were compared using a log-rank test. Univariate and multivariable survival analyses were performed using Cox regression analysis. Comparisons between 2 groups were performed using the Student’s t test. Bivariate correlations between study variables were calculated by Pearson’s correlation coefficients. Data represent the means ± SD. A P value less than 0.05 was considered statistically significant.
Study approval.
The use of human ESCC tissue specimens was evaluated and approved by the Ethical Committee of Sun Yat-sen University Cancer Center, and written informed consent was obtained from all participants or their appropriate surrogates. All animal studies were conducted with the approval of the Sun Yat-sen University IACUC and were performed in accordance with established guidelines.
Supplementary Material
Acknowledgments
This work was supported by the Natural Science Foundation of China (81071780, 81272198, and U1201121), The Science and Technology Department of Guangdong Province (S2011020002757), and The Ministry of Education of China (20100171110080).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(6):2576–2589. doi:10.1172/JCI68143.
References
- 1.Shuai K, Liu B. Regulation of JAK-STAT signalling in the immune system. Nat Rev Immunol. 2003;3(11):900–911. doi: 10.1038/nri1226. [DOI] [PubMed] [Google Scholar]
- 2.Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu H, Jove R. The STATs of cancer — new molecular targets come of age. Nat Rev Cancer. 2004;4(2):97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 4.Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, Ng IO. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell. 2011;9(1):50–63. doi: 10.1016/j.stem.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Guryanova OA, et al. Nonreceptor tyrosine kinase BMX maintains self-renewal and tumorigenic potential of glioblastoma stem cells by activating STAT3. Cancer Cell. 2011;19(4):498–511. doi: 10.1016/j.ccr.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marotta LL, et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(–) stem cell-like breast cancer cells in human tumors. J Clin Invest. 2011;121(7):2723–2735. doi: 10.1172/JCI44745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burke WM, et al. Inhibition of constitutively active Stat3 suppresses growth of human ovarian and breast cancer cells. Oncogene. 2001;20(55):7925–7934. doi: 10.1038/sj.onc.1204990. [DOI] [PubMed] [Google Scholar]
- 8.Chan KS, et al. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J Clin Invest. 2004;114(5):720–728. doi: 10.1172/JCI21032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Catlett-Falcone R, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10(1):105–115. doi: 10.1016/S1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 10.Silvennoinen O, Witthuhn BA, Quelle FW, Cleveland JL, Yi T, Ihle JN. Structure of the murine Jak2 protein-tyrosine kinase and its role in interleukin 3 signal transduction. Proc Natl Acad Sci U S A. 1993;90(18):8429–8433. doi: 10.1073/pnas.90.18.8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamaoka K, Saharinen P, Pesu M, Holt VE 3rd, Silvennoinen O, O’Shea JJ. The Janus kinases (Jaks). Genome Biol. 2004;5(12):253. doi: 10.1186/gb-2004-5-12-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saharinen P, Silvennoinen O. The pseudokinase domain is required for suppression of basal activity of Jak2 and Jak3 tyrosine kinases and for cytokine-inducible activation of signal transduction. J Biol Chem. 2002;277(49):47954–47963. doi: 10.1074/jbc.M205156200. [DOI] [PubMed] [Google Scholar]
- 13.Feener EP, Rosario F, Dunn SL, Stancheva Z, Myers MG., Jr Tyrosine phosphorylation of Jak2 in the JH2 domain inhibits cytokine signaling. Mol Cell Biol. 2004;24(11):4968–4978. doi: 10.1128/MCB.24.11.4968-4978.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo H, et al. Mutation in the Jak kinase JH2 domain hyperactivates Drosophila and mammalian Jak-Stat pathways. Mol Cell Biol. 1997;17(3):1562–1571. doi: 10.1128/mcb.17.3.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saharinen P, Takaluoma K, Silvennoinen O. Regulation of the Jak2 tyrosine kinase by its pseudokinase domain. Mol Cell Biol. 2000;20(10):3387–3395. doi: 10.1128/MCB.20.10.3387-3395.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.James C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature. 2005;434(7037):1144–1148. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 17.Baxter EJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365(9464):1054–1061. doi: 10.1016/S0140-6736(05)71142-9. [DOI] [PubMed] [Google Scholar]
