

Abstract
MicroRNAs (miRNAs) are excellent tumor biomarkers because of their cell-type specificity and abundance. However, many miRNA detection methods, such as real-time PCR, obliterate valuable visuospatial information in tissue samples. To enable miRNA visualization in formalin-fixed paraffin-embedded (FFPE) tissues, we developed multicolor miRNA FISH. As a proof of concept, we used this method to differentiate two skin tumors, basal cell carcinoma (BCC) and Merkel cell carcinoma (MCC), with overlapping histologic features but distinct cellular origins. Using sequencing-based miRNA profiling and discriminant analysis, we identified the tumor-specific miRNAs miR-205 and miR-375 in BCC and MCC, respectively. We addressed three major shortcomings in miRNA FISH, identifying optimal conditions for miRNA fixation and ribosomal RNA (rRNA) retention using model compounds and high-pressure liquid chromatography (HPLC) analyses, enhancing signal amplification and detection by increasing probe-hapten linker lengths, and improving probe specificity using shortened probes with minimal rRNA sequence complementarity. We validated our method on 4 BCC and 12 MCC tumors. Amplified miR-205 and miR-375 signals were normalized against directly detectable reference rRNA signals. Tumors were classified using predefined cutoff values, and all were correctly identified in blinded analysis. Our study establishes a reliable miRNA FISH technique for parallel visualization of differentially expressed miRNAs in FFPE tumor tissues.
Introduction
Consistent detection of tumor-specific microRNA (miRNA) expression profiles indicates that some small RNAs are valuable disease biomarkers. To date, miRNA profiling has been used to classify cancers of known and unknown primary origin (1–3), determine prognosis and disease progression (4), predict chemoresistance (5), monitor therapy (6), and screen for disease (7). Given widespread clinical utility, there is substantial need for advanced miRNA detection, quantitation, and visualization assays for molecular diagnostic purposes.
To establish miRNA-based diagnostics, it is essential to define a clinical need, reliably extract small RNAs from clinical materials, detect and quantitate miRNA expression differences among samples, and establish tractable tests for clinical laboratory use. Because miRNAs accurately identify cancer tissue origin, we sought to establish a multicolor FISH protocol to visualize differentially expressed miRNAs in formalin-fixed paraffin-embedded (FFPE) tissues. While miRNA microarray and/or real-time PCR analyses of fresh or archived materials can be used for molecular diagnostics, these approaches obliterate valuable cytoarchitectural details.
To develop miRNA FISH for tumor differential diagnosis, we selected a rare diagnostic problem in skin pathology involving basal cell carcinoma (BCC) and Merkel cell carcinoma (MCC), leveraging our preliminary findings that each tumor expresses cell lineage–specific miRNAs in high abundance. BCC and MCC frequently share overlapping histologic features and are typically differentiated from each other and similar appearing tumors on histologic grounds and/or using a panel of immunohistochemical stains (8–10). Although there is little specific need to optimize BCC and MCC diagnostics, this unusual scenario was ideal for our proof-of-concept study.
In this study, we detected miRNA expression differences among BCC, MCC, and normal skin (NS) samples using quantitative barcoded small RNA sequencing and, subsequently, developed a multicolor miRNA FISH protocol targeting tumor-specific miRNAs and ribosomal RNA (rRNA). We addressed shortcomings in (a) RNA fixation, (b) signal amplification and detection, and (c) probe design that impede short (e.g., miRNA) and long (e.g., mRNA and rRNA) RNA visualization. We also tested our samples for Merkel cell polyomavirus (MCV), as this virus causes approximately 80% of MCC cases and encodes a miRNA, potentially influencing cellular miRNA expression and sample clustering (11–13).
Results
Identification of differentially expressed miRNAs in BCC and MCC tumor tissues and cell lines.
To assess miRNA expression differences among tumors, we first extracted total RNA from 36 archived clinical materials and cultured cell lines from patients with BCC, MCC, and NS (Supplemental Table 1; supplemental material available online with this article; doi: 10.1172/JCI68760DS1). We subsequently profiled and quantitated miRNAs in all samples using our barcoded small RNA sequencing method (14). Sequence reads were annotated by RNA category (Supplemental Table 2). Total miRNA concentrations were calculated from sequence read frequencies of miRNAs relative to spike-in calibrator RNAs (Supplemental Table 3). Higher total miRNA concentrations were seen in TRIzol-extracted cell lines compared with those in MasterPure- or RecoverAll-extracted FFPE samples (Supplemental Figure 1). To minimize the effects of sample processing and/or RNA extraction method on RNA recovery (15), we compared concentrations in RecoverAll-extracted FFPE samples from sequencing run one (hereafter termed the training set). Total miRNA concentration was 2-fold higher in BCC than in MCC or NS, whereas no significant difference was seen between MCC and NS (Supplemental Figure 1). Tumor-specific differences in total miRNA concentration may reflect altered rates of miRNA biogenesis, processing, or degradation (2).
Hierarchical clustering of human miRNA cluster sequence read frequencies indicated 2 major groups corresponding to MCC and non-MCC (BCC and NS) tumors and cell lines (Figure 1). To minimize sample processing and extraction differences, we clustered training set samples only and saw the same grouping (data not shown). Interestingly, MCV-positive MCC-derived cell lines (MKL-1, MKL-2, MS-1) clustered in the MCC group, whereas MCV-negative MCC-derived cell lines (MCC13, MCC26, UISO) clustered in the non-MCC group. When compared with miRNA profiles from approximately 1,000 clinical samples in our in-house database, MKL-1, MKL-2, and MS-1 cells clustered with MCC tumors, whereas MCC13 cells clustered with liposarcoma cell lines, and MCC26 and UISO cells clustered with breast cancer cell lines, suggesting that some cell lines are either MCV-negative MCC variants or misclassified. Sample clustering was not influenced by the low number of MCV miRNA sequence reads (<30 per sample) in MCC tissues.
Figure 1. Unsupervised hierarchical clustering of miRNA expression profiles.

Unsupervised hierarchical clustering was performed using log2 relative frequencies (RF) of miRNA precursor cluster sequence reads for the given cell line and FFPE tissue samples; MCV status is also indicated where available. miRNA precursor clusters were selected from the top 85% expressed miRNA precursor clusters across all samples. The number of members per precursor cluster is indicated in parentheses following the miRNA gene name; precursor clusters are named according to Farazi et al. (30). miR-205 and miR-375 expression values for all samples are indicated by red and green arrows, respectively.
We compared miRNA expression profiles between MCC and non-MCC groups and identified tumor-specific miRNAs through discriminant analysis. These groups were accurately differentiated based on miR-205 and miR-375 expression, with no errors in the training set (sequencing run one) and one misassignment (sample NS5a) in the testing set (sequencing run two). Significant differences in miR-205 and miR-375 concentrations were seen between MCC and non-MCC groups (ANOVA normalized MCC group = 20, normalized non-MCC group = 15); miR-205 concentrations were 4.5-fold higher in non-MCC groups than in MCC groups (adjusted P value [Padj] = 0.26), and miR-375 concentrations were 60-fold higher in MCC groups than in non-MCC groups (Padj < 0.001) (Supplemental Figure 2). Significant differences were again seen when analyzing training set samples only (ANOVA Padj < 0.001, normalized MCC = 12, normalized non-MCC = 8); miR-205 concentrations were 308-fold higher in non-MCC groups than in MCC groups (Padj < 0.001), whereas miR-375 concentrations were 577-fold higher in MCC groups than in non-MCC groups (Padj < 0.001) (Supplemental Figure 2). We confirmed grouping and expression differences in a subset of MCC and NS samples using miRNA microarray and real-time RT-PCR analyses (Supplemental Figure 2).
