

Abstract
Background
While the genomes of hundreds of organisms have been sequenced and good approaches exist for finding protein encoding genes, an important remaining challenge is predicting the functions of the large fraction of genes for which there is no annotation. Large gene expression datasets from microarray experiments already exist and many of these can be used to help assign potential functions to these genes. We have applied Support Vector Machines (SVM), a sigmoid fitting function and a stratified cross‐validation approach to analyze a large microarray experiment dataset from Drosophila melanogaster in order to predict possible functions for previously un‐annotated genes. A total of approximately 5043 different genes, or about one‐third of the predicted genes in the D. melanogaster genome, are represented in the dataset and 1854 (or 37%) of these genes are un‐annotated.
Results
39 Gene Ontology Biological Process (GO‐BP) categories were found with precision value equal or larger than 0.75, when recall was fixed at the 0.4 level. For two of those categories, we have provided additional support for assigning given genes to the category by showing that the majority of transcripts for the genes belonging in a given category have a similar localization pattern during embryogenesis. Additionally, by assessing the predictions using a confidence score, we have been able to provide a putative GO‐BP term for 1422 previously un‐annotated genes or about 77% of the un‐annotated genes represented on the microarray and about 19% of all of the un‐annotated genes in the D. melanogaster genome.
Conclusions
Our study successfully employs a number of SVM classifiers, accompanied by detailed calibration and validation techniques, to generate a number of predictions for new annotations for D. melanogaster genes. The applied probabilistic analysis to SVM output improves the interpretability of the prediction results and the objectivity of the validation procedure.
Keywords: Gene ontology, Support Vector Machines, Drosophila melanogaster, Gene expression data, Gene function prediction
Background
While the genomes of hundreds of organisms have been sequenced and good approaches exist for finding protein encoding genes, an important remaining challenge is predicting the functions of the large fraction of genes for which there is no annotation. For example, for Drosophila melanogaster, approximately 28% of the 14,029 predicted genes have no Gene Ontology (GO) term (either Molecular Function, Biological Process and/or Cellular Component) associated with them (including both curated and electronic annotations) and only 58% have a GO‐BP (Biological Process) term [1]. If one excludes genes that only have an electronic annotation, then only 41% have a GO‐BP term [1]. Functional genomic data and in particular microarray mRNA expression data have been used by numerous researchers as a means to help predict the function of un‐annotated genes. The analysis of such data is based on the premise that genes involved in a particular biological, molecular, and/or biochemical process are often co‐expressed. This co‐expression is dependent on the presence of common cis‐regulatory elements of the co‐regulated genes that bind one or more common transcription factors. A common approach to examine co‐expression profiles from microarray experiment data is to use clustering analysis. In this type of analysis, genes are organized and grouped based on their expression profile, with genes having similar expression pattern being “clustered” (grouped) together. The results of clustering analysis depend (a) on the metric used for comparing the expression profiles of genes that are analyzed and (b) on the clustering algorithm used. Euclidean distance and Pearson’s correlation coefficient are two simple commonly used metrics [2]. Popular clustering algorithms are hierarchical clustering, k‐means clustering and Self Organizing Maps [3].
Although clustering provides an insightful way of exploring gene co‐expression patterns, it does not directly convey any information regarding potential functions of un‐annotated genes. For that purpose, researchers have applied a further layer of analysis to the results of clustering, making use of additional data containing annotations of genes being analyzed. According to this type of analysis, clusters that are “enriched” for a particular gene function (i.e. they contain a larger than by chance number of genes having that function) are “assigned” that function. Subsequently, and following the “guilt‐by‐association” principle, all un‐annotated genes in that cluster are predicted to have the same function [4]‐[7].
In addition to the clustering analyses of genes in an unsupervised fashion and without annotation information, a number of supervised methods have been proposed for predicting gene functions. These methods make use of available information about the annotations of genes in order to discover gene expression patterns that characterize those annotations. Then, functional predictions for un‐annotated genes are made based on how well their expression profiles are matched with “annotated” patterns. Perhaps the most popular class of supervised methods are the binary classifiers, where first, expression data are separated into two classes either having or not having a particular annotation, and then un‐annotated genes are predicted of having or not having the annotation, based on which class their expression profile falls under. Examples of applications of these methods include Support Vector Machines (SVM) [8,9], random forest [10], neural networks [11], factor analysis [12], logistic regression, linear discriminant and quadratic discriminant analysis [13]. The application of the aforementioned approaches for gene function prediction benefits from the use of large data sets where many different experimental treatments or conditions make up the microarray expression dataset (e.g. developmental time points, mutations, specific tissues, environmental conditions, drug treatments, etc.). For the most part, the studies that have tried to do large‐scale gene prediction assignment have used well‐known model organisms where large microarray datasets were available. These include studies in Saccharomyces cerevisiae[7,14], Arabidiopsis thaliana[13], D. melanogaster[15], and Mus musculus[9]. Other approaches include assembling and integrating a large number of datasets from different experimental approaches (e.g. microarray expression, genetic interaction, and protein‐protein interaction) to create a network which in turn improves the robustness of the gene function predictions. These include studies in Saccharomyces cerevisiae[16], C. elegans[17], D. melanogaster[10,18] and Mus musculus[19].
In this study we focus on the use of SVM for the prediction of D. melanogaster gene functions. SVM is a popular machine learning method for classification and regression. Its proven high performance as well as its solid theoretical basis justify its frequent use in many fields, including bioinformatics and predictions of gene functions. As a two‐class classification tool, SVM attempts to separate the data points not in the original feature space but in an “enlarged” higher‐dimensional space instead. The seemingly highly computationally expensive data transformation is not performed but instead, ingeniously, the separation is performed “implicitly” based on their distances measured with the use of a kernel function. The SVM algorithm uses optimization techniques to find the surface that gives the optimal margin between the points of the two classes [20,21].
Despite its popularity in bioinformatics along with other research areas, SVM has been used for the prediction of GO‐BP annotations for genes of various organisms (such as S. cerevisiae[8] and M. musculus[9]) but not for D. melanogaster. Our study fills this gap, investigating how well this very popular method works for D. melanogaster gene expression data, in particular when the dataset is of a specific structure imposed by the nature of time‐course experiments, as the one we use in our study.
Using microarray data from the life cycle of D. melanogaster[22], and a controlled vocabulary for annotation of biological processes associated with D. melanogaster genes from the Gene Ontology Consortium (GO‐BP) [23], in this study we propose a method of predicting gene function of un‐annotated genes in the D. melanogaster genome by using Support Vector Machines and a two‐level data splitting rotation scheme for validation (double cross‐validation). Our prediction method was evaluated also externally with the use of an independent dataset.
Using this approach we have been able to provide a putative GO‐BP term for about 77% of the un‐annotated genes represented in the dataset and about 19% of all of the un‐annotated genes in the D. melanogaster genome helping to bridge the gap for the large number of genes that have little or no annotation. In addition, this SVM approach provides a precision and probability estimate that can help guide users as to the likelihood a given gene belongs to GO annotation class.
Methods
Microarray data and annotation sources
The microarray data used in this study was obtained from the series of 138 cDNA microarrays spanning the life cycle of Drosophila melanogaster[22]. The microarrays contained probes from 6765 cDNA clones and represented 5043 genes, roughly one third of the total number of genes in the D. melanogaster genome. From those genes, 1854 were not annotated with any GO‐BP term at the time of the analysis. Data were obtained from the Stanford Microarray Database [24] and normalized using a ratio‐based method according to the original publication. The dataset can also be obtained from the Gene Expression Omnibus [25], GEO accession number GSE4347. cDNA clone names were converted to primary Flybase Gene Identifiers (FBgn ids) from release 4.2 of the D. melanogaster genome using annotation available at Flybase [26]. Biological process annotation was downloaded from the Gene Ontology Consortium [23] in February 2006. Only GO‐BP categories containing a minimum of 10 and a maximum of 999 genes in the dataset were included, and 788 categories met these criteria. Clones with duplicate computer gene (CG) numbers were purposely not removed as we were interested in investigating the consistency of the predictions across the duplicates.
