

Abstract
The objectives of this study were to develop a novel solid dutasteride formulation with improved physicochemical properties and oral bioavailability, and to examine the correlation between its in vitro dissolution and in vivo pharmacokinetic parameters. Hydroxypropyl-β-cyclodextrin (HP-β-CD) nanostructures with or without hydrophilic additives were manufactured using the supercritical antisolvent process. The dutasteride-loaded HP-β-CD nanoparticles formed aggregates with a mean particle size of less than 160 nm and a specific surface area greater than 100 m2/g. Increases in the supersaturation and dissolution rate for dutasteride were dependent on the type of additive; increases in maximum solubility and extended supersaturation were observed in dutasteride-loaded HP-β-CD nanostructures with hydroxypropylmethyl cellulose, whereas the dissolution rate was the highest for nanostructures containing d-α-tocopheryl polyethylene glycol 1000 succinate. In rats, the oral bioavailability of dutasteride increased with the supersaturation induced by the HP-β-CD nanostructures. In addition, compared with the in vitro drug release rate, the in vivo pharmacokinetic parameters were more closely correlated with in vitro parameters related to supersaturation (solubility). Further, the bioavailability of the dutasteride-loaded HP-β-CD nanostructures with hydroxypropylmethyl cellulose was similar to that of the commercially available soft gelatin capsule (Avodart®). In conclusion, preparation of dutasteride-loaded HP-β-CD nanostructures using the supercritical antisolvent process affords a viable alternative solid dosage form for dutasteride.
Keywords: solubility, bioavailability, dutasteride, nanostructure, supercritical antisolvent
Introduction
Dutasteride, a 4-azasteroid, is a potent inhibitor of 5α-reductase types 1 and 2.1 Currently, this agent is used for the treatment of benign prostatic hyperplasia and hair loss.2 However, dutasteride is insoluble in water (<0.038 ng/mL),3 and in its commercial dosage form is formulated as a soft gelatin capsule containing monoglycerides and diglycerides of caprylic/capric acid as solubilizers. Although the oral bioavailability of the soft gelatin capsule is 60% (range 40%–94%) in humans, it must be swallowed whole and not chewed or opened because the contents may irritate the oropharyngeal mucosa.4 Further, the rate of drug release from the soft gelatin capsule is reduced by gelatin crosslinking.5 We have previously evaluated various formulation strategies, including a solid dispersion, a Eudragit E nanoparticle suspension, a self-microemulsifying drug delivery system, and the Soluplus® (BASF, Ludwigshafen, Germany)-coated colloidal silica nanomatrix system, for their ability to enhance the solubility and bioavailability of dutasteride.6–9 Among these formulations studied, the Eudragit E nanoparticle suspension afforded a dutasteride bioavailability 5.5-fold higher than that afforded by a 0.3% w/v Soluplus suspension (mean particle size 1.92 μm). However, use of the Eudragit E nanoparticle suspension was limited by its stability and difficulty in scaleup.10
Supersaturatable dosage forms improve the solubility and oral bioavailability of water-insoluble drugs, and are now being widely explored in the pharmaceutical industry.11–15 Amorphous solid dispersion and lipid-based formulations (eg, the supersaturatable self-microemulsifying drug delivery system) containing polymeric precipitation inhibitors are regarded as supersaturatable formulations. The supersaturated state is a thermodynamically unstable amorphous form and eventually reverts to a stable state because of drug precipitation. However, drug precipitation inhibitors help to maintain the supersaturated state by inhibiting nucleation and crystal growth by steric stabilization, surface stabilization, and/or specific interactions with the drug.16–18
Cyclodextrin and its derivatives have gained considerable attention in drug formulation because of their ability to increase solubility and stability by forming inclusion complexes,19 as well as by forming molecular aggregates and micelle-like structures.20,21 Studies have also reported enhanced supersaturation and inhibition of drug precipitation by cyclodextrin.22,23 Commercial supersaturatable formulations such as solid dispersions are manufactured using hot-melt extrusion, spray-drying, and supercritical fluid processes.24–26 Among the supercritical fluid processes, the supercritical antisolvent process may be used for manufacturing nanoparticles and nanostructures of a solid supersaturatable composition by using carbon dioxide as a green solvent.27–29
The objectives of this study were to develop a novel solid dutasteride formulation with improved physicochemical properties and higher bioavailability, and to establish a correlation between the in vitro dissolution and in vivo pharmacokinetic parameters for orally administered dutasteride. Hydroxypropyl-β-cyclodextrin (HP-β-CD) was used as a drug precipitation inhibitor to enhance the solubility and oral bioavailability of dutasteride. Dutasteride-loaded HP-β-CD nanostructures with or without hydrophilic additives were manufactured using the supercritical antisolvent process. In vitro and in vivo evaluations were performed.
Materials and methods
Dutasteride and finasteride were purchased from Dr Reddy’s Laboratories Ltd (Andhra Pradesh, India) and Sigma-Aldrich (St Louis, MO, USA), respectively. Hydroxypropyl-P-cyclodextrin (HP-β-CD, Cavitron® W7 HP5 Pharma, ISP Pharmaceuticals, Wayne, NJ, USA), hydroxypropyl cellulose (Nippon Soda Co, Ltd, Tokyo, Japan), hydroxypropylmethyl cellulose (HPMC 2910, Shin-Etsu Chemical Co, Ltd Tokyo, Japan), polyethylene glycol (PEG 6000, Sigma-Aldrich), polyvinylpyrrolidone (PVP K30, BASF Co, Ltd Germany), Poloxamer 407 and polyvinylpyrrolidone vinyl acetate (PVP-VA 64, BASF Co, Ltd), and d-α-tocopheryl polyethylene glycol 1000 succinate (Vitamin E TPGS, Eastman Chemical Company, Kingsport, TN, USA) were used as hydrophilic additives. Avodart® soft gelatin capsules (GlaxoSmithKline, Middlesex, UK) containing 0.5 mg dutasteride were purchased from a pharmacy. The acetonitrile and methanol used were high-performance liquid chromatography grade; all other chemicals were analytical grade.
