

Abstract
TNF has remarkable antitumor activities; however, therapeutic applications have not been possible because of the systemic and lethal proinflammatory effects induced by TNF. Both the antitumor and inflammatory effects of TNF are mediated by the TNF receptor p55 (p55TNFR) (encoded by the Tnfrsf1a gene). The antitumor effect stems from an induction of cell death in tumor endothelium, but the cell type that initiates the lethal inflammatory cascade has been unclear. Using conditional Tnfrsf1a knockout or reactivation mice, we found that the expression level of p55TNFR in intestinal epithelial cells (IECs) is a crucial determinant in TNF-induced lethal inflammation. Remarkably, tumor endothelium and IECs exhibited differential sensitivities to TNF when p55TNFR levels were reduced. Tumor-bearing Tnfrsf1a+/– or IEC-specific p55TNFR-deficient mice showed resistance to TNF-induced lethality, while the tumor endothelium remained fully responsive to TNF-induced apoptosis and tumors regressed. We demonstrate proof of principle for clinical application of this approach using neutralizing anti-human p55TNFR antibodies in human TNFRSF1A knockin mice. Our results uncover an important cellular basis of TNF toxicity and reveal that IEC-specific or systemic reduction of p55TNFR mitigates TNF toxicity without loss of antitumor efficacy.
Introduction
In high doses, TNF has remarkable antitumor effects, especially when it is combined with IFN-γ and/or chemotherapeutics (1). Unfortunately, TNF also possesses strong proinflammatory properties, and its use is often accompanied by unacceptable shock symptoms, such as hypotension and organ failure (2–4). Initial studies in patients showed that the maximum tolerated dose (MTD) that can be applied systemically in humans is too low for effective tumor therapy (5). Therefore, therapeutic anticancer application of TNF is limited to local settings, such as isolated limb perfusion, which does not cause systemic toxicity but leads to a very high rate of complete regression of melanomas and soft tissue sarcomas, avoiding amputation of the extremities (6–8). Such successes illustrate that TNF has great potential as an anticancer drug, providing that its toxicity can be reduced (9).
TNF acts by binding to two different receptors, TNF receptor p55 (p55TNFR) (Tnfrsf1a) and p75TNFR (Tnfrsf1b) (10). The p55TNFR was identified as the receptor responsible for both the antitumor effects (11–14) and the shock-inducing proinflammatory signals induced by TNF (12, 15), whereas p75TNFR has been implicated mainly in immune modulation (16) and plays no significant role in TNF’s toxicity (15). The default pathway of p55TNFR signaling, activated in most cell types, leads to cell survival and induction of inflammation, but when cells fail to sufficiently induce these survival signals, an alternate pathway leading to cell death is activated (17, 18). In contrast to initial beliefs, the antitumor effects of TNF are not due to direct induction of cell death of tumor cells. Evidence suggests that TNF directly affects the tumor neovasculature, causing endothelial cell death and vascular dysfunction, leading to tumor necrosis (11, 19, 20).
While the cellular target of the TNF-induced antitumor effects has been elucidated, the mechanisms and the tissues involved in TNF-induced toxicity have only been partially characterized. Recently, a role for necroptosis in TNF-induced toxicity was uncovered (21), but earlier work stresses the importance of TNF-mediated induction of inflammation (e.g., classical antiinflammatory drugs such as steroids and indomethacin protect against TNF toxicity; refs. 22, 23) and of inflammatory mediators, such as IL-17, IL-1, IFN-β, ROS, and iNOS (24–28). Multiple organs, such as intestine, liver, and kidney, suffer from the TNF-induced effects, but it is still unclear which cell type is essential in mediating/initiating the TNF-induced toxicity (2, 29, 30). Of interest, several acute inflammatory conditions in which TNF plays a role have been attributed to effects on the intestinal epithelial cells (IECs) (31, 32).
In this study, we addressed the question of whether TNF’s anticancer and proinflammatory effects can be uncoupled, leading to safer anticancer effects. We report that reducing the expression or availability of p55TNFR strongly dampens the proinflammatory signal without affecting the induction of apoptosis and antitumor effects. Moreover, using conditional Tnfrsf1a reactivation mice, we demonstrate that p55TNFR expression in IECs is sufficient to induce lethal toxicity, while conditional Tnfrsf1a-deficient mice prove that a reduction in p55TNFR expression in IECs significantly protects against lethal TNF-induced toxicity. Our findings uncover an important cellular basis of systemic TNF toxicity, reveal an IEC-specific haploinsufficiency of p55TNFR, and lay the ground for safe and effective TNF-based antitumor treatment strategies.
Results
A 50% reduction of p55TNFR completely protects mice against acute TNF.
Three different Tnfrsf1a–/– mouse lines have been generated using different targeting strategies (12–14). All of them were extremely resistant to the lethal inflammatory effect of TNF (Table 1). Hemizygous Tnfrsf1a mice were equally resistant to a single TNF injection and easily tolerated 1,000 μg per mouse (Table 1), i.e., 40-fold more than Tnfrsf1a+/+ mice, whose LD100 was 25–30 μg. Upon injection of murine TNF at 100 μg per mouse, Tnfrsf1a+/+ mice died from inflammation within 24 hours, but no effects were observed in Tnfrsf1a+/– mice (data not shown). In addition, TNF-induced IL-6 was absent in the sera of all Tnfrsf1a–/– lines and was significantly lower (on average 32.5 fold) in Tnfrsf1a+/– mice than in Tnfrsf1a+/+ mice after TNF injection (Figure 1A). The results were independently confirmed in the 3 different types of Tnfrsf1a–/– and Tnfrsf1a+/– mouse lines. Subsequent experiments were performed on one type of Tnfrsf1a–/– line, namely the one generated by Rothe et al. (12). In contrast to Tnfrsf1a+/– and Tnfrsf1a–/– mice, Tnfrsf1a+/+ mice challenged with TNF displayed hypothermia (Figure 1B), sickness symptoms (ruffled fur, diarrhea, and physical inactivity) (Figure 1C), and liver and kidney damage (Figure 1D and Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI65624DS1) as well as increases in plasma hexosaminidase and LDH, general markers of cellular damage (Supplemental Figure 1, B and C). The intestinal barrier function, measured by leakage of orally gavaged FITC-dextran into the blood, was significantly affected in Tnfrsf1a+/+ mice, but not in Tnfrsf1a+/– mice, 8 hours after TNF challenge (Figure 1E).
Table 1.
Lethal effect of 3 different doses of i.p. TNF in 3 types of Tnfrsf1a–/– mice
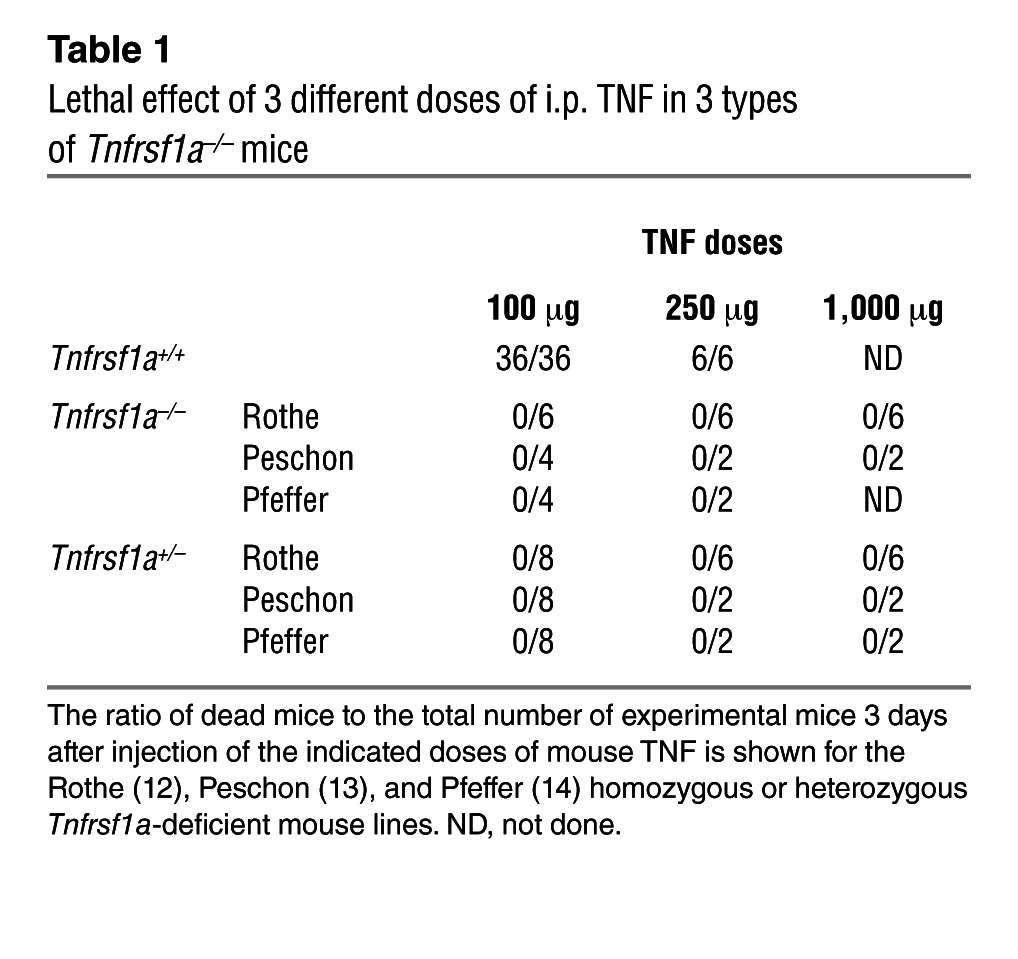
Figure 1. Resistance of Tnfrsf1a+/– mice to TNF-induced lethal inflammation.