Support Vector Machines
A Support Vector Machine (SVM) is a classification and regression method originally developed by Vapnik [21]. Given a set of p‐dimensional vector data xi and their labels yi taking the values {-1,+1}, a linear Support Vector Machine finds the optimal hyperplane that separates the “positive” from the “negative” class. This plane is maximizing the margin between the two classes. According to the mathematical formulation of the problem, the solution refers to a weight vector w and a scalar b that satisfy the optimization problem
where N is the number of samples.
When classes are overlapping and misclassifications are allowed, the above constraints become
where ξi are slack variables and C a constant. Solutions can be generated by using the “Lagrange” formulation of the problem and its “Wolfe dual” problem. Given the solution, the weight vector w, for a new point x, the function f(x)=wTx+b is calculating the discriminant value for x, which can be used for its classification to the positive or negative class. It turns out that only a number of training points are important for the determination of the solution, the support vectors.
This mechanism can be applied to problems of non‐linear separation, after mapping the data onto a higher dimensional space, with the use of a mapping Φ. Φ is determined by a kernelK(.,.) such that K(x,y)=<Φ(x),Φ(y)>. The optimization problem is similar with the linear separation case, and following the Lagrange formulation and given a solution of Lagrange multipliers αi, the discriminant function is given by , where xi,i=1,…,Ns are the support vectors. It is important to note here that the solution and discriminant function depend on the data only through the kernel, and also that the explicit expression of the mapping Φ is not needed for the solutions. This is one of the reasons that make SVM computationally attractive. More information on SVM can be found in [27].
It is evident that SVM depends on the choice of the kernel. After performing a small set of experiments with pilot runs evaluating a number of kernel choices, we decided to use a radial basis kernel,
where , over a linear or polynomial kernel. Our decision was also supported by the large popularity of this kernel (previously used, for instance, in [8,9]). We used as σ a heuristic value equal to the median value of the distances between positive training points and their nearest negative training points. To combat asymmetric classes, a suggestion from [28] is implemented, where K(x,x) is augmented by λ·|C|/N where |C| is the size of the class C that x belongs to, and λ a tuning parameter. For our calculations we use λ=m/2, where m is the median value of the diagonal of the kernel matrix. For the implementation of the SVM algorithm we use the publicly available Gist package [29].
Estimation of class membership probabilities
For the translation of the discriminant values f(x) to posterior probabilities of class membership, p(y=1|x), we use the method proposed in [30]. The author proposes a sigmoid function model, where
with parameters a,b. The estimation of the parameters a,b is done by solving a maximum likelihood problem
where
and
where N+,N- are the sizes of the positive and negative class respectively.
For the sigmoid model we use the model fitting algorithm proposed by [31], which is implemented in the Gist package.
Cross‐validation and performance evaluation
For each selected GO‐BP category a set of annotated genes consisting from the positively annotated and a subset of the negatively annotated of size equal to up to four times the size of the positively annotated is participating in the evaluation of the SVM prediction algorithm. If I is the index set of all these genes participating in the cross‐validation, a partition {I1,I2,I3,I4} is generated, where each Ij has equal size and equal number of positively labelled genes (to the degree that this is achievable). Two of those sets are used as “training set” for the SVM, one set is used for calibration and sigmoid fitting, and the last set is used as “test” set, where probability estimate values are output from SVM and compared with the known annotations of the genes for the evaluation of the method. For each permutation (α,β,γ,δ) of {1,2,3,4}, and if gene gi belongs to set Iα, we denote with pβ|γ,δ(i) the probability estimate for gene gi from an SVM classifier that was trained with training set Iγ∪Iδ and calibrated with the set Iβ. Keeping the test set fixed we can have three different ways of constructing the training and calibration sets, and therefore each gene of the test set has three different probability estimate values. For example, for gene gi in I1, we have p2|3,4(i),p4|2,3(i) and p3|4,2(i). Those three values are averaged to produce a unique probability estimate for every gene. For each test set (or fold) the probability estimates and true annotations of the genes are used for the calculation of performance measures such as precision at 20, 30 and 40. The values of those measures are then averaged to produced one final measure for the whole SVM procedure. Note here the distinction between the probability estimate generation for each gene that involves averaging over the three possible arrangements of training and calibration sets, and the calculation of the performance measure that is done independently for each one of the four folds (test sets), using each time only the genes of the test set.
For the calculation of precision and recall and the generation of the precision‐recall plots the R package ROCR [32] was used. For the precision‐recall plots, the curves corresponding to the different folds were averaged vertically (i.e. precision values were averaged for the same recall value).
Results
Gene function prediction using Support Vector Machines
The prediction of gene functions was performed with the use of a number of SVM classifiers. Annotation predictions were made independently for each of 788 GO‐BP categories selected based on their size (see Microarray data and annotation sources in Methods section), based on the results of an SVM classifier. Each SVM was trained with the use of a training set of data points of known labels (in this case, genes with Gene Ontology Biological Processes (GO‐BP) annotation). To remedy any model fitting problem caused by the high imbalance between the positive and negative classes, for every GO‐BP category and SVM classifier we limited the negative examples by randomly selecting a subset of them with a ratio of positive to negative examples being 1:4. The trained system was used for the classification of new, unlabelled data (i.e. prediction of functions for un‐annotated genes). The classification was based on the discriminant value of the data point, which is output by the SVM. As the discriminant value measures the “distance” of the data point from the hyperplane that separates the two classes, traditionally points with discriminant value larger than some threshold (typically zero) are classified as positives and the rest as negatives. Here, we refrain from translating the discriminant values to binary classifications, but, instead, we used a previously published algorithm to estimate the posterior probability of a gene membership to each GO‐BP category. The algorithm fits a sigmoid function to the discriminant values of labeled data and it uses this function to approximate posterior class probabilities for unlabelled data [31]. A flowchart describing the probability estimation procedure using SVM and the sigmoid fitting function is shown in Figure 1.
Figure 1.
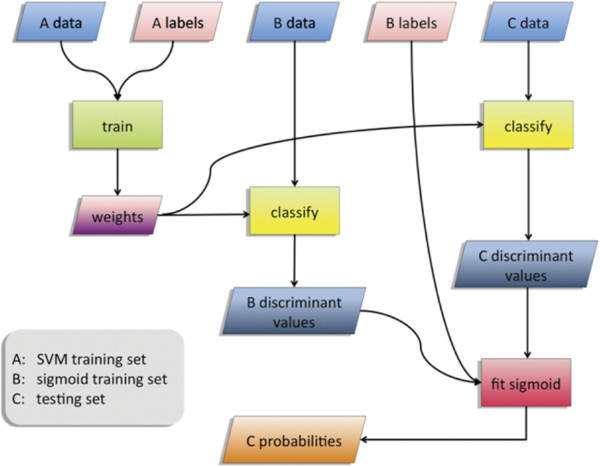
Flowchart Describing the Probability Estimation Procedure using SVM and the Sigmoid Fitting Function. First, SVM is trained using dataset A (SVM training set). Then, classification predictions (in the form of discriminant values) for dataset B (sigmoid training or tuning set) are generated. Those predictions along with the known labels of B are used for the fitting of the sigmoid function. Finally, classification results for dataset C (test set) are mapped to estimated class membership probabilities using the fitted sigmoid.