- 18.Levine RL, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 19.Bromberg J. Stat proteins and oncogenesis. J Clin Invest. 2002;109(9):1139–1142. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bektas M, Payne SG, Liu H, Goparaju S, Milstien S, Spiegel S. A novel acylglycerol kinase that produces lysophosphatidic acid modulates cross talk with EGFR in prostate cancer cells. J Cell Biol. 2005;169(5):801–811. doi: 10.1083/jcb.200407123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waggoner DW, Johnson LB, Mann PC, Morris V, Guastella J, Bajjalieh SM. MuLK, a eukaryotic multi-substrate lipid kinase. J Biol Chem. 2004;279(37):38228–38235. doi: 10.1074/jbc.M405932200. [DOI] [PubMed] [Google Scholar]
- 22.Epand RM, et al. Substrate chirality and specificity of diacylglycerol kinases and the multisubstrate lipid kinase. Biochemistry. 2007;46(49):14225–14231. doi: 10.1021/bi701584v. [DOI] [PubMed] [Google Scholar]
- 23.Nouh MA, et al. Expression of autotaxin and acylglycerol kinase in prostate cancer: association with cancer development and progression. Cancer Sci. 2009;100(9):1631–1638. doi: 10.1111/j.1349-7006.2009.01234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mootha VK, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 26.Argetsinger LS, Kouadio JL, Steen H, Stensballe A, Jensen ON, Carter-Su C. Autophosphorylation of JAK2 on tyrosines 221 and 570 regulates its activity. Mol Cell Biol. 2004;24(11):4955–4967. doi: 10.1128/MCB.24.11.4955-4967.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ungureanu D, et al. The pseudokinase domain of JAK2 is a dual-specificity protein kinase that negatively regulates cytokine signaling. Nat Struct Mol Biol. 2011;18(9):971–976. doi: 10.1038/nsmb.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao JS, et al. Tumor initiating cells in esophageal squamous cell carcinomas express high levels of CD44. PLoS One. 2011;6(6):e21419. doi: 10.1371/journal.pone.0021419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bleau AM, et al. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell. 2009;4(3):226–235. doi: 10.1016/j.stem.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ho MM, Ng AV, Lam S, Hung JY. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007;67(10):4827–4833. doi: 10.1158/0008-5472.CAN-06-3557. [DOI] [PubMed] [Google Scholar]
- 31.Xu W, Harrison SC, Eck MJ. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385(6617):595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- 32.Sicheri F, Moarefi I, Kuriyan J. Crystal structure of the Src family tyrosine kinase Hck. Nature. 1997;385(6617):602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- 33.Hirai H, Varmus HE. Mutations in src homology regions 2 and 3 of activated chicken c-src that result in preferential transformation of mouse or chicken cells. Proc Natl Acad Sci U S A. 1990;87(21):8592–8596. doi: 10.1073/pnas.87.21.8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annu Rev Med. 2007;58:267–284. doi: 10.1146/annurev.med.58.062105.204854. [DOI] [PubMed] [Google Scholar]
- 35.Frank NY, Schatton T, Frank MH. The therapeutic promise of the cancer stem cell concept. J Clin Invest. 2010;120(1):41–50. doi: 10.1172/JCI41004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsunoda S, et al. ABCG2 expression is an independent unfavorable prognostic factor in esophageal squamous cell carcinoma. Oncology. 2006;71(3–4):251–258. doi: 10.1159/000106787. [DOI] [PubMed] [Google Scholar]
- 37.Bass AJ, et al. SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat Genet. 2009;41(11):1238–1242. doi: 10.1038/ng.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang SD, et al. Self-renewal and chemotherapy resistance of p75NTR positive cells in esophageal squamous cell carcinomas. BMC Cancer. 2009;9:9. doi: 10.1186/1471-2407-9-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li B, Li YY, Tsao SW, Cheung AL. Targeting NF-κB signaling pathway suppresses tumor growth, angiogenesis, and metastasis of human esophageal cancer. Mol Cancer Ther. 2009;8(9):2635–2644. doi: 10.1158/1535-7163.MCT-09-0162. [DOI] [PubMed] [Google Scholar]
- 40.Kausar T, et al. Clinical significance of GPR56, transglutaminase 2, and NF-κB in esophageal squamous cell carcinoma. Cancer Invest. 2011;29(1):42–48. doi: 10.3109/07357907.2010.512597. [DOI] [PubMed] [Google Scholar]
