

Abstract
Patients carrying two loss-of-function (or hypomorphic) alleles of STAT1 are vulnerable to intracellular bacterial and viral diseases. Heterozygosity for loss-of-function dominant-negative mutations in STAT1 is responsible for autosomal dominant (AD) Mendelian susceptibility to mycobacterial disease (MSMD), whereas heterozygosity for gain-of-function loss-of-dephosphorylation mutations causes AD chronic mucocutaneous candidiasis (CMC). The two previously reported types of AD MSMD-causing STAT1 mutations are located in the tail domain (p.L706S) or in the DNA-binding domain (p.E320Q and p.Q463H), whereas the AD CMC-causing mutations are located in the coiled-coil domain. We identified two cases with AD-STAT1 deficiency in two unrelated patients from Japan and Saudi Arabia carrying heterozygous missense mutations affecting the SH2 domain (p.K637E and p.K673R). p.K673R is a hypomorphic mutation that impairs STAT1 tyrosine phosphorylation, whereas the p.K637E mutation is null and affects both STAT1 phosphorylation and DNA-binding activity. Both alleles are dominant-negative and result in impaired STAT1-mediated cellular responses to IFN-γ and IL-27. By contrast, STAT1-mediated cellular responses against IFN-α and IFN-λ1 were preserved at normal levels in patients’ cells. We describe here the first dominant mutations in the SH2 domain of STAT1, revealing the importance of this domain for tyrosine phosphorylation and DNA-binding, as well as for anti-mycobacterial immunity.
Keywords: MSMD, STAT1, osteomyelitis, dominant-negative effect
Introduction
Mendelian susceptibility to mycobacterial diseases (MSMD; MIM# 209950) is a rare congenital disorder characterized by susceptibility to clinical disease caused by poorly virulent mycobacteria, such as Bacillus Calmette-Guérin (BCG) or non-tuberculous mycobacteria (NTM; Casanova and Abel, 2002; Hamosh, et al., 2005). Known genetic etiologies of MSMD affect the IL-12-IFN-γ immunological circuit, with mutations in six genes (IFNGR1; MIM# 107470, IFNGR2; MIM# 147569, STAT1; MIM# 600555, IL12B; MIM# 161561, IL12RB1; MIM# 601604 and NEMO; MIM# 300248) (Supp. Table S1) (Altare, et al., 1998a; Altare, et al., 1998b; Chapgier, et al., 2006a; Chapgier, et al., 2009; de Jong, et al., 1998; Doffinger, et al., 2000; Dorman and Holland, 1998; Dupuis, et al., 2001; Dupuis, et al., 2003; Fieschi, et al., 2004; Filipe-Santos, et al., 2006a; Filipe-Santos, et al., 2006b; Jouanguy, et al., 1996; Jouanguy, et al., 2000; Jouanguy, et al., 1997; Jouanguy, et al., 1999; Kristensen, et al., 2011; Newport, et al., 1996; Vogt, et al., 2008; Vogt, et al., 2005). Furthermore, germline mutations in the CYBB (MIM# 300481) and in the IRF8 (MIM# 601565) have been recently identified as the genes which are responsible for MSMD (Bustamante, et al., 2011; Hambleton, et al., 2011), MSMD-causing mutations in STAT1 are dominant and among the least common genetic etiologies of MSMD, with only six patients from four families reported to date among the 200 patients with known molecular defects.
STAT1 is a DNA-binding factor that regulates the transcription of specific genes. IFN-γ stimulation results in the phosphorylation of STAT1 at p.Tyr701 (p.Y701), inducing its homodimerization to form gamma-activated factor (GAF), which is translocated to the nucleus. GAF binds the gamma-activated sequence (GAS) to induce the transcription of target genes. By contrast, IFN-α/β stimulation induces the phosphorylation of both STAT1 and STAT2, resulting in the formation of the interferon-stimulated genes factor-3 (ISGF3) complex. ISGF3 recognizes IFN-stimulated response element (ISRE) motifs in target genes. GAF-mediated IFN-γ responses are important for immunity against intracellular microorganisms and some viruses, whereas ISGF-3-mediated IFN-α/β responses are important for immunity against most viruses.
Six patients with an autosomal dominant (AD) form of MSMD-causing STAT1 (AD-MSMD-STAT1) deficiency have been reported. These patients suffered only from mycobacterial diseases. The c.2116T>C (p.Leu706Ser; p.L706S) mutation in the tail segment domain of STAT1 abolishes phosphorylation at p.Y701 (Dupuis, et al., 2001). Although the c.958G>C (p.Glu320Gln; p.E320Q) and c.1389G>T (p.Gln463His; p.Q463H) mutations in the DNA-binding domain (DBD) do not prevent the normal phosphorylation of STAT1, they affect the DNA-binding capacity of GAF (Chapgier, et al., 2006a). These mutant STAT1 alleles have a dominant-negative effect on IFN-γ-induced GAF-mediated immunity, but not on IFN-α-induced ISGF3-mediated immunity. On the other hand, the autosomal recessive (AR) form of human STAT1 (AR-STAT1) deficiency confers broad susceptibility to mycobacteria and viruses (Chapgier, et al., 2009; Chapgier, et al., 2006b; Dupuis, et al., 2003; Kong, et al., 2010; Kristensen, et al., 2011; Vairo, et al., 2011). Nine patients with AR-STAT1 deficiency have been reported with different severities (Supp. Fig S1A). Surprisingly, heterozygous mutations in the coiled-coil domain of STAT1 were recently identified in up to approximately 30% of patients with AD chronic mucocutaneous candidiasis (CMC) (Liu, et al., 2011; van de Veerdonk, et al., 2011). CMC-causing STAT1 mutations are gain-of-function because they impair dephosphorylation of p.Y701 phosphorylated STAT1, resulting in increased phosphorylation, GAF DNA-binding ability and GAF transcription activity in response to IFN-γ, IFN-α, and IL-27 stimulation. (Puel, et al., 2011).
Thus, germline mutations in human STAT1 can be responsible for various kinds of infectious diseases. It also appears that the impact of the mutation is largely determined by the affected domain. We report here the first two germline dominant form of mutations in the SH2 domain of STAT1. We show that the mutations are loss-of-function and dominant-negative, underlying MSMD in two unrelated patients by distinct mechanisms.