Addressing technical shortcomings in multicolor miRNA FISH.
After identifying tumor-specific biomarkers for BCC (miR-205) and MCC (miR-375) in this proof-of-concept scenario, we established a multicolor miRNA FISH protocol suitable for use on FFPE tissue sections. To achieve this goal, we revisited RNA fixation, signal detection and amplification, and oligonucleotide probe design steps. Suboptimal RNA fixation leading to short and long RNA loss by diffusion, rather than RNA degradation, is the primary problem in optimizing RNA FISH (Supplemental Figure 3). To resolve this problem, we previously reported the use of 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide (EDC), a water-soluble condensation reagent that promotes phosphoamide bond formation between the miRNA 5′ phosphate end and aliphatic amines from amino acid side chains of surrounding proteins (Supplemental Figure 4A and ref. 16).
We further studied the EDC condensation reaction in vitro by reacting 5′ AMP and modified peptides. Near-complete phosphoamide bond formation was seen following incubation at pH 8 for 10 hours at 50°C (Supplemental Figure 5). We found that the phosphate moiety predominantly reacted with the primary amine of lysine (Supplemental Figure 6). We also observed a pH-dependent hydrolysis side reaction of the EDC-miRNA intermediate that reduced the yield of crosslinking between protein and miRNA (data not shown). To further investigate the condensation and side reactions, we prepared N-(4-aminobutyl) isonicotinamide (ABINA), a highly soluble UV-absorbing model compound with a pKa similar to that of the lysine side chain amine (Supplemental Figure 7). We subsequently optimized condensation reaction conditions by reacting ABINA with AMP (Supplemental Figure 8) in the presence of various EDC derivatives (Supplemental Table 4) and heterocyclic compounds (Supplemental Table 5) that form stable reaction intermediates and reduce hydrolysis. The reaction time was reduced to 1 hour using EDC-methiodide and 5-ethylthio-1H-tetrazole (5-ETT). The use of two heterocyclic derivatives (1-methylimidazole and 5-ETT) also resulted in competing intermediate formation, enhancing nucleophilic attack of the aliphatic amine and rapid crosslinking (Supplemental Figure 4B). We ultimately selected EDC-hydrochloride (EDC-HCl) and 5-ETT in 1-methylimidazole buffer for miRNA crosslinking, because the mixture is cheaper and more stable and the short reaction time (3 hours) minimizes miRNA diffusion during EDC fixation. miRNA retention in EDC-fixed FFPE tissues was confirmed in Northern blot analyses (Supplemental Figure 3).
In addition to miRNA crosslinking, EDC promoted protein crosslinking through the formation of carboxylic acid amide bonds. We modeled this reaction using ABINA and 3-pyridylacetic acid (Supplemental Figure 9). Complete amide bond formation was observed after 6 hours, double the completion time for phosphoamide bond formation under the same reaction conditions. To assess the effects of protein crosslinking on tissue sections, we monitored the retention of abundant long RNAs. Paraffin embedding of formalin-fixed tissues is accompanied by mild RNA hydrolysis and fragmentation of RNAs into smaller pieces; hydrolysis products carrying 2′,3′ cyclic and 2′ or 3′ phosphate termini do not yield stable phosphoamide bonds (17). We detected 28S rRNA using directly labeled fluorescent locked nucleic acid–modified (LNA-modified) oligodeoxynucleotide probes complementary to rRNA sequences. As expected, the rRNA signal was predominantly cytoplasmic with the exception of nucleolar staining, corresponding to the sites of rRNA biogenesis. EDC fixation also increased the retention of partially hydrolyzed rRNAs (Supplemental Figure 10). Monitoring rRNA enabled us to assess RNA fixation; confirm RNA retention, integrity, and specificity for probe hybridization; and normalize miRNA signals.
Following EDC treatment, increased fixation of the protein matrix was anticipated to hinder access of antibody-based signal amplification reagents to the target-RNA-bound probe-conjugated haptens. miRNA detection by directly labeled probes was not possible, because these small RNAs are at least 100-fold less abundant than rRNAs, and the rRNA signal obtained from one directly labeled LNA-modified probe was only 100-fold above background (data not shown). To enable access of the target-RNA-bound probe-conjugated hapten to detection by antibodies, we systematically varied the linker length between the nucleic acid probe and hapten. Using rRNA as a target, we compared a biotin-labeled oligonucleotide probe with varying linker lengths to a set of directly labeled fluorescent probes hybridizing at distinct sites; linker lengths above 10 nm substantially improved signal amplification–based fluorescence detection, presumably by increasing the fraction of haptens displayed at the surface of the tissue section (Supplemental Figure 11).
To amplify the hybridization signal of hapten-conjugated probes, we used tyramide signal amplification and enhanced the HRP-mediated oxidative tyramide coupling reaction by adding 4-bromophenylboronic acid (18). We confirmed optimal tyramide signal amplification for our reagent set by preparing Cy3-tyramide reagents and buffers for comparison with commercial Cy3-tyramide equivalents. After optimizing the reaction, we switched to tyramides of ATTO dyes that are brighter, more stable, and water soluble.
Direct visualization of rRNA by fluorescently labeled probes enabled us to assess probe specificity. Mishybridization of LNA-modified miRNA probes to rRNA was detected through colocalization of signals to nucleoli. During our initial studies, we noticed that the probes for miR-375, and for miR-205 to a lesser degree, yielded an rRNA-like pattern, indicating cross-hybridization (Supplemental Figures 12 and 13, respectively). Upon sequence analysis, we realized that 8-nt short segments of complementarity to rRNA, especially when modified by LNA residues, were responsible for probe mishybridization to rRNA, which could be corrected through probe shortening and placing LNAs outside segments (not longer than 6–7 nt) with rRNA sequence complementarity (Supplemental Figures 12 and 13 and Supplemental Table 6).
Differentiating tumors using miRNA FISH — proof-of-concept study.