(A) Serum IL-6 6 hours after a single i.p. injection of 100 μg TNF in the Rothe (12), Pfeffer (14), and Peschon (13) Tnfrsf1a-deficient mouse lines. ***P < 0.001, compared with Tnfrsf1a+/+. (B) Body temperature (***P < 0.001, compared with Tnfrsf1a+/– and Tnfrsf1a–/–), (C) sickness score, (D) serum AST and ALT levels, and (E) intestinal permeability of Tnfrsf1a+/+, Tnfrsf1a+/–, and Tnfrsf1a–/– mice after i.p injection with 50 μg TNF i.p. (or 25 μg i.p. for permeability assay) (n = 8 for all groups). (F) p55TNFR expression, measured by ELISA, in liver samples. (G) Specific binding of 125I-hTNF to BMDMs. In D and F, levels in Tnfrsf1a–/– and Tnfrsf1a+/– were 0, so no statistical significance could be calculated toward Tnfrsf1a+/+ data. Data represent mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t test).
The amount of cell-associated p55TNFR protein in livers, lungs, and other organs of hemizygous mice was about half of that in Tnfrsf1a+/+ mice (Figure 1F and Supplemental Figure 1, D–F). These results were confirmed by qPCR, which showed that livers of Tnfrsf1a+/– mice had about half of the p55TNFR mRNA of Tnfrsf1a+/+ mice (Supplemental Figure 1G). Binding studies using I125-labeled human TNF (125I-hTNF), which binds mouse p55TNFR (mp55TNFR) but not mouse p75TNFR (33), showed that binding is reduced by about half in Tnfrsf1a+/– bone marrow–derived macrophages (BMDMs) compared with Tnfrsf1a+/+ BMDMs (Figure 1G). Finally, p55TNFR expression was measured on Tnfrsf1a+/– cells by FACS and was intermediate between Tnfrsf1a+/+ and Tnfrsf1a–/– BMDMs (Supplemental Figure 1H).
TNF-induced inflammation is strongly reduced in Tnfrsf1a+/– mice.
To explain the reduced induction of inflammation in Tnfrsf1a+/– cells, we isolated BMDMs and thioglycolate-elicited peritoneal macrophages from the 3 different genotypes, Tnfrsf1a+/+, Tnfrsf1a+/–, and Tnfrsf1a–/–, and stimulated them in vitro with TNF. In this setup, we looked at different steps of p55TNFR-mediated inflammation, such as NF-κB and MAPK activation and production of IL-6. NF-κB activation was studied by IκB degradation (Supplemental Figure 2A) and EMSA (Figure 2A and Supplemental Figure 2B). The incomplete degradation of IκB in Tnfrsf1a+/– cells was confirmed by the reduced activation and nuclear translocation of NF-κB compared with that in Tnfrsf1a+/+ cells. Similarly, phosphorylation of the MAPKs JNK2 and ERK was reduced in Tnfrsf1a+/– cells compared with that in Tnfrsf1a+/+ cells (Figure 2, B and C, and Supplemental Figure 2, C and D). Accordingly, the induction of IL-6 after TNF stimulation in Tnfrsf1a+/– mice was significantly lower than that in Tnfrsf1a+/+ mice at 24 hours (Supplemental Figure 2E).
Figure 2. Signal transduction, gene expression, and cytokine release induced by TNF in Tnfrsf1a+/– mice.

(A) Quantification of NF-κB EMSA results in nuclear extracts of BMDMs after stimulation with 10 ng/ml TNF. The graph shows intermediate activation of NF-κB in the Tnfrsf1a+/– samples compared with the Tnfrsf1a+/+ and Tnfrsf1a–/– samples. (B and C) Quantification of Western blot results showing JNK and ERK phosphorylation in BMDMs at several time points after stimulation with 10 ng/ml TNF. See also Supplemental Figure 2, B–D, for original blots. (D–F) Intermediate induction of genes coding for IKBA, IL-1β, and IFN-β in livers of mice injected i.p. with 30 μg TNF. Data for Tnfrsf1a–/– mice are not shown, because these mice are not responsive to TNF. (G–I) Intermediate IL-1β, IL-12p40, and NOx release in sera of Tnfrsf1a+/– mice injected with 30 μg TNF. (J and K) Cumulative protection against hypothermia and lethality induced by injection of 37.5 μg TNF (1.5-times LD100 in Tnfrsf1a+/+ mice). Ifnar1+/+ mice (n = 7) were compared with Ifnar1–/– mice (n = 10), which were protected by the ROS inhibitor Tempol (n = 9) or by Tempol and IL-1RA (Anakinra) (n = 10). Numbers in parentheses indicate the number of mice in each group that did not survive. Data represent mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t test in D–I and log-rank test in K).
Similar experiments performed in vivo showed that degradation of IκB in the liver was undetectable in TNF-injected Tnfrsf1a–/– mice and was reduced significantly in Tnfrsf1a+/– mice compared with that in Tnfrsf1a+/+ mice (Supplemental Figure 2F). Similar data were obtained in lungs (data not shown). To know whether this leads to reduced TNF-induced toxicity, we injected mice with a single dose of 30 μg TNF and measured the expression of NF-κB–dependent genes in the liver by qPCR 0, 1, and 6 hours after challenge. The expression of IKBA, Il1b, IFN-β, Cxcl9, c-FLIP, and Nox2 was significantly lower (~50%) in Tnfrsf1a+/– mice than that in Tnfrsf1a+/+ mice (Figure 2, D–F, and Supplemental Figure 2, G–I). The same pattern was found for a whole set of other NF-κB–inducible genes, including A20, Icam1, and Vcam1 (data not shown). We also measured serum cytokine levels after TNF stimulus and found that, while no induction was observed in Tnfrsf1a–/– mice, most of them were half-maximally induced in Tnfrsf1a+/– mice: IL-1β and IL-12p40 (Figure 2, G and H) as well as IL-1α, MIP1β, MCP-1, and CCL5/Rantes (Supplemental Figure 2, J–M). In addition, we found half the amount of NO metabolites in Tnfrsf1a+/– mice compared with that in Tnfrsf1a+/+ mice (Figure 2I).
The strong protection of Tnfrsf1a+/– mice from TNF and the general reduction of expression of genes that have previously been shown to mediate TNF toxicity is consistent with a model in which TNF toxicity is indirect and mediated by several molecules, a combined reduction of which leads to accumulating protection. To verify this hypothesis, we studied the effect of inhibition of multiple proven toxic mediators on injection of 37.5 μg TNF, a 1.5-times LD100 dose. Type I IFNs were inhibited genetically by using Ifnar1–/– animals (27), ROS were inhibited using tempol (28), and IL-1 was inhibited using IL-1RA (25). The inhibition of all 3 toxic mediators indeed resulted in a cumulative protection against TNF (Figure 2, J and K).
Beneficial functions of TNF are preserved in Tnfrsf1a+/– mice.