We validated the performance of SVM and sigmoid fitting function with the use of stratified cross‐validation, where the initial set of known data points (positively and selected negatively annotated genes) was randomly partitioned into four equal size sets, with equal number of positively annotated genes [33]. For every possible set combination, two fourths were used as “SVM‐training” set (used to train the SVM classifier), one fourth as a “sigmoid‐training” set (used for fitting the sigmoid function to the discriminant values) and one fourth as an overall “test” set, for which the membership probabilities were estimated. Predictions results for the latter set can then be validated, since the true labels (gene functions) are known. For more details in the cross‐validation procedure used see Methods section.
For any chosen probability threshold value pt we can calculate the corresponding precision value, defined as the ratio of the number of genes that have average probability estimate equal or larger than pt and belong to the class, divided by the number of genes that have a probability estimate equal or larger than pt (i.e. True Positives / Predicted Positives). Similarly, recall is defined as the proportion of the genes annotated with the term that also have average probability estimate equal or larger than the threshold (i.e. True Positives / Total Positives). While precision gives a measure of the accuracy of the prediction, recall measures the coverage or completeness. The prediction performance of SVM for a specific GO‐BP category was evaluated with the use of a measure equal to the precision value corresponding to a specific recall value (e.g. 20%, 30%, 40%). Precision at 20, 30 and 40 were calculated for each fold and averaged over all four folds, and the number of categories with high performance measure values were calculated. More specifically, the method identified 39 high‐precision categories reaching precision at 40 values equal or larger than 0.75. Table 1 presents these categories. Some of these categories are related through ancestor‐predecessor relationships and therefore have a certain amount of redundancy. (The relationship between the categories is shown in a GO graph presented in Additional file 1: Figure S1.) Taking into account these relationships as well as the size of the categories, we came up with a subset of 24 categories of minimum redundancy.
Table 1.
Selected GO‐BP categories with high precision values
| Category names | Size | Max‐prec‐40 |
|---|---|---|
| Detection of external stimulus GO 0009581 |
36 |
1.000 |
|
Detection of light stimulus GO 0009583 |
28 |
1.000 |
| Rhodopsin mediated phototransduction GO 0009586 |
14 |
1.000 |
| Detection of stimulus GO 0051606 |
38 |
0.964 |
|
Chitin metabolism GO 0006030 |
25 |
0.958 |
|
Regulation of mitosis GO 0007088 |
21 |
0.938 |
| Phototransduction GO 0007602 |
25 |
0.938 |
|
Fatty acid oxidation GO 0019395 |
20 |
0.929 |
|
Oxidative phosphorylation GO 0006119 |
68 |
0.919 |
| ATP synthesis coupled electron transport sensu Eukaryota GO 0042775 |
22 |
0.917 |
|
Cell matrix adhesion GO 0007160 |
25 |
0.909 |
| ATP synthesis coupled electron transport GO 0042773 |
22 |
0.906 |
|
Segment specification GO 0007379 |
21 |
0.900 |
|
Interphase of mitotic cell cycle GO 0051329 |
16 |
0.900 |
| Cell substrate adhesion GO 0031589 |
25 |
0.875 |
|
Homophilic cell adhesion GO 0007156 |
16 |
0.854 |
|
Regulation of exocytosis GO 0017157 |
20 |
0.850 |
| Aerobic respiration GO 0009060 |
31 |
0.845 |
| Response to light stimulus GO 0009416 |
33 |
0.839 |
|
Chromosome condensation GO 0030261 |
27 |
0.833 |
|
Sphingolipid metabolism GO 0006665 |
12 |
0.833 |
|
Neuromuscular junction development GO 0007528 |
19 |
0.833 |
|
Chromatin assembly GO 0031497 |
20 |
0.833 |
|
Nuclear import GO 0051170 |
33 |
0.817 |
| Glucosamine metabolism GO 0006041 |
27 |
0.813 |
| N acetylglucosamine metabolism GO 0006044 |
27 |
0.813 |
| SRP dependent cotranslational protein targeting to membrane GO 0006614 |
12 |
0.813 |
|
RNA transport GO 0050658 |
14 |
0.800 |
|
Membrane lipid biosynthesis GO 0046467 |
20 |
0.792 |
| DNA amplification GO 0006277 |
19 |
0.788 |
|
Chorion gene amplification GO 0007307 |
18 |
0.783 |
|
DNA dependent DNA replication GO 0006261 |
53 |
0.776 |
| response to radiation GO 0009314 |
37 |
0.774 |
|
Ribosome biogenesis GO 0007046 |
18 |
0.771 |
|
Regulation of DNA metabolism GO 0051052 |
13 |
0.771 |
|
Ventral cord development GO 0007419 |
14 |
0.767 |
| Protein targeting to membrane GO 0006612 |
12 |
0.750 |
|
Neuron recognition GO 0008038 |
12 |
0.750 |
| Protein targeting to ER GO 0045047 | 12 | 0.750 |
Each one of these 39 categories has a fold‐average precision‐at‐40 (i.e. that corresponds to recall value = 0.4) equal or larger than 0.75. The number of genes in the dataset annotated with a category is also shown. Bold fonts are used for the 24 GO‐BP categories that show minimal redundancy with genes found in other categories.
Two of the high‐precision GO‐BP categories, DNA‐dependent DNA replication and oxidative phosphorylation, were chosen for further analysis. Precision‐recall plots were generated for both categories and presented in Figures 2A and 2B.
Figure 2.
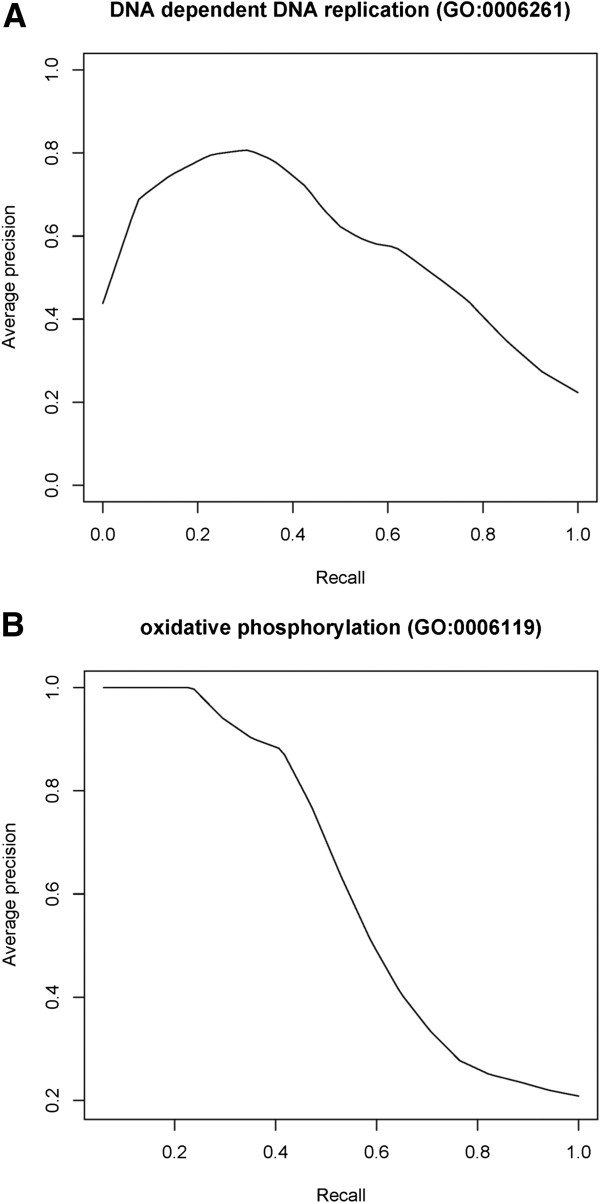
Precision‐recall Plots for Two of the GO‐ Biological Process Categories. Plots show the fold‐average precision that corresponds to a recall value for (A) DNA‐dependent DNA replication, and (B) oxidative phosphorylation GO categories. Vertical averaging method was used.
