

Abstract
Brain cellular heterogeneity may bias cell type specific DNA methylation patterns, influencing findings in psychiatric epigenetic studies. We performed fluorescence activated cell sorting (FACS) of neuronal nuclei and Illumina HM450 DNA methylation profiling in post mortem frontal cortex of 29 major depression and 29 matched controls. We identify genomic features and ontologies enriched for cell type specific epigenetic variation. Using the top cell epigenotype specific (CETS) marks, we generated a publically available R package, “CETS,” capable of quantifying neuronal proportions and generating in silico neuronal profiles from DNA methylation data. We demonstrate a significant overlap in major depression DNA methylation associations between FACS separated and CETS model generated neuronal profiles relative to bulk profiles. CETS derived neuronal proportions correlated significantly with age in the frontal cortex and cerebellum and accounted for epigenetic variation between brain regions. CETS based control of cellular heterogeneity will enable more robust hypothesis testing in the brain.
Keywords: DNA methylation, neurons, glia, fluorescence activated cell sorting, epigenetics, cellular heterogeneity, microarray, age, brain region
Introduction
Until recently, the combination of genetic risk factors in conjunction with environmental influence was believed to cause complex non-Mendelian diseases. Over recent years, a paradigm shift has occurred and an increasing number of studies have focused on a search for epigenetic differences and their contribution to disease risk; however, despite the promise of epigenetic etiology in conferring disease risk, the success of the first round of epigenomic studies in psychiatric disease has been limited.1 In the first epigenomic profiling studies performed in major psychosis, Mill et al. found moderate fold changes in prefrontal cortex DNA methylation. In the WDR18 glutamate receptor subunit gene, an 8% DNA methylation difference was detected between males with schizophrenia and controls, while female patients with bipolar disorder were 6% more methylated than controls at the RPL39 gene.2 No significant differences were found in an analysis of 50 loci in the temporal cortex of schizophrenia affected individuals.3 A recent methylome profiling study in major depressive disorder (MDD) did not identify any significant loci after correction for multiple testing; however, it successfully validated 60% of the top nominally significant differences.4 In the above studies, the brain samples interrogated consisted of “bulk” brain tissue preparations representing cellularly heterogeneous mixes of neuronal and non-neuronal cell types.
Cellular heterogeneity in the nervous system is important because DNA methylation has long been established as a distinguishing feature of differing cell types.5-8 Recent DNA methylation microarray profiling studies using the comprehensive high-throughput arrays for relative methylation (CHARM) technique9 identified tissue-specific differentially methylated regions (tDMRs) in CpG island adjacent regions called CpG island shores.10 DNA methylation in shores is capable of distinguishing bulk brain regions, as well as pluripotent stem cells from differentiated cells.10,11 Other tDMRs were identified as being responsible for myeloid vs. lymphoid cell fate decisions from hematopoietic stem cell progenitors.12 Tissue heterogeneity could therefore confound epigenetic studies in two ways.
First, heterogeneity of measurements is brought about by differing ratios of cellular subtypes in the individuals tested. This issue is of particular importance in psychiatric diseases where the morphology of various brain regions is changed. MRI imaging studies identified a reduced hippocampal volume in females with depression13 while smaller inferior frontal gyri of the dorsolateral prefrontal cortices were correlated with increased lifetime manic episodes in bipolar patients.14 Using an optical dissector, Cotter et al. demonstrated that neuronal but not oligodendroglial density was decreased in cortical layers 1 and 5 in bipolar and depression patients,15 while Urnova et al. found a reduction of oligodendroglial cells in schizophrenia, bipolar, and depression patients.16 Alternate reports suggest that neuronal inflammation may elevate levels of activated microglia,17 which could in turn skew observed levels of cell type specific epigenetic patterns. In this way, a large proportion of disease-implicated loci identified in epigenomic studies may be simply a consequence of morphological abnormalities of disease or other facets of disease state such as inflammation or neurodegeneration.
The second way cellular heterogeneity may confound psychiatric epigenetic studies is a dilution of observed disease effects by alternate cell types not exhibiting the disease effect. Using an isotropic fractionation method, Azevedo et al. determined that the human CNS contains approximately equal numbers of neurons and glia as a whole, but a ratio of 3.76/1 glia to neurons in the cerebral cortex.18 Glutamatergic and dopaminergic neurons are implicated in schizophrenia,19,20 while serotonergic systems are involved in MDD;21 however, the relative proportions of these neuronal subtypes are small relative to the cellular architecture in the CNS regions investigated. If psychiatric disease relevant epimutations are occurring within specific subtypes, decreases in the observed effect sizes may be expected if sampling is performed from bulk tissue.
In this paper, we present the generation of cell epigenotype specific (CETS) maps in the cortex and introduce a new bioinformatics model capable of quantifying the proportion of neurons to glia based on DNA methylation measures across multiple CETS markers. Furthermore, we provide a technique for transforming existing DNA methylation data sets derived from bulk tissue preparations generated on Illumina microarrays to neuronal and glial DNA methylation profiles. We demonstrate the application of these techniques to the analysis of DNA methylation differences in MDD.
Results
Identification of CETS markers
Following FACS based isolation of neuronal and non-neuronal nuclei (Fig. S1), a paired t-test between neuronal and glial DNA methylation samples per locus identified 32.3% of loci (n = 112,331 out of 347,536 trimmed loci) exhibited a DNA methylation change greater than 5% and were significantly different between neurons and glia after FDR based correction for multiple testing (Fig. 1A). While, on average, the effect size of all FDR significant cell type specific differences is quite small (0.067%), over 14% (n = 37,399) and 1% (n = 2693) of significant loci exhibit DNA methylation differences greater than 20 and 50%, respectively. An example of cell-type specific epigenetic differences detected is depicted in Figure S2. We observed a significant over-representation of loci exhibiting cell-type specific change in the same direction as previously identified differentially methylated regions (DMRs) unique to both neurons and non-neurons22 at 2,262 overlapping loci from the top 25th percentile of cell-type specific differences in our study (Fisher’s OR = 4.6, p value = 1.1 × 10−7).