We investigated whether genetic reduction of p55TNFR expression compromises the beneficial physiological functions of TNF, such as resistance to infection and immune function. First, the Tnfrsf1a+/+, Tnfrsf1a+/–, and Tnfrsf1a–/– mice were infected with Listeria monocytogenes. The Tnfrsf1a–/– mice succumbed to the infection, but all Tnfrsf1a+/+ and Tnfrsf1a+/– mice survived (Figure 3A). We then examined the structure of lymphoid organs and humoral immune responses, both dependent on Tnfrsf1a (34), in Tnfrsf1a+/– mice. Spleen sections were made from mice 15 days after immunization with sheep red blood cells (SRBCs). These sections showed that germinal centers and follicular dendritic cell networks were present in Tnfrsf1a+/+ and Tnfrsf1a+/– mice but missing in Tnfrsf1a–/– mice (Figure 3B). In addition, a significant antibody response against SRBCs was detected in Tnfrsf1a+/– mice, while no antibody response was observed in Tnfrsf1a–/– mice (Figure 3C).
Figure 3. Antimicrobial effects, immune response, and spleen germinal centers in Tnfrsf1a+/– mice.

(A) Survival of mice following i.p. challenge with 105 Listeria monocytogenes per mouse. (B) Germinal center formation and follicular dendritic cell networks in spleens of mice immunized with SRBCs are evident after staining with either PNA (blue) or CR-1 (brown). In Tnfrsf1a+/+ and Tnfrsf1a+/– mice, germinal centers and follicular dendritic cell networks are both detected, but they are absent in Tnfrsf1a–/– mice. Original magnification, ×40. (C) Serum antibody responses to SRBC immunization. Data are expressed in arbitrary units as the mean ± SEM of at least 10 mice per group and are representative of 3 independent experiments. **P < 0.01, ***P < 0.001.
p55TNFR expression in the IECs is essential to induce TNF toxicity.
To identify which cell type crucially regulates TNF-induced toxicity, we used two complementary approaches. In a first approach, we made use of mice expressing a Tnfrsf1a conditional reactivation mutant allele, Tnfrsf1aflNeo mice (35, 36). Crossing these mice with tissue-specific Cre-lines yielded Tnfrsf1a+/flNeo mice expressing Tnfrsf1a+/– levels in all cell types, except in those cells expressing Cre, in which Tnfrsf1a+/+ levels are obtained. Crosses with Tie2- and Flk1-Cre mice (endothelial cell reactivation), Albumin- and Alfp-Cre mice (hepatocyte reactivation), LysM-Cre mice (myeloid cell reactivation), and Villin-Cre mice (IEC reactivation) were performed (Supplemental Figure 5A), and the resulting mice were injected with TNF. Only the mice with Villin-Cre–induced reactivation displayed sensitivity for TNF-induced toxicity and lethality, suggesting a crucial role for gut epithelium (Figure 4A). Villin-Cre–reactivated mice, in contrast to nonreactivated Tnfrsf1a+/flNeo mice, showed loss of intestinal permeability upon TNF challenge (ref. 37 and Figure 4B) and increased intestinal Il6 and Il17a mRNA expression (Supplemental Figure 5B). Intestine-derived IL-17 has previously been linked to TNF toxicity (24). Induction of NF-κB–mediated genes by TNF, as shown before in liver (Figure 2), was strongly reduced in intestinal tissues derived from Tnfrsf1a+/– mice compared with that in Tnfrsf1a+/+ mice (Supplemental Figure 3, A–D), whereas TNF-mediated IEC death sensitivity remained unaffected (Figure 4C).
Figure 4. p55TNFR expression in the intestinal epithelium is a critical determinant for TNF-induced toxicity.

(A) Tnfrsf1a+/–, Tnfrsf1a+/flNeo, and Tnfrsf1a+/flNeo mice with tissue-specific Cre expression were injected i.p. with 150 μg TNF, and lethality was monitored. (B) Relative intestinal permeability measured by increase in FITC signal in plasma of Tnfrsf1a+/flNeo (gray) and Tnfrsf1a+/flNeo IEC reactivation mice (black) orally gavaged with 4-kDa FITC-dextran and injected with TNF (0 hour, n = 3; 8 hours, n = 8). (C) IEC death in Tnfrsf1a+/+, Tnfrsf1a+/–, Tnfrsf1a+/flNeo, and Villin-Cre+ Tnfrsf1a+/flNeo mice 2 hours after TNF challenge (30 μg, i.p.). Similar amounts of TUNEL-positive cells on ileal sections were detected in all samples (n = 4–5; scale bar: 38 μm). Multiple time points (1, 4, and 8 hours after TNF) were tested, and all showed a similar amount of TUNEL-positive cells in Tnfrsf1a+/+ and Tnfrsf1a+/– IECs. (D) Tnfrsf1afl/fl (black line), Villin-Cre Tnfrsf1afl/+ (light gray line), and Villin-Cre Tnfrsf1afl/fl (dark gray line) mice were injected i.v. with 6 μg TNF, and lethality was monitored. (E) Relative intestinal permeability of Villin-Cre Tnfrsf1afl/+ mice compared with Tnfrsf1afl/+ mice when mice were injected with 7 μg TNF and orally gavaged with 4-kDa FITC-dextran (0 hours, n = 5; 8 hours, n = 11). (F) IEC death in Tnfrsf1a–/–, Tnfrsf1a+/fl, and Villin-Cre Tnfrsf1a+/fl mice 2 hours after TNF challenge (6 μg, i.v.). Similar amounts of TUNEL-positive cells on ileal sections were detected in all samples (n = 6; scale bar: 38 μm). Data represent mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t test in B and E, and log-rank test in A and D).
To further investigate the cell-specific requirement of p55TNFR in TNF-induced toxicity, we generated Tnfrsf1a conditional knockout mice (Tnfrsf1afl/fl mice) (the generation and initial characterization of these mice are described in the Methods and in Supplemental Figure 4). Crossing the LysM-Cre, Villin-Cre, or Alfp-Cre strains into the Tnfrsf1afl/fl background resulted in p55TNFR inactivation in myeloid cells, IECs, or hepatocytes, respectively (Supplemental Figure 4, D–F). Only the Villin-Cre Tnfrsf1afl/fl mice significantly resisted TNF-induced toxicity, in contrast to the mice with hepatocyte or myeloid cell Tnfrsf1a deficiency, which remained as sensitive as the controls (Supplemental Figure 5C). Villin-Cre Tnfrsf1afl/fl mice showed reduced intestinal permeability (Supplemental Figure 5D) as well as decreased levels of Il6, Il1b, and Il17a mRNA in intestinal tissue in comparison with that in controls (Supplemental Figure 5E). Interestingly, when TNF was injected in the Villin-Cre Tnfrsf1afl/+ mice, the mice were as resistant to TNF toxicity as the Villin-Cre Tnfrsf1afl/fl mice (Figure 4, D and E). The Villin-Cre Tnfrsf1afl/+ mice exhibited a minimal barrier loss compared with non-TNF-challenged Villin-Cre Tnfrsf1afl/+ control mice, but the barrier was significantly less affected than in TNF-challenged Tnfrsf1afl/+ mice (Figure 4E). We also noticed that IEC death was not defective in Villin-Cre Tnfrsf1afl/+ mice compared with that in p55TNFR-sufficient control mice (Figure 4, C and F), suggesting that TNF-induced systemic death is related to TNF-induced loss of IEC barrier rather than to TNF-induced IEC cell death. In conclusion, these results show that p55TNFR expression levels in the IECs are crucially linked with the induction of intestinal permeability and consequent systemic lethal inflammation.
p55TNFR-dependent apoptosis induction remains functional in Tnfrsf1a+/– mice.
To characterize TNF-induced apoptosis in Tnfrsf1a+/– cells, we studied the TNF response of primary fibroblasts derived from Tnfrsf1a+/+, Tnfrsf1a+/–, and Tnfrsf1a–/– mice. When cells were treated with TNF for 24 hours, intermediate production of IL-6 in supernatant of Tnfrsf1a+/– cells was found (Figure 5A). However, when apoptosis was induced by incubation of cells with TNF and cycloheximide (CHX) for 24 hours, an equal amount of cell death induction was observed in Tnfrsf1a+/+ and Tnfrsf1a+/– cells (Figure 5B). Western blot analysis of caspase-8 activation at different time points after TNF/CHX stimulation showed that both Tnfrsf1a+/+ and Tnfrsf1a+/– fibroblasts activate similar levels of caspase-8 (Figure 5C). We then induced apoptosis in vivo by injecting TNF plus d-galactosamine (GalN). This combination leads to lethal apoptosis in hepatocytes (38). First, we tested the lethality of two different doses of TNF (0.3 and 0.5 μg) combined with 20 mg GalN. While none of the Tnfrsf1a–/– mice died from the higher dose, all of the Tnfrsf1a+/– mice and 24 out of the 26 Tnfrsf1a+/+ mice died within 72 hours (Supplemental Figure 6A). Next, mice were injected with 1 μg TNF and 20 mg GalN and sacrificed when their body temperature dropped to 30°C, and blood and liver samples were obtained. This occurred at 7 and 7.5 hours in Tnfrsf1a+/+ and Tnfrsf1a+/– mice, respectively (Figure 5D). Tnfrsf1a–/– mice were unresponsive to the treatment, and their samples were taken 8 hours after challenge. Liver damage was assessed by measurement of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in serum (Figure 5E), and apoptosis was measured by DEVDase assay in liver lysates (Figure 5F). AST and ALT levels in Tnfrsf1a+/+ and Tnfrsf1a+/– mice were not significantly different, indicating comparable amounts of liver damage. No significant differences were observed in the cleavage of the caspase-3/7 substrate DEVD, indicating comparable induction of apoptosis in Tnfrsf1a+/+ and Tnfrsf1a+/– mice. We also studied apoptosis by using the TNF/actinomycin D (TNF/ActD) model and obtained similar results (Supplemental Figure 6, B and C). These results, along with the in vivo results of p55TNFR-mediated death in the intestine (Figure 4, C and F), show that induction of apoptosis by TNF is intact in Tnfrsf1a+/– genotypes.
