

Abstract
Recent research suggests that chromatin-modifying enzymes are metabolic sensors regulating gene expression. Epigenetics is linked to metabolomics in response to the cellular microenvironment. Specific metabolites involved in this sensing mechanism include S-adenosylmethionine, acetyl-CoA, alphaketoglutarate and NAD+. Although the core metabolic pathways involving glucose have been emphasized as the source of these metabolites, the reprogramming of pathways involving non-essential amino acids may also play an important role, especially in cancer. Examples include metabolic pathways for glutamine, serine and glycine. The coupling of these pathways to the intermediates affecting epigenetic regulation occurs by “parametabolic” mechanisms. The metabolism of proline may play a special role in this parametabolic linkage between metabolism and epigenetics. Both proline degradation and biosynthesis are robustly affected by oncogenes or suppressor genes, and they can modulate intermediates involved in epigenetic regulation. A number of mechanisms in a variety of animal species have been described by our laboratory and by others. The challenge we now face is to identify the specific chromatin-modifying enzymes involved in coupling of proline metabolism to altered reprogramming of gene expression.
Keywords: metabolism and cancer, epigenetics, redox regulation, reactive oxygen species, non-essential amino acids, proline metabolism
Introduction
Over the past 30 years, the genetic basis of cancer has been firmly established, from Knudson’s two-hit hypothesis1 to the identification of numerous specific mutations characterized as oncogenes and suppressor genes.2,3 The clinical application of these seminal discoveries, however, has been disappointing. Attempts to repair or replace mutated genes, by “gene therapy” have been hindered not only by technical difficulties in delivery, but, more importantly, by the redundancy and plasticity of the cancer genome.3 In this regard, the discoveries in cancer epigenetics offer new opportunities and strategies.4-6 For this Point-of-View, epigenetic mechanisms refer to those structural and enzymatic interactions that regulate transcription independently of the DNA primary sequence. We will focus on the links between epigenetics and the metabolic mechanisms mediating adjustments to the tumor microenvironment.
The expression of genes is modulated by the interaction between DNA and chromatin, including acetylation of histones and methylation of both DNA and histones. A number of excellent reviews describe the families of histone and DNA methylases,7 and the histone acetylases and deacetylases.8 Thus, cancer genetics not only depends on mutations in the primary sequence of oncogenes and suppressor genes, but also on subsequent gene regulation. Importantly, the expression of genes downstream from oncogenes and suppressor genes are ultimately dependent on these regulated modifications of DNA and histones.
Although mutations have been found in the genes encoding DNA/histone modifying enzymes,6 targeting them for repair or replacement faces the same obstacles as therapy directed against primary oncogenes or suppressor genes. Importantly, as pointed out in recent reviews, the modulation of the epigenome by these enzymes depends on the level of metabolic intermediates and cofactors.9,10 They could be considered a minute-by-minute sensing mechanism of the cellular metabolic state.9 Examples of these linkages include the metabolism-dependent fluctuations in acetyl CoA, the substrate for histone acetyltransferases.11 On the other hand, sirtuins remove acetyl groups from lysine in histones requiring NAD+,12 a sensitive and dynamic indicator of energy metabolism. Methylation of both DNA and histones by their respective methyltransferases requires 1-carbon transfers from S-adenosylmethionine9 whereas TET2- and JMDH2-mediated demethylation depends, in large part, on α-ketoglutarate (α-KG) as substrate for α-KG dioxygenases for oxidizing methyl-cytosine to hydroxymethyl-cytosine.10 Thus, epigenetic enzymes not only depend on metabolites but they regulate genes which reprogram metabolism.
About 15 years ago, the resurgence of research interest led to the identification of reprogramming in metabolism in cancer.13,14 These metabolic changes are not due to so called “passenger genes” but, instead, are the integrated responses to “driver genes” such as c-MYC and PI3K/PTEN.14 The constellation of metabolic changes in cancer cells includes aerobic glycolysis first described by Warburg,15 changes in TCA cycle function,16 activation of the pentose phosphate pathway17 and the “addiction” to glutamine.18 An attractive explanation is the rerouting of glucose and glutamine into pathways generating substrates (amino acids, ribonucleotides and lipids) for building cell mass.19 Pyruvate is diverted to lactate to recycle NADH to NAD+ for glycolysis.20 Pharmacologic intervention of reprogrammed metabolic pathways starves tumor cells and halts tumor progression. However, the success of this strategy was limited presumably because these core pathways have numerous compensatory mechanisms, including the network of endocrine factors providing homeostatic compensation.21
Non-Essential Amino Acids
The emphasis on the core metabolic pathways for glucose and glutamine metabolism in tumor cells is understandable since “addiction” to glucose22 and glutamine18 has been demonstrated. However, recent discoveries that a number of non-essential amino acids play a critical role in cancer metabolism deserve consideration.23 Non-essential amino acids (NEAA) may not be important nutritionally, but may make critical contributions to metabolism.