Having identified tumor-specific miRNA biomarkers and optimized our miRNA FISH protocol, we validated our approach on FFPE tissue sections from 4 BCC and 12 MCC cases. We performed multicolor miRNA FISH (Supplemental Figure 14) using a cocktail of 4 ATTO-647N–conjugated probes complementary to 28S rRNA (directly labeled), fluorescein-hexalabeled miR-205 probe (detected using anti-fluorescein-HRP antibody and ATTO-532-tyramide), and biotin-hexalabeled miR-375 probe (detected using HRP-conjugated streptavidin and ATTO-488-tyramide). Specific miR-205 and miR-375 FISH signals were detected, corresponding to our small RNA sequencing results (Figure 2). miR-205 signals were present in the cytoplasm and nuclei of BCC but absent from MCC tumor cells, whereas miR-375 signals were present in the cytoplasm and nuclei of MCC but absent from BCC tumor cells; cytoplasmic and nuclear signals were attributed to targeting of mature and precursor miRNAs and/or variable tyramide coupling to cellular matrix, reflecting distinct nuclear and cytoplasmic, as well as cell type–dependent, protein composition. miRNA and rRNA signals showed partially overlapping cytoplasmic localization but were not colocalized to nucleoli. miRNA signal intensities were obtained, corrected (Supplemental Figure 15 and Supplemental Table 7), and subsequently normalized against rRNA intensities; a cutoff value was previously established to differentiate BCC or MCC tumors (Figure 3 and Supplemental Table 7). miR-205 and miR-375 probe specificities were confirmed in wild-type and knockout mouse tissues (Supplemental Figures 16 and 17). Substantially lower signals were seen for both probes when EDC fixation was omitted, yielding near background signals that no longer differentiated tumors (Supplemental Figure 10). In future studies, shorter rRNAs (5S or 5.8S) will be evaluated as reference RNA signals.
Figure 2. Multicolor miRNA FISH for BCC and MCC differential diagnosis.
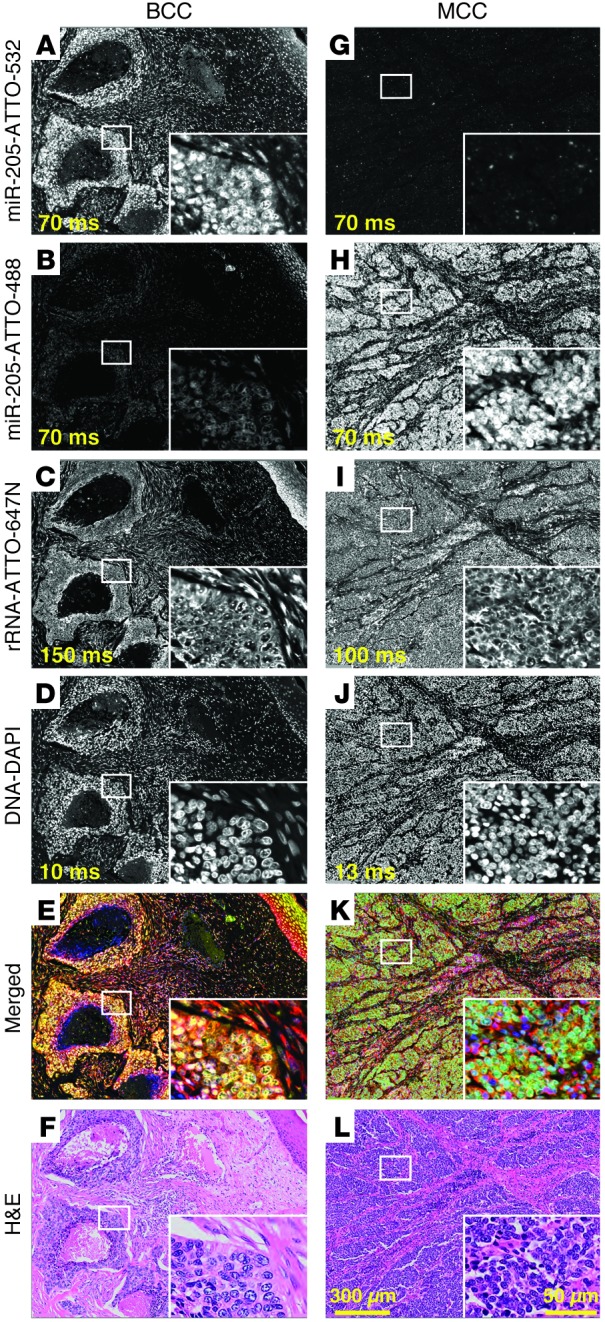
Parallel detection of miR-205, miR-375, and 28S rRNA in FFPE BCC and MCC tissue sections (samples BCC1 and MCC1) by multicolor miRNA FISH. Probes (25 nM each) were cohybridized at 55°C for 16 hours in buffer containing 50% formamide and 1.0 M NaCl (probe sequences are listed in Supplemental Table 6). miR-205 was detected using a 14-nt fluorescein-hexalabeled LNA-modified probe and anti-fluorescein-HRP antibody with and tyramide/ATTO-532 signal amplification. miR-375 was detected using a 15-nt biotin-hexalabeled LNA-modified probe with HRP-conjugated streptavidin and tyramide/ATTO-488 signal amplification. miR-205 signal intensities were higher in (A) BCC than in (G) MCC, whereas miR-375 signal intensities were conversely higher in (H) MCC than in (B) BCC, consistent with our small RNA sequencing results. (C and I) 28S rRNA was detected without signal amplification by a cocktail of 4 fluorescent ATTO-647N–conjugated probes. 28S rRNA signals were similar for both BCC and MCC and used to normalize miR-205 and miR-375 signals. (D and J) Nuclei were visualized using DAPI staining. (E and K) Merged images depict BCC in yellow and MCC in green, enabling rapid tumor identification. (F and L) H&E-stained tissue sections illustrate the potential for histologic similarity between BCC and MCC. Exposure times are indicated in ms. Representative areas, indicated by white rectangles, illustrate signal localization at higher magnification in insets. Original magnification, ×20; ×60 (insets). Scale bar: 50 μm (inset); 300 μm.
Figure 3. Differentiating BCC and MCC using normalized miRNA signal intensity ratios.

Following signal collection and correction (see Supplemental Figure 15 and Supplemental Table 7), miR-205 (black) and miR-375 (gray) signal intensities were normalized against rRNA signal intensities for each tumor. A cutoff value (0.4; indicated by red line) to differentiate BCC and MCC was first established on a test set (BCC1 and MCC1) of tumors. This cutoff value was subsequently used in blinded analysis to correctly identify 4 BCC (BCC2-5) and 12 MCC (MCC2-13) tumors.
Discussion
Our multicolor miRNA FISH method has several advantages over existing methods, including (a) increased miRNA retention through optimized EDC fixation; (b) avoidance of tissue permeabilization and related miRNA loss through longer hapten-probe linker lengths; (c) elimination of probe mishybridization to abundant RNAs through judicious design of LNA-modified DNA probes; (d) easy assessment of RNA retention, signal normalization, and probe specificity using directly labeled fluorescent rRNA probe; (e) increased multiplexing capacity through simultaneous fluorescent detection of two miRNAs and rRNA; and (f) development for use with archived FFPE tissue sections (16, 19–23).
Multicolor RNA FISH for histologic differentiation also offers advantages in rapid design and high-throughput synthetic generation of nucleic acid probes, quantitative detection of both coding and noncoding transcripts, discrimination among mRNA isoforms, and multiplexing capacity. In contrast, immunohistochemical methods for histologic differentiation rely on costly diagnostic-grade antibodies and require many months for antibody generation, validation, and adaptation for antigen retrieval in fixed tissues. It is important to note that our miRNA FISH and conventional immunohistochemical methods are incompatible because EDC-based protein crosslinking alters antigen structure (Supplemental Figure 18); however, conventional H&E staining can be readily performed upon acquisition of fluorescent images (e.g., Figure 2).