Figure 5. Induction of apoptosis in Tnfrsf1a+/– mice.

(A) IL-6 production in supernatant of Tnfrsf1a+/+, Tnfrsf1a+/–, and Tnfrsf1a–/– fibroblasts (n = 4) 24 hours after TNF stimulation. (B) Measurement of Tnfrsf1a+/+, Tnfrsf1a+/–, and Tnfrsf1a–/– fibroblast survival after stimulation with different concentrations of TNF/CHX (10 μg/ml). Both Tnfrsf1a+/+ and Tnfrsf1a+/– cells undergo apoptosis to a similar extent. (C) Caspase-8 Western blot after TNF/CHX stimulation (1,000 IU/ml and 10 μg/ml) at different time points. The intensity of cleaved caspase-8 (p43/p41) bands was normalized to actin levels. (D) Body temperature of Tnfrsf1a+/+ (n = 4), Tnfrsf1a+/– (n = 5), and Tnfrsf1a–/– (n = 3) mice after i.p. injection with 1 μg TNF plus 20 mg GalN. Tnfrsf1a+/+ and Tnfrsf1a+/– mice were euthanized for sampling when their body temperature dropped to 30°C. (E and F) Hepatocyte cell death parameters after i.p. injection with TNF (1 μg/mouse) plus GalN (20 mg/mouse). After challenge, mice were sacrificed when their body temperature dropped to 30°C. (E) Serum ALT and AST and (F) DEVDase activity in liver. (G and H) Cell death and caspase activation in CD45–CD31+PI– tumor neovascular endothelial cells. B16BL6 melanoma-bearing mice were injected s.c. paralesional with 15 μg TNF plus 5,000 IU IFN-γ or with PBS, and 24 hours later tumors were excised and (G) cell death and (H) caspase activation were measured by FACS using annexin V and Flica staining, respectively. Actual percentages of cell death in the PBS-treated animals were 63%, 26%, and 50% (G) and 55%, 55%, and 73% (H) for Tnfrsf1a+/+, Tnfrsf1a+/–, and Tnfrsf1a–/–, respectively. Data represent mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t test).
TNF/IFN-γ–induced apoptosis in tumor endothelium.
Effective TNF/IFN-γ–based antitumor therapy induces apoptosis in the neovasculature of tumors, and this is considered key to tumor destruction (7, 20). To study the induction of apoptosis in the tumor endothelium, B16BL6 tumor-bearing Tnfrsf1a+/+, Tnfrsf1a+/–, and Tnfrsf1a–/– mice were injected s.c. paralesionally with 15 μg TNF and 5,000 IU IFN-γ or with PBS, and 24 hours later tumors were excised and apoptosis and caspase-3/7 activation were measured by FACS. The percentage of apoptotic tumor endothelial cells, identified as CD31+CD45–PI– cells, was measured using annexin V and Flica staining, respectively. Figure 5, G and H shows the increase of annexin V–stained cells and Flica-stained cells in TNF/IFN-γ–injected mice compared with that in PBS-treated mice. Tnfrsf1a+/+ and Tnfrsf1a+/– mice showed comparable degrees of apoptotic annexin V–positive cells (Figure 5G) and Flica-positive cells containing active caspases (Figure 5H). No increase in annexin V– and Flica-positive cells was observed in Tnfrsf1a–/– mice.
TNF/IFN-γ therapy in Tnfrsf1a+/– mice induces tumor regression in the absence of systemic toxicity.
We first studied the toxicity profile of standard TNF/IFN-γ therapy, i.e., daily paralesional injection during 10 days, in Tnfrsf1a+/+ and Tnfrsf1a+/– tumor-free mice as well as in mice bearing a s.c. B16BL6 melanoma. The tumor regresses well in response to TNF, but it also sensitizes mice to TNF toxicity (39). The LD50 dramatically increased 12.1 fold in tumor-free mice and 8.2 fold in tumor-bearing Tnfrsf1a+/– mice relative to that in Tnfrsf1a+/+ mice (Figure 6A and Supplemental Figure 7A). This therapeutic window in Tnfrsf1a+/– mice is close to the required 10-fold factor increase in MTD that was proposed to be essential to increase the safety of TNF to an acceptable level (5). These data suggest that a full and safe antitumor effect may be expected in Tnfrsf1a+/– mice, so we performed antitumor experiments in 2 different models using high doses of TNF/IFN-γ. As shown in Figure 6, B and C, established B16BL6 and Lewis lung carcinoma (LLC) tumors did not regress in Tnfrsf1a–/– mice, and all Tnfrsf1a+/+ mice died on the second day of TNF/IFN-γ treatment due to inflammatory shock. In contrast, Tnfrsf1a+/– mice remained healthy, and tumors regressed either completely or to a very large extent after 10 days of treatment. Further proof that p55TNFR expression on host-derived cells but not on tumor cells sensu stricto is linked to the response to TNF/IFN-γ is discussed in Supplemental Figure 7B, where we show that TNF-mediated tumor regression is dependent on p55TNFR expression of the host tissue but not on the tumor cells themselves. To further explore the role of TNF-p55TNFR-mediated toxic signaling through IECs, we performed the antitumor experiment using high doses of TNF/IFN-γ in Villin-Cre Tnfrsf1afl/fl mice. Tumor regression occurred equally well in both groups of mice, but survival was significantly better in Villin-Cre Tnfrsf1afl/fl mice (Figure 6D). We conclude that a general reduction of p55TNFR levels by 50% or in a IEC-specific manner greatly reduces TNF-induced toxicity without diminishing the antitumor effects. This can make TNF therapy both safe and effective.
Figure 6. Increased MTD and anticancer effects in Tnfrsf1a+/– mice.

Antitumor effect and survival after 10 days of high-dose TNF (50 μg/mouse) plus IFN-γ treatment (5,000 IU). (A) Toxicity of TNF plus IFN-γ in Tnfrsf1a+/+ mice (black triangles) and Tnfrsf1a+/– mice (blue circles). Healthy tumor-free mice (continuous line) and B16BL6 melanoma-bearing mice (dotted line) were injected daily with different doses of TNF plus IFN-γ for 10 days. Healthy mice were injected i.p., whereas tumor-bearing mice were injected s.c. paralesional. The horizontal line represents LD50. (B) B16BL6 melanoma-bearing Tnfrsf1a+/+, Tnfrsf1a+/–, and Tnfrsf1a–/– mice were treated daily by paralesional s.c. injection with PBS (white symbols; Tnfrsf1a+/+, n = 5; Tnfrsf1a+/–, n = 3; Tnfrsf1a–/–, n = 4) or high-dose TNF (black symbols; Tnfrsf1a+/+, n = 6; Tnfrsf1a+/–, n = 10; Tnfrsf1a–/–, n = 6) plus IFN-γ. Tnfrsf1a–/– mice had to be euthanized due to large tumor size (indicated by //). (C) LLC-bearing mice were treated daily with PBS (white symbols; Tnfrsf1a+/+, n = 6; Tnfrsf1a+/–, n = 9; Tnfrsf1a–/–, n = 4) or TNF/IFN-γ (black symbols; Tnfrsf1a+/+, n = 5; Tnfrsf1a+/–, n = 13; Tnfrsf1a–/–, n = 9). (D) B16BL6 melanoma-bearing Tnfrsf1afl/fl and Villin-Cre Tnfrsf1afl/fl mice were treated daily by paralesional injection of 12 mg TNF plus IFN-γ (5,000 IU) for 10 days (Tnfrsf1afl/fl, n = 20 and Villin-Cre Tnfrsf1afl/fl, n = 16). Tnfrsf1afl/fl PBS-treated mice (n = 11) had to be euthanized because of their large tumor size (indicated by //). Data represent mean ± SEM. *P < 0.05, ***P < 0.001, Student’s t test in B and C (left) and log-rank test (right); in tumor size index (TSI) graphs, compared with PBS- and TNF/IFN-γ–treated Tnfrsf1a+/– (A–C) or TNF/IFN-γ–treated Tnfrsf1afl/fl and Villin-Cre Tnfrsf1afl/fl (D).