- 41.Brose MS, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62(23):6997–7000. [PubMed] [Google Scholar]
- 42.Corcoran RB, Dias-Santagata D, Bergethon K, Iafrate AJ, Settleman J, Engelman JA. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci Sig. 2010;3(149):ra84. doi: 10.1126/scisignal.2001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanami H, et al. Involvement of overexpressed wild-type BRAF in the growth of malignant melanoma cell lines. Oncogene. 2004;23(54):8796–8804. doi: 10.1038/sj.onc.1208152. [DOI] [PubMed] [Google Scholar]
- 44. Zhang Y, et al. Reciprocal activation between PLK1 and Stat3 contributes to survival and proliferation of esophageal cancer cells. Gastroenterology. 2012. 142 3 521 530 e523. 10.1053/j.gastro.2011.11.023 [DOI] [PubMed] [Google Scholar]
- 45.You Z, Xu D, Ji J, Guo W, Zhu W, He J. JAK/STAT signal pathway activation promotes progression and survival of human oesophageal squamous cell carcinoma. Clin Transl Oncol. 2012;14(2):143–149. doi: 10.1007/s12094-012-0774-6. [DOI] [PubMed] [Google Scholar]
- 46.Bosset JF, et al. Chemoradiotherapy followed by surgery compared with surgery alone in squamous-cell cancer of the esophagus. N Engl J Med. 1997;337(3):161–167. doi: 10.1056/NEJM199707173370304. [DOI] [PubMed] [Google Scholar]
- 47.Herskovic A, et al. Combined chemotherapy and radiotherapy compared with radiotherapy alone in patients with cancer of the esophagus. N Engl J Med. 1992;326(24):1593–1598. doi: 10.1056/NEJM199206113262403. [DOI] [PubMed] [Google Scholar]
- 48.van Hagen P, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Engl J Med. 2012;366(22):2074–2084. doi: 10.1056/NEJMoa1112088. [DOI] [PubMed] [Google Scholar]
- 49.Enzinger PC, Mayer RJ. Esophageal cancer. N Engl J Med. 2003;349(23):2241–2252. doi: 10.1056/NEJMra035010. [DOI] [PubMed] [Google Scholar]
- 50.Lightdale C J. Esophageal cancer. American College of Gastroenterology. Am J Gastroenterol. 1999;94(1):20–29. doi: 10.1111/j.1572-0241.1999.00767.x. [DOI] [PubMed] [Google Scholar]
- 51.Berger AC, et al. Complete response to neoadjuvant chemoradiotherapy in esophageal carcinoma is associated with significantly improved survival. J Clin Oncol. 2005;23(19):4330–4337. doi: 10.1200/JCO.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 52.Heath EI, et al. Phase II evaluation of preoperative chemoradiation and postoperative adjuvant chemotherapy for squamous cell and adenocarcinoma of the esophagus. J Clin Oncol. 2000;18(4):868–876. doi: 10.1200/JCO.2000.18.4.868. [DOI] [PubMed] [Google Scholar]
- 53.Faivre J, Forman D, Esteve J, Gatta G. Survival of patients with oesophageal and gastric cancers in Europe. EUROCARE Working Group. Eur J Cancer. 1998;34(14 spec no):2167–2175. doi: 10.1016/S0959-8049(98)00329-3. [DOI] [PubMed] [Google Scholar]
- 54.Mariette C, et al. Pattern of recurrence following complete resection of esophageal carcinoma and factors predictive of recurrent disease. Cancer. 2003;97(7):1616–1623. doi: 10.1002/cncr.11228. [DOI] [PubMed] [Google Scholar]
- 55.Che SM, Zhang XZ, Liu XL, Chen X, Hou L. The radiosensitization effect of NS398 on esophageal cancer stem cell-like radioresistant cells. Dis Esophagus. 2011;24(4):265–273. doi: 10.1111/j.1442-2050.2010.01138.x. [DOI] [PubMed] [Google Scholar]
- 56.Verstovsek S, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–1127. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pardanani A, et al. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011;29(7):789–796. doi: 10.1200/JCO.2010.32.8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koppikar P, et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature. 2012;489(7414):155–159. doi: 10.1038/nature11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andl CD, et al. Epidermal growth factor receptor mediates increased cell proliferation, migration, and aggregation in esophageal keratinocytes in vitro and in vivo. J Biol Chem. 2003;278(3):1824–1830. doi: 10.1074/jbc.M209148200. [DOI] [PubMed] [Google Scholar]
- 60.Shimada Y, Imamura M, Wagata T, Yamaguchi N, Tobe T. Characterization of 21 newly established esophageal cancer cell lines. Cancer. 1992;69(2):277–284. doi: 10.1002/1097-0142(19920115)69:2<277::AID-CNCR2820690202>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 61.Liu L, et al. Astrocyte elevated gene-1 upregulates matrix metalloproteinase-9 and induces human glioma invasion. Cancer Res. 2010;70(9):3750–3759. doi: 10.1158/0008-5472.CAN-09-3838. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.