Prokaryotes can synthesize every proteinogenic amino acid but, as organisms evolved to consume other organisms, this biosynthetic capability is no longer necessary.24 Certain biosynthetic pathways are evolutionarily conserved because they serve other necessary metabolic functions.24 For example, the biosynthesis of arginine is involved in the incorporation of ammonia into urea and the biosynthesis of cysteine is a product in the degradation of homocysteine, an important component of the pathway for generating S-adenosylmethionine. Alanine, aspartic acid and glutamic acid are amino acid analogs of metabolically important α keto-acids.
Recently, the syntheses of glycine and serine have been identified as essential for tumor growth.25 Glutamine is critical for de novo purine and pyrimidine synthesis as well as a source of TCA cycle intermediates.26 In the physiologic setting, glutamine is the currency for transferring carbons and nitrogens from proteolysis to central tissues for further carbon and nitrogen processing.27 The tiny genome of Cryptosporidium, a protozoan parasite found in human intestine, lacks genes for the TCA cycle and biosynthetic pathways to sugars and nucleotides, yet it has the synthetic pathways for asparagine, glutamine, glycine and proline.24 In fact, proline biosynthesis is retained from S. cerevisiae, D. discoidem to H. sapiens. The functions of these aforementioned NEAA are recognized, but “the utility of the pathway to proline is not immediately obvious.”24 However, work from our laboratory has shown important functions for proline metabolism to explain the basis for its evolutionary conservation.
To understand the regulatory functions of NEAA, we must introduce a “parametabolic” paradigm (Fig. 1).23 For most biosynthetic pathways, the enzyme is regulated to produce a specific product from a precursor. However, for those in the pathways for NEAA, the specific AA is the by-product of coupled reactions. An example of this is in the serine metabolic pathway, where the conversion of serine and glycine transfers a carbon group to tetrahydrofolate and 5-methyltetrahydrofolate regenerates methionine from homocysteine. In an ATP-dependent reaction, methionine is converted to S-adenosylmethionine, which is the principal methyl donor for biosynthesis of a variety of intermediates as well as the methylation of DNA.28 In the case of NEAA participating in transaminations, one endpoint is the transfer of reducing potential across mitochondrial membranes; for example, the malate-aspartate shuttle.29
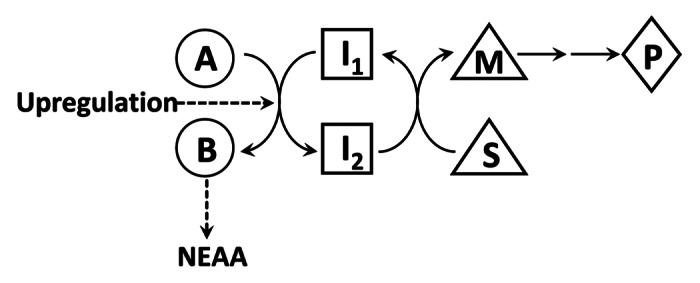
Figure 1. Proposed Parametabolic Regulation by NEAA Metabolism This cartoon is a representation of a generic parametabolic scheme. The conversion of A to B yields a non-essential amino acid and this step may be upregulated by cancer reprogramming. I1 and I2 are in a metabolic interlock with the conversion of S to M, which is then used for epigenetic modification of DNA or chromatin. For example, if A is serine and B is glycine, the reaction is coupled to the conversion of tetrahydrofolate (THF) to 5,10-methylene THF, which as 5-methyl THF, transfers methyl groups to homocysteine forming methionine. In the presence of ATP, S-adenosylmethionine is formed for DNA methylation. In another example, if pyrroline-5-carboxylate, A, is converted to proline, B, NADH, I1, is oxidized to NAD+, I2, which can accept acetyl groups hydrolyzed from histone lysines by sirtuins to form acetyl-ADP-ribose.