Preparation of drug-loaded HP-β-CD nanostructures
Dutasteride-loaded HP-β-CD nanostructures with or without hydrophilic additives were prepared using SAS200 (Thar Technologies, Pittsburgh, PA, USA).30 First, dutasteride/excipient solutions (1.0%, w/v) were prepared by dissolving the dutasteride with excipients in ethanol or a 50/50 (v/v) mixture of ethanol and dichloromethane. Compositions of the dutasteride-loaded HP-β-CD nanostructures are shown in Table 1. The flow rates for the supercritical carbon dioxide and dutasteride/excipient solutions were 11 g per minute and 1.0 mL per minute, respectively. During formation of the particles, the operating pressure and temperature were fixed at 40°C and 15 mPa, respectively, based on preliminary experiments (data not shown). The precipitated dutasteride-loaded HP-β-CD nanostructures were obtained from the particle collection basket. For comparison, physical mixtures were also prepared by simply mixing the drug with the excipients in a glass vial.
Table 1.
Composition and physicochemical properties of dutasteride-loaded HP-β-CD nanostructures with or without hydrophilic additives prepared using the supercritical antisolvent process
| Formulation (weight ratio) | Drug content (%) | Mean particle size (nm) | Specific surface area (m2/g) |
|---|---|---|---|
| DT:HP-β-CD, 1:13.3 | 98.5 ± 1.1 | 140.5 ± 15.6 | 107.52 ± 1.59 |
| DT:HP-β-CD, 1:26.6 | 97.9 ± 1.9 | 155.3 ± 22.5 | 110.63 ± 1.28 |
| DT:HP-β-CD, 1:39.9 | 99.1 ± 0.9 | 150.9 ± 20.5 | 102.52 ± 1.09 |
| DT:HP-β-CD:HPC, 1:26.6:13.3 | 98.5 ± 1.6 | 201.6 ± 22.3 | 85.38 ± 1.10 |
| DT:HP-β-CD:HPMC 2910, 1:26.6:13.3 | 99.5 ± 1.3 | 209.8 ± 25.6 | 82.15 ± 0.89 |
| DT:HP-β-CD:PVP K30, 1:26.6:13.3 | 98.2 ± 1.1 | 189.5 ± 15.6 | 90.31 ± 1.01 |
| DT:HP-β-CD:PVP-VA 64, 1:26.6:13.3 | 98.5 ± 1.5 | 215.3 ± 26.7 | 87.21 ± 0.75 |
| DT:HP-β-CD:PEG 6000, 1:26.6:13.3 | 97.6 ± 2.0 | 225.8 ± 20.2 | 71.51 ± 0.68 |
| DT:HP-β-CD:Poloxamer 407, 1:26.6:13.3 | 99.6 ± 2.1 | 230.2 ± 35.2 | 60.71 ± 0.59 |
| DT:HP-β-CD:Ryotoester L1695, 1:26.6:13.3 | 98.6 ± 0.9 | 278.5 ± 50.5 | 66.61 ± 0.58 |
| DT:HP-β-CD:SLS, 1:26.6:13.3 | 99.2 ± 0.9 | 170.2 ± 15.9 | 96.51 ± 0.89 |
| DT:HP-β-CD:TPGS, 1:26.6:13.3 | 98.5 ± 1.1 | ND | 48.59 ± 0.47 |
Notes: Drug content (%) = weight of loaded drug/weight of feeding drug × 100. Data are expressed as the mean ± standard deviation (n = 3).
Abbreviations: DT, dutasteride; HP-β-CD, hydroxypropyl-β-cyclodextrin; HPC, hydroxypropyl cellulose; HPMC, hydroxypropylmethyl cellulose; ND, not determined; PEG, polyethylene glycol; PVP, polyvinylpyrrolidone; PVP-VA, polyvinylpyrrolidone-vinyl acetate; SAS, supercritical antisolvent; SLS, sodium lauryl sulfate; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate.
Characterization of drug-loaded HP-β-CD nanostructures
The morphology of the dutasteride-loaded HP-β-CD nanostructures was observed using scanning electron microscopy (JSM-7000F, JEOL Ltd, Tokyo, Japan). The size of the dutasteride-loaded HP-β-CD nanostructures was determined using a laser particle size analyzer (BI-9000, Brookhaven, Upton, NY, USA). The samples were dispersed in mineral oil and sonicated for 10 minutes. The specific surface area of the dutasteride-loaded HP-β-CD nanostructures was determined by nitrogen adsorption using a surface area analyzer (ASAP 2010, Micromeritics Instrument Corporation, Norcross, GA, USA). The crystal structure of the dutasteride within the HP-β-CD nanostructure was analyzed using a D8 Advance x-ray diffraction system (Bruker AXS GmbH, Karlsruhe, Germany). The scanning speed was 3°C per minute from 5°C to 50°C with a step size of 0.02°C. The drug content in the HP-β-CD nanostructure was determined using high-performance liquid chromatography (an LC 10ADvp pump with an SPD-10ADvp UV detector, Shimadzu Corporation, Kyoto, Japan). A sample of approximately 20 mg was dissolved in 100 mL of methanol. An analytical Luna C18(2) column (5 μm, 250 × 4.6 mm, Phenomenex, Torrance, CA, USA) was used and the mobile phase consisted of a mixture of acetonitrile and water (60/40, v/v). A flow rate of 1.0 mL per minute was used. The injection volume was 20 μL, and detection was performed at 210 nm.
Supersaturated dissolution tests were performed using a USP rotating paddle apparatus (USP apparatus II; VK 7000, Vankel, Cary, NC, USA). Different HP-β-CD nanostructures equivalent to 15 mg of dutasteride were added into a dissolution vessel containing 300 mL of simulated gastric fluids without pepsin at a pH of 1.2, maintained at 37°C ± 0.5°C, and stirred at 100 rpm. At different time intervals, 3 mL samples were drawn and filtered using a 0.11 μm nylon syringe filter. The filtrate was diluted with methanol, and the concentration of dutasteride was analyzed using high-performance liquid chromatography.