Materials and Methods
Case Report
Kindred A
The subject (P1) was a Japanese boy born to nonconsanguineous parents (Fig. 1A). His sister had a history of regional lymph node enlargement after BCG vaccination in infancy. Two months later, the enlarged lymph node was cured spontaneously. Except for episode of BCGitis, she had no remarkable infectious episodes. Neither of his parents had any clinical signs suggestive of immunodeficiency. As shown in Supp. Table S2, the laboratory findings were normal in his sister and father. The patient had a history of regional lymph node enlargement after BCG vaccination. Although the lymph node had a draining abscess, it was effectively treated by local anti-infective agents and cured in three months. At six years of age, he developed multifocal osteomyelitis on the left humerus, three vertebrae and pelvic bones. Histological analysis of the left humerus showed structures resembling tuberculoid granulomas, but we could not exclude the possibility of granulomatous change due to sarcoidosis. No pathogenic bacteria, including Mycobacterium, were detected by PCR or culture of the tissues. The white blood cell, lymphocyte, and NK cell counts were normal, including those of the subpopulations with CD3, CD4, CD8, CD19 and CD16/56. The serum immunoglobulin levels were within normal ranges (Supp. Table S2). Furthermore, the generation of specific antibodies against measles, rubella and varicella after vaccination was observed to be normal. The leukocytes of the patient displayed a normal oxidative burst and normal proliferation in response to stimulation with PHA and ConA. At the beginning, the patient was treated with glucocorticoid, based on the suspected diagnosis of sarcoidosis. In spite of the treatment, his symptoms gradually progressed. No change was found in exon 4 of CARD15, which is known to be responsible for early-onset sarcoidosis and Blau syndrome. The patient was thus diagnosed with MSMD.
Figure 1.

Identification of STAT1 mutations and cytokine production.(A, B) The heterozygous p.K673R mutation was detected in the patient (A.II.2), his father (A.I.1) and his sister (A.II.1) in Kindred A from Japan. The heterozygous p.K637E mutation was identified in the patient (B.II.1) in Kindred B from Saudi Arabia. Closed symbols indicate the affected individual and open symbols indicate a healthy family member. Genetically affected individuals with no clinical phenotype are indicated by a vertical line. This mutation was not detected in 445 healthy individuals. (C) The PBMCs were incubated in the presence of LPS (100 ng/ml) and various concentrations of IFN-γ for 48 hours and TNF-α production was analyzed with OptEIA ELISA Set. PBMCs from the patient (A.II.2), his elder sister (A.II.1) and his father (A.I.1), carrying the heterozygous p.K673R STAT1 mutation displayed impaired TNF-α production. (D) Cytokine levels in the supernatant of whole blood from P2, healthy controls (local controls) and travel controls. P2 displayed a poor response to stimulation with BCG+IFN-γ. Open circle, no stimulation; closed circle, BCG stimulation; closed triangle, BCG+IFN-γ stimulation. Local control; the controls analyzed immediately after blood collection, travel control; the controls which were collected and transferred under the same conditions as the patients’blood.
Kindred B
The patient (P2) was a girl from Saudi Arabia born to consanguineous parents (Fig. 1B). Neither of her parents had ever presented any clinical manifestations that suggested immunodeficiency. She developed BCGosis at the age of five months and presented skin erosion around the site of BCG vaccination and on her back. Osteomyelitis was also observed in her right fibula and right zygomatic bone. Skin biopsy revealed non caseating granulomatous dermatitis. M. tuberculosis complex was isolated, by culture, from a bone biopsy specimen taken from the right fibula. The leukocytes of the patient displayed a normal oxidative burst and normal proliferation in response to stimulation with PHA, ConA and PWM. The patient was diagnosed with MSMD and successfully treated with antimycobacterial agents, including isoniazid, rifampicin and ciprofloxacin.
Blood samples were obtained from the patients, their relatives and healthy adult controls, after informed consent was given. This study was approved by the Ethics Committee/Internal Review Boards of Hiroshima University and Rockefeller University.
Cytokine measurements
Kindred A: Peripheral blood mononuclear cells (PBMCs) were dispensed into 96-wellplates (4×105/well) in RPMI 1640 supplemented with 10% fetal bovine serum (FBS). They were stimulated with 100 ng/ml LPS (Invivogen) and various concentrations of IFN-γ (PeproTech) for 48 hours. The concentration of TNF-α was determined in duplicate with the OptEIA ELISA kit (BD). Experiments were performed in triplicate. Kindred B: Whole-blood assays were performed as previously described (Feinberg, et al., 2004).
Molecular genetic analysis
Genomic DNA was extracted from peripheral blood leukocytes. Exon 22 of STAT1 and its flanking introns were amplified by PCR with primer set-1, and sequenced (Supp. Table S3).
Total RNA was extracted from the PBMCs, and cDNA was synthesized. PCR of the wild-type (WT), p.K637E, and p.K673R mutant alleles was performed with primer set-2. The PCR products were cloned into pGEM-T Easy vector (Promega) and their sequences were confirmed with primer set-3. We generated the p.L706S and p.Q463H mutations of STAT1, by PCR-based mutagenesis of the WT construct with mismatched primers (primer set-4, and set-5). The resulting fragments were inserted in-frame into pcDNA-C-Flag or pcDNA-C-Myc mammalian expression vectors, via the BamHI and EcoRV sites.
Analysis of gene expression and immunoprecipitation
EBV-transformed B lymphocytes (EBV-B cells) were maintained in RPMI 1640 with 10% FBS. They were stimulated by incubation with 105 IU/ml of IFN-γ or IFN-α (IFNs) for 20 min and subjected to immunoblot analysis.
HEK293 cells and U3C STAT1 null osteosarcoma cells (Chapgier, et al., 2006a) maintained in DMEM supplemented with 10% FBS in 100 mm culture dishes. Plasmid DNA (5 μg/dish for HEK293 cells, 20 μg/dish for U3C cells) was introduced into the cells by calcium phosphate-mediated transfection (Invitrogen). After 36 hours, the cells were incubated with 105 IU/ml of IFNs or 100 ng/ml of IL-27 (R&D Systems) for 30 min and subjected to immunoblot or immunoprecipitation analysis, as previously described (Ohno, et al., 2010; Okada, et al., 2007). Information about the antibodies used is provided in Supp. Table S4.
Immunostaining
Immunostaining techniques are described in the Supp. Methods section.
Luciferase reporter assay
Luciferase assay was performed as previously described (Liu, et al., 2011). U3C transfectants were stimulated with 103 IU/ml of IFN-γ (16 h) or IFN-α (12 h) and subjected to analysis. The data are expressed in relative luciferase units (RLU) or as fold inductions with respect to unstimulated cells. Experiments were performed in triplicate.
Quantitative real-time-PCR
EBV-B cells were stimulated with 20 ng/mL IFN-λ1 (1h, 2h, and 6h) or 105 IU/ml of IFNs (2h and 6h) and analyzed as previously described (Chapgier, et al., 2006a; Kong, et al., 2010). The results were normalized with respect to the values obtained for the endogenous GAPDH cDNA.
Electrophoretic mobility shift assay (EMSA)
U3C transfectants or EBV-B cells were stimulated with 105 IU/ml IFNs or 100 ng/ml IL-27 for 20 min and subjected to nuclear extraction. We incubated nuclear extract with 32P-labeled (α-dATP) GAS (from the FCGR1 promoter) or ISRE (from the ISG15 promoter) probe for 30 min. Then, they are subjected it to EMSA as previously described (Dupuis, et al., 2001; Dupuis, et al., 2003).