Acetylation of histones is dependent on the availability of acetyl-CoA, and levels of acetyl-CoA can fluctuate as much as 10-fold;11,30 its metabolism primarily reflects the abundance of glucose and the flux through glucose metabolic pathways. On the other hand, the production of serine from 3-phosphoglycerate is promoted by the shift of pyruvate kinase isozymes from PK to PKM2.19,31 The latter has much lower catalytic activity, allowing the buildup of phosphoenolpyruvate and 3-phosphoglycerate. Thus, the augmented production of serine would accommodate the substrates for production of SAM and methylation. These observations suggest that for some of the NEAA, their metabolic pathway serves as a parametabolic link between metabolism and epigenetics.
The Regulatory Role of Proline Metabolism
An intriguing question is whether the metabolism of the non-essential amino acid proline also plays such a parametabolic role and whether the proline regulatory axis plays a role not only in the metabolism of core substrates, such as glucose or glutamine, but also in regulating epigenetics. With its α-nitrogen sequestered within a pyrrolidine ring, proline is not a substrate for generic amino acid metabolizing enzymes. Instead, it has its own metabolic system, which has been recently reviewed.32,33 Of special interest, proline dehydrogenase (oxidase) is bound to mitochondrial inner membranes. Although the catalytic product of the reaction pyrroline-5-carboxylate (P5C) can be sequentially converted to glutamate and α-KG, an epigenetic metabolite, electrons from proline are donated to the mitochondrial electron transport chain accompanying the production of P5C.34 In addition, this enzyme is under diverse regulation making it an ideal candidate as an epigenetic regulator. It is robustly and rapidly induced by p5335-37 and by PPARγ,38 leading to ROS generation,39,40 cell cycle arrest,41 apoptosis39,42 and, depending on the cellular context, prosurvival autophagy.43 The enzyme is upregulated by AMPK43,44 but downregulated by a specific microRNA, miR-23b*,45 which in turn is modulated by c-MYC.46 Furthermore, the discovery that CoQ is the acceptor for proline-derived electrons(ref. 47 and Hancock, unpublished results) enabled redox signaling proposed by others through complex III.48 As first pointed out by Adams, P5C is not only the committed precursor for proline but also its immediate degradative product.49 Thus, the interconversions of proline and P5C constitute a redox cycle.50 The cycling was thought to function in a metabolic interlock with the oxidative arm of the pentose phosphate pathway,50 but recent findings by our lab and others have added several additional functions to the original hypothesis (Liu et al., unpublished results). Mutations in PRODH in humans may be associated with hyperprolinemia and neuropsychiatric disorders, but the clinical reports are controversial.51 Metabolic findings may be masked by the overlap in redox function between PRODH/POX and hydroxyproline oxidase PRODH2/HYPOX.52
Proline Biosynthesis
We have shown that the enzymes in the pathway from glutamine to proline catalyzed sequentially by glutaminase (GLS), pyrroline-5-carboxylate synthase (P5CS)46,53 and pyrroline-5 carboxylate reductase 1 (PYCR1)46 are markedly increased by the oncogene c-MYC. This response occurred in the face of physiologic concentrations of medium proline; others have shown the net synthesis of proline by cultured cells resulting in an increase in medium proline concentration.54 Therefore, the upregulation of the proline synthetic pathway by c-MYC is not to meet demands for protein synthesis. These findings are consistent with the observations of Windmueller et al., which demonstrate intestines containing proliferating cells primarily use glutamine and a major product is proline.55 Our findings showed that the enzymes of this pathway are markedly upregulated by c-MYC with correspondingly increased flux demonstrated by stable isotope-resolved metabolomics.46
Effects of Proline Metabolism
Humans with PYCR1 mutation have altered mitochondrial function and progeroid changes in connective tissues, including cutis laxa.56 These findings cannot be explained on the basis of inadequate proline for protein synthesis. Interestingly, the crystal structure for human PYCR1 has been solved: the native enzyme is a decamer with a conformation suggesting transporter or chaperone functions.57 Furthermore, others have characterized 3 isozymes of PYCR in cultured melanoma cells.58 They showed that PYCR1/2 prefer NADH as a cofactor whereas PYCRL mainly uses NADPH. PYCR1 appears associated with mitochondrial outer membranes whereas PYCR2 is in the matrix and PYCRL is in the cytosol. Although the localization and properties of these isozymes were shown in melanoma cells and need generalization to other tumors as well as normal tissue, they provide versatility for redox transfers for the proline regulatory axis originating from either proline or from glutamine. P5CS catalyzes the first step of proline biosynthesis from glutamate. Deficiency in humans59 has profound manifestations with progressive neurodegeneration, mental retardation, peripheral neuropathy, joint laxity and cataracts. Metabolically, these patients have hypoprolinemia and hypoarginemia with hyperammonemia. How the phenotype relates to metabolic or epigenetic mechanisms remains unknown.