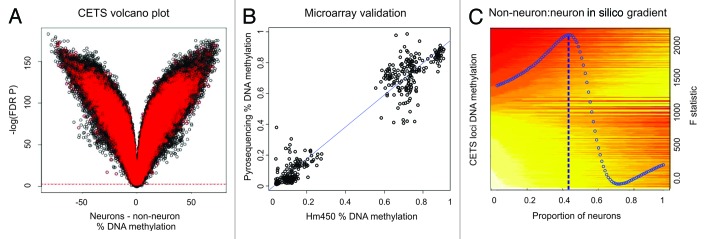
Figure 1. CETS model generation. (A) Volcano plot of DNA methylation vs. –log of FDR significance for neuronal vs. glial DNA methylation profiles. Almost the entire microarray identifies FDR significant changes across cell types. Red spots represent loci significant at a nominal p value of 0.05 in a comparison of MDD vs. control individuals that are excluded from CETS model generation. Green boxes represent top 10,000 CETS markers. (B) Scatterplot of DNA methylation as obtained by HM450 microarrays (x-axis) and by independent pyrosequencing assays (y-axis) at five loci within the top1000 CETS markers. (C) Heat map depicting the in silico virtual gradient of neuronal to glial DNA methylation at the top 10,000 CETS markers generated across our sample of 58 individuals. Yellow and red denote β values of methylated and unmethyated DNA, respectively. Linear modeling F-statistic for each neuronal proportion is overlaid (blue). Blue dashed line depicts the model prediction of 43%.
In order to generate CETS markers independent of the effects of disease status and various medication influences, we selected CpGs significantly different between neurons and non-neurons after FDR based correction for multiple testing that were not significant in a separate case-control analysis of the MDD phenotype in either neurons or non-neurons at a raw significance threshold of 5%, resulting in the exclusion of 21,290 loci. We performed independent validation of five loci located within the top 10,000 CETS markers and obtained a significant correlation with microarray values (R2 = 0.95, p = 2.2 × 10−16) (Fig. 1B).
Genomic features associated with neuronal and glial DNA methylation differences
DNA methylation differences distinguishing between neurons and non-neurons were evaluated at CpG positions associated with various gene regulatory features. For each analysis, the distribution of the absolute cell-type specific difference within a specific category was compared with that of all CpG loci not falling within that category. CpG islands (CGI) exhibited significantly less cell-type specific differences (Wilcoxon Rank Sum test, CGI = 0.040 ± 1.1 × 10−6, non-CGI = 0.085 ± 7.2 × 10−7, Bonferroni p < 1 × 10−220), while CGI shores exhibited significantly more (Wilcoxon Rank Sum test, CGI shore = 0.075 ± 1.9 × 10−6, non-CGI shore = 0.07 ± 6 × 10−7, Bonferroni p = 1.7 × 10−211). CpG loci within 5′ untranslated regions (UTR), first exons (Wilcoxon Rank Sum test, first exon = 0.035 ± 2.0 × 10−6, non-first exon = 0.066 ± 2.6 × 10−7, Bonferroni p < 1 × 10−220), and upstream of transcription start sites (TSS) (TSS = 0.047 ± 6.1 × 10−7, non-TSS = 0.07 ± 3.7 × 10−7, Bonferroni p < 1 × 10−220) exhibited significantly lower cell-type specific DNA methylation differences. Conversely, CpGs located within 3′UTRs (Wilcoxon Rank Sum test, 3′UTR = 0.087 ± 5.7 × 10−6, non-3′UTR = 0.062 ± 2.5 × 10−7, Bonferroni p < 1 × 10−220), gene bodies (Wilcoxon Rank Sum test, gene body = 0.082 ± 6.7 × 10−7, non-gene body = 0.05 ± 3.5 × 10−7, Bonferroni p < 1 × 10−220), and enhancer sequences (Wilcoxon Rank Sum test, enhancers = 0.10 + 2.6 × 10−6, non-enhancers = 0.062 + 5.4 × 10−7, p < 1 × 10−220) exhibited significantly higher DNA methylation differences between neurons and non-neurons.
Gene ontology analysis of top CETS markers
We selected a stringent set of the top 1,000 CETS markers maximizing the DNA methylation difference between neurons and glia, corresponding roughly to those loci with a FDR p value less than the lower 0.5th percentile of p-values within the CETS marker group. The average DNA methylation difference in this group was 56 ± 9.8 × 10-5%. Molecular function of genes associated with the top 1,000 CETS markers was investigated using the g.Profiler analysis suite.23 A number of significantly over-represented gene ontology (GO) categories dealing with neuron development and function were identified such as central nervous system development (GO:0007417), cell morphogenesis involved in neuron differentiation (GO:0048667), axonogenesis (GO:0007409), axon guidance (GO:0007411), dendritic spine (GO:0043197), synapse (GO:0045202) and post synaptic density (GO:0014069). The full list of over-represented GO categories can be viewed in Table S1.
A model for quantifying neuronal and glial proportions from methylation data
Using the top 10000 CETS markers, corresponding roughly to the top 5th percentile of CETS loci, we developed a method capable of quantifying the proportions of neurons and glia based on generalized and disease non-specific cell type epigenetic markers as a proxy for cellular proportions. Mean neuronal and glial DNA methylation profiles were used to generate an in silico gradient of DNA methylation mixes from 100% glia to 100% neurons at our top CETS markers (Fig. 1C). To quantify the neuronal proportion, we fit a linear slope model of the observed DNA methylation in bulk tissue at the top 10,000 CETS loci to the predicted values for each neuronal percentage of the in silico gradient. The proportion of neurons, N, is determined by the in silico gradient percentage corresponding to the best fit (largest F-statistic). The p value of the linear model at the optimal neuronal proportion is taken as a measure of model performance for a given sample after Bonferroni correction for multiple testing.
Model performance was evaluated using a number of metrics. A reconstitution experiment was performed by mixing neuron and glial derived DNA from a single individual in 10 percent increments from 0–100% for a total of 11 mixes and followed by microarray hybridization (Fig. S3). The accuracy of mixed proportions was confirmed by comparing the Euclidean distance between arrays and the expected proportion of DNA methylation difference between mixes (R2 = 1, p = 4.9 × 10−13). CETS model predictions of proportions of neurons to non-neurons matched the empirical mixes to a high degree of accuracy (R2 = 0.99, p = 2.7 × 10−11) (Fig. 2A). As an independent measure, we correlated the CETS model generated neuronal proportions against the ratio of a neuronal-nuclei specific nuclear protein, NeuN, positive: NeuN negative cell counts as determined by FACS in the 20 bulk samples and observed a significant correlation (R2 = 0.6, p = 4.2 × 10−5) (Fig. 2B). We next downloaded data generated in a recent analysis of human prefrontal cortex DNA methylation and gene expression across the lifespan (n = 100)24 and compared the mean gene expression at probes representative of RBFOX3, which encodes the NeuN protein, with neuronal proportions predicted using Illumina Infinium Human Methylation 27 (HM27) microarray loci and observed a significant correlation (R2 = 0.17, p = 3 × 10−5) (Fig. 2C).