Results
Cytokine production upon IFN-γ stimulation in peripheral blood cells
PBMCs from the members of Kindred A were incubated in the presence of LPS and various concentrations of IFN-γ for 48 hours and TNF-α production was analyzed. TNF-α production was severely impaired in PBMCs from P1 (Fig. 1C), and impairment of TNF-α production was also observed in the patient’s sister and father. We carried out whole-blood cytokine analysis for Kindred B. IL-12p40 production in response to BCG and IFN-γ was partially impaired in cells from P2 (Fig. 1D). Thus, a poor response to IFN-γ stimulation was observed in peripheral blood cells from P1, P2 and two close relatives from Kindred A (A.II.1 and A.I.1).
Identification of STAT1 mutations
We identified a previously unknown heterozygous mutation, c.2018A>G (p.Lys673Arg; p.K673R), in exon 22 of STAT1 in P1 (Fig. 1A). The same mutation was also detected in the father (A.I.1) and elder sister (A.II.1) of this patient. The elder sister had a history of regional lymph node enlargement after BCG vaccination in infancy. The patient’s father was vaccinated with BCG without complications in infancy and presented no clinical signs suggestive of immunodeficiency.
P2, from Kindred B, was born into a consanguineous family. Neither of her parents have clinical signs suggestive of immunodeficiency, so P2 was suspected to have an autosomal recessive disorder. However, a previously unknown heterozygous mutation, c.1909A>G (p.Lys637Glu; p.K637E), was identified in exon 22 of STAT1 in P2 (Fig. 1B). It was not possible to test the parents of P2.
Both the p.K637E and p.K673R mutations affect the SH2 domain of STAT1. Alignment of the human STAT1 sequence with those from seven animal species showed that the p.K637 and p.K673 residues have been strictly conserved throughout evolution (Supp. Fig. S1B). We excluded the possibility of these mutations being common or irrelevant polymorphisms, by sequencing 445 healthy controls, including 79 Japanese and 49 Middle Eastern populations.
STAT1 expression, phosphorylation and dimerization
Expression of the p.K637E and p.K673R STAT1 alleles was demonstrated by analysis of the mRNA produced in EBV-B cells (data not shown). We then carried out immunoblotting to analyze the production and phosphorylation of STAT1 protein, using EBV-B cells from one healthy control, P1, P2, two members of Kindred A carrying the same STAT1 mutation and two other patients with partial (p.L706S/WT) and complete STAT1 deficiency. Similar levels of STAT1 protein were found in all the EBV-B cells analyzed, other than those from a patient with complete STAT1 deficiency, in analyses with an antibody recognizing the C-terminal part of the STAT1α isoform (Supp. Fig. S2A, S2B). However, the EBV-B cells of P1 and P2 displayed levels of STAT1 phosphorylation in response to stimulation with IFN-α and IFN-γ intermediate between those of control and p.L706S/WT cells (Supp. Fig. 2A). The impairment of STAT1 phosphorylation was highly reproducible and was also observed in EBV-B cells from the other family members of Kindred A, AI.1 and AII.1, demonstrating cosegregation of the genotype with the cellular phenotype (Supp. Fig. S2B). We further evaluated STAT1 phosphorylation by transiently transfecting U3C cells with WT and mutant STAT1 constructs and analyzing the phosphorylation of the resulting STAT1 protein in response to stimulation with IFN-α or IFN-γ. The phosphorylation of both p.K637E STAT1 and p.K673R STAT1 was severely impaired, but not entirely abolished, as it is in the p.L706S mutant, demonstrating that the two mutant alleles were severely hypomorphic for STAT1 tyrosine phosphorylation (Fig. 2A).
Figure 2.

STAT1 phosphorylation and GAF DNA binding activity. (A) U3C cells transiently expressing WT or mutant STAT1 genes were stimulated by incubation with IFNs for 30 minutes and analyzed. The p.Q463H STAT1 mutant was normally phosphorylated like the WT protein. By contrast, no phosphorylation of the p.L706S STAT1 mutant was observed. Both p.K637E STAT1 and p.K673R STAT1 displayed low levels of phosphorylation. Experiments were performed at least twice, independently. (B) U3C cells transiently expressing WT or mutant STAT1 constructs were used for EMSA assays. IFN-γ stimulation abolished the ability of GAF to bind DNA, for the p.Q463H, p.K637E and p.L706S STAT1 proteins. By contrast, residual binding of DNA by GAF was observed with the p.K673R STAT1 protein. Similar impairment was also observed when cells were stimulated with IL-27. (C) Cotransfection experiments with U3C cells. All the mutant proteins had strong negative effects on the WT protein, and these effects were particularly strong for GAF DNA binding and the L706S STAT1 protein. Experiments were performed at least twice, independently.
We then assessed STAT1 homodimerization before and after IFN-γ stimulation, by carrying out co-immunoprecipitation with Myc- or Flag-tagged STAT1. In both conditions, the interaction between WT and mutant STAT1 was normal (Supp. Fig. S3A, S3B). In summary, the p.K637E and p.K673R STAT1 alleles are expressed normally to generate mutant STAT1 protein. Unlike the p.L706S mutation, they impair but do not abolish phosphorylation in response to IFN stimulation, and they form homodimers with WT STAT1 before and after IFN-γ stimulation.
Nuclear translocation and DNA-binding of STAT1 upon IFN-γ stimulation
U2OS stable transfectants were stimulated with IFN-γ and the nuclear translocation of STAT1 was analyzed. Both WT and mutant STAT1 proteins were found mostly in the cytoplasm before stimulation (Fig. 3A). After IFN-γ stimulation, STAT1 translocation to the nucleus was observed in cells expressing WT, p.Q463H, p.K637E and p.K673R STAT1 (Fig. 3B). However, p.K637E STAT1 and p.K673R STAT1 were also detected in the cytoplasm, suggesting that nuclear translocation was incomplete for these two mutants. Nuclear transduction was abolished in p.L706S STAT1-expressing cells. These results suggest that mutant STAT1 proteins can be translocated to the nucleus if they are phosphorylated.
Figure 3.

Immunostaining of WT and mutant STAT1-expressing cells. U2OS cells stably expressing constructs encoding Flag-tagged WT or mutant STAT1 were incubated with (B) or without (A) IFN-γ for 30 min and stained with FITC-tagged anti-Flag antibody and Hoechst 34442. (A) WT and mutant STAT1 were found mostly in the cytoplasm before IFN-γ stimulation. (B) After IFN-γ stimulation, translocation of STAT1 to the nucleus was observed in cells expressing WT, p.Q463H, p.K637E and p.K673R STAT1 constructs. However, p.K637E STAT1 and p.K673R STAT1 were also detected in the cytoplasm, suggesting that nuclear translocation was incomplete for these two mutants. This process was impaired in cells expressing the p.L706S construct. Experiments were performed at least twice, independently.