Proline Metabolism and Collagen
Collagen, the most abundant protein (by weight) in the body with 25% of its amino acid residues either as proline or hydroxyproline, can serve as reservoir or metabolic dump for proline.32 Cells under metabolic stress upregulate metalloproteinases and PRODH/POX. Cells starved for substrates increase proline degradation through AMPK43,44 to provide an alternative mitochondrial substrate and generate ROS signals to activate prosurvival autophagy.43 Under conditions of rapid proliferation induced by growth factors and MYC activation, proline degradation is downregulated46 whereas robust induction of P5CS and PYCR mediates increased turnover of NAD(P)H to NAD(P)+ (ref. 46 and Liu et al., unpublished results). The increased availability of NADP+ and NAD+ provides oxidizing potential for critical steps in the oxidative arm of the pentose phosphate pathway and perhaps in glycolysis, respectively. The latter not only spares pyruvate for conversion to other vital intermediates, but NAD+ also supports the activity of sirtuins in deacylating histones. The resultant proline can be incorporated into collagen thereby eliminating it from the metabolome, much as excreted lactate results in the recycling of cellular NADH to NAD+ for glycolysis. Breast cancer provides an example of such coupling between proliferation and collagen synthesis. Although mechano-regulation between collagen fibrils and tumor cells has been proposed,60 the robust increase in collagen production also provides metabolic advantages for tumor growth. These metabolic features of proline and collagen metabolism may not be provided by a single cell type. In fact, it is likely that asymmetry in metabolism between tumor and stromal cells are a necessary feature. Metabolic/signaling collaboration between tissues and/or different cell types have been proposed61,62 and an intercellular complementation in proline metabolism between erythrocytes and hepatocytes based on the asymmetry of cellular enzymes has been described.63,64
The redox turnover of pyridine nucleotides has been suggested, albeit direct demonstration of the effect requires further studies. Since the steady-state ratio of NAD(P)H to NAD(P)+ is difficult to ascertain, the demonstration of turnover can be inferred by flux through pathways in metabolic interlocks. For example with induction of PRODH/POX, the flux through the oxidative arm of the pentose phosphate pathway is markedly increased.44,65 Similarly, with MYC-induction of P5CS and PYCR1, glycolysis is markedly increased by c-MYC expression.46 However, in the face of this Warburg effect, the knockdown of PYCR1 or P5CS markedly inhibited the response (Liu et al., unpublished results). These findings suggest that the proline regulatory axis is providing the redox turnover to optimally activate the c-MYC activation of glycolysis. Thus, from both proline degradation and proline biosynthesis, we have robust, albeit indirect, evidence of the recycling of pyridine nucleotides and the parametabolic regulation of glucose metabolism.
Evidence for Cellular Response to Epigenetic Modulation by Proline
Is there experimental evidence that the parametabolic effect of the proline regulatory axis influences epigenetic mechanisms? Certainly, epigenetic changes induced by ROS have been demonstrated and reviewed.66 Others have shown that treatment of cultured hepatocellular carcinoma cells with hydrogen peroxide caused methylation of the E-cadherin promoter.67 We have shown that generation of ROS due to upregulation of PRODH/POX can activate a parametabolic response or program ranging from downregulation of COX-2 to the activation of prosurvival autophagy depending on the metabolic context.23 There are a number of published findings from other investigators suggesting that the proline regulatory axis is involved in epigenetics. First, Washington et al., using cultured mouse embryonic stem (ES) cells, showed that of all media constituents, proline uniquely regulated pluripotency.68 This finding was corroborated by Casalino et al.,69 who showed that the effect of proline in maintaining the metastability of ES cells required proline metabolism. Another recent example of proline-dependent epigenetic regulation was described by Zarse et al.70,71 Their studies in C. elegans and in MEFs showed the life span extension with defective insulin/IGF1 (daf-2 in C. elegans) signaling was dependent on the catabolism of proline. Their interpretation is that the ROS generated by proline degradation induces the expression of a genetic program including antioxidant enzymes protecting against ROS so that lifespan is extended.