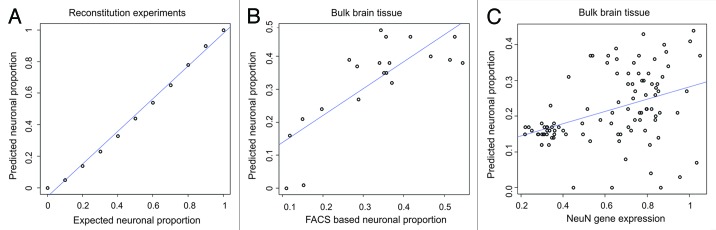
Figure 2. CETS model validation. (A) Scatter plot of predicted neuronal proportions vs. empirical mixes of neuronal and glial DNA in increments of 10%. (B) Scatter plot of predicted neuronal proportion vs. FACS based estimate in 20 bulk tissue samples. (C) Scatter plot of predicted neuronal proportion vs. NeuN gene (RBFOX3) expression (2 log(fold change)) in 100 bulk brain tissue samples evaluated on Illumina HM27 microarrays and custom gene expression platforms.24
We next sought to compare the performance of the CETS algorithm to that of a recently published quadratic programming based algorithm for quantification of cell type proportion from DNA methylation data in blood.25 Across the three test data sets above, the quadratic algorithm performed similarly to the CETS algorithm in evaluating the proper proportion of mixes (R2 = 1, p = 2.1 × 10−13), FACS based neuronal percentages in bulk tissue (R2 = 0.59, p = 8.1 × 10−5), and NeuN expression (R2 = 0.17, p = 1.9 × 10−5). A major difference between the two algorithms was identified in regards to the robustness of neuronal prediction to batch effects within a given microarray experiment. This was tested by generating in silico batch effects at random proportions of probes within the top 10,000 CETS markers and predicting neuronal proportion using both algorithms. We inserted batch effects by multiplying DNA methylation values at random probes by a range of percentages from 10–100% at a range of randomly selected probes within half of the reconstitution data set, leaving the other half untouched (Fig. S4A and B). Five iterations were performed at each level for a total of 500 comparisons. Quadratic neuronal predictions were found to be highly influenced by the proportion of probes affected by the batch effect (R2 = 0.99, p = 2.2 × 10−16) (Fig. S4C). In the CETS model, the predicted neuronal proportions across all permutations were virtually identical to the original predictions without in silico induced batch effects (mean R2 = 1 ± 1.9 × 10−6) (Fig. S4C).
As the algorithm uses the relative proportions of DNA methylation across CETS markers, the input data required is not limited to data generated on the Illumina Infinium Human Methylation450 Beadchip (HM450) platform. We performed 100 permutations of randomly selected loci within the CETS marker set and determined that numerous combinations of CETS markers of different sizes within the top 10,000 CETS set are capable of accurately determining the correct proportions of mixed neuronal and non-neuronal input DNA (Table 1; Fig. S5). To confirm this, we applied CETS model quantification to DNA methylation profiles of three NeuN positive and negative prefrontal cortical samples profiled by Iwamoto et al. on Affymetrix Human Promoter 1.0R arrays.22 The ratio of methyl-enriched to non-enriched signals at 3,841 probes overlapping with the top 10,000 CETS marker locations were used for quantification and generated predictions of 100% and 0% neuronal proportions for all NeuN positive and negative samples, respectively (AUC = 1). These analyses suggest that while the neuronal proportion prediction accuracy is optimized with large sets of CETS markers, the performance of the algorithm in subset of these markers is adequate to generate accurate predictions and is dependent only on relative methylation as opposed to absolute methylation signals.
Table 1. Random probe combination performance.
| # Probes | Mean R2 | Standard deviation |
|---|---|---|
| 5 |
0.93 |
1.0 × 10−1 |
| 10 |
0.98 |
1.2 × 10−2 |
| 25 |
0.99 |
9.7 × 10−3 |
| 50 |
0.99 |
4.6 × 10−3 |
| 75 |
0.99 |
3.6 × 10−3 |
| 100 |
0.99 |
2.5 × 10−3 |
| 200 |
0.99 |
2.1 × 10−3 |
| 300 |
0.99 |
1.6 × 10−3 |
| 400 |
0.99 |
1.4 × 10−3 |
| 500 |
0.99 |
1.3 × 10−3 |
| 600 |
0.99 |
1.3 × 10−3 |
| 700 |
0.99 |
1.1 × 10−3 |
| 800 |
0.99 |
9.5 × 10−4 |
| 900 |
0.99 |
8.2 × 10−4 |
| 1,000 |
0.99 |
8.6 × 10−4 |
| 2,000 |
0.99 |
6.1 × 10−4 |
| 3,000 |
0.99 |
5.3 x10−4 |
| 4,000 |
0.99 |
4.6 × 10−4 |
| 5,000 |
0.99 |
3.5 × 10−4 |
| 6,000 |
0.99 |
3.0 × 10−4 |
| 7,000 |
0.99 |
2.6 × 10−4 |
| 8,000 |
0.99 |
1.5 × 10−4 |
| 9,000 |
0.99 |
1.3 × 10−4 |
| 10,000 | 0.99 | NA |
Transformation of bulk tissue profiles to neuronal and glial specific profiles
We generated an algorithm capable of transforming bulk tissue derived DNA methylation profiles to an expected neuronal and glial profile. The method uses the estimated neuronal proportions generated in the above section to determine the amount of neuronal or glial signal to subtract from each data point generalized at CETS markers. If a data point at a given locus is not significant at an FDR level of 5%, no transformation is performed. While the quantification algorithm presented above is applicable to data generated on multiple microarray platforms, this algorithm is applicable only to Illumina HM450 or HM27 platforms. We evaluated the algorithm’s capability to generate neuronal or glial specific profiles in the empirically mixed sample by correlating the 100% neuron DNA methylation profile to the bioinformatically generated neuronal profile for each 10% mix (Table 2 and Fig. 3A and B). The model accounted for over 93% of the variance across the spectrum of potential neuron to glia ratios (Table 2 and Fig. 3A and B).