We assessed the DNA-binding ability of the mutant STAT1 proteins by stimulating EBV-B cells with IFN-γ and carrying out EMSA. EBV-B cells carrying heterozygous p.K637E, p.K673R or p.L706S mutations displayed markedly lower levels of GAF binding to DNA (Supp. Fig. S4A). Impaired GAF binding to DNA was also observed in relatives carrying heterozygous p.K673R mutations (Supp. Fig. S4B). We assessed the intrinsic DNA-binding capacity of mutant STAT1 protein by introducing WT and mutant STAT1 into U3C cells and analyzing the binding of GAF to DNA (Fig. 2B). p.K637E STAT1, like p.Q463H and p.L706S STAT1, was a loss-of-function mutation, resulting in the abolition of GAF binding to DNA. By contrast, p.K673R STAT1, like p.E320Q STAT1, displayed residual DNA binding (approximately 19%), resulting in hypomorphic features (Chapgier, et al., 2006a). Next, we carried out a cotransfection experiment to determine whether or not these mutants had a dominant-negative effect on WT STAT1. All the mutants had a dose-dependent negative effect on WT STAT1 (Fig. 2C). Thus, these STAT1 mutants have a dominant-negative effect, possibly through their interaction with WT STAT1, on IFN-γ-induced WT STAT1-mediated GAF binding to DNA.
ISGF3 formation and DNA-binding in response to IFN-α
We investigated the impact of STAT1 mutations on IFN-α-induced signaling by assessing ISGF3 DNA-binding activity. Some impairment was suspected in the patients’ EBV-B cells, but it was much milder than that observed for DNA binding by GAF (Supp. Fig. S4A). Similar results were obtained for the cells of siblings carrying the heterozygous K673R mutation (Supp. Fig. S4B). Supershift experiments showed that these GAF or ISGF3 DNA-binding proteins contained STAT1 (data not shown).
Both K637E and K673R STAT1 exert a dominant-negative effect on GAS activation, but not on ISRE activation
The p.Q463H, p.K637E and p.L706S STAT1 proteins abolished the transcriptional activity of GAS in response to IFN-γ stimulation (Fig. 4A). However, p.K673R STAT1 retained a low level (approximately 14%) of GAS transcriptional activity. Cotransfection experiments revealed that each of the mutant forms of STAT1 had a negative effect on WT STAT1-mediated GAS transcriptional activity (Fig. 4C). Furthermore, a dose-dependent negative effect on the WT protein was observed for the p.K637E and p.K673R STAT1 proteins, respectively. We also analyzed GAS transcription activity in response to IFN-α stimulation (Supp. Fig. S5). WT STAT1 introduced cells induced GAS transcription activity in response to IFN-α stimulation. However, induction level was weak compared with IFN-γ stimulation. Both p.K637E and p.K673R STAT1 introduced cells showed severe impairment in GAS transcription activity.
Figure 4.

Analysis of transcription activity. GAS (A, C) or ISRE (B, D) reporter assay. Reporter plasmids and plasmids carrying the WT and/or mutant STAT1 genes were introduced into U3C cells by lipofection. The amounts of plasmid used are shown below the bar. The cells were stimulated, 24 hours after transfection, with IFN-γ (16 hours) or IFN-α (12 hours), and used for luciferase assays with the Dual-Glo luciferase assay system. (A) p.Q463H, p.K637E and p.L706S STAT1 abolished GAS transcriptional activity. With p.K673R STAT1, GAS activity was about 14% of that obtained with the WT protein. (B) p.L706S STAT1 abolished ISRE activation. By contrast, p.Q463H, p.K637E and p.K673R STAT1 proteins had residual ISRE activity. (C) Cotransfection experiments revealed that U3C cells expressing mutant STAT1 constructs had lower levels of GAS transcriptional activity than observed with the WT protein. A dose-dependent negative effect was observed with the p.Q463H, p.K637E, p.K673R and p.L706S STAT1 proteins. (D) Some negative effect on WT activity was observed with the p.Q463H, p.K637E and p.L706S proteins, in a cotransfection experiment. However, the decrease in transcriptional activity observed was smaller for ISRE than for GAS. No clear dose-dependent negative effect was observed. Experiments were performed at least three times, independently. Error bars represent SD of one experiment done in triplicate.
We then investigated IFN-α-induced ISRE transcription activity. The p.Q463H, p.K637E, and p.K673R STAT1 proteins retained some residual ISRE transcriptional activity, whereas ISRE transcriptional activity was abolished in p.L706S STAT1 mutants (Fig. 4B). GAF binds the GAS element via the DNA-binding domain of STAT1 (Reich, 2007), but ISGF3 binds the ISRE element via the DNA-binding domain of IRF-9 (Taniguchi, et al., 2001). These different mechanisms of DNA binding may account for the differences in the results obtained for GAF and ISGF3. Furthermore, these results are consistent with previous reports showing that phosphorylated WT and p.Q463H STAT1 proteins interact with Tyr690-phosphorylated STAT2 following IFN-α stimulation, whereas no such interaction is observed with p.L706S STAT1 (Chapgier, et al., 2006a). Cotransfection experiments revealed that the p.Q463H, p.K637E and p.L706S STAT1 mutations had some negative effects on the WT protein (Fig. 4D). However, these effects were much milder than those on GAS transcriptional activity. No clear dose-dependent negative effect was observed for any of these mutants. These results and those for EMSA suggest that the p.K637E mutation is a loss-of-function mutation and that the p.K673R mutation is a severely hypomorphic mutation affecting the transcriptional activity of GAF. These mutant STAT1 proteins probably have a dominant-negative effect on GAS activation, but not on ISRE activation.
Downstream gene induction in response to IFN-γ and IFN-α
We stimulated EBV-B cells with IFNs and investigated IFN-γ and/or IFN-α inducible genes, CXCL9, IRF1 and ISG15, by quantitative PCR analysis. EBV-B cells carrying p.K637E, p.K673E and p.L706S mutation showed severe impairment in CXCL9 induction after IFN-γ stimulation (Fig. 5A). The EBV-B cells also showed mild impairment in IRF1 transcription upon IFN-γ stimulation (Fig. 5B). We then analyzed the induction of ISG15, one of the IFN-α-induced ISRE-regulated genes, after IFN-α stimulation. Induction of ISG15 in patients’ cells was comparable to those from healthy controls (Fig. 5C). Since the induction of CXCL9, IRF1 and ISG15 are STAT1-dependent, we used STAT1 null EBV-B cells as a negative control and indeed, induction of these genes was not observed in these cells.
Figure 5.