In summary, we propose that the metabolism of NEAA not only regulates core metabolism, but also produces substrates or cofactors participating in epigenetic mechanisms. Thus, the metabolism of NEAA acts as a coupling mechanism between metabolomics and epigenetics in cancer. Several of the NEAA mechanisms involve the cycling of substrates, cofactors or transfer mechanisms necessary for epigenetic regulation. We also introduce parametabolic regulation to understand the metabolic interlocks between NEAA and critical core pathways, i.e., glucose metabolism and ribonucleotides synthesis. We propose that the regulation of the enzymes of the proline regulatory axis provides plasticity in the production of various intermediates as well as in the recycling of pyridine nucleotides (Table 1). We hope that this regulatory axis will offer novel therapeutic targets for the treatment of cancer.
Table 1. Proline axis: regulatory effects on metabolic reprogramming and epigenetics .
| Pathway | Enzyme | Regulation | Metabolic Effect | Clinical, Cellular Findings Epigenetic Mechanism |
|---|---|---|---|---|
| Degradative |
PRODH (POX) |
Upregulated by p53;35-37,42 Increased by AMPK43,44 under glucose deprivation and/or hypoxia |
ROS39–40 Increased α-KG;41 Decreased glycolysis Decreased Ox. Phos (Hancock, unpublished results) |
Apoptosis39-41 Blockade Cell Cycle41 Stem cell pluripotency68,69 Ins/IGF1 prosurvival signaling dependent on proline metabolism70,71 |
| |
|
Decreased by miR-23b*45 and c-MYC46 |
|
permissive effect on cell proliferation46 |
| |
|
Mutations in PRODH51 |
Hyperprolinemia51 |
Neuropsychiatric disorders |
| Synthetic |
P5CS |
Induced by c-MYC46 |
Proline inreased Involved in c-MYC- stimulation of glycolysis (Warburg Effect) (Liu et al., unpublished results) |
necessary for c-MYC stimulated proliferation;46 essential for T-cell activation53 |
| |
|
Inborn error59 |
Hypoprolinemia, Hypoornithinemia Hypoammonemia59 |
Neurodegeneration, mental deficiency, neuropathy connective tissue disorder59 |
| |
PYCR1 |
Induced by c-MYC46 |
As for P5CS (Liu et al., unpublished results) |
As for P5CS (Liu et al., unpublished results) Knockdown in xenograft tumors inhibits tumor growth25 |
| Inborn error56 | More sensitive to peroxide56 | Multiple genetic defects Cutis laxa, progeroid56 |
Acknowledgments
This work was supported by the Intyrtamural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. This project also has been funded in part with federal funds from the National Cancer Institute, NIH, under contract no. HHSN27612080001. The content of this review does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US. Government. We gratefully acknowledge Gregory Borchert and Dr Kyle Christian for their helpful discussions and Meredith Harman for reading the manuscript.
Glossary
Abbreviations:
- PRODH
proline dehydrogenase
- P5C
pyrroline-5-carboxylate
- P5CS
pyrroline-5-carboxylate synthase
- PYCR
pyrroline-5-carboxylate reductase
- NEAA
non-essential amino acids
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/24042
References
- 1.Knudson AG., Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. 1993;9:138–41. doi: 10.1016/0168-9525(93)90209-Z. [DOI] [PubMed] [Google Scholar]
- 3.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 4.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–59. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 5.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–34. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 7.Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- 8.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459:108–12. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yun J, Johnson JL, Hanigan CL, Locasale JW. Interactions between epigenetics and metabolism in cancers. Front Oncol. 2012;2:163. doi: 10.3389/fonc.2012.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu C, Thompson CB. Metabolic regulation of epigenetics. Cell Metab. 2012;16:9–17. doi: 10.1016/j.cmet.