Table 2. Comparison of FACS based vs. bioinformatically transformed neuronal profile.
| % Neuron DNA | % Glia DNA | R non-transformed | R transformed |
|---|---|---|---|
| 10 |
90 |
-0.46 |
0.98 |
| 20 |
80 |
-0.42 |
0.98 |
| 30 |
70 |
-0.32 |
0.97 |
| 40 |
60 |
-0.18 |
0.97 |
| 50 |
50 |
0.03 |
0.97 |
| 60 |
40 |
0.29 |
0.97 |
| 70 |
30 |
0.62 |
0.97 |
| 80 |
20 |
0.88 |
0.98 |
| 90 | 10 | 0.98 | 0.99 |
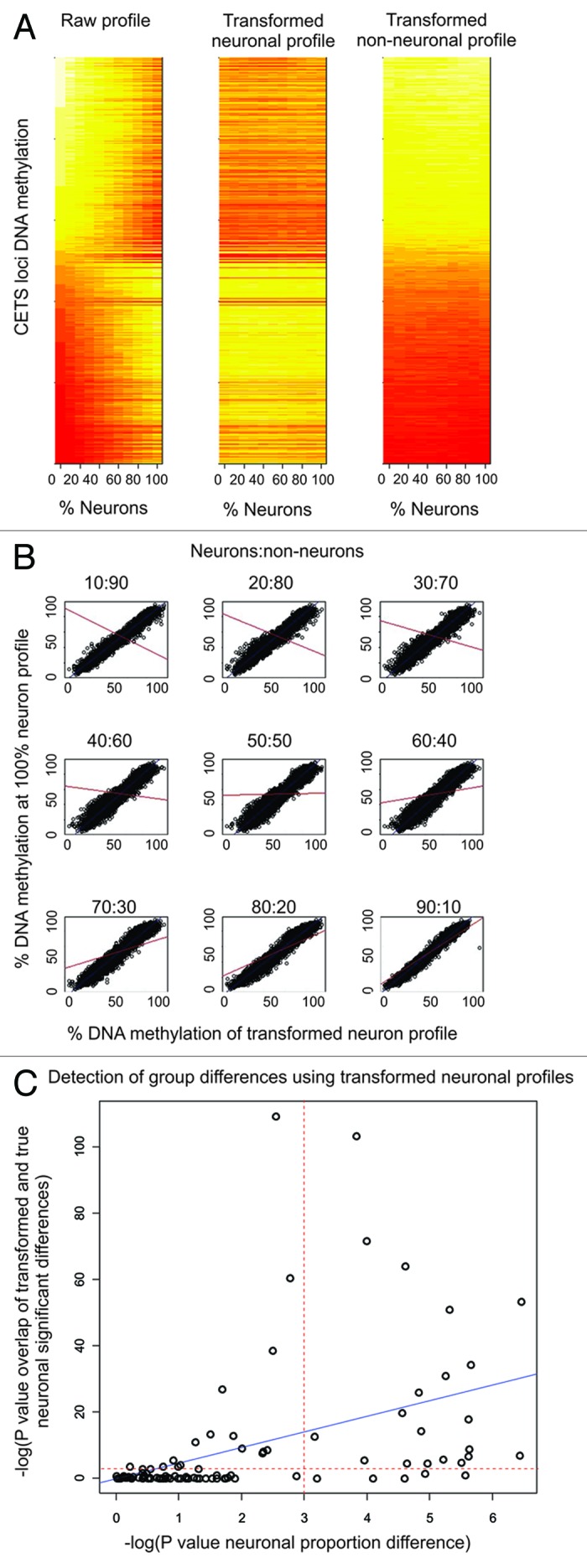
Figure 3. Transformation of bulk tissue derived data to in silico neuronal and non-neuronal profiles. (A) Heat Maps of raw non-transformed, transformed neuronal, and transformed glial DNA methylation values at the top 10,000 CETS markers across the empirically mixed sample range. (B) Scatter plots of the raw 100% neuronal vs. the transformed neuronal DNA methylation profile at the top 10000 CETS markers across the empirically mixed sample range with the blue line depicting the line of best fit. Red lines represent the line of best fit for the correlation between 100% neuronal vs. non-transformed DNA methylation profile across the empirically mixed sample range. (C) Scatter plots of the –log of the p-values generated from 100 iterations of randomly shuffling 20 bulk tissue samples. The –log(p value) of a Fisher’s exact test evaluating the degree of overlap between FACS derived neuronal profiles and the transformed and non-transformed bulk tissues in each of the 100 pair wise comparisons (y-axis) is plotted as a function of the –log(p value) of a test evaluating the group-wise neuronal proportion differences (x-axis) for each of the 100 random comparisons.
Identification of phenotype associations using transformed data
The ability of transformed neuronal profiles to identify cell type specific disease associations was investigated in 20 bulk tissue preparations hybridized to the HM450 microarray and compared with FACS isolated neuronal cell preparations. Each sample was assigned to one of two groups, followed by statistical comparison of the mean neuronal proportion per group and a spot-wise t-test based association to the group classifier in the non-transformed bulk tissue and the CETS model transformed cell type specific profile. The degree of overlap observed between loci achieving a nominal significance of 5% in the FACS separated neurons was compared between the transformed and non-transformed bulk tissue data by Fisher’s exact test. We randomly permuted group assignments and iterated the test 100 times. We observed that the larger the difference in neuronal proportion between groups, the CETS model derived neuronal profile identifies significantly higher overlap with neuron specific group associations as compared with the bulk derived data (R = 0.48, p = 4.7 × 10−7) (Fig. 3C). Similarly, evaluating significance with a linear model incorporating neuronal proportion as a covariate generates significantly higher overlap than that of bulk data (R = 0.57, p = 5.2 × 10−10). These analyses demonstrate the efficacy of the model for identifying neuron specific DNA methylation associations in data generated from bulk tissue hybridizations.
A comparison of FACS based vs. CETS model transformed MDD DNA methylation associations
CETS based quantification was applied to a data set investigating DNA methylation associations in MDD data from the Stanley Medical Research Institute (SMRI).4 Smoothed DNA methylation levels derived from the CHARM package at 8,130 probes overlapping the top 10,000 CETS loci were used for quantification of neuronal proportions. A significantly higher proportion of neurons were observed in the MDD group (mean proportion controls = 0.51 ± 0.0034, mean proportion depression = 0.55 ± 0.0028, Wilcoxon rank sum p = 0.05) (Fig. 4A). A re-analysis of CHARM data was performed following incorporation of neuronal proportion information. CHARM data was transformed by taking the residuals of a linear model of probe fold change information and neuronal proportion for each locus, followed by DMR identification and quantification using the CHARM package standard functions. Of the 420 DMRs originally identified as significant below a nominal p-value of 0.05, 33% were identified as nominally significant in the CETS model corrected data. While the original analysis did not return any hits significant after correction for multiple testing, CETS modeled data identified three DMRs located proximal to the LASS2, PCTK1 and FAM20B genes. A total of 77 DMRs adjacent to genes exhibited a trend toward significant association to MDD at a significance level below 10%. A full list of nominally significant DMRs generated in the CETS modeled data appears in Table S2.