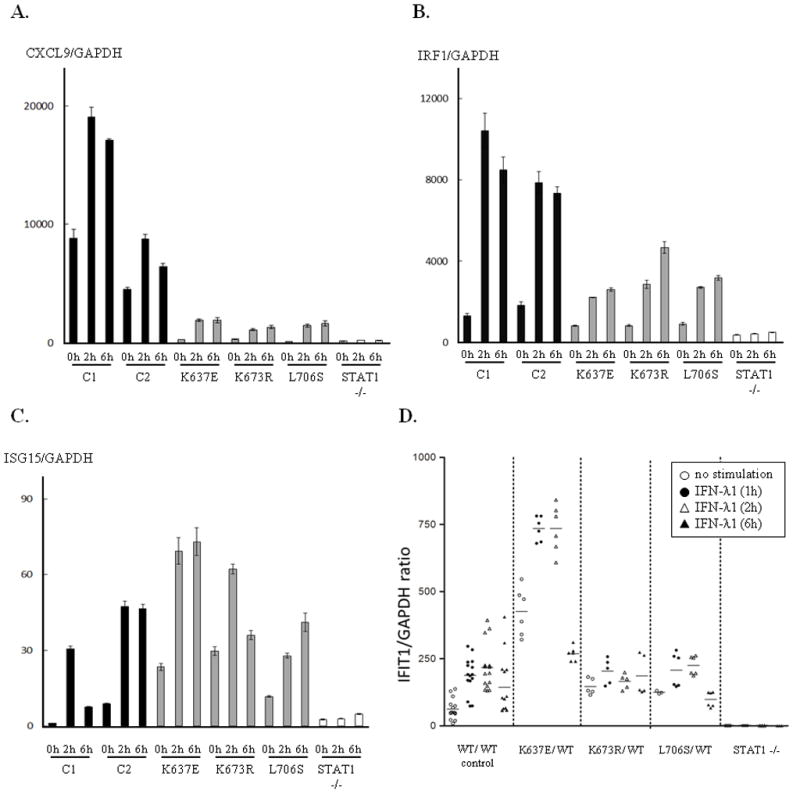
Selective impairment of downstream gene induction. (A, B) The induction of CXCL9 and IRF1 were investigated 2 h and 6 h after IFN-γ stimulation. EBV-B cells carrying p.K637E, p.K673R and p.L706S STAT1 showed a severe impairment in CXCL9 induction. The EBV-B cells also showed a mild impairment in IRF1 induction following IFN-γ stimulation. (C) Induction of ISG15 after IFN-α stimulation. The induction of ISG15 was preserved in patients’ EBV-B cells to an almost normal level. Error bars represent SD of one experiment performed in triplicate. (D) IFIT1 induction in response to IFN-λ1 stimulation. Almost normal levels of IFIT1 mRNA were observed in EBV-B cells carrying the p/K673R or p.L706S mutation. By contrast, high levels of IFIT1 mRNA were observed in cells carrying the p.K637E mutation. IFIT1 mRNA levels were assessed before (open circle) IFN-λ1 stimulation and 1 h (closed circle), 2 h (open triangle) and 6 h (closed triangle) after IFN-λ1 stimulation. These results were normalized with respect to those for GAPDH. Two independent experiments were performed.
Impact of STAT1 mutations on IL-27 and IL-29 signaling
IL-27 is known to induce both STAT1 and STAT3 phosphorylation. IL-27 activates GAF and IFN-λ1 activates ISGF3 via STAT1 activation. We assessed phosphorylation of STAT1 and STAT3 in response to IL-27 stimulation in U3C transient transfectants (Supp. Fig. S6A). Both p.K637E and p.K673R STAT1 showed partial impairment in STAT1 phosphorylation in response to IL-27 stimulation. In contrast, introduced STAT1 did not affect STAT3 phosphorylation in U3C transfectants. These results are consistent with previous reports showing that both WT and CMC-causing STAT1 mutations do not affect STAT3 phosphorylation (Liu, et al., 2011). This comparable level of STAT3 phosphorylation upon IL-27 stimulation was also confirmed in EBV-B cells from the patients (Supp. Fig. S6B). From these experiments, it is clear that MSMD-causing STAT1 mutations did not affect STAT3 phosphorylation after IL-27 stimulation. Next, we tested the GAF DNA-binding ability following IL-27 stimulation. EBV-B cells from the patients carrying heterozygous p.K637E, p.K673R or p.L706S mutations displayed low levels of DNA binding by GAF (Supp. Fig. S4A). Furthermore, GAF binding to DNA was abolished in U3C cells transiently expressing p.Q463H, p.K637E, and p.L706S STAT1, whereas residual binding was observed in cells transiently expressing the p.K673R form (Fig. 2B). We then investigated the induction of IFIT1 mRNA in response to IFN-λ1 stimulation. EBV-B cells carrying the p.K673R or p.L706S mutation produced similar levels of IFIT1 mRNA after IFN-λ1 stimulation, whereas EBV-B cells carrying the p.K637E mutation produced larger amounts of IFIT1 mRNA (Fig. 5D). By contrast, STAT1-null EBV-B cells produced no IFIT1 mRNA, suggesting that STAT1 plays a key role in IFIT1 induction after IFN-λ1 stimulation. Thus, EBV-B cells from the patients with AD-MSMD-STAT1 deficiency produced IFIT1 as efficiently as WT cells in response to IFN-λ1 stimulation.
Discussion
We describe here a novel molecular mechanism of AD-MSMD-STAT1 deficiency underlying MSMD, caused by mutations affecting the SH2 domain of STAT1. The two SH2 domain mutations are intrinsically deleterious in conditions of IFN-γ stimulation: p.K673R is hypomorphic and impairs the Y701 phosphorylation of STAT1; p.K637E is null and, surprisingly, impairs both STAT1 phosphorylation and the binding of GAF to DNA (summarized in Table 1). These mutations exert a dominant negative effect on the WT STAT1, impairing cellular responses to IFN-γ and MSMD. By contrast, IFN-α-induced ISRE activation was preserved in the cells of the patients. The previously described p.L706S mutation in the TS domain, which leads to a defect in STAT1 phosphorylation, was identified as the first molecular mechanism to cause AD-MSMD-STAT1 deficiency (Dupuis, et al., 2001). The p.E320Q and p.Q463H mutations in the DBD cause this disorder in the second molecular mechanism, impair DNA binding by GAF, despite normal phosphorylation (Chapgier, et al., 2006a). The p.K637E and p.K673R mutations, the first dominant mutations to be identified in the SH2 domain of STAT1, define a third molecular mechanism of AD-MSMD-STAT1 deficiency. The clinical symptoms caused by these three different molecular mechanisms seem to be indistinguishable, with all patients presenting mild MSMD. One of the typical clinical features of AD-MSMD-STAT1 deficiency is its incomplete penetrance. Only nine of the 14 heterozygous patients from six kindreds identified, including our patients, have presented a clinical phenotype of MSMD. However, asymptomatic carriers also displayed functional defects. Incomplete penetrance may be accompanied by a mild clinical phenotype of this disorder. Actually, incomplete penetrance has also been reported in patients with partial IFNGR1 and IL12RB1 deficiencies, who have a clinical phenotype milder than that of many other MSMD patients (Casanova and Abel, 2007; de Beaucoudrey, et al., 2010; Dorman, et al., 2004).
Table 1.