Developmental gene expression profiles for the genes in these two categories are shown in Figure 3. For all of the expression profiles, the gene expression ratio for a given gene at a given time point is calculated by dividing the intensity signal for that gene at that time point by the average intensity for that gene throughout development. For the genes belonging to the DNA‐dependent DNA replication category, there is a strong up‐regulation of these transcripts from the earliest time points in development (0.5‐1.5h embryos) through approximately mid‐embryogenesis (11‐12h embryos) (Figure 3A). By way of comparison, the gene expression profiles of an equivalent number of genes selected at random are shown in the lower half of the figure and in general, the pattern seen for the DNA‐dependent DNA replication genes is not observed for other genes. The genes belonging to the oxidative phosphorylation category have a very different transcriptional profile. They are generally expressed at low levels during early embryogenesis and begin to show increased expression beginning in late embryogenesis and throughout the three larval stages. At late third instar (L 96h and 105h), prepupal and the majority of pupal stages (M00h‐M48h) these genes are down‐regulated and then are dramatically up‐regulated in late metamorphosis (M60h) and stay high until early adulthood (Figure 3B). It should be noted that during embryogenesis and metamorphosis the organisms are not feeding or moving but during the larval and adult stages they are. Interestingly, the oxidative phorphorylation genes are up‐regulated well in advance of the organisms moving or feeding, suggesting that the organism is preparing itself for the next stage of the development that will require the enzyme activities to carry out this process.
Figure 3.

Developmental Transcription Profiles for Two of the GO‐ Biological Process Categories. The gene expression profiles of two of the GO‐BP categories: (A) DNA‐dependent DNA replication, and (B) oxidative phosphorylation are shown. The top portion of each of the category figures contains the expression profiles throughout development for each gene in the category in the same order as they appear in either Table 2 or 3 respectively. The bottom portion of each category represents an equal number of genes randomly selected from the expression profiles in the entire data set. Red denotes genes having up‐regulated transcription at a given time point and green down‐regulated genes. The scale at the top of the figure indicates the degree of up‐ (in red) and down‐ (in green) regulation (in fold change).
Gene function predictions for un‐annotated genes
One approach of declaring an un‐annotated gene as predicted to be annotated with some GO‐BP term, is to ascertain if its probability estimate output from the SVM specific for this term was larger than some threshold value (e.g. 0.5) and then assign the un‐annotated gene with that term. However, drawbacks to this approach include the arbitrary choice of the threshold and the fact that it ignores the evaluation of the performance of the classifier, as measured by cross‐validation. In this approach, any gene that has probability estimate score larger than 0.5 (or any other pre‐specified threshold value) for two categories A and B, will be predicted to belong to both A and B, regardless how well SVM is performing for these categories. A third problem with this method is that it assumes that probability estimate values are comparable between different GO‐BP categories. Although the purpose of the sigmoid function fit method is to provide with comparable estimates, one cannot be certain that this is necessarily obtained.
We have chosen an approach that takes into account the predictive behaviour of SVM for a specific GO‐BP term, when a prediction is made for an un‐annotated gene, as well as it addresses the other issues mentioned above. For each gene‐GO category pair a “gene precision” score is assigned to be equal to the maximum precision that can be achieved using thresholds smaller or equal to the probability estimate of the gene (output from SVM). If the gene is to be included in the “positive” predictions from SVM (and therefore to be assigned the GO‐BP function), the threshold has to be such that it will give a precision value for the classifier not larger than the “gene precision” score for the gene‐GO category pair. This score measures how probable is for the gene to truly belong to GO‐BP, given its probability estimate output from SVM, and also given the precision value of SVM for the particular category. This gene precision score is calculated separately for each one of the four folds (since for each fold probability estimates are generated for all un‐annotated genes) and the four fold‐specific scores are averaged.
We can then declare as gene function predictions all the gene‐category pairs that have “gene precision” score equal or larger than some threshold value. More specifically, 9887 new predictions representing 1422 genes (CG numbers) were generated corresponding to a gene precision score larger than 0.75. A total of approximately 5043 different genes, or about one‐third of the predicted genes in the D. melanogaster genome, are represented in the dataset. 1854 (or 37%) of these genes are un‐annotated and therefore this method is providing gene function predictions for about 77% (1422/1854) of the un‐annotated genes represented on the microarray and about 19% of all of the un‐annotated genes in the D. melanogaster genome. A graphical representation of these results is shown in Figure 4. The complete list of the these predictions can be found in Additional file 2: Table S1.
Figure 4.
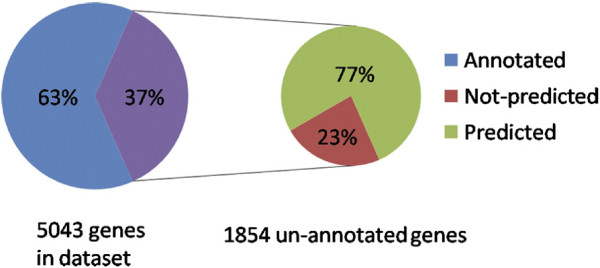
The Proportion of genes that are Annotated, Un‐annotated, and Predicted by the SVM to Belong to a GO‐BP Category. Genes from the developmental time course data set that had no GO‐BP annotation were broken down into genes that were predicted with high confidence to belong to a GO‐BP category (predicted), and those that had low prediction values (not predicted).
Experimental support of predicted gene functions
The un‐annotated genes contained in two of the high‐precision GO‐BP categories, DNA‐dependent DNA replication and oxidative phosphorylation are listed in Tables 2 and 3. For each gene, the gene precision score is indicated. The transcripts for genes involved in the same function are often co‐localized within cells. Each of the genes (i.e. CG numbers) on each list was searched in a D. melanogaster FISH database [34,35] to visually examine the spatial and temporal mRNA expression for that gene during early embryogenesis. Genes that are marked with a (+) or (-) had images present in the database. For the genes with images present in the FISH database, their spatial and temporal pattern were compared with those of known genes annotated with the category. Genes that had patterns that largely matched that of known genes in the category are marked with a (+). If either their temporal or their spatial pattern did not match the known gene pattern, they are marked with (-).
Table 2.