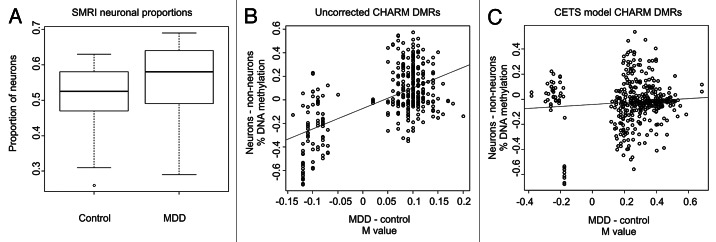
Figure 4. Identification and correction of cell heterogeneity in MDD. (A) Boxplots of the proportion of CETS model predicted neuronal proportions for control and MDD cases. (B) Scatterplot of the log2 fold change (M value) between MDD and controls in non-corrected CHARM data (x-axis) vs. the percentage of DNA methylation difference in FACS separated neuronal and glial nuclei (y-axis) at overlapping loci between the CHARM and HM450 microarray platforms. (C) Scatterplot of the M value between MDD and controls in CETS model corrected CHARM data (x-axis) vs. the percentage of DNA methylation difference in FACS separated neuronal and glial nuclei (y-axis) at overlapping loci between the CHARM and HM450 microarray platforms.
We cross-referenced nominally significant DMRs from both analyses with FDR significant cell-type specific DNA methylation differences generated on the HM450 microarrays above. A significant correlation was observed between nominally significant DMRs from the non-corrected CHARM data and overlapping loci with FDR significant cell-type specific DNA methylation differences from our FACS based experiments above (R2 = 0.33, p = 2.2 × 10−16) (Fig. 4B), suggesting a portion of the identified DMRs are resultant from the differences in neuronal proportion between groups. Conversely, DMRs identified in CETS model transformed data did not demonstrate a relationship to cell-type specific differences (R2 = 0.006, p = 0.07) (Fig. 4C).
Independently, we generated MDD specific DNA methylation associations in our FACS sorted neuronal and non-neuronal nuclear fractions within the Caucasian population of the NICHD sample. No loci exhibited significance after correction for multiple testing in either comparison. To assess the replicability of MDD associations across cohorts, the direction of DNA methylation change between MDD and controls was compared at nominally significant DMRs identified from the SMRI CHARM analyses and NICHD analyses. DMRs identified in the CETS modeled SMRI data exhibited a significantly higher proportion of DNA methylation changes in the same direction as the NICHD neuronal data set as compared with the non-transformed CHARM data (CETS modeled overlap = 92%, non-modeled overlap = 36%, Fisher’s OR = 17.3, p = 0.0053). Similarly in non-neuronal nuclei, a significantly higher agreement between NICHD and SMRI CETS modeled data was observed (CETS modeled overlap = 39%, non-modeled overlap = 11%, Fisher’s OR = 5.1, p = 3 × 10−4). While the degree of MDD specific DNA methylation associations identified in each cohort was not high, these analyses suggest that the CETS model transformation leads to a higher cross cohort agreement and that removal off cellular heterogeneity based confounds can improve the potential for cross cohort replication of disease specific findings.
Model based comparison of different brain regions and age
Using the CETS model, we quantified neuronal proportion across 506 individuals and across four brain regions using DNA methylation profiles generated on Illumina HM27 beadchip microarrays by Gibbs et al.26 We observed significant variation in neuronal proportion across brain regions relative to the frontal cortex. Frontal cortex was significantly different from pons (neuronal proportion pons = 0.093 ± 0.00029, frontal cortex = 0.31 ± 0.00076, p = 1.6 × 10−33) and cerebellum (neuronal proportion cerebellum = 0.44 ± 0.00062, frontal cortex = 0.31 ± 0.00076, p = 7.5 × 10−32), while the temporal cortex was not significantly different (neuronal proportion temporal cortex = 0.32 ± 0.00082, frontal cortex = 0.31 ± 0.00076, p = 0.12) (Fig. 5A). The degree of difference is consistent with hierarchical clustering of DNA methylation data presented by Gibbs et al. We observe a significant correlation between the relative Euclidean distance of between microarrays and the distance between neuronal proportions (Spearman’s Rho = 0.69, p = 2.2 × 10−16) (Fig. 5B). We performed spot-wise t-tests across tissues and observed highly significant FDR corrected differences similar to that of our neuron vs. non-neuron comparison in Figure 1A with FDR p values ranging as low as 3.1 × 10−126 (Fig. 5C). Correcting for neuronal proportion results in no FDR significant differences. Cumulatively, this data suggests that a majority of variation in DNA methylation previously reported between brain regions is due to differing proportions of neuronal to non-neuronal cells.
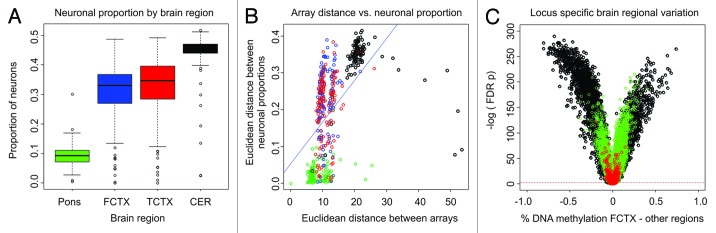
Figure 5. Brain region specific epigenetic variation is a function of neuronal proportion. (A) Boxplots of the CETS model predicted neuronal proportion (y-axis) in Pons (green), frontal cortex (FCTX, blue), temporal cortex (TCTX, red), and cerebellum (CER, black) (x-axis). (B) A plot of the neuronal proportion (y-axis) vs. euclidean distance of each array (x-axis). Color coding is the same as in A. (C) Volcano plots depicting the –log FDR significance of FCTX vs. pons (green), TCTX (red), and CER (black).
We next investigated the relationship between predicted neuronal proportion and age per brain region. No correlations were observed in the pons (Spearman's Rho = -0.11, p = 0.21) or temporal cortex (Spearman's Rho = 0.078, p = 0.38), while neuronal proportion was found to increase with age in the frontal cortex (Spearman's Rho = 0.2, p = 0.021) and cerebellum (Spearman's Rho = 0.53, p = 2.9 × 10−10) (Fig. 6).
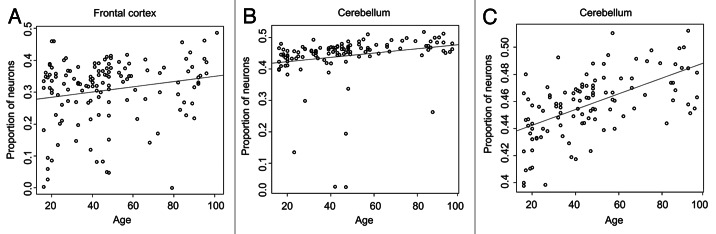
Figure 6. Variation of neuronal proportion by age. CETS model predicted neuronal proportion (y-axis) vs. age in years (x-axis) for frontal cortex (A), cerebellum (B), and cerebellum excluding outlier neuronal predictions (C). Outliers were those predictions located beyond the 9th and 91st percentile of the predicted neuronal distribution.
