

Abstract
Rett syndrome (RTT) is a neurodevelopmental disorder with neurological symptoms, such as motor disorders and mental retardation. In most cases, RTT is caused by mutations in the DNA binding protein MeCP2. In mice, MeCP2 gene deletion has been reported to result in genome-wide increased histone acetylation. Transcriptional regulation of neurotrophic factor BDNF and transcription factor DLX5, essential for proper neurogenesis, is further altered in MeCP2-deleted animals. We therefore investigated the chromatin environment of MeCP2 target genes BDNF and DLX5 in lymphocytes from RTT patients and human controls, and analyzed the density of histones H3, H2B and H1, as well as the levels of methylation and acetylation on selected lysines of histone H3. Notably, we found a general increase in the density of histone H3 in RTT patients’ lymphocytes compared with controls, and decreased levels of trimethylation of lysine 4 on histone H3 (H3K4me3), a modification associated with transcriptional activation. The levels of acetylation of lysine 9 (H3K9ac) and 27 (H3K27ac) did not show any statistically significant changes when normalized to the decreased histone H3 levels; nevertheless, an average decrease in acetylation was noted. Our results reveal an unexpected alteration of the chromatin state of established MeCP2 target genes in lymphocytes of human subjects with RTT.
Keywords: Rett syndrome, MeCP2, RTT, lymphocytes, histone H3
Introduction
Rett syndrome (RTT) is a developmental disorder manifested by primarily neurological symptoms, such as motor disabilities, mental retardation, communication problems, epilepsy and breathing disturbances. Although the diagnosis is clinical and not genetic, about 95% of patients with classical RTT have mutations in the MeCP2 gene.1 MeCP2 binds preferentially to methylated CpG motifs in the genome2 and genome-wide profiling of MeCP2 binding showed MeCP2 only on methylated DNA.3 In the study by Skene et al., it was also suggested that MeCP2 was present at levels equaling histones in neurons, while MeCP2 expression was absent in glia cells. Contradicting this finding are several studies showing MeCP2 expression in astrocytes and microglia and that MeCP2 functions in astrocytes and microglia affect the phenotype in mouse models of RTT.4-8
A major effort has been made to find specific target genes regulated by MeCP2.9,10 One direct target, BDNF, is a growth factor important for survival, growth and differentiation of neural cells. The BDNF IV promoter can be bound directly by MeCP2 and repress transcription.11 However, neuronal depolarization that triggers Ca2+ dependent phosphorylation of MeCP2 has been reported to relieve this repression. In RTT patients, as well as in MeCP2 gene deleted mice, BDNF is paradoxically downregulated and, when overexpressed in mice, can partially compensate for the MeCP2 mutant phenotype.12 The downregulation of BDNF in MeCP2 mutants could be an indirect effect of loss of neuronal activity.13
Imprinting of genes to ensure expression from only the maternal or paternal allele is maintained by DNA methylation. Since MeCP2 is binding preferentially to methylated DNA, its involvement in genomic imprinting has been studied. DLX5 is one gene that has been shown to lose imprinting in lymphoblastoid cells from RTT patients as well as in MeCP2 null mouse brains.14 DLX5 is, among other things, a regulator of the expression of the enzymes that synthesize GABA, GAD1 and GAD2. It has further been shown that MeCP2 may have more general effects on chromatin structure. On methylated DNA, MeCP2 binds chromatin as a dimer to nucleosome linker DNA in a manner that is very similar to the linker histone H1, and it has therefore been suggested that MeCP2 compete with histone H1 for DNA binding.15
Despite molecular insights into MeCP2 mutations underlying RTT and a large number of studies of MeCP2 function, there is still a lack of connection between how MeCP2 functions on the DNA and the phenotypic outcome at the cell and organism level. In this study, we aimed at investigating alterations of the chromatin environment on MeCP2 target genes in human samples from patients with RTT compared with controls. Our long-term goal is to develop protocols to assay the state of the chromatin environment as readout of therapeutic interventions. Since earlier studies have reported effects on histone acetylation in MeCP2 knockout mice, as well as expression changes on BDNF and DLX5, we aimed at specifically studying basic chromatin modifications on these MeCP2 target genes. We used chromatin immunoprecipitation (ChIP) to study the occupancy of histones and histone marks on promoters of MeCP2 target genes BDNF and DLX5 in lymphocytes from RTT patients and controls. Unexpectedly, we found that, while no significant effects were detected in histone H3 lysine acetylation (H3K9 and H3K27), histone H3 was enriched in BDNF and DLX5 promoters of lymphocytes from RTT patients compared with controls. In these promoters, the transcriptional activation mark H3K4me3 was also reduced.