Our long-term goal is to develop RNA FISH for molecular diagnostic purposes. Combining RNA sequencing and FISH technologies provides a powerful platform for rapidly identifying and visualizing disease-specific biomarkers. Although we focused on two miRNAs in two skin cancers for this proof-of-principle study, we expect our method to be widely applicable, because EDC fixation pertains to all miRNAs in any FFPE tissue sample, probe design (typically aiming for a temperature of 60°C in 50% formamide by varying probe length and the number of incorporated LNAs) and signal detection and amplification steps are flexible, and experimental conditions (i.e., 50% formamide for complete duplex denaturation, fixed hybridization temperature at 50°C) are constant. Our next step is to explore the clinical utility of RNA FISH in larger sample collections. Although the present proof-of-concept study is small, it demonstrates the potential and relative ease of implementing RNA FISH in clinical laboratories.
Methods
Clinical materials, cell lines, and knockout tissues.
Thirty-six clinical samples and cell culture aliquots were obtained for miRNA expression profiling by barcoded small RNA sequencing (Supplemental Table 1). Tissue punches from FFPE biopsy specimens of MCC (n = 12), BCC (n = 4), and NS (n = 4) were collected in the Department of Pathology and Molecular Medicine, Kingston General Hospital, Kingston, Ontario, Canada. Tissue rolls from FFPE biopsy specimens of MCC (n = 2), BCC (n = 1), and NS (n = 1) were obtained from the Department of Pathology, Fox Chase Cancer Center. Biopsy samples were fixed in 10% neutral buffered formalin for 6 to 24 hours before processing to paraffin using a standard protocol. FFPE specimens were stored at room temperature for an average of 2.5 years (range: 1–8 years) prior to nucleic acid extraction.
Aliquots of 6 MCC-derived cell lines were obtained from the Tumor Virology Laboratory, University of Pittsburgh Cancer Institute. MCV status was established using PCR, Southern blot, and immunohistochemical stains for MCV T antigen (24).
Wild-type and miR-205 knockout mouse skin tissues were provided by Rui Yi, University of Colorado, Boulder, Colorado, USA. Wild-type and miR-375 knockout mouse pancreatic tissues were provided by Markus Stoffel, Swiss Federal Institute of Technology (ETH), Zurich, Switzerland.
RNA extraction and RNA integrity assessment.
Total RNA was extracted from FFPE tissue punches and rolls, respectively, using the RecoverAll Total Nucleic Acid Isolation Kit (Ambion) or the MasterPure Complete DNA and RNA Purification Kit (Epicenter Biotechnologies) according to the manufacturer’s guidelines. Total RNA was extracted from cultured cell lines using TRIzol Reagent (Invitrogen) following the manufacturer’s instructions. Total RNA concentrations were determined using a Nanodrop spectrophotometer ND-1000 or a Bio-Rad SmartSpec Plus Spectrophotometer. Concentrations of recovered total RNA ranged from 0.28 to 2.44 μg/μl, 0.01 to 1.80 μg/μl, and 0.81 to 3.65 μg/μl for RecoverAll, MasterPure, and TRIzol methods, respectively. Low RNA yields for samples NS5a and NS5b likely resulted from difficulty in dissolving paraffin from tissue rolls. RNA integrity was assessed by visual inspection of 28S and 18S rRNA bands following electrophoresis of 1 μg total RNA on an agarose gel stained with ethidium bromide.
Sequencing-based miRNA expression profiling, quantitation, and sample clustering.
Barcoded small RNA sequencing was performed as described previously (14); total RNA input was 2 μg per sample, except where limited sample (NS5a, NS5b) was available. Barcoded sequence reads were annotated as reported previously (25). We excluded 1 sample (MCC14b) from further analyses due to the low (<20,000) number of miRNA sequence reads. miRNA expression profiles were generated from relative counts of different miRNAs within a sample. Total miRNA concentrations were calculated for each sample by multiplying the ratios of miRNA to calibrator read frequencies by the amount of input calibration marker per μg of total RNA input. Hierarchical clustering was performed as described previously (25), and miRNAs were presented as precursor clusters according to our ongoing human miRNA reannotation studies (26). The sequencing data discussed in this publication have been deposited in the NCBI’s Gene Expression Omnibus (accessible through GEO series accession no. GSE34137; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE34137).
Microarray-based miRNA expression profiling and data analysis.
We profiled miRNA expression in FFPE tissues from 6 MCC and 2 NS samples using Agilent human miRNA arrays (27). Briefly, 100 ng total RNA was dephosphorylated, ligated to pCp-Cy3 using T4 RNA ligase 1, purified, and hybridized to an Agilent Human miRNA Microarray (v2), consisting of 8 identical subarrays with probes for 723 human miRNAs. Following scanning with the Agilent microarray scanner (Agilent), images were acquired and analyzed using Agilent feature extraction software version 9.5.3. GeneSpring software was used to normalize and log transform raw data and perform unsupervised hierarchical clustering. Differential expression between MCC and non-MCC groups was assessed by an unpaired t test. P values were adjusted for multiplicity using the Benjamini-Hochberg approach to control the false discovery rate. miRNAs were considered differentially expressed if the false discovery rate was <0.05 and the fold change was ≥2. The array data discussed in this publication have been deposited in the NCBI’s Gene Expression Omnibus (accessible through GEO series accession no. GSE-45146; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE45146).
miRNA real-time PCR.
For comparison with sequencing-derived miRNA concentration estimates, we measured miR-375 expression in total RNA extracted from FFPE tissue punches from 11 MCC and 4 NS samples using TaqMan MicroRNA Assays (Applied Biosystems) according to the manufacturer’s guidelines; miR-205 expression was not studied due to a paucity of material. Briefly, miRNAs were reversed transcribed using miRNA-specific stem-loop RT primers (Applied Biosystems). Subsequently, real-time PCR reactions were performed using the Eppendorf Realplex System (Eppendorf). PCR reactions were incubated in a 96-well plate at 95°C for 10 minutes, followed by 40 cycles at 95°C for 15 seconds and 60°C for 1 minute. All samples were assayed in triplicate, and data were normalized to endogenous RNU6B. Relative miRNA expression levels were calculated using the ΔΔCt method.
Synthesis of ABINA.
ABINA was prepared by refluxing methylisonicotinate (5.5 g, 40.0 mmol, Sigma-Aldrich, catalog no. M52950) and 1,4-diaminobutane (3.5 g, 40 mmol, Sigma-Aldrich, catalog no. D13208) in deionized water (2 ml) for 20 hours (28). Following removal of water and volatiles, the reaction was purified by silica flash column chromatography (gradient, 2:2:1:95 to 32:32:16:20, methanol/acetonitrile/triethylamine/diethylether) to yield ABINA (2.5 g, 32%) as a yellowish oil. ABINA purity was analyzed by thin-layer chromatography (silica gel; methanol/acetonitrile/triethylamine/diethylether [32:32:16:20]; Rf [methylisonicotinate] = 0.90, Rf [ABINA] = 0.10) and HPLC (only a single peak was detected).
Tissue sectioning.