Dissolution tests, using two different under-sink conditions, were performed in simulated gastric fluids (pH 1.2) containing sodium lauryl sulfate 0.1% or 2%. The sodium lauryl sulfate concentration was determined by the phase solubility study. The dissolution medium was maintained at 37°C with continuous stirring at 50 rpm. Accurately weighed nanostructures containing the equivalent of 0.5 mg dutasteride and the commercial product were added to the dissolution vessel. Next, 3 mL samples were taken at different time intervals, filtered using a 0.11 μm nylon syringe filter, diluted using methanol, and analyzed using high-performance liquid chromatography. The phase solubility analysis was performed by adding excess amounts of dutasteride into a capped glass vial containing 5 mL of simulated gastric fluids (pH 1.2) with different concentrations of sodium lauryl sulfate. The samples were placed on a shaking water bath and agitated (60 rpm) at 37°C for 24 hours, which was previously determined to be an adequate time for equilibration. At the end of this period an aliquot of the sample was collected and filtered through a 0.11 μm nylon syringe filter. The filtrate was diluted with methanol, and the concentration of dutasteride was analyzed using high-performance liquid chromatography.
Pharmacokinetic study
The animal experiments were approved by the Committee on the Care and Use of Laboratory Animals at Inje University and performed according to the guidelines of the university. Male Sprague-Dawley rats (240–260 g) were purchased from Samtaco Bio Korea Inc (Osan-si, Korea) and randomly divided into six groups of four animals each. The rats were fasted for 24 hours prior to the study, and the jugular veins cannulated using a 23-gauge polyethylene cannula under zolazepam anesthesia (25 mg/kg, intramuscularly). The animals were allowed access to food four hours after dosing. Each group was given dutasteride-loaded HP-β-CD nanostructures or the commercial product by oral gavage at a dose of 2 mg/kg. The dutasteride-loaded HP-β-CD nanostructures or nanostructures modified with PVP-VA 64, TPGS, or HPMC were dispersed in 1 mL of water immediately prior to oral dosing; the selection of additives was based on the results of an in vitro supersaturation study. After administration of the liquid content of the commercial product (equivalent to 2 mg/kg), the animals immediately received 1 mL of water. Blood samples (approximately 350 μL) were collected into heparinized tubes at indicated time intervals. Plasma samples were obtained from blood by centrifugation at 12,000 rpm (16,582 × g) for five minutes. Dutasteride was extracted from the plasma matrix by protein precipitation, and drug concentrations in the plasma samples were analyzed using liquid chromatography with tandem mass spectrometry as previously reported.6 The pharmacokinetic parameters, ie, AUC0→24h (area under the concentration-time curve), Cmax (peak concentration), and Tmax (time to peak concentration), were calculated using noncompartmental analysis. Relative bioavailability was calculated using the following equation, with the commercial product used as a reference: relative bioavailability (%) = AUCtest/AUCreference × 100.
Statistical analysis
The data were analyzed by one-way analysis of variance followed by the least squares difference and Student-Newman-Keuls test using Statistical Package for Social Sciences version 19.0 software (IBM Corporation, Armonk, NY, USA). P < 0.05 was considered to be statistically significant.
Results and discussion
Scanning electron microscopic images showed that the dutasteride-loaded HP-β-CD nanostructures (Figure 1A–C) were composed of aggregated nanoparticles with a mean particle size of less than 160 nm, resulting in a very high specific surface area greater than 100 m2/g (Table 1). Hydrophilic additives decreased the mean particle size and increased the specific surface area of the nano structures (Table 1). In particular, Poloxamer 407 or TPGS caused high particle aggregation and fusion (Figure 1G and J), which might be attributed to the lower melting temperatures of the surfactant used. The melting temperatures of Poloxamer 407 and TPGS were approximately 55°C and 37°C, respectively. Similar results were observed in previous studies.15,28,31 Dutasteride was not degraded during the supercritical antisolvent process, and the drug content of all the prepared nanostructures was at least 98% (Table 1). X-ray diffraction patterns for the nanostructures did not show any characteristic diffraction peaks corresponding to the raw material (dutasteride), suggesting that dutasteride existed in an amorphous form within the HP-β-CD nanostructures (Figure 2).
Figure 1.

Scanning electron microscopic images of dutasteride-loaded HP-β-CD nanostructures with or without hydrophilic additives prepared using the supercritical antisolvent process. DT:HP-β-CD, 1:39.9 (A), DT:HP-β-CD:HPC (B), DT:HP-β-CD:HPMC 2910 (C), DT:HP-β-CD:PVP K30 (D), DT:HP-β-CD:PVP-VA 64 (E), DT:HP-β-CD:PEG 6000 (F), DT:HP-β-CD:Poloxamer 407 (G), DT:HP-β-CD:Ryotoester L1695 (H), DT:HP-β-CD:SLS (I), and DT:HP-β-CD:TPGS (J).
Abbreviations: DT, dutasteride; HP-β-CD, hydroxypropyl-β-cyclodextrin; HPC, hydroxypropyl cellulose; HPMC, hydroxypropylmethyl cellulose; PEG, polyethylene glycol; PVP, polyvinylpyrrolidone; PVP-VA, polyvinylpyrrolidone-vinyl acetate; SLS, sodium lauryl sulfate; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate.
Figure 2.

X-ray diffraction patterns of raw material and dutasteride-loaded HP-β-CD nanostructures with or without hydrophilic additives prepared using the supercritical antisolvent process.
Abbreviations: DT, dutasteride; HP-β-CD, hydroxypropyl-β-cyclodextrin; HPC, hydroxypropyl cellulose; HPMC, hydroxypropylmethyl cellulose; PEG, polyethylene glycol; PVP, polyvinylpyrrolidone; PVP-VA, polyvinylpyrrolidone-vinyl acetate; SLS, sodium lauryl sulfate; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate.