Results
We used lymphocytes purified from blood samples of RTT patients or control patients and performed ChIP on chromatin from around 8,000 cells/subject using antibodies against specific histone modifications to investigate whether there was any correlation in histone modifications on previously characterized downstream gene targets of MeCP2 in humans with the disease compared with healthy controls. We chose to study the histone modifications on promoters of BDNF (Fig. 1) and DLX5, two genes that have been implicated as targets of MeCP2, the products of which play important roles in normal neuronal development. In addition to being essential for neuronal development, both BDNF and DLX5 are expressed also in lymphocytes, allowing us to study the consequences of RTT on these promoters in these blood cells. We also used two control loci, Myglobin exon 2, which should not be expressed in lymphocytes, and the promoter of C-FOS, which should be expressed in most proliferating cell types.
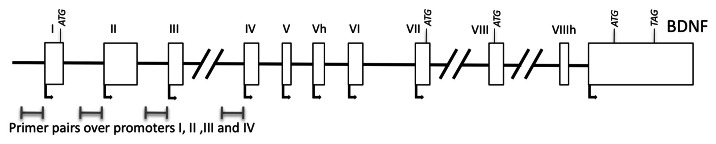
Figure 1. Model of the BDNF locus with primers marked over promoters I-IV
The density of histone H3 was significantly higher in the myoglobin and C-FOS loci of RTT patient samples compared with controls (Fig. 2A). When all analyzed loci were taken into account and tested statistically with a mixed model analysis, there was a significant difference between RTT samples and controls, showing that H3 density throughout the tested regions was consistently higher in RTT patient samples. In order to test whether this was a reflection of a general increase in histone density, we further examined the density of histone H2B but did not find a reproducible increase in the RTT patient samples on any loci, and a slight trend toward higher histone H2B occupancy did not reach statistical significance (Fig. 3B).

Figure 2. RTT syndrome is associated with higher density of histone H3 and lower levels of histone H3 methylation on promoters in lymphocytes. ChIPs of histone H3 (A), histone H3, trimethylated on lysine 4 (H3K4me3) (B), histone H3 acetylated on lysine 9 (H3K9ac) (C), and histone H3 acetylated on lysine 27 (H3K27ac) (D) on BDNF promoters, DLX5 promoter, C-FOS promoter and Myglobin exon 2. H3K4me3 was significantly lower in RTT patient lymphocytes than control subjects whereas H3K9ac and H3K27ac modification ratio was not significantly altered. A trend toward lower acetylation ratio in RTT patients was noted.

Figure 3. No significant differences were seen in density of histones H1 and H2B between RTT patient lymphocytes and control subjects. No significant differences were seen in occupancy of S5P-RNAP II or occupancy of MeCP2 between RTT patient lymphocytes and control subjects. ChIPs of histone H1 (A), histone H2B (B), S5P-RNAP II (C) and MeCP2 (D) on BDNF promoters, DLX5 promoter, C-FOS promoter and Myglobin exon 2.
In accordance with the elevated histone H3 levels in the RTT samples, the modified versions of histone H3 investigated—histone H3 trimethylated on lysine 4 (H3K4me3), histone H3 acetylated on lysine 9 (H3K9ac) and histone H3 acetylated on lysine 27 (H3K27ac)—showed a trend toward higher levels of occupancy in the RTT samples with the only statistically significant change being an increase in H3K9ac on the myoglobin exon (data not shown). None of the modifications showed a significant difference in the mixed model analysis when comparing RTT samples and controls on all loci.
However, when compensated for the general increase in histone H3 levels by normalizing over H3, a significant decrease in H3K4me3 in RTT samples compared with control was evident (Fig. 2B). Putative changes in the levels of acetylation on the other lysines were found not to be statistically significant (Fig. 2C and D). Our data suggest that RTT samples display a higher histone H3 density than control samples and a lower proportion of histone H3 is modified.
We further analyzed whether the density of linker histone H1 was altered in the RTT patient samples, as previously suggested in brain samples from mice with RTT-like phenotype.3 However, we did not detect any significant differences in histone H1 occupancy between RTT patients and controls on the analyzed loci (Fig. 3A). In order to assess whether the observed changes in histone H3 occupation and modifications resulted in direct effects on transcription, we performed ChIP against RNA polymerase II phosphorylated on serine 5 (S5P-RNAP II), an established mark of active transcription. However, there was no significant difference between RTT patient samples and control samples in these experiments (Fig. 3C). Lastly, we tested whether there was any significant difference in the occupancy of MeCP2 itself that could be due to altered DNA methylation or other secondary events. There were minor variations between patients as well as control subjects, but no significant difference between the groups was detected. This result suggests that there are no major general differences in DNA methylation affecting the recruitment of MeCP2 to DNA (Fig. 3D).