Gene Lists and Fluorescent In Situ Hybridization (FISH) analysis for DNA‐dependent DNA replication GO‐BP category
|
GeneID |
Sample |
FISH |
Gene |
Prob. |
GeneID |
Sample |
FISH |
Gene |
Prob. |
|---|---|---|---|---|---|---|---|---|---|
| match | prec. | score | match | prec. | score | ||||
| CG5924 |
LD38710 |
|
0.923 |
0.705 |
CG1109 |
LD26389 |
|
0.798 |
0.592 |
| CG1109 |
LD27350 |
|
0.923 |
0.736 |
CG8290 |
LD37351 |
+ |
0.798 |
0.575 |
| CG7663 |
LD46979 |
+ |
0.923 |
0.724 |
CG2910 |
GH11110 |
|
0.798 |
0.598 |
| CG7384 |
LD46023 |
|
0.923 |
0.76 |
CG10364 |
LD32040 |
- |
0.798 |
0.58 |
| CG14464 |
LD29015 |
|
0.923 |
0.707 |
CG5877 |
LD29352 |
|
0.79 |
0.572 |
| CG16892 |
LD26813 |
+ |
0.923 |
0.728 |
CG10625 |
LD39545 |
- |
0.79 |
0.568 |
| CG1578 |
LD28359 |
+ |
0.923 |
0.755 |
CG17509 |
GH12788 |
|
0.787 |
0.568 |
| CG16892 |
LD42637 |
|
0.923 |
0.739 |
CG11409 |
LD40802 |
|
0.787 |
0.607 |
| CG11122 |
LD29040 |
|
0.86 |
0.644 |
CG30007 |
LD29335 |
|
0.787 |
0.58 |
| CG9300 |
LD21924 |
|
0.86 |
0.666 |
CG17681 |
LD30009 |
|
0.787 |
0.573 |
| CG3287 |
SD03445 |
|
0.86 |
0.643 |
CG18622 |
LD26416 |
|
0.787 |
0.539 |
| CG1960 |
GH21591 |
|
0.86 |
0.66 |
CG31152 |
LD29477 |
+ |
0.787 |
0.545 |
| CG1024 |
LD28076 |
|
0.86 |
0.652 |
CG11990 |
LD47989 |
+ |
0.787 |
0.549 |
| CG13096 |
SD03546 |
|
0.86 |
0.664 |
CG6724 |
LD40657 |
|
0.783 |
0.589 |
| CG11596 |
LD45925 |
|
0.86 |
0.682 |
CG32069 |
LD47413 |
|
0.783 |
0.565 |
| CG4857 |
LD29423 |
+ |
0.86 |
0.638 |
CG2962 |
LD27487 |
|
0.783 |
0.578 |
| CG4949 |
LD46305 |
+ |
0.86 |
0.669 |
CG6049 |
LD27763 |
|
0.776 |
0.558 |
| CG11943 |
SD04935 |
|
0.86 |
0.703 |
CG2260 |
LD30339 |
|
0.772 |
0.563 |
| CG2469 |
LD30285 |
+ |
0.86 |
0.677 |
CG3735 |
LD35854 |
|
0.771 |
0.553 |
| CG11596 |
LD42227 |
+ |
0.839 |
0.66 |
CG7110 |
LD39933 |
- |
0.771 |
0.577 |
| CG12785 |
LD27528 |
|
0.839 |
0.616 |
CG12202 |
LD30511 |
+ |
0.771 |
0.584 |
| CG11329 |
LD26217 |
|
0.839 |
0.619 |
CG9591 |
LD26057 |
+ |
0.771 |
0.554 |
| CG6066 |
LD27582 |
|
0.839 |
0.621 |
CG12340 |
LD26050 |
|
0.771 |
0.552 |
| CG17050 |
LD35611 |
|
0.839 |
0.647 |
CG30020 |
LD40262 |
|
0.771 |
0.549 |
| CG18004 |
LD27741 |
|
0.839 |
0.662 |
CG12050 |
LD30416 |
+ |
0.771 |
0.561 |
| CG1647 |
LD30287 |
|
0.839 |
0.591 |
CG6151 |
LD28933 |
|
0.766 |
0.567 |
| CG31697 |
SD02518 |
|
0.839 |
0.618 |
CG14657 |
LD28447 |
|
0.766 |
0.556 |
| CG15736 |
LD33780 |
+ |
0.83 |
0.619 |
CG4203 |
LD29184 |
|
0.761 |
0.537 |
| CG2691 |
LD46946 |
|
0.829 |
0.61 |
CG4281 |
SD03946 |
|
0.76 |
0.548 |
| CG7728 |
LD39680 |
+ |
0.812 |
0.592 |
CG14005 |
LD30293 |
|
0.76 |
0.566 |
| CG31163 |
SD09611 |
+ |
0.812 |
0.598 |
CG9028 |
LD27194 |
+ |
0.76 |
0.555 |
| CG3680 |
LD27862 |
|
0.81 |
0.63 |
CG7824 |
LD26655 |
|
0.76 |
0.542 |
| CG3362 |
LD28544 |
|
0.81 |
0.595 |
CG7407 |
LD29166 |
- |
0.758 |
0.548 |
| NA ∗ |
LD42550 |
|
0.81 |
0.612 |
CG3338 |
LD27356 |
+ |
0.756 |
0.524 |
| CG11906 | LD27134 | 0.798 | 0.566 |
∗CG number not known
DNA‐dependent DNA replication GO‐BP category (along with oxidative phosphorylation) was selected for further biological validation using fluorescent in situ hybridization (FISH) analysis of early D. melanogaster embryos. In this table, genes that are predicted to belong to this category are shown along with information regarding their presence in the FISH database. Each of the genes (i.e. CG numbers) in the list was searched in a D. melanogaster FISH database to visually examine the spatial and temporal mRNA expression for that gene during early embryogenesis. Genes that are marked with (+) or (-) sign had images present in the database. Those that had patterns that largely matched that of known genes in the category are marked with a (+). If either their temporal or their spatial pattern did not match the known gene pattern, they are marked with (-). For each gene the “gene‐precision” score and the average probability estimate output from SVM are reported.
Table 3.