Systemic administration of anti-p55TNFR antibody strongly protects against TNF-induced toxicity and leads to safe antitumor therapy.
To assess clinical applicability, we used two approaches based on using antibodies to neutralize p55TNFR. First, an anti-mp55TNFR monoclonal antibody protected mice against acute TNF lethality in a dose-responsive way (Figure 7A) and protected B16BL6 tumor-bearing mice against toxicity of TNF/IFN-γ therapy without loss of the anticancer effect, while mice treated with a control antibody showed no significant difference compared with PBS-treated animals. (Figure 7, B and C). Elaborate dose-response studies (see Methods) showed an increase of LD50 of 4 times using cotreatment with the antibody compared with the control antibody (Figure 7D and Supplemental Figure 7A). Second, we used a newly developed human TNFRSF1A knockin humanized mouse model (Supplemental Figure 8) and examined the therapeutic efficacy of an anti-human p55TNFR (anti-hp55TNFR) PEGylated Fab’ fragment that neutralizes human but not mp55TNFR. Administration of this antibody (10 mg/kg) prevented acute TNF lethality in human TNFRSF1A knockin mice (Figure 7E) but not in wild-type mice (data not shown). The specific protective effect of coadministration of the antibody in tumor treatment settings was evaluated using the B16BL6 tumor model. Notably, the antibody conferred complete protection against lethality, while the antitumor effect was fully preserved (Figure 7, F and G). The magnitude of the protection studied in an elaborate and standard LD50 experiment was 5 times (Figure 7H). This was a slight improvement over the anti-mp55TNFR antibody, possibly because of better pharmacokinetics of the PEGylated Fab′ fragment compared with the classical anti-mp55TNFR antibody (Supplemental Figure 7A).
Figure 7. Antibodies against p55TNFR protect against TNF toxicity, increase MTD, and allow safe anticancer therapy.

(A) TNF (30 mg) was administered to mice pretreated i.p. with different doses of anti-mp55TNFR monoclonal antibodies (10 mg/ml, n = 5; 5 mg/ml, n = 5; 2.5 mg/ml, n = 4) or PBS (n = 7) or control hamster IgG1 (10 mg/ml, n = 5). (B) Antitumor effects and (C) survival in antitumor experiments on Tnfrsf1a+/+ mice treated for 10 days paralesionally with TNF (25 mg) plus IFN-γ (5,000 IU) (black symbols) or PBS (white symbols) and cotreated i.p. daily with PBS (black), control (red), or anti-mp55TNFR antibodies (gray). (D) Toxicity study of TNF/IFN-γ in B16BL6 melanoma-bearing Tnfrsf1a+/+ mice cotreated i.p. with PBS, control antibodies, or anti-mp55TNFR antibody for 10 days. (E) Human TNFRSF1A knockin mice treated i.p. with anti-hp55TNFR monoclonal antibodies (10 mg/ml, n = 6 or 4 mg/ml, n = 6) or control antibody (10 mg/ml, n = 6) and injected with 50 mg TNF or PBS (n = 6). (F) Antitumor effects and (G) survival in antitumor experiments in human TNFRSF1A knockin mice treated with PBS (white symbols) or TNF/IFN-γ (black symbols) and daily i.p. coadministration of PBS (n = 6), control (n = 6), or anti-hp55TNFR antibodies (n = 12). (H) Toxicity study of TNF/IFN-γ in B16BL6 melanoma-bearing human TNFRSF1A knockin mice i.p. cotreated with PBS, control antibody, or anti hp55TNFR antibody. The horizontal line in D and H represents LD50. In the TSI graphs, “***” is between PBS and TNF/IFN-γ–treated p55TNFR antibody groups. All data represent mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 (Student’s t test in B and F and log-rank test in A, C, E, and G).
Discussion
TNF was first discovered as a cytokine that can cause death of tumor cells and regression of established, solid tumors (4, 40). Shortly afterward, TNF was rediscovered as cachectin, a cytokine responsible for cachexia during chronic infection (41). The dilemma that TNF has strong anticancer effects but is toxic was confirmed in the first clinical trials (42). The effective anticancer dose of TNF was found to be high, close to the MTD, and to be associated with liver toxicity, shock, and bowel necrosis. Lowering the dose of TNF reduced side effects but also the anticancer effect (1). Hence, systemic use with high doses of TNF seemed impossible (2, 4, 42) unless a more substantial increase in MTD (up to 10 fold, as proposed; ref. 5) could be obtained.
We demonstrate here that deletion of one functional Tnfrsf1a allele leads to complete protection against challenge with a dose of TNF that is at least 40 times of LD100. This protective haploinsufficiency is likely mediated by dampening the proinflammatory effect of TNF. Hemizygous mice challenged with TNF exhibited significantly reduced inflammation, as shown by a 32-fold reduction in serum IL-6 levels, compared with Tnfrsf1a+/+ mice. This decreased inflammation seems to be due to reduced expression of NF-κB–dependent genes, which was also reflected in downmodulation of a number of cytokines in serum as well as reduction of NO metabolites. These results suggest that the TNF resistance of Tnfrsf1a+/– animals can be explained by a combined reduction of multiple toxic mediators, a hypothesis that we confirmed by inhibiting 3 such toxic mediators (namely IL-1, ROS, and type I IFNs), which led to cumulative protection against TNF. Interestingly, reduction of p55TNFR levels by 50% does not affect physiological functions of TNF, such as antibacterial resistance and secondary lymphoid organ structure and function.
The anticancer effect of TNF has been shown to be essentially directed to the neovascular endothelium of tumors, causing endothelial cell death and vascular dysfunction (11, 19, 20). In order to understand and interfere with the mechanism of systemic toxicity of TNF, it is critical to identify cell types that are essential in this process. TNF-induced toxicity previously has been shown to be associated with hypotension (43), liver damage (44), and severe bowel damage (31). In this study, we have made use of 2 informative genetic approaches (i.e., Tnfrsf1a conditional knockout and reactivation mice), which basically provided us with the novel finding that expression of p55TNFR on IECs (and not on endothelial, hepatocyte, or myeloid cells) is essential to cause TNF-induced lethality: mice expressing Tnfrsf1a+/+ levels in IECs in a Tnfrsf1a+/– background regain TNF sensitivity, while IEC-specific Tnfrsf1a–/– mice become TNF resistant. We found that TNF-induced inflammatory gene expression in intestinal epithelium is strongly reduced in IEC-specific Tnfrsf1a–/– mice as well as in Tnfrsf1a+/– mice and observed a clear association between lethality and loss of intestinal epithelial permeability, which might lead to influx of gut bacteria or bacterial antigens in the system, as previously suggested (31, 32, 45).
p55TNFR-dependent apoptosis was addressed both in vitro by TNF/CHX and in vivo by using the TNF/GalN and TNF/ActD models, in which massive liver apoptosis is induced (38). Fibroblasts derived from Tnfrsf1a+/– mice were equally sensitive to TNF/CHX–induced apoptosis as cells from Tnfrsf1a+/+ mice and showed no reduced caspase-8 cleavage. In the same Tnfrsf1a+/– fibroblasts, inflammatory gene induction was strongly reduced compared with that in Tnfrsf1a+/+ cells, as was found in macrophages. Clearly, the receptor-stimulated activation of transcription factors (as NF-κB) and kinases (MAPK), leading to inflammation, was sensitive to p55TNFR levels, while the activation of caspase-8, leading to apoptosis, was not. Also, in the in vivo models, Tnfrsf1a+/+ and Tnfrsf1a+/– cells appeared equally responsive and showed comparable levels of caspase activation, although the response in the TNF/GalN and TNF/ActD models appeared slightly delayed in Tnfrsf1a+/– mice. Also, a similar degree of apoptosis was measured in the typically TNF-sensitive IECs of Tnfrsf1a+/+ and Tnfrsf1a+/– mice and in the tumor-associated neovascular endothelial cells after treatment with TNF/IFN-γ, since Tnfrsf1a+/+ and Tnfrsf1a+/– mice showed comparable induction of apoptosis, which is essential for induction of tumor regression (7, 20).