FFPE tissue blocks were cooled on ice prior to sectioning; 5- or 20-μm tissue sections were cut using a Leica RM2255 Rotary Microtome. For mounting sections on glass slides, sliced tissues were manually unfolded in a room temperature water bath and then transferred to a 42°C water bath to allow tissue stretching. To minimizing miRNA diffusion, incubation times at 42°C were less than 10 seconds. Tissue sections were immediately mounted on Colorfrost Plus slides (Fisher Scientific, catalog no. 12-550-18), air dried for 1 hour, and incubated in an oven at 56°C for 1 hour to attach the tissue slice to the slide upon melting of the paraffin.
Synthesis of ATTO-488 and ATTO-532 tyramides.
ATTO-488-NHS (5 mg, ATTO-TEC GmbH, catalog no. AD 488-35) was dissolved in 500 μl anhydrous N,N-dimethylformamide (DMF, Solulink, catalog no. S-4001-005) to obtain an active ester solution A (10 mg/ml). Tyramine-HCl solution B was prepared by dissolving 10 mg tyramine-HCl in 1 ml DMF (10 mg/ml) to which a 1.25-fold equimolar amount of triethylamine (10 μl, Sigma-Aldrich, catalog no. 90335) was added. 500 μl solution A was mixed with 84 μl solution B, and the reaction mixture was left at ambient temperature in the dark for 2 hours. After the completion of synthesis, 5.9 μl ethanolamine solution C (dissolve 10 μl ethanolamine in 990 μl DMF) was added to the reaction mixture and stirred for 5 minutes to quench unreacted ATTO-488-NHS ester. The synthesized tyramide conjugate was diluted with 4.4 ml anhydrous DMSO (Sigma-Aldrich, catalog no. D2438) to obtain a final volume of 5 ml.
We prepared ATTO-532 solution A (10 mg/ml) by dissolving 5 mg ATTO-532-NHS (ATTO-TEC GmbH, catalog no. AD 532-35) in 500 μl anhydrous DMF. Solutions B and C were prepared as above. 500 μl solution A was mixed with 76 μl solution B, incubated in the dark for 2 hours at 25°C, quenched by 5.3 μl ethanolamine solution C, and brought to a final 5 ml volume by addition of 4.4 ml anhydrous DMSO. ATTO-tyramide solutions were aliquoted into light-protected 1.5 ml Eppendorf tubes.
miRNA FISH.
miRNA FISH was performed using an extensively modified version of our existing protocol (16). Briefly, we (a) optimized EDC condensation reaction conditions for crosslinking miRNAs to surrounding proteins; (b) eliminated proteinase K treatment for tissue permeabilization, which reduced RNA diffusion; (c) designed probes with increased linker lengths to increase accessibility to HRP-mediated amplification systems; and (d) synthesized fluorescent tyramide/dye derivatives as described previously (29), developing an amplification buffer for performing tyramide deposition containing 4-bromoboronic acid to control the oxidation process (18).
Antisense LNA-modified oligodeoxynucleotide probes targeting miR-205 and miR-375 were designed using mature miRNA sequences from miRBase (http:/ www.mirbase.org). To minimize rRNA cross-hybridization, probe sequences with greater than 6 consecutive nucleotide matches were avoided and shortened to 14 nt and 15 nt for miR-205 and miR-375, respectively (Supplemental Table 6). Hairpin formation and self-dimerization of probe sequences were predicted using MFold ( http://mfold.rna.albany.edu), and LNA modifications were placed in regions with no secondary structure or self-hybridization. LNA probes were synthesized at 1.0-μmol scale on an ABI 3400 DNA synthesizer, deprotected, and quantified, and their UV profiles were determined as previously published (16).
miR-205 probe was synthesized with a linker that enabled conjugation of 6 fluorescein moieties: 5′GGTGGAAtgaAgga-(L)3-F-L-F-L-F-L-F-L-F-(F-CPG), where uppercase letters indicate DNA nucleotides, lowercase letters indicate LNA modification, L represents spacer 18 (GlenResearch, catalog no. 10-1918-02), F represents 6-fluorescein serinol (GlenResearch, catalog no. 10-1994-02), and F-CPG represents 3′-6-fluorescein serinol CPG (GlenResearch, catalog no. 20-2994-10). miR-375 probe was synthesized with a linker that enabled conjugation of 6 biotin moieties: 5′AGCCGaaCGaAcaaA-(L)3-B-L-B-L-B-L-B-L-B-(B-CPG), where B represents protected biotinLC serinol (GlenResearch, catalog no. 10-1995-02) and B-CPG represents 3′-protected biotinLC serinol CPG (GlenResearch, catalog no. 20-2995-10).
Following synthesis, the CPG was transferred to a 1.5 ml screw cap tube and incubated with 1.2 ml 28%–30% aqueous ammonium hydroxide solution (EMD, catalog no. AX1303-6) for 16 hours at 55°C. The tube was placed on ice for 5 minutes, and the supernatant was transferred to a 13-ml centrifugation tube. Ten milliliters 1-butanol was added and vigorously mixed, and the LNA pellet was collected by centrifugation in a Sorvall RC5C Plus centrifuge (SS-34 rotor) at 20,000 g at 4°C for 20 minutes. The supernatant was removed completely, and the pellet was dried in an Eppendorf Vacufuge concentrator and redissolved in 400 μl water. After deprotection and precipitation, probes were directly used in miRNA FISH approach, without further denaturing PAGE gel purification.
Four rRNA antisense 3′-amino-modified LNA probes (Supplemental Table 6) were synthesized using 3′-amino-modifier C7 CPG (500 Å) solid glass support (GlenResearch, catalog no. 20-2957-10) and directly labeled using ATTO-647N-NHS (ATTO-TEC GmbH, catalog no. AD 647N-31) using an established protocol (16). All directly labeled probes were purified on a denaturing 18% PAGE gel (16).
Five-μm FFPE tissue sections from BCC and MCC samples were mounted on glass slides and deparaffinized with Histo-Clear II (National Diagnostics, catalog no. HS-202) twice for 6 minutes, rehydrated with ethanol (successive washes with 95%, 70%, and 50% ethanol) for 1 minute each, and rinsed in water. Sections were fixed in 4% PFA (Electron Microscopy Sciences, catalog no. 15710) in TBS (100 mM NaCl, 10 mM Tris-HCl, pH 7.4) for 45 minutes and washed in TBS for 5 minutes. Sections were subsequently incubated in 1-methylimidazole buffer (0.1 M 1-methylimidazole [Sigma-Aldrich, catalog no. 336092] adjusted to pH 8.0 using 1 M HCl, 0.3 M NaCl) for 3 minutes, followed by incubation in 500 μl EDC fixative solution (0.1 M EDC-HCl [Fluka, catalog no. 03450], 0.1 M 5-ETT [AIC, catalog no. 89797-98], 2X Denhardt’s solution [Applichem, catalog no. A3792], 0.1 M 1-methylimidazole buffer, readjusted to pH 8.0 using 10 M NaOH) in a humidified chamber at 50°C for 3 hours. Slides were rinsed in TBS for 3 minutes. After washing, sections were incubated in 50 ml freshly prepared acetylation solution (100 mM triethanolamine [Sigma-Aldrich, catalog no. T1377], 52.9 mM [0.5% v/v] acetic anhydride [Acros, catalog no. 149490010] in TBS) at 25°C for 10 minutes. Slides were rinsed again as above. Endogenous biotin was sequentially blocked with avidin and biotin solution from the biotin-blocking system (Dako, catalog no. X0590) at 25°C for 10 minutes. Following each blocking step, slides were rinsed in 50 ml TBS at 25°C for 3 minutes.