Gene Lists and Fluorescent In Situ Hybridization (FISH) analysis for oxidative phosphorylation GO category
|
GeneID |
Sample |
FISH |
Gene |
Prob. |
GeneID |
Sample |
FISH |
Gene |
Prob. |
|---|---|---|---|---|---|---|---|---|---|
| match | prec. | score | match | prec. | score | ||||
| CG12934 |
LP05346 |
|
1 |
0.814 |
CG5608 |
LD32461 |
|
0.898 |
0.537 |
| CG1715 |
LD33960 |
+ |
1 |
0.81 |
CG8401 |
GH01937 |
|
0.897 |
0.548 |
| CG33316 |
SD08735 |
|
0.975 |
0.656 |
CG6094 |
GH26345 |
|
0.897 |
0.553 |
| CG5523 |
GH14535 |
|
0.975 |
0.754 |
CG9813 |
GH04365 |
|
0.892 |
0.522 |
| CG10675 |
GH14673 |
|
0.975 |
0.79 |
CG13220 |
GH06079 |
+ |
0.885 |
0.538 |
| CG9921 |
GH07174 |
|
0.975 |
0.714 |
CG9056 |
GH11503 |
|
0.885 |
0.559 |
| CG8486 |
GH04578 |
|
0.975 |
0.594 |
CG4577 |
GH23863 |
|
0.875 |
0.535 |
| CG8086 |
GH25625 |
|
0.975 |
0.752 |
CG5325 |
GM14611 |
|
0.871 |
0.539 |
| CG10075 |
GH25609 |
+ |
0.975 |
0.679 |
CG7710 |
LP03578 |
|
0.871 |
0.544 |
| CG30116 |
GH04922 |
|
0.975 |
0.677 |
CG14125 |
GH07601 |
|
0.868 |
0.502 |
| CG5532 |
GH01442 |
+ |
0.975 |
0.662 |
CG1859 |
GH26443 |
|
0.868 |
0.52 |
| CG12239 |
GH14380 |
|
0.975 |
0.66 |
CG5325 |
GH03076 |
|
0.829 |
0.505 |
| CG8740 |
GH05582 |
+ |
0.975 |
0.658 |
CG1135 |
GH01794 |
|
0.829 |
0.489 |
| CG18616 |
GH04932 |
+ |
0.975 |
0.737 |
CG4757 |
SD01814 |
|
0.826 |
0.496 |
| CG3420 |
GH11502 |
+ |
0.975 |
0.682 |
CG5989 |
GH26459 |
+ |
0.826 |
0.481 |
| CG15669 |
GH02495 |
|
0.975 |
0.629 |
CG3153 |
GH04701 |
|
0.812 |
0.468 |
| CG11203 |
GH26638 |
|
0.975 |
0.691 |
CG1927 |
GH11112 |
|
0.812 |
0.462 |
| CG6044 |
GH12587 |
|
0.975 |
0.61 |
CG7217 |
LD45324 |
|
0.812 |
0.457 |
| CG5903 |
GH13386 |
- |
0.975 |
0.768 |
CG6123 |
GH13094 |
|
0.812 |
0.47 |
| CG14823 |
GH02020 |
|
0.975 |
0.656 |
CG2269 |
GH06015 |
- |
0.809 |
0.48 |
| CG13367 |
GH14959 |
|
0.975 |
0.7 |
CG4589 |
LP05955 |
|
0.807 |
0.489 |
| CG6424 |
GH08256 |
|
0.975 |
0.743 |
CG14438 |
GH25521 |
|
0.8 |
0.47 |
| CG7083 |
GH27162 |
|
0.947 |
0.68 |
CG8206 |
GH04557 |
|
0.794 |
0.437 |
| CG3631 |
LD29155 |
+ |
0.925 |
0.655 |
CG9336 |
GH22472 |
|
0.794 |
0.453 |
| CG4281 |
GH10944 |
+ |
0.925 |
0.604 |
CG7570 |
GH27163 |
|
0.794 |
0.459 |
| CG12706 |
GH14695 |
|
0.925 |
0.648 |
CG10973 |
LD28549 |
|
0.792 |
0.452 |
| CG10249 |
GH11802 |
|
0.925 |
0.56 |
CG7506 |
GH02466 |
|
0.788 |
0.467 |
| CG14292 |
GH14813 |
|
0.912 |
0.592 |
CG6455 |
GH04666 |
|
0.786 |
0.463 |
| CG4972 |
GH14975 |
|
0.912 |
0.592 |
CG17828 |
GH04984 |
+ |
0.778 |
0.432 |
| CG32795 |
HL08104 |
- |
0.912 |
0.532 |
CG15765 |
GH28601 |
|
0.778 |
0.447 |
| CG4975 |
GH18454 |
|
0.912 |
0.638 |
CG10585 |
GH23839 |
+ |
0.775 |
0.432 |
| CG6550 |
GH28477 |
+ |
0.912 |
0.609 |
CG10039 |
GH11404 |
|
0.775 |
0.436 |
| CG3971 |
GH11554 |
+ |
0.912 |
0.549 |
CG14817 |
GH01621 |
|
0.771 |
0.433 |
| CG6659 |
LD45943 |
+ |
0.912 |
0.578 |
CG17666 |
GH08313 |
|
0.762 |
0.42 |
| CG15386 |
GH19557 |
|
0.906 |
0.516 |
CG11737 |
GH22337 |
+ |
0.761 |
0.439 |
| CG1231 |
GH01151 |
|
0.906 |
0.582 |
CG7358 |
GH14795 |
|
0.759 |
0.436 |
| CG15067 |
GH14961 |
|
0.906 |
0.545 |
CG5773 |
GH07612 |
|
0.758 |
0.432 |
| CG6008 | GH05862 | 0.898 | 0.557 |
This table contains the genes that were predicted to belong to oxidative phospholylation GO‐BP category along with information regarding the presence in the FISH database. Notation is similar to Table 2.
One annotated gene from the dataset known to belong to the GO‐BP and one un‐annotated gene from each of the two GO‐BP categories listed in each of Tables 2 and 3 were chosen and FISH images for those genes was retrieved from the FlyFISH database. Four different stage categories of early embryogenesis are shown in Figure 5. Green fluorescence represents the mRNA localization pattern for that transcript/gene and red fluorescence is showing the position of nuclei within the organism at that stage. For the known gene belonging to the GO‐BP DNA‐dependent DNA replication, the annotation for gene CG1584 (Gene name = Orc6, i.e. Origin recognition complex subunit 6) has a molecular function described as DNA binding. It is involved in the following biological processes: DNA replication initiation; DNA‐dependent DNA replication; chromatin silencing. The un‐annotated gene, CG1578 (from Table 2), had a very similar in situ hybridization pattern (Figure 5A). For the oxidative phophorylation GO‐BP category, the known gene is CG1970, annotated as having NADH dehydrogenase (ubiquinone) activity. It is involved in the biological process mitochondrial electron transport. The un‐annotated gene, CG1715 (from Table 3), has an in situ hybridization pattern closely resembling the pattern seen for CG1970.
Figure 5.

Fluorescent In Situ Hybridization (FISH) Images of Annotated and Un‐annotated Gene mRNAs. One annotated gene and one un‐annotated gene from each of the two GO‐BP categories shown in Tables 2 and 3 were chosen and FISH images for those genes was retrieved from the FlyFISH database. Four different stage categories of early embryogenesis are shown. Green fluorescence represents the mRNA localization pattern for that transcript/gene and red fluorescence is showing the position of nuclei within the organism at that stage. CG1584 = Orc6, Origin recognition complex subunit 6. Its molecular function is described as DNA binding and it is involved in the biological processes: DNA replication initiation; DNA‐dependent DNA replication; chromatin silencing. CG1970‐ NADH dehydrogenase (ubiquinone) activity. It is involved in the biological process mitochondrial electron transport.
Of the un‐annotated genes that are predicted to belong to the DNA‐dependent DNA‐replication GO‐BP genes, 22 had images in the FlyFISH database and 18 out of 22 (82%) had FISH images that were the same or very similar to the known genes belonging to the category (Table 2). Similarly, of the un‐annotated genes that are predicted to belong to the oxidative phosphorylation GO‐BP category, 19 had images in the FlyFISH database and 16 out of 19 (84%) had FISH images that were the same or very similar to the known genes belonging to the category (Table 3).
Discussion
We have applied Support Vector Machines (SVM) and a stratified cross‐validation approach to analyze a large microarray experiment dataset from D. melanogaster in order to predict possible functions for previously un‐annotated genes. This approach successfully generated a preliminary GO Biological Process annotation for a large number of these genes.
The SVM analysis, employing a sigmoid fitting function, generated annotation probability values for all the genes in the dataset. Using a recall value of 40%, 39 high‐precision GO‐BP categories were identified and 77% of the un‐annotated genes in the dataset have been assigned to one or more of these GO‐BP categories. Assigning new annotations based on gene‐precision scores takes into account both the overall precision of the GO‐BP category, as well as the individual probability of the gene, outcome of SVM and the sigmoid function. In that way, gene predictions of high confidence can be considered even for GO‐BP categories that have an overall poor precision performance.