Discussion
We have generated a novel set of bioinformatics tools designed to identify and correct for cellular heterogeneity based bias in genome-scale DNA methylation studies in the brain. Recently, techniques have been developed for the bioinformatics adjustment of cell subfraction heterogeneity in peripheral blood cells based on DNA methylation proxies for FACS sorted cell-types;25 however, to our knowledge, our study represents the first attempt to generate such a model in brain. DNA methylation profiling has been performed previously in FAC sorted neuronal and non-neuronal nuclear populations using Affymetrix tiling microarrays;22 however, the relatively small sample size of three individuals for this study calls into question the generalizability of these markers for neuronal quantification in the population. Our results confirm and expand upon the original findings by Iwamoto et al. and allow us to define more generalizable cell-type specific DNA methylation differences between neurons and glia that are independent of psychiatric phenotype. The cohort investigated in our study is significantly larger and derives from both healthy control brains and those with a psychiatric disease. The inclusion of MDD cases in the sample allowed for the refinement of a more robust set of CETS markers through exclusion of disease associated loci that may exhibit variation in DNA methylation due to disease or medication status.
Enriched GO categories within the top CETS markers appear to be related to cell fate commitment and generation of neurons. These results are consistent with the interpretation that DNA methylation marks capable of distinguishing neurons from non-neurons also define their cellular identity. We identified enriched neuronal vs. non-neuronal DNA methylation differences in specific genomic categories, including CGI shores, gene bodies, and 3′UTRs. Conversely, CGIs and promoters exhibited significantly less cell-type specific differences. These findings are consistent with previous reports identifying DNA methylation variation at CGI shores as important regulators of tissue identity.10 Gene body methylation, specifically at exonic sequences, has been shown to direct alternative transcriptional splicing.27,28 Neuronal cell fate determination is largely influenced by alternative splicing mechanisms during neuronal development,29 while in mature neurons alternative splicing variation contributes to a number of key neuronal functions including axonal guidance, synaptic vesicle release,30,36 synaptic remodeling, and long-term potentiation,31 among others.29,32 Cell-type specific DNA methylation differences in these regions may be related to the GO categories identified in the top CETS loci such as axon guidance, dendritic spine, and postsynaptic density.
We validated the neuronal proportion quantification algorithm using multiple metrics. The technique performed best in the reconstitution experiment. The second method was a comparison to FACS based measurements of neuronal proportion in a set of 20 bulk tissue samples run on the microarrays. In general, quantification of cortical neuronal populations is a well-accepted technique and has been found to be more accurate than fluorescent microscopy quantifications using a Neubauer chamber.33 A possible source of higher variation relative to the reconstitution experiments was that microarrays and FACS based quantification were run on the same individual, but on different preparations of the bulk tissue such that variation in the cellular composition of the sectioned tissue per individual may have added to the experimental noise. A second possibility is that variation was induced at the level of selection of FACS gate parameters used to define the neuronal population. The comparison of predicted neuronal proportion to the average gene expression of the RBFOX3 gene, which encodes the NeuN protein, demonstrated the weakest correlation. The strength of the correlation may have been affected by variation of factors know to affect gene expression measurements such as brain pH34 and post mortem interval.35 Importantly, we observed an average predicted proportion of 32.2% neurons to non-neurons in the NICHD cortical population and 31.1% and 32.1% in the frontal and temporal cortical samples from the Gibbs et al. study. These values are similar to the published 32.4% and 27% gray matter and total cortical neuronal content, respectively, as determined by Azevedo et al. using isotropic fractionation.18
The approach designed for quantifying neuronal proportions was enabled by the large DNA methylation difference and small variance between neuronal and non-neuronal DNA methylation profiles at top CETS loci. The technique measures neuronal proportions by determining the best match to a gradient of DNA methylation profiles for each sample and as such, variation within an individual sample will be tested equally across the in silico gradient, allowing for an even chance of proper neuronal prediction per sample. These features make the predictions robust to batch effects, as demonstrated above. Importantly, we modeled batch effects by induced systematic increases in detected DNA methylation values across randomized proportions of probes across the CETS marker set and demonstrated that prediction values are independent of these factors and out-perform quadratic programming alternatives. The design of the CETS algorithm also makes it applicable across multiple experimental platforms that contain overlapping data points with the top CETS loci, as evidenced by our perfect classification of NeuN positive and negative samples from Iwamoto et al.22 This feature makes CETS applicable not only to data generated on tiling arrays but also next generation sequencing platforms. The performance of a given platform may vary depending on the distribution of overlaps with top CETS loci; however, the neuronal proportions calculated will remain relative across a given experiment, allowing for the quantification of cellular heterogeneity bias.
The technique allows for the correction of cell heterogeneity bias through transformation of the data in two ways, depending on the platform used. For Illumina platforms containing the same probes, the method will generate a transformed neuronal and glial profile for the data, allowing cell-type specific analyses. For other platforms, the inclusion of neuronal proportion information as a covariate in a linear model is recommended. Our permutation analysis of pair-wise differences suggests that both methods perform equally well at generating cell-type specific findings when the proportion of neurons is unequal between the groups being tested. When using non-Illumina data; however, the distinction of whether the identified phenotype associations are resultant from neuronal or non-neuronal cells cannot be determined. However, a comparison of the pair-wise analysis output from neuronal proportion corrected to non-corrected data may indicate the presence of cell type specific associations and may direct future analyses such as laser capture microdisection (LCM) experiments.
We provide a proof of principle analysis of recently published MDD specific DNA methylation profiling data generated in the SMRI cohort using the CHARM algorithm. These analyses demonstrated that a large portion of the originally identified DMRs was positively correlated with cell-type specific DNA methylation differences. This observation is expected, as the proportions of neurons to non-neurons were significantly different between MDD and controls. Application of CETS model correction identified novel DMRs that did not correlate with cell-type specific epigenetic differences and are therefore more likely to be associated with MDD independent of cell-type heterogeneity. Importantly, a number of the originally identified DMRs remained significant after CETS model correction and portion of these that had previously not survived multiple correction testing now passed genome-wide significance. Among these genes were LASS2, which was validated and discussed in the original study,4 FAM20B and PCTK1. Epigenetic variation at PCTK1 could be important for MDD, as overexpression of this gene in rats has been demonstrated to result in impaired spatial working memory and cognitive function.36 The degree of overlap across the results generated in the CETS modeled SMRI data and the FAC sorted NICHD data was higher than the overlap with non-corrected data, suggesting that the statistical removal of neuronal to non-neuronal heterogeneity in this sample improved the replicability of identified findings across cohorts and may thus be more relevant to the disease phenotype in the population.