As shown in Figure 3 and Table 2, the dutasteride-loaded HP-β-CD nano structures at a ratio of 1:13.3 showed a maximum solubility of 7.77 μg/mL and rapid subsequent drug precipitation. The solubility of dutasteride was maintained above 20 μg/mL for 24 hours in dutasteride-loaded HP-β-CD nanostructures at ratios of 1:26.6 and 1:39.9; maximum solubility was not different at these ratios (P > 0.05). In fact, as an inhibitor of drug precipitation, the HP-β-CD nanostructure provided a high degree of supersaturation and extended supersaturation of dutasteride, suggesting that mechanisms other than simple complex formation were responsible for the stabilization effects, such as inhibition of nucleation and/or crystal growth from a highly supersaturated state. In general, drug precipitation inhibitors inhibit nucleation and/or crystal growth by steric stabilization, surface stabilization by adsorption onto an active surface, and/or specific interactions with a drug (eg, by hydrogen bonding or hydrophobic interactions).32,33 Cyclodextrin can solubilize and stabilize drugs by forming an inclusion complex with a drug and/or by forming molecular aggregates and micelle-like structures.34,35 Recently, Makhlof et al reported that cyclodextrin could act as a surface stabilizer of nanocrystals by forming a cyclodextrin network via intermolecular interaction of cyclodextrin molecules.36
Figure 3.

Supersaturated dissolution of a physical mixture and dutasteride-loaded HP-β-CD nanostructures with or without hydrophilic additives prepared using the supercritical antisolvent process.
Note: Data are expressed as the mean ± standard deviation (n = 3).
Abbreviations: DT, dutasteride; HP-β-CD, hydroxypropyl-β-cyclodextrin; HPC, hydroxypropyl cellulose; HPMC, hydroxypropylmethyl cellulose; PEG, polyethylene glycol; PVP, polyvinylpyrrolidone; PVP-VA, polyvinylpyrrolidone-vinyl acetate; SLS, sodium lauryl sulfate; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate.
Table 2.
Supersaturated dissolution data for dutasteride-loaded HP-β-CD nanostructures with or without hydrophilic additives prepared using the supercritical antisolvent process
| Formulation (weight ratio) | HP-β-CD nanostructure
|
Physical mixture
|
|||
|---|---|---|---|---|---|
| Maximum solubility (μg/mL) | Degree of supersaturationa | Relative AUC (%)b | Relative Cmax (%)c | Equilibrium solubility (μg/mL) | |
| DT:HP-β-CD, 1:13.3 | 7.77 ± 1.23 | 38.85 | 3.5 | 21.6 | 0.21 ± 0.01 |
| DT:HP-β-CD, 1:26.6 | 31.25 ± 3.21 | 89.29 | 84.0 | 86.8 | 0.35 ± 0.03 |
| DT:HP-β-CD, 1:39.9 | 35.99 ± 4.52 | 76.57 | 100.0 | 100.0 | 0.47 ± 0.03 |
| DT:HP-β-CD:HPC, 1:26.6:13.3 | 13.70 ± 2.30 | 32.61 | 16.4 | 38.1 | 0.42 ± 0.01 |
| DT:HP-β-CD:HPMC 2910, 1:26.6:13.3 | 47.10 ± 3.05 | 104.88 | 140.2 | 130.9 | 0.45 ± 0.01 |
| DT:HP-β-CD:PVP K30, 1:26.6:13.3 | 10.59 ± 3.52 | 25.83 | 6.7 | 29.4 | 0.41 ± 0.02 |
| DT:HP-β-CD:PVP-VA 64, 1:26.6:13.3 | 16.16 ± 3.41 | 34.39 | 12.2 | 44.9 | 0.47 ± 0.03 |
| DT:HP-β-CD:PEG 6000, 1:26.6:13.3 | 6.89 ± 1.23 | 17.7 | 4.3 | 19.1 | 0.39 ± 0.01 |
| DT:HP-β-CD:Poloxamer 407, 1:26.6:13.3 | 15.30 ± 3.16 | 7.97 | 44.6 | 42.5 | 1.92 ± 0.23 |
| DT:HP-β-CD:Ryotoester L1695, 1:26.6:13.3 | 8.94 ± 3.12 | 10.34 | 14.6 | 24.8 | 0.86 ± 0.11 |
| DT:HP-β-CD:SLS, 1:26.6:13.3 | 13.52 ± 2.58 | 6.44 | 42.7 | 37.6 | 2.11 ± 0.31 |
| DT:HP-β-CD:TPGS, 1:26.6:13.3 | 30.83 ± 3.58 | 19.51 | 61.5 | 85.7 | 1.58 ± 0.19 |
Notes:
Degree of supersaturation is defined as the maximum solubility of dutasteride (HP-β-CD nanostructure) in the dissolution medium divided by the equilibrium solubility of dutasteride (physical mixture) within medium;
relative AUC (%) = AUC0→6h of sample/AUC0→6h of the HP-β-CD nanostructure (1:39.9) × 100. The AUC0→6h was calculated using noncompartmental analysis from supersaturated dissolution data
relative Cmax(%) = Cmax of sample/Cmax of HP-β-CD nanostructure (1:39.9) × 100. Data are expressed as the mean ± standard deviation (n = 3).
Abbreviations: AUC, area under the concentration-time curve; Cmax, peak concentration; DT, dutasteride; HP-β-CD, hydroxypropyl-β-cyclodextrin; HPC, hydroxypropyl cellulose; HPMC, hydroxypropylmethyl cellulose; PEG, polyethylene glycol; PVP, polyvinylpyrrolidone; PVP-VA, polyvinylpyrrolidone-vinyl acetate; SLS, sodium lauryl sulfate; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate.