Sections were prehybridized in a humidified chamber at 25°C for 1 hour in 500 μl hybridization buffer composed of 50% formamide (Sigma-Aldrich, catalog no. F7503), 1.0 M NaCl, 75 mM Tris-HCl (pH 8.5), 1X Denhardt’s solution, 250 μg/ml baker’s yeast tRNA (Sigma-Aldrich, catalog no. R8759–2KU), 500 μg/ml salmon sperm DNA (Applichem, catalog no. A2159), 2.5 mM CHAPS (Sigma-Aldrich, catalog no. C3023), and 0.5% (v/v) Tween 20 (Sigma-Aldrich, catalog no. p1379). Hybridization buffer was replaced with a hybridization solution, containing (a) antisense miR-205 probe conjugated to 6 fluorescein moieties, (b) antisense miR-375 probe conjugated to 6 biotin moieties, and (c) 4 antisense rRNA probes directly labeled with ATTO-647N, each at 25 nM in hybridization buffer, and hybridization proceeded at 55°C for 16 hours.
Following hybridization, slides were washed once in wash buffer 1 (50% [v/v] formamide, 250 mM NaCl, 75 mM Tris-HCl [pH 8.5], and 0.1% [v/v] Tween 20) for 10 minutes at 25°C, again in wash buffer 1 for 10 minutes at 33°C, once in wash buffer 2 (50 mM NaCl, 75 mM Tris-HCl [pH 8.5], and 0.1% [v/v] Tween 20) for 5 minutes at 25°C, and once in TBS-T (TBS with 0.1% [v/v] Tween 20) for 3 minutes at 25°C. Sections were next incubated in 0.3% hydrogen peroxide (Fisher, catalog no. H325) in TBS-T for 20 minutes at 25°C and washed 3 times in TBS-T at 25°C.
The following signal amplification steps were carried out at 25°C. Slides were immersed in antibody “blocking” solution (5% goat serum [Sigma-Aldrich, catalog no. G9023] in TBS-T) for 30 minutes. Sections were incubated with 500 μl HRP-conjugated anti-fluorescein antibody diluted 1:250 in blocking solution in a humidified chamber for 1 hour. Slides were washed twice in TBS-T for 10 minutes before incubating each slide in 400 μl tyramide-ATTO-532 solution (tyramide dye conjugate was diluted 1:50 in PC amplification buffer [50 mM Tris-HCl {pH 8.0}, 2 M NaCl, 200 μg/ml 4-bromophenylboronic acid {Sigma-Aldrich, catalog no. B75956}, and 0.015% hydrogen peroxide]) for 30 minutes in a dark, humidified chamber. Slides were washed 3 times in TBS-T prior to incubation in 0.3% hydrogen peroxide solution. Each section was then incubated with 500 μl streptavidin conjugated to HRP diluted 1:1,000 for 1 hour in a humidified chamber. Slides were washed twice in TBS-T for 10 minutes before applying 400 μl tyramide-ATTO-488 solution, prepared and incubated as above. Slides were washed 3 times in TBS-T for 3 minutes, counterstained with 500 μl DAPI (5 μg/ml in TBS-T, Sigma-Aldrich, catalog no. D8417) in a humidified chamber for 10 minutes, and washed twice in TBS-T for 3 minutes. Each slide was mounted with MOWIOL mounting solution (25% [w/v] glycerol, 10% [w/v] MOWIOL [Polysciences Inc., catalog no. 17951]) and 0.1 M Tris-HCl, pH 6.8, and a glass coverslip. Prior to imaging, slides were dried in the dark for 30 minutes.
Microscopy, image processing, and signal normalization.
Images in Figure 2 and Supplemental Figures 10, 11, 16, and 17 were captured using the Olympus VS110 Virtual Microscopy System using ×20 and ×60 UPlanSApo objectives. For fluorescent imaging, we used ATTO dye combinations and the 86000v2 Sedat Quad filter set (Chroma), including filters for DAPI, Cy2, Cy3, and Cy5 (Supplemental Table 8). Images in Supplemental Figures 12 and 13 were captured on an Olympus BX50 microscope equipped with a DP70 camera and Olympus DP controller software. For fluorescent imaging, we used the following filter sets: U-MWU2 (Olympus) for DAPI, 41001 HQ (Chroma) for Alexa Fluor 488, 49004 ET (Chroma) for Cy3, and 49006 ET (Chroma) for Cy5.
Coded tissue sections from 16 BCC and MCC cases were provided by the Department of Pathology and Laboratory Medicine (Queen’s University), and miRNA FISH was performed in the Laboratory of RNA Molecular Biology (The Rockefeller University). Under blinded conditions, miR-205, miR-375, and rRNA signal intensities were measured on areas with at least 85% tumor composition. miR-205, miR-375, and rRNA signal intensity histograms were obtained to delineate specific RNA and background signals (Supplemental Figure 15). Following background removal, pixel intensities for miR-205, miR-375, and rRNA were multiplied by the corresponding sum of pixels, and these values were subsequently used to normalize miRNA against reference RNA signals. Based on a pilot test of representative tumors (BCC1 and MCC1), a cutoff value of 0.4 was established to differentiate BCC from MCC. Following data collection and interpretation by a blinded tester, sample codes were broken and analyzed.
Immunohistochemistry on EDC-fixed and unfixed tissues.
Ten-μm FFPE tissue sections of human breast cancer and 5-μm FFPE tissue sections of mouse embryonic liver, human gastrointestinal stromal tumor, and human Sloan-Kettering ovarian cancer cell line 3 (SKOV3) xenografted to mouse, mouse embryonic brain, and human placenta were mounted on Colorfrost Plus Microscope Slides (Fisher Scientific, catalog no. 12-550-18). Sections were deparaffinized with Histo-Clear II twice for 6 minutes, rehydrated with ethanol (successive washes with 95%, 70%, and 50% ethanol) for 1 minute each, and rinsed in water. Sections were fixed in 4% PFA in TBS for 45 minutes and washed in TBS for 5 minutes. Sections were subsequently processed with and without EDC fixation. For EDC fixation, sections were incubated in 1-methylimidazole buffer for 3 minutes, followed by incubation in 500 μl EDC fixative solution in a humidified chamber at 50°C for 3 hours. Slides were rinsed in TBS for 3 minutes.