Full antitumor activity was obtained in tumor-bearing mice by daily injections of TNF in combination with IFN-γ for 10 days (1). We investigated the efficacy and potential toxicity of TNF/IFN-γ for 10 days as a treatment for cancer in mice bearing s.c. B16BL6 melanoma. LD50 of a 10-day treatment was increased in Tnfrsf1a+/– mice by more than 8 fold. This therapeutic window is close to the required 10-fold factor proposed by Lejeune and colleagues (6), who concluded that protecting mice or human patients against a 10-fold toxic dose with retention of anticancer effects should be sufficient to allow systemic therapy with TNF (5). Our results indicate that TNF/IFN-γ treatment is effective against tumors in Tnfrsf1a+/– mice. We tested this hypothesis in cancer models (B16BL6 and LLC) and found that TNF/IFN-γ therapy indeed led to highly significant tumor regression as well as survival. We conclude that reduction of p55TNFR levels by 50% greatly reduces TNF-induced toxicity without diminishing the antitumor effects. To provide proof of principle for pharmacologic treatment based on our approach, we inhibited p55TNFR both in normal mice (with anti-mp55TNFR antibodies) and in newly generated mutant mice humanized for the TNFRSF1A gene (combined with anti-hp55TNFR antibodies). Systemic inhibition of p55TNFR was clearly very protective against TNF toxicity, while the antitumor activity remained functional and led to an increase of 4 to 5 times of LD50.
The dosing of p55TNFR-inhibiting antibodies in these experiments is crucial and should lead to a Tnfrsf1a+/–-like phenotype. Treatment with an insufficient amount of antibody might lead to lethal toxicity, while excessive antibody may block TNF-induced antitumor responses. In order to translate our findings to the clinic, it may be necessary to generate a reliable prediction method to quantify the available or signaling-competent p55TNFR. Future studies should investigate the stability and range of p55TNFR expression in human samples and correlate antibody dosing with TNF-induced responses.
To provide further mechanistic explanation for our findings, we suggest a model in which p55TNFR-induced inflammation is a dynamic system, which, in response to a given TNF stimulus, leads to an adequate activation of NF-κB and MAPK signaling. The magnitude of an inflammatory response not only depends on TNF dosage but also on the amount of available p55TNFR, a mechanism which is also applied in pathophysiology by shedding of the p55TNFR (46). Halving the amount of p55TNFR leads to a halving of the concentration of multiple (n) toxic inflammatory mediators, such as IL-1, IFN-β, IL-17 and others, identified previously (24–28), leading to a robust (2n) combined protection against TNF in Tnfrsf1a+/– mice. In contrast, TNF-induced cell death of TNF-sensitive cells (IECs, tumor blood vessels) behaves like a binary system, which can still induce the necessary threshold when p55TNFR levels are halved.
In conclusion, we have found a way to increase the safety of the potent anticancer effect of TNF, and we provide evidence that this approach may be directly translatable to the clinic. Our results uncover the cellular basis of systemic TNF toxicity and reveal an IEC-specific haploinsufficiency of p55TNFR, which allows mitigation of TNF toxicity without loss of antitumor efficacy. Our results also highlight the differential quantitative requirements for the p55TNFR in diverse physiological phenomena and provide a framework for the development of more selective therapeutic interventions using the TNF/TNFR system.
Methods
Animals.
Tnfrsf1a knockout mice generated by M. Rothe (12) were a gift of H. Bleuthmann (Hoffmann-La Roche, Basel, Switzerland). Other mice deficient in the Tnfrsf1a gene, generated by gene targeting (13, 14), were purchased from The Jackson Laboratories. Most of the experiments were performed on the Tnfrsf1a–/– mice generated by M. Rothe. There was no specific reason for this choice. Crosses with C57BL/6J mice were performed to obtain Tnfrsf1a+/– F1 mice. Ifnar1 knockout mice were provided by D. Bonaparte (Gulbenkian Institute of Science, Oeiras, Portugal). nu/nu mice were purchased from Janvier. Mice were maintained in conventional temperature-controlled, air-conditioned animal houses with a 14- to 10-hour-light/dark cycle and received food and water ad libitum. Experiments with nu/nu and all other mice were performed in the SPF VIB animal house, and experiments with the Tnfrsf1afl and human TNFRSF1A knockin mice (described in Supplemental Figures 4 and 8, respectively) were performed in the Alexander Fleming Centre animal house. Experiments were performed when the mice were 8–12 weeks old. Conditional Tnfrsf1a reactivation mutant mice (Tnfrsf1aflNeo mice) were previously described (35). In short, the Tnfrsf1aflNeo mouse strain was generated by insertion of a floxed neo into intron 5 of the locus to create this conditional gain-of-function allele. The presence of the neo prevents expression of the gene.
Generation of Tnfrsf1a conditional knockout mice.
The Tnfrsf1afl/fl mouse strain was generated using standard gene-targeting techniques. In brief, a targeting vector has been generated to induce homologous recombination in ES cells, with exons 2–5 flanked by loxP sites, which upon recombination will result in a frame shift mutation. Upstream of the first loxP site, a flipped neo cassette was inserted to allow for antibiotic resistance selection, and flanking sequences of 3.6 and 2.8 kb 5′ and 3′ completed the vector, fulfilling the homologous recombination requirements. All sequences of the mouse Tnfrsf1a gene were derived by PCR amplification from the respective BAC clone. All exons have been sequenced, and integrity of the sequence was confirmed (Supplemental Figure 4A). The vector was introduced in 129/ola ES cells, and clones having successfully recombined were used for the generation of chimaeras (Supplemental Figure 4B). Chimeric mice were crossed to C57BL/6J mice, and offspring having germline transmission of the mutated allele were used for expansion. The mice were crossed to FLP deleter mice for the neo cassette to be removed. Then, Tnfrsf1afl/fl mice were crossed with Cre deleter mice for the generation of Tnfrsf1 complete knockout mice, designated as Tnfrsf1aΔ/Δ mice. The neo cassette removal was successfully. The Tnfrsf1afl/fl mouse line was expanded, and the mice were backcrossed to C57BL/6J for at least 6 generations. After crossing the Tnfrsf1afl/fl mice with deleter cre mice, the deficiency in p55TNFR expression in Tnfrsf1aΔ/Δ mice was confirmed in BMDMs by FACS analysis (Supplemental Figure 4C) as previously described (47).
Generation of human TNFRSF1A knockin mice.
Human TNFRSF1A knockin mice were generated by gene targeting in C57BL/6 ES cells. A targeting vector containing human and mouse Tnfrsf1a genomic sequences was engineered for the expression of a recombinant transcript consisting of the mouse 5′ UTR, signal peptide, and 3′ UTR sequences flanking the human Tnfrsf1a-coding sequences and expressing the mature hp55TNFR protein. The transgene was knocked-in into the mouse locus, thereby inactivating the mouse gene and expressing the human gene under the mouse regulatory sequences. hp55TNFR in these mice was present and functional in all tissues and assays tested, giving to these “humanized” mice a normal TNFRSF1A+/+ phenotype (Supplemental Figure 8 and data not shown).
Cytokines, tempol, IL-1RA, anti-mp55TNFR and anti-hp55TNFR antibodies.
Recombinant mouse TNF and IFN-γ were expressed in Escherichia coli and purified in our laboratories. TNF had a specific activity of 1.0 × 109 IU/mg. IFN-γ had a specific activity of 1.1 × 109 IU/mg. Endotoxin in the cytokine preparations was below the detection limit of a Limulus amoebocyte lysate assay. Cytokines and LPS were diluted in endotoxin-free PBS (GIBCO, Invitrogen) immediately before injection. Tempol, 6 mg per mouse (Sigma-Aldrich) was injected 45 minutes before TNF injection (28). IL-1RA (Anakinra), 650 μg per mouse, was injected together with TNF and again 4 and 8 hours after TNF. The neutralizing hamster anti-mp55TNFR IgG1 antibody (55R170) was bought from R&D Systems. It was pure and contained no detectable LPS contamination. A control hamster IgG1 antibody was bought from Innovative Research. The anti-mp55TNFR was specific for the p55TNFR, because it protected Tnfrsf1a+/+ mice against LPS but not Tnfrsf1a–/– mice against LPS (data not shown). The ultrapure neutralizing anti-hp55TNFR antibody m5R16 was generated and provided by UCB. This antibody is a PEGylated F(ab)′ fragment and is highly specific for the hp55TNFR, as indicated by its ability to block TNF-induced biological activities (e.g., TNF lethality) in human TNFRSF1A knockin mice but not in Tnfrsf1a+/+ mice (see Figure 7E and data not shown). As a control, a purified PEGylated control F(ab)′ fragment (mA33) was provided by UCB.