Discussion
A general effect of MeCP2 gene deletion and mutation has been suggested through the demonstration of an increase in H3 acetylation throughout the genome, indicating that MeCP2 is generally associated with decreased global H3 acetylation levels when it is present.3 Notably, these results were achieved when levels of acetylated histone H3 were plotted without accounting for total levels of histone H3. Remarkably, our results for H3K9ac on the promoters of BDNF, DLX5 and C-FOS and an exon of myoglobin show striking similarities to the genome-wide ChIP sequencing results from Skene et al. when normalization was performed toward total input. In this study, we demonstrate a major increase of histone H3 density in the analyzed promoters of samples from RTT patients compared with controls. Thus, when histone H3 levels were chosen for normalization of histone H3 modifications, it became evident that the ratio of acetylated histone H3 (H3K9 and H3K27) is actually lower in lymphocytes from RTT patients than in control subjects. Similar analysis in neuronal and neural cells is definitely required, but we strongly suggest that future studies take into account potential aberrant occupancies of histone H3 when analyzing histone modifications in models of RTT.
MeCP2 has been reported to bind directly to the BDNF IV promoter,11 whereas BDNF I and II promoters are regulated by REST, which in turn is regulated by MeCP2.16 Therefore, we hypothesized that the effect of lower MeCP2 activity would have different effects on the different promoters of BDNF. However, the results from RTT patient samples and control samples were similar on the four tested promoters regarding the analyzed modifications analyzed and histone H3 accumulation.
DLX5 is an imprinted gene that is normally only expressed from the maternal allele. Imprinting of DLX5 has been shown to be lost in MeCP2 gene deleted mice as well as in lymphoblastoid cells from RTT patients.14 Similarly, we found effects on histone H3 density as well as on histone H3 modifications at the DLX5 locus, but did not detect any differences specific to this imprinted region. The effects on histone H3 occupancy showed a similar trend on all tested loci, including both active promoters and myoglobin exon (which should not be expressed in lymphocytes), suggesting that the effects we noted may be general.
Even though the occupancy of histones and histone modifications throughout the genome has not been extensively studied in RTT patients, there have been studies of histone levels using immunoblotting of proteins. In clonal T cells, no significant change in histone acetylation was detected.17 However, in primary lymphocytes, histone H3 levels were indeed reported to be higher in RTT patients than controls, whereas the levels of acetylated H3K9 were found to be lower.18 These findings are similar to our results using ChIP on specific MeCP2 target genes. In MeCP2 mutant astrocytes from mice, levels of acetylated histone H3 were slightly elevated when similar amounts of histone H3 were analyzed.4 It has further been reported that histone H1 display higher levels in neurons from MeCP2 gene deleted mice than in control.3 When we analyzed the occupancy of histone H1, we did not see any significant changes between RTT patient samples and controls (Fig. 3A). It should be noted that Skene et al. only found a significant change in histone H1 levels when they used sorted neurons but not when using whole brain samples. Thus, although our results from human lymphocytes indeed show clear similarities to the phenotype seen in mouse neurons, cell-specific effects of loss of MeCP2 activity should be taken into account.
Since the MeCP2 gene is present on the X chromosome and either the mutated or the wild type X chromosome can be inactivated, there may be varying contributions of the different X chromosomes in different samples. Nevertheless, in samples of mixed lymphocytes, enough are expressing MeCP2 from the mutated X chromosome to show the marked differences we have observed. When expression changes due to MeCP2 inactivation have been studied previously, small expression changes have been observed on many genes making it hard to find bona fide targets of MeCP2.19-21 We assessed transcription by determining the occupancy of serine 5SP-RNAP II and, although there was a trend of higher accumulation in RTT patient samples, there was no statistically significant difference between the groups, suggesting that the differences in histone H3 occupancy and modification do not lead to obvious changes in RNAP II recruitment and onset of transcription.