Automated immunohistochemistry was performed using the Discovery XT System (Ventana Medical Systems), and sections were processed in parallel using the same protocol. Briefly, sections were subjected to standard deparaffinization and cell conditioning steps. Samples were blocked for 30 minutes in 10% normal goat serum, followed by incubation with rabbit primary antibody of glucose transporter 1 (Glut1, rabbit, Chemicon, catalog no. AB1340, 0.5 μg/ml), human epidermal growth factor receptor 2 (HER2, rabbit, Enzo, catalog no. alx-810-227, 5 μg/ml), phospho-histone-H3 (Phh3, rabbit, Millipore, catalog no. 06-570, 1 μg/ml), mouse primary antibody of pan-keratin (mouse, Abcam, catalog no. ab8068, 0.5 μg/ml), smooth muscle actin (SMA, mouse, Sigma-Aldrich, catalog no. A5228, 1 μg/ml), or vimentin (VIM, mouse, Ventana Medical Systems, catalog no. 790-2917, 2.5 μg/ml) for 4 hours. After washing, samples were incubated in buffer containing biotinylated goat anti-rabbit IgG (1:200 dilution, ABC Vectastain Kit, Vector Laboratories, catalog no. PK-4001) and biotinylated horse anti-mouse IgG (1:200 dilution, MOM Basic Kit, Vector Laboratories, catalog no. BMK-2202) at 25°C for 1 hour. For VIM immunohistochemical detection, universal secondary antibody (Ventana Medical Systems, catalog no. 760-4205, prediluted) was used. Antigen detection was completed using the DAB-MAP Chromogenic Detection Kit (Ventana Medical Systems, catalog no. 760-124). Images in Supplemental Figure 18 were captured using the Olympus VS110 Virtual Microscopy System (Olympus) using the ×20 objective (Olympus).
Quantitative Northern blot analyses.
To compare the effects of EDC fixation on miRNA diffusion and retention, we prepared FFPE tissue blocks from macaque brain, obtained 10-μm sections, performed miRNA FISH with and without EDC fixation, recovered total RNA from hybridization buffers and treated tissues, and performed quantitative Northern blot analyses for miR-124 as described previously (16).
FFPE tissues were prepared by immersing blocks of macaque brain tissue in 40 ml of 4% PFA at 4°C for 24 hours prior to standard paraffin embedding. One hundred and fifty milligrams of 10-μm sections of FFPE tissue was transferred into a SigmaPrep Spin Column (Sigma-Aldrich, catalog no. SC1000) to facilitate separation of solutions from tissue by centrifugation. Tissue sections were repeatedly incubated in 600 μl Histo-Clear II until the flow through was clear. Tissues were rehydrated by successive incubation in 100%, 95%, 70%, and 50% ethanol, each for 1 minute, prior to 3 washes in water. Tissues were incubated in 600 μl of 4% PFA for 45 minutes at 25°C and washed briefly in 600 μl TBS. Tissues were subsequently incubated in either 600 μl 1X TBS for 3 hours at 25°C or 600 μl 1-methylimidazole buffer for 3 minutes prior to fixation in 600 μl EDC fixative solution for 3 hours at 50°C and were washed twice consecutively in TBS for 3 minutes at 25°C. EDC-fixed and unfixed tissues were subsequently incubated in modified hybridization buffer (50% formamide, 1.0 M NaCl, 75 mM Tris-HCl, pH 8.5, 2.5 mM CHAPS, and 0.5% [v/v] Tween 20, omitting Denhardt’s solution, Baker’s yeast tRNA, and salmon sperm DNA to facilitate miRNA recovery) for 16 hours at 50°C. Hybridization solutions were collected (ca. 500 μl), diluted with 250 μl of 3 M NaCl, 1.8 ml Millipore water, and 5 μl spermine (30 mg/ml; Sigma-Aldrich, catalog no. 85590), and RNAs were precipitated upon addition of 7.5 ml 100% EtOH; RNA pellets were dissolved in 40 μl water. Residual tissues were incubated in 700 μl Proteinase K solution (100 μg/μl; 0.2 M Tris-HCl, pH 7.4, 0.3 M NaCl, 2% SDS [w/v], and 25 mM EDTA) for 5 hours at 50°C. Total RNA was extracted from liquids containing protease-digested tissues using standard phenol-chloroform extraction and ethanol precipitation protocols. Each pellet was resuspended in 40 μl H2O, and total RNA concentration was measured using a Bio-Rad SmartSpec Plus Spectrophotometer.
Statistics.
Statistical analyses were conducted using R language ( http://www.r-project.org/) and contributed packages. A P value of less than 0.05 was considered statistically significant. We tested normality assumptions using the Kolmogorov-Smirnov (KS) test. All P values are 2 tailed unless otherwise specified.
We assessed differences in total miRNA concentrations following different RNA extraction methods using Kruskal-Wallis rank-sum and ANOVA tests; due to the similarity of results only ANOVA test results are presented. Normality assumption was not rejected (KS P = 0.168 and 0.285). When comparing data among BCC, MCC, and NS groups in sequencing run one, pairwise differences were assessed using Tukey’s honest significant difference method.
Differences in miR-205 and miR-375 concentrations and miR-375 real-time PCR values between MCC and non-MCC groups were assessed using ANOVA after log2 transformation. In this scale, the normality assumption was not rejected (P = 0.197, 0.219, and 0.318, respectively). Correlations between miR-205 and miR-375 concentrations were assessed with Pearson correlation for log2-transformed values and Spearman correlation.
Discriminant analyses between MCC and non-MCC groups were performed using linear discriminant analysis (lda package from R). Sequencing run one was used as the training set to construct a classifier, and sequencing run two was used as the testing set to evaluate the performance of the classifier within an independent sample set. Identical results were obtained using the nearest shrunken centroid (pamr package) and support vector machine (svm). The addition of other miRNAs did not improve the performance of the classifier.
Differences in miR-205 and miR-375 fluorescence intensities between BCC and MCC groups were assessed using ANOVA models after log2 transformation. In this scale, the normality assumption was not rejected (P = 0.145, 0.783, and 0.318, respectively).
Study approval.
The Rockefeller University Institutional Review Board approved the use of deidentified human FFPE tissues from pathology archives in this study, and informed consent was not required.
Supplementary Material
Acknowledgments
N. Renwick is supported through a K08 award (NS072235) from the National Institute of Neurological Disorders and Stroke. M. Hafner is supported by a fellowship from the Charles H. Revson Jr. Foundation. P.S. Moore and Y. Chang are supported as American Cancer Society Professors and through R01 funding from NIH CA136363 and CA120726. T. Tuschl is an HHMI investigator and supported through R01 funding from NIH CA159227 and MH080442 and a grant by the Starr Cancer Consortium. The project described was supported through The Rockefeller University Bridges to Better Medicine Technology Innovation Fund. The project was also partially supported by grant award number (UL1RR024143) from the National Center for Research Resources (NCRR), a component of the NIH and NIH Roadmap for Medical Research, and its contents are solely the responsibility of the authors and do not necessarily represent the official view of NCRR or NIH. We thank S. Dewell (Genomics Resource Center, The Rockefeller University) for Solexa sequencing, H. Zebroski (Proteomics Core Facility, The Rockefeller University) for peptide synthesis, K. Manova and M. Turkekul (Pathology Core Facility, Memorial Sloan-Kettering Cancer Center) for H&E staining and immunohistochemistry, P. Berninger (EMBL Grenoble) and M. Khorshid (Biozentrum, University of Basel) for additional bioinformatic support, R. Yi (University of Colorado) for wild-type and miR-205 knockout skin tissues, and M. Stoffel (ETH Zürich, Switzerland) for wild-type and miR-375 knockout pancreatic tissue.