The functional defects caused by a dominant form of STAT1 mutations
| WT | Q463H | K637E | K673R | L706S | |
|---|---|---|---|---|---|
| STAT1 expression | Normal | Normal | Normal | Normal | Normal |
| Phosphorylation | Normal | Normal | Partially impaired | Partially impaired | Abolished |
| Dimerization before IFN-γ stimulation | Yes | Yes | Yes | Yes | Yes |
| Dimerization after IFN-γ stimulation | Yes | Yes | Yes | Yes | Yes |
| Nuclear translocation | Normal | Normal | Partially impaired | Partially impaired | Abolished |
| GAF DNA-binding ability | Normal | Abolished | Abolished | Residual ability (19 %) | Abolished |
| GAS transcription activity | Normal | Abolished | Abolished | Residual ability (14%) | Abolished |
| ISRE transcription ability | Normal | Residual activity (34%) | Residual ability (62%) | Residual ability (71%) | Abolished |
| Dominant negative effects against WT STAT1 induced GAS activation | Yes | ||||
| Dominant negative effects against WT STAT1 induced ISRE activation | No | ||||
These results were derived from cells from heterozygous patients and gene expression experiments using STAT1-deficient cells.
Determination of the crystal structure of the STAT1-DNA complex has shown that dimeric interactions between SH2 domains are crucial for the formation of a DNA-binding clamp that wraps itself almost entirely around the duplex (Chen, et al., 1998). The K637E mutation replaces a positively charged residue by a negatively charged residue, and this clearly affects the interaction of the protein with DNA (Supp. Fig. S1C). The p.K673 residue is not correctly positioned for interaction with the DNA, accounting for the low levels of residual binding of GAF to DNA observed for the mutant STAT1 p.K673R protein. However, the effect of the residue change in itself may be less damaging than the potential effect on SH2 domain conformation. This mutation may have effects on SH2 folding and p.Y701 phosphorylation. Posttranslational modifications are known to play an important role in the regulation of signaling pathways. STAT1 has a large number of sites for posttranslational modification, with phosphorylation occurring at p.Y701 and p.S727, acetylation at p.K410 and p.K413, and SUMOylation at p.K703 (Kim and Lee, 2007). No ubiquitination sites have yet been identified, but polyubiquitination has been observed in many cell lines (Kim and Maniatis, 1996). In addition, proteasome-mediated STAT1 degradation has been reported (Ungureanu and Silvennoinen, 2005). The two mutations studied here affect lysine residues in the SH2 domain. However, STAT1 levels were similar in EBV-B cells from patients and controls, suggesting an absence of change in STAT1 ubiquitination.
The SH2 domain is known to play an important role in signal transduction and nuclear import, through phosphorylation-induced dimerization (Lim and Cao, 2006). Mutations in STAT3, another member of the STAT1 family, are known to cause an autosomal dominant form of hyper IgE syndrome (Minegishi, et al., 2007). Most of the mutations identified are missense mutations affecting the DBD or SH2 domain (Renner, et al., 2008). DBD mutations have dominant-negative effects on DNA binding by WT STAT3 homodimers (Minegishi, et al., 2007). By contrast, SH2 domain mutations impair phosphorylation upon IL-6 stimulation in many situations. The effects of these disease-causing SH2 domain mutations of STAT3 on DNA binding have not been investigated, but several different molecular mechanisms resulting in a similar presentation have been proposed to underlie hyper IgE syndrome (Renner, et al., 2008). Thus, the SH2 domain may also play an important role, at least in phosphorylation of the Y705 residue of STAT3. These data and our description of two new mutations affecting the SH2 domain of STAT1 confirm the important role of the SH2 domain of STAT proteins not only in tyrosine phosphorylation, but also in subsequent DNA binding.
We also studied the impact of STAT1 mutations on IFN-λ1- and IL-27-induced STAT1-mediated signaling, in the first investigation of the effect of these cytokines in MSMD patients with AD-MSMD-STAT1 deficiency. IFN-λ1 is thought to inhibit the proliferation of various viruses (Doyle, et al., 2006; Kotenko, et al., 2003; Marcello, et al., 2006; Sheppard, et al., 2003; Zhang, et al., 2008). An impaired response to IFN-λ1 has been observed in patients with AR-STAT1 deficiency presenting severe viral infections (Chapgier, et al., 2009). In this study, IFN-λ1-induced STAT1-mediated signaling was maintained at similar levels in the cells of patients and healthy individuals. These findings were consistent with the normal viral phenotype of patients with AD-MSMD-STAT1 deficiency. IL-27 belongs to the IL-12 family of cytokines and plays an important role in the regulation of helper T-cell differentiation (Yoshida and Miyazaki, 2008). We show here that IL-27-induced GAF binding to DNA is impaired in cells from the patients and in cells into which mutant STAT1 constructs have been introduced. The impairment of IL-27-induced STAT1-mediated signaling has also been reported in patients with AR-STAT1 deficiencies, and is thought to be related to host susceptibility to Salmonella (Chapgier, et al., 2009). Neither P1 nor P2 has presented severe Salmonella infections. However, we cannot rule out the effect of the impaired IL-27 pathway on the clinical phenotype of AD-MSMD-STAT1 deficiency, because this pathway has been shown to have STAT1-dependent proinflammatory effects on human monocytes (Yoshida and Miyazaki, 2008). Thus, impairment of the response to IL-27 is likely to be a common cellular phenotype in patients with AD-MSMD-STAT1 deficiency. Further studies will be required to clarify the role of IL-27 in the clinical phenotype of patients with AD-MSMD-STAT1 deficiencies.
Supplementary Material
Acknowledgments
Grant Sponsor
This study was supported in part by Grants in Aid for Scientific Research from the Japan Society for the Promotion of Science [20790731 to S.O] and by Grants-in Aid for Scientific Research from the Ministry of Education, Culture, Sport, Science and Technology of Japan [22591161 to M.K.]. This study was also supported in part by Research on Measures for Intractable Diseases funding from the Japanese Ministry of Health, Labor and Welfare [H22-Nanchi-ippan-078 to M.K.]. The Laboratory of Human Genetics of Infectious Diseases is supported by grants from the Bill and Melinda Gates Foundation, the St. Giles Foundation, the Jeffrey Modell Foundation and Talecris Biotherapeutics, the Rockefeller University Center for Clinical and Translational Science [5UL1RR024143-03], the Rockefeller University, and the National Institute of Allergy and Infectious Diseases [1R01AI089970-01].
We thank Vanessa Bryant, PhD, Magali Audry, PhD, Dusan Bogunovic, PhD, Carolina Prando, MD, Alexandra Kreins, MD, and Marcela Moncada, BSc for helpful discussions and critical reading. Sequence analysis was supported by the Analysis Center of Life Science, Hiroshima University.