Measuring tumor neovascular endothelial cell caspase activation and cell death.
Eight-week-old mice were injected with 6 × 105 B16BL6 cells in the shaved right thigh. Tumors were allowed to grow for 12 to 14 days until they reached a tumor size index of about 80 mmβ, at which time the mice received a single injection of 15 μg TNF plus 5,000 IU IFN-γ. The tumors were isolated 24 hours later and processed mechanically (by cutting and passing through a 19 G needle) and enzymatically (30 minutes 37°C in a collagenase solution) to obtain single cell suspensions. Red blood cells were lysed in an ACK lysis buffer for 5 minutes. Staining was done on 5 × 105 cells of each sample. For annexin V staining, cells were incubated for 30 minutes at room temperature with anti-CD31, anti-CD45, and anti-annexin V. 7-AAD staining was always negative (data not shown). For the Flica staining (AbD Serotec), cells were incubated for 1 hour at 37°C in 200 μl DMEM, 25% FCS, and 1 μl of the 150× Flica stock. The cells were then washed and stained with anti-CD31 and anti-CD45 for 30 minutes. Endothelial cells in these cell suspensions were identified as CD45 negative, and CD31-positive cells and SYTOX Red-positive cells were excluded. This population was analyzed for annexin V and Flica staining by FACS.
Determination of IL-6 and other cytokines in serum.
IL-6 was measured by using an IL-6–dependent 7TD1 hybridoma cell line (48). Levels of IL-1α, IL-1β, MCP-1, MIP1b, and Rantes were measured with the Bio-plex system (Bio-Rad) according to the manufacturer’s instructions.
Gene induction experiments.
Liver and intestine samples were collected in RNA Later (Qiagen). RNA was isolated with the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. RNA concentration was measured with the Nanodrop1000 (Thermo Scientific), and 1 μg RNA was used to prepare cDNA with SuperScript II (Invitrogen). qPCR was performed using the SYBR Green 1 Master Mix (Roche) and the LightCycler 480 (Roche). The best-performing housekeeping genes for each tissue were determined with Genorm (49). Values are presented as relative expression normalized to the geometric mean of the 2 selected housekeeping genes.
Detection of membrane p55TNFR by flow cytometry.
BMDMs were collected, washed, and incubated with antibodies in phosphate-buffered saline supplemented with 0.2% sodium azide-BDH and 10% FBS. The antibodies were 0.5 μg primary hamster anti-mp55TNFR antibody (clone 55R-286, BD Pharmingen) and 0.75 μg biotinylated goat anti-hamster IgG (Vector Laboratories), followed by 0.5 μg streptavidin SAv-PE (BD Pharmingen), each of which was incubated for 30 minutes at 4°C. p55TNFR expression was measured by flow cytometry on a Coulter EPICS XL-MCL Flow Cytometer (Coulter Co.) and analyzed with the WinMDI 2.8 software.
TNF-binding assay.
BMDMs (2 × 106 cells per tube) were incubated for at least 3 hours at 4°C with radiolabeled 125I-hTNF (Amersham), which binds only to p55TNFR. As a control for nonspecific binding, the same experiment was performed in the presence of a 500-fold excess of unlabeled hTNF. After incubation, the cells were washed 4 times with PBS at 4°C to remove the unbound ligand, and radioactivity of the cell pellet was measured with a gamma counter (Beckman). Nonspecific binding was subtracted from the total activity.
p55TNFR ELISA.
Whole organ samples were homogenized in ice-cold buffer (PBS, 0.5% CHAPS, protease inhibitors [Complete, Roche]). Homogenates were centrifuged for 30 minutes at 20,000 g and 4°C, after which the supernatant was collected and stored at –80°C.
Protein concentration was determined by the Bradford method (Bio-Rad). p55TNFR levels were determined with the Quantikine sp55TNFR ELISA Kit (R&D Systems). The levels shown are normalized to the Tnfrsf1a+/+ levels, which were set as 100%.
Statistics.
Survival curves (Kaplan-Meier plots) were compared using a log-rank test. Results (mean ± SEM) were compared with a 2-tailed Student’s t test. Final mortality data were compared using a Fisher’s exact test. P values of less than 0.05 were considered significant.
Study approval.
All mice were used with the approval of and in accordance with the guidance of the institutional animal care and use committee of Biomedical Sciences Research Center Alexander Fleming and the institutional ethics committee for animal welfare of the Faculty of Sciences, Ghent University.
Supplementary Material
Acknowledgments
This work was supported by grants to G. Kollias from the Greek General Secretariat for Research and Technology, NSFR 2007-2013 (GR-ERC-06), European Commission FP7 programs INFLACARE (contract no. 223151), and IMI project BTCure (grant agreement no. 115142) and by grants to C. Libert from the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT-Vlaanderen), Vlaamse Liga tegen kanker (VLK), BOF-UGent, the Research Foundation Flanders (FWO Vlaanderen), and the Interuniversity Attraction Poles Program of the Belgian Science Policy. The authors wish to thank Amin Bredan for editing the manuscript. We thank M. Rothe for providing mice and UCB for providing the m5R16 antibody. We thank Wilma Burm, Koen Breyne, Panos Athanasakis, and Spyros Lalos for technical help.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2013;123(6):2590–2603. doi:10.1172/JCI65624.
Sonja Loges’s present address is: University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Jan Staelens’s present address is: VIB headquarters, VIB, Ghent, Belgium.
References
- 1.Brouckaert PG, Leroux-Roels GG, Guisez Y, Tavernier J, Fiers W. In vivo anti-tumour activity of recombinant human and murine TNF, alone and in combination with murine IFN-γ, on a syngeneic murine melanoma. Int J Cancer. 1986;38(5):763–769. doi: 10.1002/ijc.2910380521. [DOI] [PubMed] [Google Scholar]
- 2.Tracey KJ, et al. Shock and tissue injury induced by recombinant human cachectin. Science. 1986;234(4775):470–474. doi: 10.1126/science.3764421. [DOI] [PubMed] [Google Scholar]
- 3.Wielockx B, et al. Inhibition of matrix metalloproteinases blocks lethal hepatitis and apoptosis induced by tumor necrosis factor and allows safe antitumor therapy. Nat Med. 2001;7(11):1202–1208. doi: 10.1038/nm1101-1202. [DOI] [PubMed] [Google Scholar]
- 4.Balkwill F. Tumour necrosis factor and cancer. Nat Rev Cancer. 2009;9(5):361–371. doi: 10.1038/nrc2628. [DOI] [PubMed] [Google Scholar]
- 5.Lejeune F, et al. Administration of high-dose tumor necrosis factor alpha by isolation perfusion of the limbs. Rationale and results. J Infus Chemother. 1995;5(2):73–81. [PubMed] [Google Scholar]
- 6.Lienard D, Ewalenko P, Delmotte JJ, Renard N, Lejeune FJ. High-dose recombinant tumor necrosis factor alpha in combination with interferon gamma and melphalan in isolation perfusion of the limbs for melanoma and sarcoma. J Clin Oncol. 1992;10(1):52–60. doi: 10.1200/JCO.1992.10.1.52. [DOI] [PubMed] [Google Scholar]
- 7.Lejeune FJ, Ruegg C, Lienard D. Clinical applications of TNF-alpha in cancer. Curr Opin Immunol. 1998;10(5):573–580. doi: 10.1016/S0952-7915(98)80226-4. [DOI] [PubMed] [Google Scholar]
- 8.Grunhagen DJ, de Wilt JH, ten Hagen TL, Eggermont AM. Technology insight: Utility of TNF-alpha-based isolated limb perfusion to avoid amputation of irresectable tumors of the extremities. Nat Clin Pract Oncol. 2006;3(2):94–103. doi: 10.1038/ncponc0426. [DOI] [PubMed] [Google Scholar]
- 9.Hundsberger H, et al. TNF: a moonlighting protein at the interface between cancer and infection. Front Biosci. 2008;13:5374–5386. doi: 10.2741/3087. [DOI] [PubMed] [Google Scholar]
- 10.Vandenabeele P, Declercq W, Beyaert R, Fiers W. Two tumour necrosis factor receptors: structure and function. Trends Cell Biol. 1995;5(10):392–399. doi: 10.1016/S0962-8924(00)89088-1. [DOI] [PubMed] [Google Scholar]
- 11.Stoelcker B, et al. Tumor necrosis factor induces tumor necrosis via tumor necrosis factor receptor type 1-expressing endothelial cells of the tumor vasculature. Am J Pathol. 2000;156(4):1171–1176. doi: 10.1016/S0002-9440(10)64986-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rothe J, et al. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes. Nature. 1993;364(6440):798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- 13.Peschon JJ, et al. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160(2):943–952. [PubMed] [Google Scholar]