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai L, Sutter BM, Li B, Tu BP. Acetyl-CoA induces cell growth and proliferation by promoting the acetylation of histones at growth genes. Mol Cell. 2011;42:426–37. doi: 10.1016/j.molcel.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–8. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 13.DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20. doi: 10.1016/j.cmet.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008;13:472–82. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 15.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 16.Cardaci S, Ciriolo MR. TCA Cycle Defects and Cancer: When Metabolism Tunes Redox State. Int J Cell Biol. 2012;2012:161837. doi: 10.1155/2012/161837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- 18.Dang CV. MYC, microRNAs and glutamine addiction in cancers. Cell Cycle. 2009;8:3243–5. doi: 10.4161/cc.8.20.9522. [DOI] [PubMed] [Google Scholar]
- 19.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Locasale JW, Cantley LC. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011;14:443–51. doi: 10.1016/j.cmet.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meier U, Gressner AM. Endocrine regulation of energy metabolism: review of pathobiochemical and clinical chemical aspects of leptin, ghrelin, adiponectin, and resistin. Clin Chem. 2004;50:1511–25. doi: 10.1373/clinchem.2004.032482. [DOI] [PubMed] [Google Scholar]
- 22.Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose metabolism of cancers. J Nucl Med. 2008;49(Suppl 2):24S–42S. doi: 10.2967/jnumed.107.047258. [DOI] [PubMed] [Google Scholar]
- 23.Phang JM, Liu W, Hancock C, Christian KJ. The proline regulatory axis and cancer. Front Oncol. 2012;2:60. doi: 10.3389/fonc.2012.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Payne SH, Loomis WF. Retention and loss of amino acid biosynthetic pathways based on analysis of whole-genome sequences. Eukaryot Cell. 2006;5:272–6. doi: 10.1128/EC.5.2.272-276.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–50. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15:110–21. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Darmaun D, Matthews DE, Bier DM. Physiological hypercortisolemia increases proteolysis, glutamine, and alanine production. Am J Physiol. 1988;255:E366–73. doi: 10.1152/ajpendo.1988.255.3.E366. [DOI] [PubMed] [Google Scholar]
- 28.Chiang PK, Gordon RK, Tal J, Zeng GC, Doctor BP, Pardhasaradhi K, et al. S-Adenosylmethionine and methylation. FASEB J. 1996;10:471–80. [PubMed] [Google Scholar]
- 29.Barron JT, Gu L, Parrillo JE. Malate-aspartate shuttle, cytoplasmic NADH redox potential, and energetics in vascular smooth muscle. J Mol Cell Cardiol. 1998;30:1571–9. doi: 10.1006/jmcc.1998.0722. [DOI] [PubMed] [Google Scholar]
- 30.Takamura Y, Nomura G. Changes in the intracellular concentration of acetyl-CoA and malonyl-CoA in relation to the carbon and energy metabolism of Escherichia coli K12. J Gen Microbiol. 1988;134:2249–53. doi: 10.1099/00221287-134-8-2249. [DOI] [PubMed] [Google Scholar]
- 31.Vander Heiden MG, Lunt SY, Dayton TL, Fiske BP, Israelsen WJ, Mattaini KR, et al. Metabolic Pathway Alterations that Support Cell Proliferation. Cold Spring Harb Symp Quant Biol 2012. [DOI] [PubMed] [Google Scholar]
- 32.Phang JM, Donald SP, Pandhare J, Liu Y. The metabolism of proline, a stress substrate, modulates carcinogenic pathways. Amino Acids. 2008;35:681–90. doi: 10.1007/s00726-008-0063-4. [DOI] [PubMed] [Google Scholar]
- 33.Phang JM, Liu W. Proline metabolism and cancer. Front Biosci. 2012;17:1835–45. doi: 10.2741/4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adams E, Frank L. Metabolism of proline and the hydroxyprolines. Annu Rev Biochem. 1980;49:1005–61. doi: 10.1146/annurev.bi.49.070180.005041. [DOI] [PubMed] [Google Scholar]
- 35.Polyak K, Xia Y, Zweier JL, Kinzler KW, Vogelstein B. A model for p53-induced apoptosis. Nature. 1997;389:300–5. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 36.Donald SP, Sun XY, Hu CA, Yu J, Mei JM, Valle D, et al. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res. 2001;61:1810–5. [PubMed] [Google Scholar]