We chose two of the high‐precision GO‐BP categories to validate using an independent data set (the FlyFISH database). These two categories, DNA‐dependent DNA replication and oxidative phosphorylation, were selected for a number of reasons but chiefly because each contained a fairly large number of previously unannotated genes allowing for a larger number of genes that could be cross‐checked in the FlyFISH database and that the GO‐BP description was fairly precise allowing for the potential to examine whether the unannotated genes are actually involved in the biological process they had been assigned to. While it could be argued that the use of FlyFISH data is perhaps too similar of a data type to be useful (i.e. is also gene transcription data), our main goal was to use data that: was from a completely independent source; had at least some data that differed in type from the original microarray data (in this case spatial data in addition to temporal data); where the raw data could be inspected independently at the gene level so that any computer or user assigned functional classification of the genes would not bias the validation. Given the high degree of precision that some of the unannotated genes have for belonging to a specific GO‐BP category, especially for the oxidative phosphorylation category, we feel quite confident that a large number of these genes will be shown in the future to be directly or indirectly involved in these biological processes. Gene expression pattern data has been used in the past to uncover the function of a gene. In a study by Hughes and coworkers [14], a large compendium of yeast gene expression data was examined for co‐expression patterns using hierarchical clustering and among several findings, they discovered that a previously unannotated gene, YER044c (now ERG28), had a gene expression pattern that was highly similar to genes that were known to be involved in ergosterol biosynthesis. Further biochemical and genetic experiments supported that the gene was indeed involved in this process [14].
In the original Arbeitman and coworkers study [22], the authors performed different hierarchical clustering analyses to identify groups of genes that were temporally co‐regulated and/or restricted to specific biological sample type (e.g. mutant or sex). The authors also identified several GO classes that could be assigned to subsets of the co‐regulated genes. While many of the GO classes reported were fairly low (i.e. general) in the GO annotation hierarchy (e.g. enzyme, metabolism) others were more detailed. Five of the more detailed GO categories reported by Arbeitman and coworkers [22] overlap with the 39 GO biological categories identified in this study including: mitochondria/electron transport; ribosome; cell cycle; cell adhesion; and phototransduction.
We attempted to compare our results with those from the study of Yan and co‐workers [10], who also assigned GO terms to groups of similarly behaving genes to a dataset which in part included the data we used in our study. The authors of that study apply a different computational method (Random Forest) to a number of data sources (including gene expression data) for the prediction of GO‐BP annotations. For the comparison with our study, first, predictions based on expression data only have to be extracted and, subsequently, precision and recall measures need to be calculated using methods similar to those used in our study. There were a few issues that made the comparison between the two studies difficult. First, Yan and co‐workers do not apply stratification in their 10‐fold cross‐validation. As a result, for some of the GO‐BP categories (and the associated classifiers) there are a number of folds that contain only negative classes, and therefore classification (and calculation of precision) cannot be performed. Estimates of measures such as precision at 40 could be generated based on the remaining folds, but this practice is generally not recommended as it may introduce bias [33]. Second, the number of categories that are represented in both studies (273) is much smaller than those that are represented in each study individually. If we exclude those that give folds without positively labeled representatives, we are left out with a very small number of GO‐BP categories for comparison. Due to these issues a strict comparison of the results from the two studies is not particularly valuable. For reference purposes we mention here that in Yan et al. study, 8 GO‐BP categories gave a precision at 40 value equal or larger than 0.75 after excluding the GO terms with less than 5 folds representing both classes, while the number of selected categories dropped to 1 if we exclude those with less than 8 folds representing both classes. Also, the reported new predictions in Yan et al. involve 2062 genes at the confidence level of 0.2, 213 genes at the confidence level of 0.5 and 11 genes at the confidence level of 0.75. It should be noted, though, that the confidence level is Yan et al. study was computed differently from the gene precision score in our study and the two are not necessarily comparable.
We believe that the methods used in this study can be applied to other microarray and RNA‐seq datasets. Our own experience trying this approach on other data suggests that the robustness of the results is largely dependent on both the number of array experiments that are in the dataset as well as on the type of experiment(s) the data represent. In this case, a developmental time course with numerous time points provided the ability to identify a large number of co‐expressed genes.
Conclusions
In this study we have applied Support Vector Machines for the prediction of GO‐BP annotation terms for D. melanogaster genes, using published time‐course gene expression data. We have assessed the predictive ability of the method using an elaborate stratified double cross‐validation procedure, involving the fitting of a sigmoid function on the raw output from SVM. Precision and recall values for each GO‐BP function were calculated, describing in detail the performance of the prediction. On a second level, our results were validated externally using independent data from FlyFISH database. Finally, we applied SVM for the prediction of GO‐BP annotations of un‐annotated genes, providing certainty estimates for those predictions, based on a method that uses the results from the cross‐validation and the predictive performance of SVM for the specific GO‐BP category.
We believe that this is the first study that investigates the performance of SVM for GO‐BP prediction for D. melanogaster genes and provides a comprehensive list of new annotation predictions that can be further researched experimentally. We believe that our study is of great value to researchers interested in computational annotation of genes, as well as to the community of D. melanogaster researchers.
Abbreviations
GO‐BP: Gene Ontology Biological Processes; SVM: Support Vector Machine
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NM participated in the conception and design of the study, performed the analysis of the data, participated in the creation of the figures and tables and wrote part of the manuscript. ZR pre‐processed the data and participated in the creation of the figures and tables. ME participated in the conception and design of the study and revised the manuscript for critically important content. TW participated in the conception and design of the study, participated in the creation of figures and tables, wrote part of the manuscript and revised the manuscript for critically important content. All authors read and approved the final manuscript.
Supplementary Material
The Parent‐Child Relationship Between Ontology Biological Process Categories. Figure showing a joined Gene Ontology graph providing an overview of the relationship between the 39 Gene Ontology Biological Process (GO‐BP) categories that were identified from the output of the SVM as having a precision‐at‐40 value equal or larger than 0.75, as indicated in the highlighted polygonal nodes.
Selected GO‐BP Predictions for Previously Unannotated Genes. Table showing all new predictions with average gene‐precision score equal or larger than 0.75. Detailed information on gene‐precision scores and probability estimates for each one of the four folds is shown.
Contributor Information
Nicholas Mitsakakis, Email: n.mitsakakis@utoronto.ca.
Zak Razak, Email: z.razak.mail@gmail.com.
Michael Escobar, Email: m.escobar@utoronto.ca.
J Timothy Westwood, Email: t.westwood@utoronto.ca.
Acknowledgements
This work was supported by CIHR Emerging Teams Grant to M.E., J.T.W. and others. J.T.W. is also supported by an NSERC Discovery Grant. We would like to thank Sarah Gonsalves (Department of Cell and Systems Biology, University of Toronto) for helping prepare Figure 3, Eric Lècuyer and Henry Krause (Banting and Best Department of Medical Research, University of Toronto) for the use of the Fly‐FISH data found in Tables 2 and 3 and Figure 5, Quaid Morris (Banting and Best Department of Medical Research, University of Toronto) for scripts to parse data from Gene Ontology, Han Yan for making available the data used in the Yan et al. study [10] and William Noble (Departments of Genome Sciences and of Computer Science and Engineering, University of Washington) for personal communication about the SVM algorithm.
References
- Tweedie S, Ashburner M, Falls K, Leyland P, McQuilton P, Marygold S, Millburn G, Osumi‐Sutherland D, Schroeder A, Seal R, Zhang H. the Fly Base Consortium. FlyBase: enhancing Drosophila Gene Ontology annotations. Nucl Acids Res. 2009;37:D555–D559. doi: 10.1093/nar/gkn788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome‐wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani R, Hastie T, Eisen M, Ross D, Botstein D, Brown P. Clustering methods for the analysis of DNA microarray data. Tech. rep., Department of Statistics, Stanford University, Stanford, California; 1999.