Hydrophilic additives significantly influenced the maximum solubility and supersaturation of dutasteride. HPMC was the most effective hydrophilic additive examined, followed by TPGS, and PVP-VA 64. Of the hydrophilic additives used, only HPMC had a synergistic effect on the maximum solubility and extended supersaturation of the dutasteride-loaded HP-β-CD nanostructures. Conversely, compared with HP-β-CD alone, the other hydrophilic additives showed an antagonistic effect, resulting in a decreased degree of supersaturation and/or an increase in drug precipitation from the supersaturated state. In particular, rapid drug precipitation was observed in HP-β-CD nanostructures with PEG 6000, with a maximum solubility of 6.89 μg/mL. HP-β-CD nanostructures with TPGS also showed rapid drug precipitation compared with the HP-β-CD nanostructures alone at ratios of 1:26.6 and 1:39.9, but no difference in maximum solubility was observed (P > 0.05). Lastly, HP-β-CD nanostructures with sodium lauryl sulfate or Poloxamer 407 showed a decrease in degree of supersaturation, although it was maintained above 10 μg/mL for 24 hours. Therefore, the surfactants added, with the exception of TPGS, inhibited supersaturation of the HP-β-CD nanostructures. Conversely, the equilibrium solubility of the physical mixtures consisting of HP-β-CD and hydrophilic polymers was increased compared with that of the physical mixture with HP-β-CD and drug at a ratio of 1:26.6. Moreover, the hydrophilic additives, with the exception of TPGS and HPMC, inhibited supersaturation of the HP-β-CD nanostructures, but increased the equilibrium solubility of dutasteride. The effect of the hydrophilic additives on the degree of supersaturation might be attributed to specific and/or nonspecific interactions between dutasteride, HP-β-CD, and the hydrophilic additives, and/or the miscibility of the HP-β-CD and hydrophilic additives within the nanostructure matrix. According to classical nucleation theory, the nucleation rate would depend on the interfacial tension and degree of supersaturation.37 The hydrophilic additives also affected the interfacial tension of the HP-β-CD nanostructures. However, the synergistic or antagonistic effects of the hydrophilic additives on the supersaturation of dutasteride require further study.
The dissolution rate of the dutasteride-loaded HP-β-CD nanostructures was determined in simulated gastric fluids (pH 1.2) containing 2% sodium lauryl sulfate. The dissolution test was performed according to US Pharmacopeia guidelines.38 Dutasteride did not dissolve in the dissolution medium in the absence of sodium lauryl sulfate because it is insoluble in water (less than 0.038 ng/mL). As shown in Figure 4A, approximately 90% of the dutasteride from all the HP-β-CD nanostructures tested dissolved within nine minutes in the dissolution medium containing 2% sodium lauryl sulfate. Because the nanostructures showed a rapid dissolution rate in this condition, a phase solubility study was performed to modify the dissolution medium for comparison between the formulations. As shown in Figure 5, the equilibrium solubility of dutasteride increased linearly with increasing concentration of sodium lauryl sulfate because of micellar solubilization. Dissolution studies were performed in simulated gastric fluids (pH 1.2) containing 0.1% sodium lauryl sulfate for the sink condition (C = 11.86 μg/mL, Ct < 0.1 Cs); the in vitro dissolution parameter, drug release rate (k), and t50% (time necessary to release 50% of the drug) were calculated using the Hixson-Crowell equation (Table 3). The most rapid dissolution rate was observed in HP-β-CD nanostructures with TPGS, followed by HP-β-CD nanostructures at a ratio of 1:39.9. Further, the percentage of dutasteride released from the HP-β-CD nanostructures was significantly higher than that of the commercial product. Drug release from the commercial product was dependent on the sodium lauryl sulfate concentration in the dissolution medium, which might be attributed to emulsification of the liquid oil content, ie, monoglycerides and diglycerides of caprylic/capric acid and butylated hydroxytoluene,3 from the soft capsule by sodium lauryl sulfate.
Figure 4.

Dissolution profiles for the commercial product and dutasteride-loaded HP-β-CD nanostructures with or without hydrophilic additives prepared using the supercritical antisolvent process in a pH 1.2 dissolution medium containing two different concentrations of sodium lauryl sulfate, ie, (A) 2% and (B) 0.1%.
Note: Data are expressed as the mean ± standard deviation (n = 3).
Abbreviations: DT, dutasteride; HP-β-CD, hydroxypropyl-β-cyclodextrin; HPMC, hydroxypropylmethyl cellulose; PVP-VA, polyvinylpyrrolidone-vinyl acetate; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate.
Figure 5.
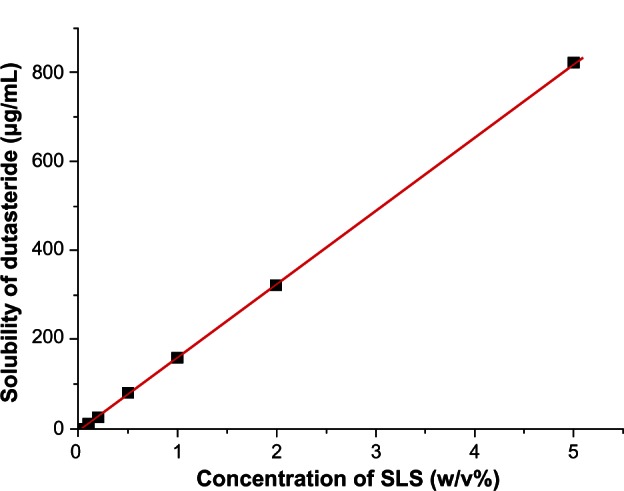
Effect of sodium lauryl sulfate concentration on the solubility of dutasteride.
Note: Data are expressed as the mean ± standard deviation (n = 3).
Abbreviation: SLS, sodium lauryl sulfate.
Table 3.
Dissolution kinetic parameters for dutasteride-loaded HP-β-CD nanostructures with or without hydrophilic additives prepared using the supercritical antisolvent process
| Formulation (weight ratio) | k (mg1/3 per minute) | t50% (minutes) |
|---|---|---|
| DT:HP-β-CD, 1:13.3 | 0.0184 ± 0.0017 | 9.22 ± 0.68 |
| DT:HP-β-CD, 1:39.9 | 0.0259 ± 0.0025 | 7.43 ± 0.61 |
| DT:HP-β-CD:PVP-VA 64, 1:26.6:13.3 | 0.0213 ± 0.0021 | 8.52 ± 0.78 |
| DT:HP-β-CD:TPGS, 1:26.6:13.3 | 0.0290 ± 0.0031 | 6.59 ± 0.55 |
| DT:HP-β-CD:HPMC 2910, 1:26.6:13.3 | 0.0233 ± 0.0020 | 8.27 ± 0.71 |
Notes: Drug release rate was determined using the Hixson-Crowell equation: where Wo is the initial amount of drug, Wt is the remaining amount of drug at time t, k is the drug release rate, and C is a constant. The t50% parameter corresponds to the time necessary for release of 50% of the drug and is calculated using the Hixson-Crowell equation. Data are expressed as the mean ± standard deviation (n = 4).