A number of reports have identified correlation of DNA methylation in the brain at numerous loci with age.24,37,38 Recently, Horvath et al. identified an age related co-methylation module in brain and blood38 using many of the same samples investigated above. Our analysis identified positive correlations between CETS model predicted neuronal proportion and age in both the frontal cortex and cerebellum, suggesting that a portion of these findings may be due to variation in cellular content over the course of aging. A recent study incorporating the samples from Gibbs et al. identified a series of genes where expression levels correlated with age in both the frontal cortex and cerebellum.39 After isolation of Purkinje neurons through laser capture microdisection, ~8% (5 out of 60) candidates validated, corroborating our interpretation that a majority of age related associations may be due to cell heterogeneity and may be corrected through CETS model transformation.
DNA methylation at the CETS markers should be robust to age related variation in order to accurately quantify neuronal proportions over a range of ages. The age of the samples used to generate the CETS loci in our study ranged from 13 to 79 y old and did not demonstrate age related variation of neuronal or non-neuronal profiles at the CETS loci. The CETS model is therefore appropriate to evaluate neuronal proportion in brain samples above these ages, such as the study by Gibbs et al.39 The performance of CETS based quantification of neuronal proportions in samples from younger ages will depend on the relative conservation of epigenetic patterns at CETS markers during development. As highlighted in the Numata et al. study, DNA methylation in the prefrontal cortex varies at different developmental time periods and exhibits rapid changes both prenatally and in the early postnatal years, specifically in neural developmental genes.24 Despite this, an analysis of the Numata et al. data demonstrated a significant correlation at the 521 CETS markers present on the HM27 array between the mean DNA methylation profiles of 44 samples less than 13 y old, including 29 prenatal samples, to the remaining 56 samples ranging from 13 to 78 y old (R = 0.95, p = 2.2 × 10−16). While, neuronal proportion quantification in samples younger than age 13 should be interpreted with caution, these analyses suggest that at least a portion of CETS patterns vary only minimally over earlier age ranges and may be appropriate for neuronal quantification in younger samples. Future studies will be necessary to refine specific CETS markers robust to early developmental changes and enable reliable quantification of neuronal proportions in younger samples.
The accuracy of CETS model prediction of neuronal to non-neuronal proportion in non-cortical brain regions will be influenced by the conservation of neuron specific epigenetic marks across regions. Prediction of cerebellar neuronal content was lower than the ~77% reported by Azevedo et al.,18 which is explained by the fact that primarily granule neurons in the cerebellum express NeuN, while Purkinje cells do not.40,41 The above correlation between age and cerebellar neuronal proportion above is therefore most likely reflective of age related changes in ratios of undetected neuronal cell types. This interpretation is consistent with observations that Purkinje neuron levels and ratios of stelate and basket neurons relative to granule neurons have been observed to decrease with age in mice.42-44
Future studies targeting distinct brain regions or cell types within a brain region will be necessary to refine CETS models. Importantly, neuronal content predictions were demonstrated to adequately account for the observed degree of DNA methylation variation across brain regions. Numerous previous studies report large scale DNA methylation differences between brain regions.26,45,46 On both the global and locus specific scale, our analyses demonstrate both a strong correlation between brain region specific DNA methylation differences and cellular proportion, and a drastic reduction in locus specific differences identified between brain regions after adjusting for neuronal to non-neuronal proportions per individual. Together, these findings suggest that the extreme differences between neurons and non-neurons are the primary driver of the brain region epigenetic differences identified and that a majority of the previously identified epigenetic variation between brain regions may be largely due to differences in neuron to non-neuron ratios. Control of this detectable heterogeneity will allow for the identification of more subtle epigenetic changes in future studies.
We have generated a publically available R package called “CETS” capable of performing the above analyses in DNA methylation data sets (http://psychiatry.igm.jhmi.edu/kaminsky/software.htm). This tool will not only allow for the generation of novel data independent of cell heterogeneity based bias, but also allowing for a re-analysis of existing data sets. Application of CETS modeling to genome-wide DNA methylation data may lead to new level of understanding of epigenetic regulation in the brain and holds the potential to identify to novel discoveries related to the epigenetic basis of neurological and psychiatric phenotypes.
Materials and Methods
Sample
Post mortem cortical tissue from MDD (n = 29) and matched control (n = 29) samples were obtained from the NICHD Brain Bank of Developmental Disorders. Demographic information appears in Table 3.
Table 3. Sample demographics.
| Cohort | Diagnosis | Sample size | Age (years) | Sex (M:F) | PMI (hours) |
|---|---|---|---|---|---|
| NICHD |
CON |
29 |
32.6 ± 16.1 |
14:15 |
16.31 ± 4.96 |
| MDD | 29 | 32.5 ± 15.9 | 14:15 | 18.1 ± 1.09 |
Isolation of neuronal and non-neuronal nuclei by fluorescence activated cell sorting (FACS)
Neuronal nuclei were isolated from prefrontal cortical tissue as described previously.47 Briefly, 250–750 mg of frozen tissue was homogenized in lysis buffer (0.32 M sucrose, 5 mM calcium chloride, 3 mM magnesium acetate, 0.1 mM EDTA, 10 mM dithiothreitol, 0.1% Triton X-100) and nuclei were isolated via sucrose cushion ultracentrifugation (1.8 M sucrose, 3 mM magnesium acetate, 1 mM dithiothreitol, 10 mM TRIS-HCl, pH 8). Ultracentrifugation was performed at 25,000 rpm for 2.5 h at 4°C (Beckman, L-90K ultracentrifuge, SW32 rotor). For nuclei immnofluorescence staining, anti-NeuN (Ms) and anti-Ms IgG antibodies were incubated together at room temperature before nuclei were added and incubated further in the dark at 4°C for 45–60 min before Fluorescence Activated Cell Sorting (FACS). FACS was performed at the Johns Hopkins Flow Cytometry Core Facility (FACSAria II, BD Biosciences). The sorting primary gate was set to properly capture nuclei based on size and density, while secondary gates were set based on fluorescence signals. Immunonegative (NeuN-) nuclei were counted, collected, and processed in parallel with the fraction of neuronal (NeuN+) nuclei. All nuclei were sorted with an efficiency of greater than 90%. After FACS, nuclei were pelleted and frozen at -80°C in Tissue and Cell Lysis Buffer (MasterPure DNA Purification Kit, Epicenter Biotechnologies) until DNA extraction. All genomic DNA from nuclei was isolated with the MasterPure DNA Purification Kit (Epicenter Biotechnologies) according to the manufacturer’s instructions.