While the RTT patients displayed an increase in histone H3 occupancy on several loci, this increase was not followed by a parallel increase in histone H2B occupancy. It is therefore not likely that our observation is a result of a general increase in nucleosome density. The histone H3 antibody used in the present study recognizes the 100 amino acids in the C-terminus, and this sequence is not a major region for histone modifications or variants. It is thus less likely that a change in the expression of histone H3 variant/s would result in the observed change. Transcriptional repression indeed affects histone H3 occupancy over the repressed promoter.22 It is thus possible that lack of functional MeCP2 affect transcriptional repression mechanisms and thereby histone H3 occupancy. To further address this and other questions raised by our results, current investigations are addressing the chromatin state in cell samples from RTT patients vs. healthy controls on a genome wide level.
Methods
Clinical data and subjects
The parents of patients with Rett syndrome (8 patients, age 7–23 y) and healthy controls (7 controls, age 2–14 y) of roughly similar age in Stockholm County were asked to take part in the present study. The control samples were taken from girls with no previous or present neurological or medical disease but in whom a blood sample was clinically motivated due to surgical treatment. The clinical evaluation took place at the neuropediatric department at Karolinska University Hospital and the patient was seen by two or three neuropediatricians. The RTT clinical diagnosis was based on distinct clinical criteria23,24 and patients were genotyped for mutations in MeCP2.
Ethics
The subjects and their parents received oral and written information about the procedure, and informed consent was obtained from the parents. The experimental protocol used in the present study was approved by the Ethics Committee of Karolinska Institutet, Stockholm, Sweden.
Isolation of lymphocytes from peripheral blood sample
Ficoll-Paque PLUS (GE Healthcare) was used according to instructions to purify lymphocytes from patient and control blood samples.
Chromatin Immunoprecipitation (ChIP)
Cells were fixed in 1% formaldehyde for 10 min. Chromatin shearing by sonication was performed using a Bioruptor UCD 200 (Diagenode). Chromatin immuno precipitation was performed according to instructions using the “Low cell #” kit from Diagenode using chromatin from around 8000 cells per reaction (50ng DNA). H3K4me3 antibody 9751 and H3K9ac 9671 was from Cell Signaling, H3K27ac antibody ab4729, H3 antibody 1791, H2B antibody 1790 and Pol II ser5pho 5131 was from Abcam, H1 antibody 05-457 was from Millipore MeCP2 pAb 052 was from Diagenode and control IgG was included in the ChIP kit. The ChIP was evaluated by qPCR using “platinum SYBR Green qPCR SuperMix-UDG” from Invitrogen. Results were normalized to 1% input and H3 levels (Table 1).
Table 1. Primers used.
| Gene | Forward | Reverse |
|---|---|---|
|
BDNF I promoter |
CCCTCCCCCATCATGACTA |
CCATTTGATCATCACTCACGA |
|
BDNF II promoter |
ATCGCCCGGATTACACAC |
TGGAAGAAACCGTCTAGAGCA |
|
BDNF III promoter |
ACCCAGAAAGAAGCATCCAG |
CTCCATCCCTCCCTCATTCT |
|
BDNF IV promoter |
TGCACGAATTACCAGAATCAA |
GCTGGAAGTGAAAACATCTGC |
|
Myoglobin exon 2 |
Diagenode primers |
|
|
C-FOS promoter |
Diagenode primers |
|
| DLX5 promoter | GGAGACTGGGAGTCGTGAAG | GGCCAATAGAACCAGATCCA |
Statistical analysis
Differences between RTT and control samples on individual loci were compared using students T test. Differences between RTT and control samples on all loci for different antibodies was compared using a mixed linear model with one within group factor, the different loci tested (7 levels) and one between group factor Group (patient and control) was used to analyze the data. Different covariance models were tested and the covariance structure with the smallest value of the Akaike’s Information Criterion (AICC and BIC) was considered to best fit the data. The distribution of the variables was positively skewed and before the formal analyses the variables were log-transformed or square root transformed. p < 0.05 was considered statistically significant. Software used: SAS® System 9.1, SAS Institute Inc.