Abbreviations: DT, dutasteride; HP-β-CD, hydroxypropyl-β-cyclodextrin; HPMC, hydroxypropylmethyl cellulose; PVP-VA, polyvinylpyrrolidone-vinyl acetate; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate.
The plasma concentration-time profiles and pharmacokinetic parameters (AUC0→24h, Cmax, and Tmax) are shown in Figure 6 and Table 4, respectively. Bioavailability of the drug from the dutasteride-loaded HP-β-CD nanostructures with HPMC was signifcantly improved compared with that from the other formulations. After oral administration of the dutasteride-loaded HP-β-CD nanostructures with HPMC, the AUC0→24h, Cmax, and relative bioavailability were 3570.5 ± 443.3 ng ⋅ h/mL, 255.5 ± 27.9 ng/ mL, and 101.3%, respectively. Further, bioavailability of the dutasteride-loaded HP-β-CD nanostructures with HPMC was similar to that of the commercial soft gelatin capsule, indicating that dutasteride-loaded HP-β-CD nanostructures manufactured using the supercritical antisolvent process could help achieve a novel supersaturatable solid formulation with a high oral bioavailability of dutasteride. This differed from the oil-based solubilization principle of the commercial product. Thus, the soft gelatin capsule could be switched to a solid dosage form, such as a tablet.
Figure 6.

Plasma concentration-time profile for dutasteride in rats after oral administration of the commercial product and HP-β-CD nanostructures with or without hydrophilic additives prepared using the supercritical antisolvent process.
Note: Data are expressed as the mean ± standard deviation (n = 4).
Abbreviations: DT, dutasteride; HP-β-CD, hydroxypropyl-β-cyclodextrin; HPMC, hydroxypropylmethyl cellulose; PVP-VA, polyvinylpyrrolidone-vinyl acetate; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate.
Table 4.
Pharmacokinetic parameters for the dutasteride-loaded HP-β-CD nanostructures with or without hydrophilic additives prepared using the supercritical antisolvent process
| Formulation (weight ratio) | AUC0→24h (ng ⋅ hour/mL) | Cmax (ng/mL) | Tmax (hours) | Relative AUC (%) | Relative Cmax (%) | Relative BA (%) |
|---|---|---|---|---|---|---|
| DT:HP-β-CD, 1:13.3 | 1367.9 ± 286.0 | 124.0 ± 22.7 | 4.3 ± 2.1 | 49.9 | 59.8 | 38.8 |
| DT:HP-β-CD, 1:39.9 | 2738.7 ± 413.7a–c | 207.3 ± 24.7ab | 5.5 ± 4.5 | 100.0 | 100 | 77.7 |
| DT:HP-β-CD:PVP-VA 64, 1:26.6:13.3 | 1746.4 ± 290.9a | 161.6 ± 20.1a | 3.8 ± 2.2 | 63.8 | 78.0 | 49.6 |
| DT:HP-β-CD:TPGS, 1:26.6:13.3 | 2137.6 ± 387.2a | 195.7 ± 19.1a | 3.5 ± 2.4 | 78.1 | 94.4 | 60.7 |
| DT:HP-β-CD:HPMC 2910, 1:26.6:13.3 | 3570.5 ± 443.3a–b | 255.5 ± 27.9a–e | 3.8 ± 1.0 | 130.4 | 123.3 | 101.3 |
| Commercial product | 3523.9 ± 402.1a–b | 210.1 ± 29.5ab | 8.3 ± 2.5 | 128.7 | 101.4 | 100 |
Notes:
Significant at P < 0.05 versus DT:HP-β-CD = 1:13.3;
significant at P < 0.05 versus DT:HP-β-CD:PVP-VA 64 = 1:26.6:13.3;
significant at P < 0.05 versus DT:HP-β-CD:TPGS = 1:26.6:13.3;
significant at P < 0.05 versus DT:HP-β-CD = 1:39.9;
significant at P < 0.05 versus commercial product. Relative AUC (%) = AUC0→24h of sample/ AUC0→24h of HP-β-CD nanostructure (1:39.9) × 100. Relative Cmax (%) = Cmax of sample/Cmax of HP-β-CD nanostructure (1:39.9) × 100. Relative bioavailability was calculated using the following equation, with the commercial product used as a reference: relative bioavailability (%) = AUCtest/AUCreference × 100. Data are expressed as the mean ± standard deviation (n = 4).
Abbreviations: AUC, area under the concentration-time curve; BA, bioavailability; Cmax, peak plasma concentration; DT, dutasteride; HP-β-CD, hydroxypropyl-β-cyclodextrin; HPMC, hydroxypropylmethyl cellulose; PVP-VA polyvinylpyrrolidone-vinyl acetate; Tmax, time to peak concentration; TPGS, d-α-tocopheryl polyethylene glycol 1000 succinate.
The relationship between the in vitro dissolution and in vivo pharmacokinetic parameters was investigated for the various formulations of dutasteride-loaded HP-β-CD nanostructures. A good linear correlation was observed between the relative AUC in vitro and the relative AUC parameters in vivo (Figure 7A), and for the relative Cmax parameters (Figure 7B). Interestingly, the oral bioavailability of dutasteride increased with maximum solubility, ie, supersaturation. After oral administration of the dutasteride-loaded HP-β-CD nanostructures, a highly supersaturated solution was generated within the gastrointestinal tract, which resulted in a higher local concentration, and consequently, higher movement through the gastrointestinal epithelial membrane.39,40 However, a poor correlation was observed between the drug release rate and relative bioavailability (Figure 7C). Therefore, these data suggest that it is more important to enhance maximum supersaturation by prolonged supersaturation than the dissolution rate under sink conditions to achieve an effective solid dosage form of dutasteride with high bioavailability.
Figure 7.

Correlation between in vitro dissolution and in vivo pharmacokinetic parameters. (A) relative AUC, (B) relative Cmax, and (C) drug release rate.
Abbreviations: AUC, area under the concentration-time curve; Cmax, peak concentration; R2, coefficient of determination.