Footnotes
Supporting Information for this preprint is available from the Human Mutation editorial office upon request (humu@wiley.com)
References
- Altare F, Durandy A, Lammas D, Emile JF, Lamhamedi S, Le Deist F, Drysdale P, Jouanguy E, Doffinger R, Bernaudin F, et al. Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science. 1998a;280(5368):1432–5. doi: 10.1126/science.280.5368.1432. [DOI] [PubMed] [Google Scholar]
- Altare F, Lammas D, Revy P, Jouanguy E, Doffinger R, Lamhamedi S, Drysdale P, Scheel-Toellner D, Girdlestone J, Darbyshire P, et al. Inherited interleukin 12 deficiency in a child with bacille Calmette-Guerin and Salmonella enteritidis disseminated infection. J Clin Invest. 1998b;102(12):2035–40. doi: 10.1172/JCI4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustamante J, Arias AA, Vogt G, Picard C, Galicia LB, Prando C, Grant AV, Marchal CC, Hubeau M, Chapgier A, et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol. 2011;12(3):213–21. doi: 10.1038/ni.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova JL, Abel L. Genetic dissection of immunity to mycobacteria: the human model. Annu Rev Immunol. 2002;20:581–620. doi: 10.1146/annurev.immunol.20.081501.125851. [DOI] [PubMed] [Google Scholar]
- Casanova JL, Abel L. Primary immunodeficiencies: a field in its infancy. Science. 2007;317(5838):617–9. doi: 10.1126/science.1142963. [DOI] [PubMed] [Google Scholar]
- Chapgier A, Boisson-Dupuis S, Jouanguy E, Vogt G, Feinberg J, Prochnicka-Chalufour A, Casrouge A, Yang K, Soudais C, Fieschi C, et al. Novel STAT1 alleles in otherwise healthy patients with mycobacterial disease. PLoS Genet. 2006a;2(8):e131. doi: 10.1371/journal.pgen.0020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapgier A, Kong XF, Boisson-Dupuis S, Jouanguy E, Averbuch D, Feinberg J, Zhang SY, Bustamante J, Vogt G, Lejeune J, et al. A partial form of recessive STAT1 deficiency in humans. J Clin Invest. 2009;119(6):1502–14. doi: 10.1172/JCI37083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapgier A, Wynn RF, Jouanguy E, Filipe-Santos O, Zhang S, Feinberg J, Hawkins K, Casanova JL, Arkwright PD. Human complete Stat-1 deficiency is associated with defective type I and II IFN responses in vitro but immunity to some low virulence viruses in vivo. J Immunol. 2006b;176(8):5078–83. doi: 10.4049/jimmunol.176.8.5078. [DOI] [PubMed] [Google Scholar]
- Chen X, Vinkemeier U, Zhao Y, Jeruzalmi D, Darnell JE, Jr, Kuriyan J. Crystal structure of a tyrosine phosphorylated STAT-1 dimer bound to DNA. Cell. 1998;93(5):827–39. doi: 10.1016/s0092-8674(00)81443-9. [DOI] [PubMed] [Google Scholar]
- de Beaucoudrey L, Samarina A, Bustamante J, Cobat A, Boisson-Dupuis S, Feinberg J, Al-Muhsen S, Janniere L, Rose Y, de Suremain M, et al. Revisiting Human IL-12Rbeta1 Deficiency: A Survey of 141 Patients From 30 Countries. Medicine (Baltimore) 2010;89(6):381–402. doi: 10.1097/MD.0b013e3181fdd832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong R, Altare F, Haagen IA, Elferink DG, Boer T, van Breda Vriesman PJ, Kabel PJ, Draaisma JM, van Dissel JT, Kroon FP, et al. Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science. 1998;280(5368):1435–8. doi: 10.1126/science.280.5368.1435. [DOI] [PubMed] [Google Scholar]
- Doffinger R, Jouanguy E, Dupuis S, Fondaneche MC, Stephan JL, Emile JF, Lamhamedi-Cherradi S, Altare F, Pallier A, Barcenas-Morales G, et al. Partial interferon-gamma receptor signaling chain deficiency in a patient with bacille Calmette-Guerin and Mycobacterium abscessus infection. J Infect Dis. 2000;181(1):379–84. doi: 10.1086/315197. [DOI] [PubMed] [Google Scholar]
- Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest. 1998;101(11):2364–9. doi: 10.1172/JCI2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman SE, Picard C, Lammas D, Heyne K, van Dissel JT, Baretto R, Rosenzweig SD, Newport M, Levin M, Roesler J, et al. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet. 2004;364(9451):2113–21. doi: 10.1016/S0140-6736(04)17552-1. [DOI] [PubMed] [Google Scholar]
- Doyle SE, Schreckhise H, Khuu-Duong K, Henderson K, Rosler R, Storey H, Yao L, Liu H, Barahmand-pour F, Sivakumar P, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology. 2006;44(4):896–906. doi: 10.1002/hep.21312. [DOI] [PubMed] [Google Scholar]
- Dupuis S, Dargemont C, Fieschi C, Thomassin N, Rosenzweig S, Harris J, Holland SM, Schreiber RD, Casanova JL. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science. 2001;293(5528):300–3. doi: 10.1126/science.1061154. [DOI] [PubMed] [Google Scholar]
- Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, Yang K, Chapgier A, Eidenschenk C, Eid P, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33(3):388–91. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- Feinberg J, Fieschi C, Doffinger R, Feinberg M, Leclerc T, Boisson-Dupuis S, Picard C, Bustamante J, Chapgier A, Filipe-Santos O, et al. Bacillus Calmette Guerin triggers the IL-12/IFN-gamma axis by an IRAK-4- and NEMO-dependent, non-cognate interaction between monocytes, NK, and T lymphocytes. Eur J Immunol. 2004;34(11):3276–84. doi: 10.1002/eji.200425221. [DOI] [PubMed] [Google Scholar]
- Fieschi C, Bosticardo M, de Beaucoudrey L, Boisson-Dupuis S, Feinberg J, Santos OF, Bustamante J, Levy J, Candotti F, Casanova JL. A novel form of complete IL-12/IL-23 receptor beta1 deficiency with cell surface-expressed nonfunctional receptors. Blood. 2004;104(7):2095–101. doi: 10.1182/blood-2004-02-0584. [DOI] [PubMed] [Google Scholar]
- Filipe-Santos O, Bustamante J, Chapgier A, Vogt G, de Beaucoudrey L, Feinberg J, Jouanguy E, Boisson-Dupuis S, Fieschi C, Picard C, et al. Inborn errors of IL-12/23- and IFN-gamma-mediated immunity: molecular, cellular, and clinical features. Semin Immunol. 2006a;18(6):347–61. doi: 10.1016/j.smim.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Filipe-Santos O, Bustamante J, Haverkamp MH, Vinolo E, Ku CL, Puel A, Frucht DM, Christel K, von Bernuth H, Jouanguy E, et al. X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production. J Exp Med. 2006b;203(7):1745–59. doi: 10.1084/jem.20060085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambleton S, Salem S, Bustamante J, Bigley V, Boisson-Dupuis S, Azevedo J, Fortin A, Haniffa M, Ceron-Gutierrez L, Bacon CM, et al. IRF8 mutations and human dendritic-cell immunodeficiency. The New England journal of medicine. 2011;365(2):127–38. doi: 10.1056/NEJMoa1100066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33(Database issue):D514–7. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouanguy E, Altare F, Lamhamedi S, Revy P, Emile JF, Newport M, Levin M, Blanche S, Seboun E, Fischer A, et al. Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guerin infection. N Engl J Med. 1996;335(26):1956–61. doi: 10.1056/NEJM199612263352604. [DOI] [PubMed] [Google Scholar]