- 37.Maxwell SA, Rivera A. Proline oxidase induces apoptosis in tumor cells, and its expression is frequently absent or reduced in renal carcinomas. J Biol Chem. 2003;278:9784–9. doi: 10.1074/jbc.M210012200. [DOI] [PubMed] [Google Scholar]
- 38.Pandhare J, Cooper SK, Phang JM. Proline oxidase, a proapoptotic gene, is induced by troglitazone: evidence for both peroxisome proliferator-activated receptor gamma-dependent and -independent mechanisms. J Biol Chem. 2006;281:2044–52. doi: 10.1074/jbc.M507867200. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y, Borchert GL, Surazynski A, Hu CA, Phang JM. Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: the role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene. 2006;25:5640–7. doi: 10.1038/sj.onc.1209564. [DOI] [PubMed] [Google Scholar]
- 40.Liu Y, Borchert GL, Donald SP, Surazynski A, Hu CA, Weydert CJ, et al. MnSOD inhibits proline oxidase-induced apoptosis in colorectal cancer cells. Carcinogenesis. 2005;26:1335–42. doi: 10.1093/carcin/bgi083. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y, Borchert GL, Donald SP, Diwan BA, Anver M, Phang JM. Proline oxidase functions as a mitochondrial tumor suppressor in human cancers. Cancer Res. 2009;69:6414–22. doi: 10.1158/0008-5472.CAN-09-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu CA, Donald SP, Yu J, Lin WW, Liu Z, Steel G, et al. Overexpression of proline oxidase induces proline-dependent and mitochondria-mediated apoptosis. Mol Cell Biochem. 2007;295:85–92. doi: 10.1007/s11010-006-9276-6. [DOI] [PubMed] [Google Scholar]
- 43.Liu W, Glunde K, Bhujwalla ZM, Raman V, Sharma A, Phang JM. Proline oxidase promotes tumor cell survival in hypoxic tumor microenvironments. Cancer Res. 2012;72:3677–86. doi: 10.1158/0008-5472.CAN-12-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pandhare J, Donald SP, Cooper SK, Phang JM. Regulation and function of proline oxidase under nutrient stress. J Cell Biochem. 2009;107:759–68. doi: 10.1002/jcb.22174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu W, Zabirnyk O, Wang H, Shiao YH, Nickerson ML, Khalil S, et al. miR-23b targets proline oxidase, a novel tumor suppressor protein in renal cancer. Oncogene. 2010;29:4914–24. doi: 10.1038/onc.2010.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu W, Le A, Hancock C, Lane AN, Dang CV, Fan TW, et al. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc Natl Acad Sci U S A. 2012;109:8983–8. doi: 10.1073/pnas.1203244109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wanduragala S, Sanyal N, Liang X, Becker DF. Purification and characterization of Put1p from Saccharomyces cerevisiae. Arch Biochem Biophys. 2010;498:136–42. doi: 10.1016/j.abb.2010.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chandel NS. Mitochondrial complex III: an essential component of universal oxygen sensing machinery? Respir Physiol Neurobiol. 2010;174:175–81. doi: 10.1016/j.resp.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adams E. Metabolism of proline and of hydroxyproline. Int Rev Connect Tissue Res. 1970;5:1–91. doi: 10.1016/b978-0-12-363705-5.50007-5. [DOI] [PubMed] [Google Scholar]
- 50.Phang JM. The regulatory functions of proline and pyrroline-5-carboxylic acid. Curr Top Cell Regul. 1985;25:91–132. doi: 10.1016/b978-0-12-152825-6.50008-4. [DOI] [PubMed] [Google Scholar]
- 51.Willis A, Bender HU, Steel G, Valle D. PRODH variants and risk for schizophrenia. Amino Acids. 2008;35:673–9. doi: 10.1007/s00726-008-0111-0. [DOI] [PubMed] [Google Scholar]
- 52.Cooper SK, Pandhare J, Donald SP, Phang JM. A novel function for hydroxyproline oxidase in apoptosis through generation of reactive oxygen species. J Biol Chem. 2008;283:10485–92. doi: 10.1074/jbc.M702181200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35:871–82. doi: 10.1016/j.immuni.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stoner GD, Merchant DJ. Amino acid utilization by L-M strain mouse cells in a chemically defined medium. In Vitro. 1972;7:330–43. doi: 10.1007/BF02661723. [DOI] [PubMed] [Google Scholar]