Footnotes
Conflict of interest: The methods described herein are the subject of a recent provisional patent application titled “Methods for fixing and detecting RNA” (provisional US patent application no. 61/512,228). Thomas Tuschl is cofounder of Alnylam Pharmaceuticals and is on the scientific advisory board of Regulus Therapeutics.
Citation for this article: J Clin Invest. 2013;123(6):2694–2702. doi:10.1172/JCI68760.
See the related Commentary beginning on page 2350.
References
- 1.Calin GA, et al. MicroRNA profiling reveals distinct signatures in B cell chronic lymphocytic leukemias. Proc Natl Acad Sci USA. 2004;101(32):11755–11760. doi: 10.1073/pnas.0404432101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu J, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 3.Rosenfeld N, et al. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol. 2008;26(4):462–469. doi: 10.1038/nbt1392. [DOI] [PubMed] [Google Scholar]
- 4.Calin GA, et al. A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353(17):1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 5.Eitan R, et al. Tumor microRNA expression patterns associated with resistance to platinum based chemotherapy and survival in ovarian cancer patients. Gynecol Oncol. 2009;114(2):253–259. doi: 10.1016/j.ygyno.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 6.Ji J, et al. MicroRNA expression, survival, and response to interferon in liver cancer. N Engl J Med. 2009;361(15):1437–1447. doi: 10.1056/NEJMoa0901282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ahmed FE, et al. Diagnostic microRNA markers for screening sporadic human colon cancer and active ulcerative colitis in stool and tissue. Cancer Genomics Proteomics. 2009;6(5):281–295. [PubMed] [Google Scholar]
- 8.Ball NJ, Tanhuanco-Kho G. Merkel cell carcinoma frequently shows histologic features of basal cell carcinoma: a study of 30 cases. J Cutan Pathol. 2007;34(8):612–619. doi: 10.1111/j.1600-0560.2006.00674.x. [DOI] [PubMed] [Google Scholar]
- 9.Thakur S, Chalioulias K, Hayes M, While A. Bilateral primary Merkel cell carcinoma of the upper lid misdiagnosed as Basal cell carcinoma. Orbit. 2008;27(2):139–141. doi: 10.1080/01676830701556715. [DOI] [PubMed] [Google Scholar]
- 10.Bobos M, Hytiroglou P, Kostopoulos I, Karkavelas G, Papadimitriou CS. Immunohistochemical distinction between Merkel cell carcinoma and small cell carcinoma of the lung. Am J Dermatopathol. 2006;28(2):99–104. doi: 10.1097/01.dad.0000183701.67366.c7. [DOI] [PubMed] [Google Scholar]
- 11.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319(5866):1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seo GJ, Chen CJ, Sullivan CS. Merkel cell polyomavirus encodes a microRNA with the ability to autoregulate viral gene expression. Virology. 2009;383(2):183–187. doi: 10.1016/j.virol.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 13.Chang Y, Moore PS. Merkel cell carcinoma: a virus-induced human cancer. Annu Rev Pathol. 2012;7:123–44. doi: 10.1146/annurev-pathol-011110-130227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hafner M, et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA. 2011;17(9):1697–1712. doi: 10.1261/rna.2799511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaarz A, et al. Bead array-based microrna expression profiling of peripheral blood and the impact of different RNA isolation approaches. J Mol Diagn. 2010;12(3):335–344. doi: 10.2353/jmoldx.2010.090116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pena JT, et al. miRNA in situ hybridization in formaldehyde and EDC-fixed tissues. Nat Methods. 2009;6(2):139–141. doi: 10.1038/nmeth.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomson JB, Patel BK, Jimenez V, Eckart K, Eckstein F. Synthesis and properties of diuridine phosphate analogues containing thio and amino modifications. J Org Chem. 1996;61(18):6273–6281. doi: 10.1021/jo960795l. [DOI] [PubMed] [Google Scholar]
- 18.Kricka LJ, Cooper M, Ji X. Synthesis and characterization of 4-iodophenylboronic acid: a new enhancer for the horseradish peroxidase-catalyzed chemiluminescent oxidation of luminol. Anal Biochem. 1996;240(1):119–125. doi: 10.1006/abio.1996.0338. [DOI] [PubMed] [Google Scholar]
- 19.Nuovo GJ, et al. A methodology for the combined in situ analyses and mature forms of microRNAs and correlation with their putative targets. Nat Protoc. 2009;4(1):107–115. doi: 10.1038/nprot.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nuovo GJ. In situ detection of microRNAs in paraffin embedded, formalin fixed tissues and the co-localization of their putative targets. Methods. 2010;52(4):307–315. doi: 10.1016/j.ymeth.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 21.Jorgensen S, Baker A, Moller S, Nielsen BS. Robust one-day in situ hybridization protocol for detection of microRNAs in paraffin samples using LNA probes. Methods. 2010;52(4):375–381. doi: 10.1016/j.ymeth.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 22.de Planell-Saguer M, Rodicio MC, Mourelatos Z. Rapid in situ codetection of noncoding RNAs and proteins in cells and formalin-fixed paraffin-embedded tissue sections without protease treatment. Nat Protoc. 2010;5(6):1061–1073. doi: 10.1038/nprot.2010.62. [DOI] [PubMed] [Google Scholar]
- 23.Kaur S, Lassus H, Butzow R. Chromogenic in situ detection of microRNAs in paraffin embedded tissue microarrays by locked nucleic acid probes. Protocol Exchange. 2012 doi:10.1038/protex.2012.030. [Google Scholar]
- 24.Shuda M, et al. Human Merkel cell polyomavirus infection I. MCV T antigen expression in Merkel cell carcinoma, lymphoid tissues and lymphoid tumors. Int J Cancer. 2009;125(6):1243–1249. doi: 10.1002/ijc.24510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berninger P, Gaidatzis D, van Nimwegen E, Zavolan M. Computational analysis of small RNA cloning data. Methods. 2008;44(1):13–21. doi: 10.1016/j.ymeth.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 26.Landgraf P, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129(7):1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, Chen J, Radcliffe T, Lebrun DP, Tron VA, Feilotter H. An array-based analysis of microRNA expression comparing matched frozen and formalin-fixed paraffin-embedded human tissue samples. J Mol Diagn. 2008;10(6):513–519. doi: 10.2353/jmoldx.2008.080077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang W, Fang S. Mono-acylation of symmetric diamines in the presence of water. Tetrahedron Lett. 2008;49:6003–6006. doi: 10.1016/j.tetlet.2008.07.174. [DOI] [Google Scholar]
- 29.Hopman AH, Ramaekers FC, Speel EJ. Rapid synthesis of biotin-, digoxigenin-, trinitrophenyl-, and fluorochrome-labeled tyramides and their application for In situ hybridization using CARD amplification. J Histochem Cytochem. 1998;46(6):771–777. doi: 10.1177/002215549804600611. [DOI] [PubMed] [Google Scholar]
- 30.Farazi TA, et al. MicroRNA sequence and expression analysis in breast tumors by deep sequencing. Cancer Res. 2011;71(13):4443–4453. doi: 10.1158/0008-5472.CAN-11-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.