- Jouanguy E, Dupuis S, Pallier A, Doffinger R, Fondaneche MC, Fieschi C, Lamhamedi-Cherradi S, Altare F, Emile JF, Lutz P, et al. In a novel form of IFN-gamma receptor 1 deficiency, cell surface receptors fail to bind IFN-gamma. J Clin Invest. 2000;105(10):1429–36. doi: 10.1172/JCI9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouanguy E, Lamhamedi-Cherradi S, Altare F, Fondaneche MC, Tuerlinckx D, Blanche S, Emile JF, Gaillard JL, Schreiber R, Levin M, et al. Partial interferon-gamma receptor 1 deficiency in a child with tuberculoid bacillus Calmette-Guerin infection and a sibling with clinical tuberculosis. J Clin Invest. 1997;100(11):2658–64. doi: 10.1172/JCI119810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouanguy E, Lamhamedi-Cherradi S, Lammas D, Dorman SE, Fondaneche MC, Dupuis S, Doffinger R, Altare F, Girdlestone J, Emile JF, et al. A human IFNGR1 small deletion hotspot associated with dominant susceptibility to mycobacterial infection. Nat Genet. 1999;21(4):370–8. doi: 10.1038/7701. [DOI] [PubMed] [Google Scholar]
- Kim HS, Lee MS. STAT1 as a key modulator of cell death. Cell Signal. 2007;19(3):454–65. doi: 10.1016/j.cellsig.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Kim TK, Maniatis T. Regulation of interferon-gamma-activated STAT1 by the ubiquitin-proteasome pathway. Science. 1996;273(5282):1717–9. doi: 10.1126/science.273.5282.1717. [DOI] [PubMed] [Google Scholar]
- Kong XF, Ciancanelli M, Al-Hajjar S, Alsina L, Zumwalt T, Bustamante J, Feinberg J, Audry M, Prando C, Bryant V, et al. A novel form of human STAT1 deficiency impairing early but not late responses to interferons. Blood. 2010;116(26):5895–906. doi: 10.1182/blood-2010-04-280586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4(1):69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- Kristensen IA, Veirum JE, Moller BK, Christiansen M. Novel STAT1 Alleles in a Patient with Impaired Resistance to Mycobacteria. Journal of clinical immunology. 2011;31(2):265–71. doi: 10.1007/s10875-010-9480-8. [DOI] [PubMed] [Google Scholar]
- Lim CP, Cao X. Structure, function, and regulation of STAT proteins. Mol Biosyst. 2006;2(11):536–50. doi: 10.1039/b606246f. [DOI] [PubMed] [Google Scholar]
- Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. The Journal of experimental medicine. 2011;208(8):1635–48. doi: 10.1084/jem.20110958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcello T, Grakoui A, Barba-Spaeth G, Machlin ES, Kotenko SV, MacDonald MR, Rice CM. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131(6):1887–98. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007;448(7157):1058–62. doi: 10.1038/nature06096. [DOI] [PubMed] [Google Scholar]
- Newport MJ, Huxley CM, Huston S, Hawrylowicz CM, Oostra BA, Williamson R, Levin M. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med. 1996;335(26):1941–9. doi: 10.1056/NEJM199612263352602. [DOI] [PubMed] [Google Scholar]
- Ohno Y, Yasunaga S, Ohtsubo M, Mori S, Tsumura M, Okada S, Ohta T, Ohtani K, Kobayashi M, Takihara Y. Hoxb4 transduction down-regulates Geminin protein, providing hematopoietic stem and progenitor cells with proliferation potential. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(50):21529–34. doi: 10.1073/pnas.1011054107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Ishikawa N, Shirao K, Kawaguchi H, Tsumura M, Ohno Y, Yasunaga S, Ohtsubo M, Takihara Y, Kobayashi M. The novel IFNGR1 mutation 774del4 produces a truncated form of interferon-gamma receptor 1 and has a dominant-negative effect on interferon-gamma signal transduction. J Med Genet. 2007;44(8):485–91. doi: 10.1136/jmg.2007.049635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puel A, Cypowyj S, Bustamante J, Wright JF, Liu L, Lim HK, Migaud M, Israel L, Chrabieh M, Audry M, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332(6025):65–8. doi: 10.1126/science.1200439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich NC. STAT dynamics. Cytokine Growth Factor Rev. 2007;18(5–6):511–8. doi: 10.1016/j.cytogfr.2007.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner ED, Rylaarsdam S, Anover-Sombke S, Rack AL, Reichenbach J, Carey JC, Zhu Q, Jansson AF, Barboza J, Schimke LF, et al. Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J Allergy Clin Immunol. 2008;122(1):181–7. doi: 10.1016/j.jaci.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, Kuestner R, Garrigues U, Birks C, Roraback J, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4(1):63–8. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Ogasawara K, Takaoka A, Tanaka N. IRF family of transcription factors as regulators of host defense. Annu Rev Immunol. 2001;19:623–55. doi: 10.1146/annurev.immunol.19.1.623. [DOI] [PubMed] [Google Scholar]
- Ungureanu D, Silvennoinen O. SLIM trims STATs: ubiquitin E3 ligases provide insights for specificity in the regulation of cytokine signaling. Sci STKE. 2005;2005(304):pe49. doi: 10.1126/stke.3042005pe49. [DOI] [PubMed] [Google Scholar]
- Vairo D, Tassone L, Tabellini G, Tamassia N, Gasperini S, Bazzoni F, Plebani A, Porta F, Notarangelo LD, Parolini S, et al. Severe impairment of IFN-gamma and IFN-alpha responses in cells of a patient with a novel STAT1 splicing mutation. Blood. 2011;118(7):1806–17. doi: 10.1182/blood-2011-01-330571. [DOI] [PubMed] [Google Scholar]
- van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, Arts P, Rosentul DC, Carmichael AJ, Smits-van der Graaf CA, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. The New England journal of medicine. 2011;365(1):54–61. doi: 10.1056/NEJMoa1100102. [DOI] [PubMed] [Google Scholar]
- Vogt G, Bustamante J, Chapgier A, Feinberg J, Boisson Dupuis S, Picard C, Mahlaoui N, Gineau L, Alcais A, Lamaze C, et al. Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J Exp Med. 2008;205(8):1729–37. doi: 10.1084/jem.20071987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt G, Chapgier A, Yang K, Chuzhanova N, Feinberg J, Fieschi C, Boisson-Dupuis S, Alcais A, Filipe-Santos O, Bustamante J, et al. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat Genet. 2005;37(7):692–700. doi: 10.1038/ng1581. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Miyazaki Y. Regulation of immune responses by interleukin-27. Immunol Rev. 2008;226:234–47. doi: 10.1111/j.1600-065X.2008.00710.x. [DOI] [PubMed] [Google Scholar]
- Zhang SY, Boisson-Dupuis S, Chapgier A, Yang K, Bustamante J, Puel A, Picard C, Abel L, Jouanguy E, Casanova JL. Inborn errors of interferon (IFN)-mediated immunity in humans: insights into the respective roles of IFN-alpha/beta, IFN-gamma, and IFN-lambda in host defense. Immunol Rev. 2008;226:29–40. doi: 10.1111/j.1600-065X.2008.00698.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.