Conclusion
The supersaturation and dissolution rate of dutasteride were significantly increased by preparing dutasteride-loaded HP-β-CD nanostructures using the supercritical antisolvent process. The maximum supersaturation and drug precipitation rate of dutasteride depends on the type of hydrophilic additive used. Among these, HPMC conferred a high maximum solubility with prolonged supersaturation. The oral bioavailability of dutasteride in rats increased with the supersaturation induced by the HP-β-CD nanostructures with or without hydrophilic additives. Furthermore, in vivo pharmacokinetic parameters were better correlated with in vitro parameters related to supersaturation (solubility) than with in vitro drug release rate. Further, the bioavailability of the dutasteride-loaded HP-β-CD nanostructures with HPMC was similar to that of the commercial soft capsule formulation. In conclusion, preparation of dutasteride-loaded HP-β-CD nanostructures using the supercritical antisolvent process is a feasible strategy for developing a new solid dosage form containing dutasteride.
Footnotes
Disclosure
The author reports no conflicts of interest in this work.
References
- 1.Gisleskog PO, Hermann D, Hammarlund-Udenaes M, Karlsson MO. The pharmacokinetic modelling of GI198745 (dutasteride), a compound with parallel linear and nonlinear elimination. Br J Clin Pharmacol. 1999;47:53–58. doi: 10.1046/j.1365-2125.1999.00843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Evans HC, Goa KL. Dutasteride. Drugs Aging. 2003;20:905–916. doi: 10.2165/00002512-200320120-00005. [DOI] [PubMed] [Google Scholar]
- 3.US Food Drug Administration Center for Drug Evaluation and Research. Application Number 21–319 Clinical pharmacology and biopharmaceutics review Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/21319_Duagen_biopharmr_P1.pdfAccessed March 4, 2013
- 4.Avodart® (product monograph) Middlesex, UK: GlaxoSmithKline; Available from: http://www.gsk.ca/english/docs-pdf/product-monographs/Avodart.pdfAccessed March 4, 2013 [Google Scholar]
- 5.Ofner CM, 3rd, Zhang YE, Jobeck VC, Bowman BJ. Crosslinking studies in gelatin capsules treated with formaldehyde and in capsules exposed to elevated temperature and humidity. J Pharm Sci. 2001;90:79–88. doi: 10.1002/1520-6017(200101)90:1<79::aid-jps9>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 6.Beak IH, Kim MS. Improved supersaturation and oral absorption of dutasteride by amorphous solid dispersions. Chem Pharm Bull. 2012;60:1468–1473. doi: 10.1248/cpb.c12-00563. [DOI] [PubMed] [Google Scholar]
- 7.Park SJ, Choo GH, Hwang SJ, Kim MS. Quality by design: screening of critical variables and formulation optimization of Eudragit E nanoparticles containing dutasteride. Arch Pharm Res. 2013 Feb 28; doi: 10.1007/s12272-013-0064-z. [Epub ahead of print.] [DOI] [PubMed] [Google Scholar]
- 8.Choo GH, Park SJ, Hwang SJ, Kim MS. Formulation and in vivo evaluation of a self-microemulsifying drug delivery system of dutasteride. Drug Res (Stuttg) 2013;63:203–209. doi: 10.1055/s-0033-1334965. [DOI] [PubMed] [Google Scholar]
- 9.Kim MS. Soluplus-coated colloidal silica nanomatrix system for enhanced supersaturation and oral absorption of poorly water-soluble drugs. Artif Cells Nanomed Biotechnol. 2013 Jan 22; doi: 10.3109/21691401.2012.762369. [Epub ahead of print.] [DOI] [PubMed] [Google Scholar]
- 10.Wang XQ, Fan JM, Liu YO, et al. Bioavailability and pharmacokinetics of sorafenib suspension, nanoparticles and nanomatrix for oral administration to rat. Int J Pharm. 2011;419:339–346. doi: 10.1016/j.ijpharm.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Kawakami K. Modification of physicochemical characteristics of active pharmaceutical ingredients and application of supersaturatable dosage forms for improving bioavailability of poorly absorbed drugs. Adv Drug Deliv Rev. 2012;64:480–495. doi: 10.1016/j.addr.2011.10.009. [DOI] [PubMed] [Google Scholar]
- 12.Ozaki S, Minamisono T, Yamashita T, Kato T, Kushida I. Supersaturation-nucleation behavior of poorly soluble drugs and its impact on the oral absorption of drugs in thermodynamically high-energy forms. J Pharm Sci. 2012;101:214–222. doi: 10.1002/jps.22760. [DOI] [PubMed] [Google Scholar]
- 13.Frank KJ, Rosenblatt KM, Westedt U, et al. Amorphous solid dispersion enhances permeation of poorly soluble ABT-102: true supersaturation vs apparent solubility enhancement. Int J Pharm. 2012;437:288–293. doi: 10.1016/j.ijpharm.2012.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Frank KJ, Westedt U, Rosenblatt KM, et al. The amorphous solid dispersion of the poorly soluble ABT-102 forms nano/microparticulate structures in aqueous medium: impact on solubility. Int J Nanomedicine. 2012;7:5757–5768. doi: 10.2147/IJN.S36571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim MS, Kim JS, Cho WK, et al. Supersaturatable formulations for the enhanced oral absorption of sirolimus. Int J Pharm. 2013;445:108–116. doi: 10.1016/j.ijpharm.2013.01.067. [DOI] [PubMed] [Google Scholar]
- 16.Overhoff KA, McConville JT, Yang W, et al. Effect of stabilizer on the maximum degree and extent of supersaturation and oral absorption of tacrolimus made by ultra-rapid freezing. Pharm Res. 2008;25:167–175. doi: 10.1007/s11095-007-9417-y. [DOI] [PubMed] [Google Scholar]