Acknowledgments
The authors would like to express our gratitude to the families who have participated in this study. This study was supported by Sällskapet Barnavård, The Sven Jerring foundation, Samariten, ALF, the Swedish Heart and Lung Foundation (to M.R. and H.L.), VR-MH, DBRM, KI TEMA, the Swedish Childhood Cancer Foundation (BCF), and the Swedish Cancer Society (CF) (to O.H.).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/23752
References
- 1.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 2.Percy AK. Rett syndrome: recent research progress. J Child Neurol. 2008;23:543–9. doi: 10.1177/0883073807309786. [DOI] [PubMed] [Google Scholar]
- 3.Skene PJ, Illingworth RS, Webb S, Kerr AR, James KD, Turner DJ, et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–68. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ballas N, Lioy DT, Grunseich C, Mandel G. Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat Neurosci. 2009;12:311–7. doi: 10.1038/nn.2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lioy DT, Garg SK, Monaghan CE, Raber J, Foust KD, Kaspar BK, et al. A role for glia in the progression of Rett’s syndrome. Nature. 2011;475:497–500. doi: 10.1038/nature10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. 2010;30:5346–56. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maezawa I, Swanberg S, Harvey D, LaSalle JM, Jin LW. Rett syndrome astrocytes are abnormal and spread MeCP2 deficiency through gap junctions. J Neurosci. 2009;29:5051–61. doi: 10.1523/JNEUROSCI.0324-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Derecki NC, Cronk JC, Lu Z, Xu E, Abbott SB, Guyenet PG, et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature. 2012;484:105–9. doi: 10.1038/nature10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vecsler M, Simon AJ, Amariglio N, Rechavi G, Gak E. MeCP2 deficiency downregulates specific nuclear proteins that could be partially recovered by valproic acid in vitro. Epigenetics. 2010;5:61–7. doi: 10.4161/epi.5.1.10630. [DOI] [PubMed] [Google Scholar]
- 10.Stancheva I, Collins AL, Van den Veyver IB, Zoghbi H, Meehan RR. A mutant form of MeCP2 protein associated with human Rett syndrome cannot be displaced from methylated DNA by notch in Xenopus embryos. Mol Cell. 2003;12:425–35. doi: 10.1016/S1097-2765(03)00276-4. [DOI] [PubMed] [Google Scholar]
- 11.Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, et al. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–9. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 12.Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–8. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 13.Chadwick LH, Wade PA. MeCP2 in Rett syndrome: transcriptional repressor or chromatin architectural protein? Curr Opin Genet Dev. 2007;17:121–5. doi: 10.1016/j.gde.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 14.Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet. 2005;37:31–40. doi: 10.1038/ng1491. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh RP, Horowitz-Scherer RA, Nikitina T, Shlyakhtenko LS, Woodcock CL. MeCP2 binds cooperatively to its substrate and competes with histone H1 for chromatin binding sites. Mol Cell Biol. 2010;30:4656–70. doi: 10.1128/MCB.00379-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abuhatzira L, Makedonski K, Kaufman Y, Razin A, Shemer R. MeCP2 deficiency in the brain decreases BDNF levels by REST/CoREST-mediated repression and increases TRKB production. Epigenetics. 2007;2:214–22. doi: 10.4161/epi.2.4.5212. [DOI] [PubMed] [Google Scholar]
- 17.Balmer D, Arredondo J, Samaco RC, LaSalle JM. MECP2 mutations in Rett syndrome adversely affect lymphocyte growth, but do not affect imprinted gene expression in blood or brain. Hum Genet. 2002;110:545–52. doi: 10.1007/s00439-002-0724-4. [DOI] [PubMed] [Google Scholar]
- 18.Kaufmann WE, Jarrar MH, Wang JS, Lee YJ, Reddy S, Bibat G, et al. Histone modifications in Rett syndrome lymphocytes: a preliminary evaluation. Brain Dev. 2005;27:331–9. doi: 10.1016/j.braindev.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 19.Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–9. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delgado IJ, Kim DS, Thatcher KN, LaSalle JM, Van den Veyver IB. Expression profiling of clonal lymphocyte cell cultures from Rett syndrome patients. BMC Med Genet. 2006;7:61. doi: 10.1186/1471-2350-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Urdinguio RG, Lopez-Serra L, Lopez-Nieva P, Alaminos M, Diaz-Uriarte R, Fernandez AF, et al. Mecp2-null mice provide new neuronal targets for Rett syndrome. PLoS One. 2008;3:e3669. doi: 10.1371/journal.pone.0003669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li LM, Arnosti DN. Long- and short-range transcriptional repressors induce distinct chromatin states on repressed genes. Curr Biol. 2011;21:406–12. doi: 10.1016/j.cub.2011.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagberg B. Condensed points for diagnostic criteria and stages in Rett syndrome. Eur Child Adolesc Psychiatry. 1997;6(Suppl 1):2–4. [PubMed] [Google Scholar]
- 24.Neul JL, Kaufmann WE, Glaze DG, Christodoulou J, Clarke AJ, Bahi-Buisson N, et al. RettSearch Consortium Rett syndrome: revised diagnostic criteria and nomenclature. Ann Neurol. 2010;68:944–50. doi: 10.1002/ana.22124. [DOI] [PMC free article] [PubMed] [Google Scholar]