- Marcotte EM, Pellegrini M, Thompson MJ, Yeates TO, Eisenberg D. A combined algorithm for genome‐wide prediction of protein function. Nature. 1999;402:83–86. doi: 10.1038/47048. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. Systematic determination of genetic network architecture. Nat Genet. 1999;22:281–285. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- Wu LF, Hughes TR, Davierwala AP, Robinson MD, Stoughton R, Altschuler S. Large‐scale prediction of Saccharomyces cerevisiae gene function using overlapping transcriptional clusters. Nat Genet. 2002;31:255–265. doi: 10.1038/ng906. [DOI] [PubMed] [Google Scholar]
- Brown MP, Grundy WN, Lin D, Cristianini N, Sugnet CW, Furey TS, M Ares J, Haussler D. Knowledge‐based analysis of microarray gene expression data by using support vector machines. Proc Natl Acad Sci USA. 2000;97:262–267. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Morris QD, Chang R, Shai O, Bakowski MA, Mitsakakis N, Mohammad N, Robinson MD, Zirngibl R, Somogyi E, Laurin N, Eftekharpour E, Sat E, Grigull J, Pan Q, Peng WT, Krogan N, Greenblatt J, Fehlings M, van der Kooy D, Aubin J, Bruneau BG, Rossant J, Blencowe BJ, Frey BJ, Hughes TR. The functional landscape of mouse gene expression. J Biol. 2004;3:21. doi: 10.1186/jbiol16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Venkatesan K, Beaver J, Klitgord N, Yildirim M, Hao T, Hill D, Cusick M, Perrimon N, Roth F, Vidal M. A genome‐wide gene function prediction resource for Drosophila melanogaster. PLoS ONE. 2010;5:e12139. doi: 10.1371/journal.pone.0012139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos A, Dopazo J, Jansen R, Tu Y, Gerstein M, Stolovitzky G. Systematic learning of gene functional classes from DNA array expression data by using multilayer perceptrons. Genome Res. 2002;12:1703–1715. doi: 10.1101/gr.192502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kustra R, Shioda R, Zhu M. A factor analysis model for functional genomics. BMC Bioinformatics. 2006;7:216. doi: 10.1186/1471-2105-7-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan H, Carson R, Provart NJ, Bonner AJ. Combining classifiers to predict gene function in Arabidopsis thaliana using large‐scale gene expression measurements. Bioinformatics. 2007;8:358. doi: 10.1186/1471-2105-8-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD, Kidd MJ, King AM, Meyer MR, Slade D, Lum PY, Stepaniants SB, Shoemaker DD, Gachotte D, Chakraburtty K, Simon J, Bard M, Friend SH. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. doi: 10.1016/S0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Zhang W, Zou S, Song J. Term‐tissue specific models for prediction of gene ontology biological processes using transcriptional profiles of aging in Drosophila melanogaster. BMC Bioinformatics. 2008;9:129. doi: 10.1186/1471-2105-9-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Li Z, Marcotte EM. An improved, bias‐reduced probabilistic functional gene network of baker’s yeast, Saccharomyces cerevisiae. PLoS ONE. 2007;2:e988. doi: 10.1371/journal.pone.0000988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Lehner B, Crombie C, Wong W, Fraser A, Marcotte E. A single gene network accurately predicts phenotypic effects of gene perturbation in Caenorhabditis elegans. Nat Genet. 2008;40:181–188. doi: 10.1038/ng.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello JC, Dalkilic MM, Beason SM, Gehlhausen JR, Patwardhan R, Middha S, Eads BD, Andrews JR. Gene networks in Drosophila melanogaster: integrating experimental data to predict gene function. Genome Biol. 2009;10:R97. doi: 10.1186/gb-2009-10-9-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena‐Castillo L, Tasan M, Myers CL, Lee H, Joshi T, Zhang C, Guan Y, Leone M, Pagnani A, Kim WK, Krumpelman C, Tian W, Obozinski G, Qi Y, Mostafavi S, Lin GN, Berriz GF, Gibbons FD, Lanckriet G, Qiu J, Grant C, Barutcuoglu Z, Hill DP, Warde‐Farley D, Grouios C, Ray D, Blake JA, Deng M, Jordan MI, Noble WS, Morris Q, Klein‐Seetharaman J, Bar‐Joseph Z, Chen T, Sun F, Troyanskaya OG, Marcotte EM, Xu D, Hughes TR, Roth F. A critical assessment of Mus musculus gene function prediction using integrated genomic evidence. Genome Biol. 2008;9:S2. doi: 10.1186/gb-2008-9-s1-s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble WS. What is a support vector machine? Nat Biotechnol. 2006;24:1565–1567. doi: 10.1038/nbt1206-1565. [DOI] [PubMed] [Google Scholar]
- Vapnik V. Statistical Learning Theory. New York: Wiley‐Interscience; 1998. [Google Scholar]
- Arbeitman MN, Furlong EE, Imam F, Johnson E, Null BH, Baker BS, Krasnow MA, Scott MP, Davis RW, White KP. Gene expression during the life cycle of Drosophila melanogaster. Science. 2002;297:2270–2275. doi: 10.1126/science.1072152. [DOI] [PubMed] [Google Scholar]
- The Gene Ontology Consortium. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [ http://www.geneontology.org] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford Microarray Database. [ http://smd.stanford.edu]
- Gene Expression Omnibus. [ http://www.ncbi.nlm.nih.gov/geo/]
- FlyBase: A Database of Drosophila Genes & Genomes. [ http://flybase.org]
- Burges CJC. A tutorial on support vector machines for pattern recognition. Data Min Knowl Dis. 1998;2:121–167. doi: 10.1023/A:1009715923555. [DOI] [Google Scholar]
- Veropoulos K, Campbell C, Cristianini N. Proceedings of the International Joint Conference on AI. San Francisco: Morgan Kaufmann Publishers; 1999. Controlling the sensitivity of support vector machines; pp. 55–60. [Google Scholar]
- Gist: Support vector machine and kernel principal components analysis, Version 2.0.9. [ http://www.bioinformatics.ubc.ca/gist]
- Platt J. In: Advances in Large Margin Classifiers. Smola A, Bartlett P, Schölkopf B, Schuurmans D, editor. Cambridge, MA: The MIT Press; 2000. Probabilistic outputs for support vector machines and comparison to regularized likelihood methods. [Google Scholar]
- Lin HT, Lin CJ, Weng RC. A note on Platt’s probabilistic outputs for support vector machines. Mach Learn. 2007;68:267–276. doi: 10.1007/s10994-007-5018-6. [DOI] [Google Scholar]
- Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21:3940–3941. doi: 10.1093/bioinformatics/bti623. [DOI] [PubMed] [Google Scholar]
- Forman G, Scholz M. Apples‐to‐apples in cross‐validation studies: pitfalls in classifier performance measurement. ACM SIGKDD Explorations Newsl. 2010;12:49–57. doi: 10.1145/1882471.1882479. [DOI] [Google Scholar]
- Fly‐FISH: A database of Drosophila embryo mRNA localization patterns. [ http://fly‐fish.ccbr.utoronto.ca]
- Lécuyer E, Yoshida H, Parthasarathy N, Alm C, Babak T, Cerovina T, Hughes TR, Tomancak P, Krause HM. Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell. 2007;131:174–187. doi: 10.1016/j.cell.2007.08.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The Parent‐Child Relationship Between Ontology Biological Process Categories. Figure showing a joined Gene Ontology graph providing an overview of the relationship between the 39 Gene Ontology Biological Process (GO‐BP) categories that were identified from the output of the SVM as having a precision‐at‐40 value equal or larger than 0.75, as indicated in the highlighted polygonal nodes.
Selected GO‐BP Predictions for Previously Unannotated Genes. Table showing all new predictions with average gene‐precision score equal or larger than 0.75. Detailed information on gene‐precision scores and probability estimates for each one of the four folds is shown.