- 17.Vandecruys R, Peeters J, Verreck G, Brewster ME. Use of a screening method to determine excipients which optimize the extent and stability of supersaturated drug solutions and application of this system to solid formulation design. Int J Pharm. 2007;342:168–175. doi: 10.1016/j.ijpharm.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 18.Gao P, Shi Y. Characterization of supersaturatable formulations for improved absorption of poorly soluble drugs. AAPS J. 2012;14:703–713. doi: 10.1208/s12248-012-9389-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park JB, Lee GH, Kang JW, et al. Improvement of photostability and dissolution profile of isradipine using inclusion complex. J Pharm Investig. 2013;43:55–61. [Google Scholar]
- 20.Loftsson T, Másson M, Brewster ME. Self-association of cyclodextrins and cyclodextrin complexes. J Pharm Sci. 2004;93:1091–1099. doi: 10.1002/jps.20047. [DOI] [PubMed] [Google Scholar]
- 21.Loftsson T, Sigurdsson H, Masson M, Schipper N. Preparation of solid drug/cyclodextrin complexes of acidic and basic drugs. Pharmazie. 2004;59:25–29. [PubMed] [Google Scholar]
- 22.Xiang TX, Anderson BD. Stable supersaturated aqueous solutions of silatecan 7-t-butyldimethylsilyl-10-hydroxycamptothecin via chemical conversion in the presence of a chemically modified beta-cyclodextrin. Pharm Res. 2002;19:1215–1222. doi: 10.1023/a:1019862629357. [DOI] [PubMed] [Google Scholar]
- 23.Brewster ME, Vandecruys R, Peeters J, et al. Comparative interaction of 2-hydroxypropyl-beta-cyclodextrin and sulfobutylether-beta-cyclodextrin with itraconazole: phase-solubility behavior and stabilization of supersaturated drug solutions. Eur J Pharm Sci. 2008;34:94–103. doi: 10.1016/j.ejps.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 24.Cha KH, Cho KJ, Kim MS, et al. Enhancement of the dissolution rate and bioavailability of fenofibrate by a melt-adsorption method using supercritical carbon dioxide. Int J Nanomedicine. 2012;7:5565–5575. doi: 10.2147/IJN.S36939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JS, Kim MS, Park HJ, et al. Physicochemical properties and oral bioavailability of amorphous atorvastatin hemi-calcium using spraydrying and SAS process. Int J Pharm. 2008;359:211–219. doi: 10.1016/j.ijpharm.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 26.Kalivoda A, Fischbach M, Kleinebudde P. Application of mixtures of polymeric carriers for dissolution enhancement of oxeglitazar using hot-melt extrusion. Int J Pharm. 2012;439:145–156. doi: 10.1016/j.ijpharm.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 27.Kim MS, Jin SJ, Kim JS, et al. Preparation, characterization and in vivo evaluation of amorphous atorvastatin calcium nanoparticles using supercritical antisolvent (SAS) process. Eur J Pharm Biopharm. 2008;69:454–465. doi: 10.1016/j.ejpb.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 28.Kim MS, Kim JS, Park HJ, et al. Enhanced bioavailability of sirolimus via preparation of solid dispersion nanoparticles using a supercritical antisolvent process. Int J Nanomedicine. 2011;6:2997–3009. doi: 10.2147/IJN.S26546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim MS, Song HS, Park HJ, Hwang SJ. Effect of solvent type on the nanoparticle formation of atorvastatin calcium by the supercritical antisolvent process. Chem Pharm Bull. 2012;60:543–547. doi: 10.1248/cpb.60.543. [DOI] [PubMed] [Google Scholar]
- 30.Montes A, Gordillo MD, Pereyra C, Martínez de la Ossa EJ. Polymer and ampicillin co-precipitation by supercritical antisolvent process. J Supercrit Fluids. 2012;63:92–98. [Google Scholar]
- 31.Kim MS, Kim JS, Hwang SJ. Enhancement of wettability and dissolution properties of cilostazol using the supercritical antisolvent process: effect of various additives. Chem Pharm Bull. 2010;58:230–233. doi: 10.1248/cpb.58.230. [DOI] [PubMed] [Google Scholar]
- 32.Wen H, Morris KR, Park K. Hydrogen bonding interactions between adsorbed polymer molecules and crystal surface of acetaminophen. J Colloid Interface Sci. 2005;290:325–335. doi: 10.1016/j.jcis.2005.04.049. [DOI] [PubMed] [Google Scholar]
- 33.Kojima T, Higashi K, Suzuki T, et al. Stabilization of a supersaturated solution of mefenamic acid from a solid dispersion with Eudragit(®) EPO. Pharm Res. 2012;29:2777–2791. doi: 10.1007/s11095-011-0655-7. [DOI] [PubMed] [Google Scholar]
- 34.Magnusdottir A, Másson M, Loftsson T. Self association and cyclodextrin solubilization of NSAIDs. J Incl Phenom Macrocycl Chem. 2002;44:213–218. [Google Scholar]
- 35.Loftsson T, Magnúsdóttir A, Másson M, Sigurjónsdóttir J F. Self-association and cyclodextrin solubilization of drugs. J Pharm Sci. 2002;91:2307–2316. doi: 10.1002/jps.10226. [DOI] [PubMed] [Google Scholar]
- 36.Makhlof A, Miyazaki Y, Tozuka Y, Takeuchi H. Cyclodextrins as stabilizers for the preparation of drug nanocrystals by the emulsion solvent diffusion method. Int J Pharm. 2008;357:280–285. doi: 10.1016/j.ijpharm.2008.01.025. [DOI] [PubMed] [Google Scholar]
- 37.Turnbull D, Fischer JC. Rate of nucleation in condensed systems. J Chem Phys. 1949;17:71–73. [Google Scholar]
- 38.US Food Drug Administration Recommended dissolution methods Available from: http://www.accessdata.fda.gov/scripts/cder/dissolution/index.cfmAccessed on March 4, 2013
- 39.Gao P, Guyton ME, Huang T, et al. Enhanced oral bioavailability of a poorly water soluble drug PNU-91325 by supersaturatable formulations. Drug Dev Ind Pharm. 2004;30:221–229. doi: 10.1081/ddc-120028718. [DOI] [PubMed] [Google Scholar]
- 40.Moes JJ, Koolen SL, Huitema AD, et al. Pharmaceutical development and preliminary clinical testing of an oral solid dispersion formulation of docetaxel (ModraDoc001) Int J Pharm. 2011;420:244–250. doi: 10.1016/j.ijpharm.2011.08.041. [DOI] [PubMed] [Google Scholar]