- 55.Windmueller HG, Spaeth AE. Uptake and metabolism of plasma glutamine by the small intestine. J Biol Chem. 1974;249:5070–9. [PubMed] [Google Scholar]
- 56.Reversade B, Escande-Beillard N, Dimopoulou A, Fischer B, Chng SC, Li Y, et al. Mutations in PYCR1 cause cutis laxa with progeroid features. Nat Genet. 2009;41:1016–21. doi: 10.1038/ng.413. [DOI] [PubMed] [Google Scholar]
- 57.Meng Z, Lou Z, Liu Z, Li M, Zhao X, Bartlam M, et al. Crystal structure of human pyrroline-5-carboxylate reductase. J Mol Biol. 2006;359:1364–77. doi: 10.1016/j.jmb.2006.04.053. [DOI] [PubMed] [Google Scholar]
- 58.De Ingeniis J, Ratnikov B, Richardson AD, Scott DA, Aza-Blanc P, De SK, et al. Functional specialization in proline biosynthesis of melanoma. PLoS One. 2012;7:e45190. doi: 10.1371/journal.pone.0045190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baumgartner MR, Hu CA, Almashanu S, Steel G, Obie C, Aral B, et al. Hyperammonemia with reduced ornithine, citrulline, arginine and proline: a new inborn error caused by a mutation in the gene encoding delta(1)-pyrroline-5-carboxylate synthase. Hum Mol Genet. 2000;9:2853–8. doi: 10.1093/hmg/9.19.2853. [DOI] [PubMed] [Google Scholar]
- 60.Provenzano PP, Inman DR, Eliceiri KW, Keely PJ. Matrix density-induced mechanoregulation of breast cell phenotype, signaling and gene expression through a FAK-ERK linkage. Oncogene. 2009;28:4326–43. doi: 10.1038/onc.2009.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fujimoto WY, Subak-Sharpe JH, Seegmiller JE. Hypoxanthine-guanine phosphoribosyltransferase deficiency: chemical agents selective for mutant or normal cultured fibroblasts in mixed and heterozygote cultures. Proc Natl Acad Sci U S A. 1971;68:1516–9. doi: 10.1073/pnas.68.7.1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012;72:5130–40. doi: 10.1158/0008-5472.CAN-12-1949. [DOI] [PubMed] [Google Scholar]
- 63.Phang JM, Yeh GC, Hagedorn CH. The intercellular proline cycle. Life Sci. 1981;28:53–8. doi: 10.1016/0024-3205(81)90365-9. [DOI] [PubMed] [Google Scholar]
- 64.Hagedorn CH, Yeh GC, Phang JM. Transfer of 1-pyrroline-5-carboxylate as oxidizing potential from hepatocytes to erythrocytes. Biochem J. 1982;202:31–9. doi: 10.1042/bj2020031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Phang JM, Downing SJ, Yeh GC, Smith RJ, Williams JA, Hagedorn CH. Stimulation of the hexosemonophosphate-pentose pathway by pyrroline-5-carboxylate in cultured cells. J Cell Physiol. 1982;110:255–61. doi: 10.1002/jcp.1041100306. [DOI] [PubMed] [Google Scholar]
- 66.Cyr AR, Domann FE. The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Signal. 2011;15:551–89. doi: 10.1089/ars.2010.3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lim SO, Gu JM, Kim MS, Kim HS, Park YN, Park CK, et al. Epigenetic changes induced by reactive oxygen species in hepatocellular carcinoma: methylation of the E-cadherin promoter. Gastroenterology. 2008;135:2128–40, 2140, e1-8. doi: 10.1053/j.gastro.2008.07.027. [DOI] [PubMed] [Google Scholar]
- 68.Washington JM, Rathjen J, Felquer F, Lonic A, Bettess MD, Hamra N, et al. L-Proline induces differentiation of ES cells: a novel role for an amino acid in the regulation of pluripotent cells in culture. Am J Physiol Cell Physiol. 2010;298:C982–92. doi: 10.1152/ajpcell.00498.2009. [DOI] [PubMed] [Google Scholar]
- 69.Casalino L, Comes S, Lambazzi G, De Stefano B, Filosa S, De Falco S, et al. Control of embryonic stem cell metastability by L-proline catabolism. J Mol Cell Biol. 2011;3:108–22. doi: 10.1093/jmcb/mjr001. [DOI] [PubMed] [Google Scholar]
- 70.Zarse K, Schmeisser S, Groth M, Priebe S, Beuster G, Kuhlow D, et al. Impaired insulin/IGF1 signaling extends life span by promoting mitochondrial L-proline catabolism to induce a transient ROS signal. Cell Metab. 2012;15:451–65. doi: 10.1016/j.cmet.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schroeder EA, Shadel GS. Alternative mitochondrial fuel extends life span. Cell Metab. 2012;15:417–8. doi: 10.1016/j.cmet.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]