- 14.Pfeffer K, et al. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73(3):457–467. doi: 10.1016/0092-8674(93)90134-C. [DOI] [PubMed] [Google Scholar]
- 15.Ameloot P, et al. Bioavailability of recombinant tumor necrosis factor determines its lethality in mice. Eur J Immunol. 2002;32(10):2759–2765. doi: 10.1002/1521-4141(2002010)32:10<2759::AID-IMMU2759>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 16.Kollias G, Douni E, Kassiotis G, Kontoyiannis D. On the role of tumor necrosis factor and receptors in models of multiorgan failure, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. Immunol Rev. 1999;169:175–194. doi: 10.1111/j.1600-065X.1999.tb01315.x. [DOI] [PubMed] [Google Scholar]
- 17.Van Antwerp DJ, Martin SJ, Verma IM, Green DR. Inhibition of TNF-induced apoptosis by NF-kappa B. Trends Cell Biol. 1998;8(3):107–111. doi: 10.1016/S0962-8924(97)01215-4. [DOI] [PubMed] [Google Scholar]
- 18.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114(2):181–190. doi: 10.1016/S0092-8674(03)00521-X. [DOI] [PubMed] [Google Scholar]
- 19.Lejeune FJ, Ruegg C. Recombinant human tumor necrosis factor: an efficient agent for cancer treatment. Bull Cancer. 2006;93(8):E90–E100. [PubMed] [Google Scholar]
- 20.Ruegg C, et al. Evidence for the involvement of endothelial cell integrin αVβ3 in the disruption of the tumor vasculature induced by TNF and IFN-γ. Nat Med. 1998;4(4):408–414. doi: 10.1038/nm0498-408. [DOI] [PubMed] [Google Scholar]
- 21.Duprez L, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2012;35(6):908–918. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- 22.Libert C, Van Bladel S, Brouckaert P, Fiers W. The influence of modulating substances on tumor necrosis factor and interleukin-6 levels after injection of murine tumor necrosis factor or lipopolysaccharide in mice. J Immunother. 1991;10(4):227–235. doi: 10.1097/00002371-199108000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Kettelhut IC, Fiers W, Goldberg AL. The toxic effects of tumor necrosis factor in vivo and their prevention by cyclooxygenase inhibitors. Proc Natl Acad Sci U S A. 1987;84(12):4273–4277. doi: 10.1073/pnas.84.12.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi N, et al. IL-17 produced by Paneth cells drives TNF-induced shock. J Exp Med. 2008;205(8):1755–1761. doi: 10.1084/jem.20080588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Everaerdt B, Brouckaert P, Fiers W. Recombinant IL-1 receptor antagonist protects against TNF-induced lethality in mice. J Immunol. 1994;152(10):5041–5049. [PubMed] [Google Scholar]
- 26.Kilbourn RG, et al. NG-methyl-L-arginine inhibits tumor necrosis factor-induced hypotension: implications for the involvement of nitric oxide. Proc Natl Acad Sci U S A. 1990;87(9):3629–3632. doi: 10.1073/pnas.87.9.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huys L, et al. Type I interferon drives tumor necrosis factor-induced lethal shock. J Exp Med. 2009;206(9):1873–1882. doi: 10.1084/jem.20090213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cauwels A, Rogge E, Janssen B, Brouckaert P. Reactive oxygen species and small-conductance calcium-dependent potassium channels are key mediators of inflammation-induced hypotension and shock. J Mol Med (Berl). 2010;88(9):921–930. doi: 10.1007/s00109-010-0633-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vassalli P. The pathophysiology of tumor necrosis factors. Annu Rev Immunol. 1992;10:411–452. doi: 10.1146/annurev.iy.10.040192.002211. [DOI] [PubMed] [Google Scholar]
- 30.Piguet PF, Vesin C, Guo J, Donati Y, Barazzone C. TNF-induced enterocyte apoptosis in mice is mediated by the TNF receptor 1 and does not require p53. Eur J Immunol. 1998;28(11):3499–3505. doi: 10.1002/(SICI)1521-4141(199811)28:11<3499::AID-IMMU3499>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 31.Vereecke L, et al. Enterocyte-specific A20 deficiency sensitizes to tumor necrosis factor-induced toxicity and experimental colitis. J Exp Med. 2010;207(7):1513–1523. doi: 10.1084/jem.20092474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guma M, et al. Constitutive intestinal NF-κB does not trigger destructive inflammation unless accompanied by MAPK activation. J Exp Med. 2011;208(9):1889–1900. doi: 10.1084/jem.20110242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lewis M, et al. Cloning and expression of cDNAs for 2 distinct murine tumor necrosis factor receptors demonstrate one receptor is species specific. Proc Natl Acad Sci U S A. 1991;88(7):2830–2834. doi: 10.1073/pnas.88.7.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Hir M, et al. Differentiation of follicular dendritic cells and full antibody responses require tumor necrosis factor receptor-1 signaling. J Exp Med. 1996;183(5):2367–2372. doi: 10.1084/jem.183.5.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Victoratos P, et al. FDC-specific functions of p55TNFR and IKK2 in the development of FDC networks and of antibody responses. Immunity. 2006;24(1):65–77. doi: 10.1016/j.immuni.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 36.Roulis M, Armaka M, Manoloukos M, Apostolaki M, Kollias G. Intestinal epithelial cells as producers but not targets of chronic TNF suffice to cause murine Crohn-like pathology. Proc Natl Acad Sci U S A. 2011;108(13):5396–5401. doi: 10.1073/pnas.1007811108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gibson PR. Increased gut permeability in Crohn’s disease: is TNF the link? Gut. 2004;53(12):1724–1725. doi: 10.1136/gut.2004.047092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Molle W, Libert C, Fiers W, Brouckaert P. Alpha 1-acid glycoprotein and alpha 1-antitrypsin inhibit TNF-induced but not anti-Fas-induced apoptosis of hepatocytes in mice. J Immunol. 1997;159(7):3555–3564. [PubMed] [Google Scholar]
- 39.Cauwels A, et al. Involvement of IFN-gamma in Bacillus Calmette-Guerin-induced but not in tumor-induced sensitization to TNF-induced lethality. J Immunol. 1995;154(6):2753–2763. [PubMed] [Google Scholar]
- 40.Carswell EA, et al. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975;72(9):3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beutler B, Cerami A. Cachectin and tumour necrosis factor as two sides of the same biological coin. Nature. 1986;320(6063):584–588. doi: 10.1038/320584a0. [DOI] [PubMed] [Google Scholar]
- 42.Smith JW 2nd, et al. Phase I evaluation of recombinant tumor necrosis factor given in combination with recombinant interferon-gamma. J Immunother. 1991;10(5):355–362. doi: 10.1097/00002371-199110000-00007. [DOI] [PubMed] [Google Scholar]
- 43.Cauwels A, Brouckaert P. Survival of TNF toxicity: dependence on caspases and NO. Arch Biochem Biophys. 2007;462(2):132–139. doi: 10.1016/j.abb.2007.01.021. [DOI] [PubMed] [Google Scholar]
- 44.Schwabe RF, Brenner DA. Mechanisms of Liver Injury. I. TNF-alpha-induced liver injury: role of IKK, JNK, and ROS pathways. Am J Physiol Gastrointest Liver Physiol. 2006;290(4):G583–G589. doi: 10.1152/ajpgi.00422.2005. [DOI] [PubMed] [Google Scholar]
- 45.Brenchley JM, Douek DC. Microbial translocation across the GI tract (*). Annu Rev Immunol. 2012;30:149–173. doi: 10.1146/annurev-immunol-020711-075001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xanthoulea S, et al. Tumor necrosis factor (TNF) receptor shedding controls thresholds of innate immune activation that balance opposing TNF functions in infectious and inflammatory diseases. J Exp Med. 2004;200(3):367–376. doi: 10.1084/jem.20040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Armaka M, et al. Mesenchymal cell targeting by TNF as a common pathogenic principle in chronic inflammatory joint and intestinal diseases. J Exp Med. 2008;205(2):331–337. doi: 10.1084/jem.20070906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Snick J, et al. Purification and NH2-terminal amino acid sequence of a T-cell-derived lymphokine with growth factor activity for B-cell hybridomas. Proc Natl Acad Sci U S A. 1986;83(24):9679–9683. doi: 10.1073/pnas.83.24.9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vandesompele J, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.