Illumina microarray analysis
Samples quality assessment and microarray analysis were conducted at The Sidney Kimmel Cancer Center Microarray Core Facility at Johns Hopkins University, supported by NIH grant P30 CA006973 entitled Regional Oncology Research Center.
Genomic DNA quality assessment
Genomic DNA quality was assessed by low concentration agarose gel (0.6%) electrophoresis and spectrometry of OD260/280 and OD 260/230 ratio.
Sodium bisulfite conversion
DNA bisulfite conversion was performed using EZ DNA Methylation Kit (Zymo Research) by following manufacturer’s manual with modifications for Illumina Infinium Methylation Assay. Briefly, 0.5–1.0 µg of genomic DNA was first mixed with 5 µl of M-Dilution Buffer and incubate at 37°C for 15 min and then mixed with 100 µl of CT Conversion Reagent prepared as instructed in the kit’s manual. Mixtures were incubated in a thermocycler with 16 thermal cycles at 95°C for 30 sec and 50°C for 1 h. Bisulfite-converted DNA samples were loaded onto 96-column plates provided in the kit for desulphonation and purification. Concentration of eluted DNA was measured using Nanodrop-1000 spectrometer.
Infinium chip assay
Bisulfite-converted DNA was analyzed using Illumina’s Infinium Human Methylation450 Beadchip Kit (WG-314-1001) by following manufacturer’s manual. Beadchip contains 485,577 CpG loci in human genome. Briefly, 4 µl of bisulfite-converted DNA was added to a 0.8 ml 96-well storage plate (Thermo Scientific), denatured in 0.014 N sodium hydroxide, neutralized and amplified with kit-provided reagents and buffer at 37°C for 20–24 h. Samples were fragmented using kit-provided reagents and buffer at 37°C for one hour and precipitated by adding 2-propanol. Re-suspended samples were denatured in a 96-well plate heat block at 95°C for 20 min. Twelve microliters of each sample was loaded onto a 12-sample chip and the chips were assembled into hybridization chamber as instructed in the manual. After incubation at 48°C for 16–20 h, chips were briefly washed and then assembled and placed in a fluid flow-through station for primer-extension and staining procedures. Polymer-coated chips were image-processed in Illumina’s iScan scanner.
Data acquisition
Data were extracted using Methylation Module of GenomeStudio v1.0 Software. Raw microarray signal intensity data was first corrected on Illumina probe type, followed by individual methyl and non-methyl channel quantile normalization using the Limma package in R/Bioconductor. Methylation status of each CpG site was then calculated as β value based on following definition:
β value = (signal intensity of methylation-detection probe)/(signal intensity of methylation-detection probe + signal intensity of non-methylation-detection probe + 100).
Pyrosequencing validation of microarray targets
Validation of microarray results was performed using sodium bisulfite pyrosequencing on genomic DNA from sorted nuclei. Target genes (A2BP1, PRMD16, S100B, SH3TC2 and TRIM2) were chosen from the top 1,000 CETS loci. Bisulfite conversion was performed using EZ DNA Methylation Gold Kit (Zymo Research) according to the manufacturer’s instructions. Two primer sets were designed to amplify each locus interrogated on the array for each of the five genes. Primer sequences and their respective microarray IDs can be found in Table 4. Nested PCR amplifications were performed with a standard PCR protocol in 25 ml volume reactions containing 3–4 µl of sodium-bisulfite-treated DNA, 0.2 µM primers, and master mix containing Taq DNA polymerase (Sigma Aldrich). After agarose gel electrophoresis to ensure successful amplification and specificity, PCR amplicons were processed for pyrosequencing analysis according to the manufacturer’s standard protocol (Qiagen). All pyrosequencing was performed using a PyroMark MD system (QIAGEN) with Pyro Q-CpGt 1.0.9 software (QIAGEN) for CpG methylation quantification.
Table 4. Primers for Validation Experiments.
| Gene | Probe ID | Primer name | Primer sequence 5′-3′ |
|---|---|---|---|
| |
|
|
|
|
PRMD16 |
cg14217558 |
PRMD_F_out |
GAATAAGGGGTTAAATGTTGAGTAA |
| |
|
PRMD_R_out |
TAAAAAAAAACTAAAATAAAAACACCCCTA |
| |
|
PRMD_F_in |
TTATAGATTTATTTATTTTTTTAGTTTTTATTAATTGG |
| |
|
PRMD_R_in |
biotin-AACCTACACCTTCCAAACTAACAATA |
| |
|
PRMD_Pyro1 |
TGTATAGGTTAGAGTAAGTTA |
| |
|
|
|
|
S100B |
cg18433784 |
S100B_F_out |
TTTTAGGGTGATATTAATATTTATGTAATA |
| |
|
S100B_R_out |
CCTAATACAACCCACTACAAAC |
| |
|
S100B_F_in |
biotin-TAGTTTATGTGTAATTTTGGTTTTTAGTA |
| |
|
S100B_R_in |
ACCAAACCTACACACAAAACTAC |
| |
|
S100B_Pyro1 |
AAACTACATTAACCAAACAACCCC |
| |
|
|
|
|
TRIM2 |
cg22471112 |
TRIM_F_out |
GGTAGTGATTTAAATTTTTTAATTTGGA |
| |
|
TRIM_R_out |
AAAATATAAACAACTAATCAACACAAAC |
| |
|
TRIM_F_in |
biotin-GGGTTGTAAAGATGATAATTTAGTTTTTAT |
| |
|
TRIM_R_in |
ACCAATAATCAACCTCTTTAAACTCTAT |
| |
|
TRIM_Pyro1 |
TACTTAAAATATTATATTTCATTCC |
| |
|
|
|
|
SH3TC2 |
cg01965939 |
SH3_F_out |
GTAGAAGTAGTTATTTATGTGTGTATTA |
| |
|
SH3_R_out |
TATAAACCTACAAACTAAACTAAACTAC |
| |
|
SH3_F_in |
biotin-TTAGAAGAGTGTTGTAAGGGATAGT |
| |
|
SH3_R_in |
CTCCCTAAACCTCCTACTATAAC |
| |
|
SH3_Pyro1 |
AACTACATTATACATTAAACAAACCT |
| |
|
|
|
|
A2BP1 |
cg00480381 |
A2B_F_out |
TGAGAATTAAATTAATGAAGAGTATTTG |
| |
|
A2B_R_out |
CCTCTAAAACTCAACACATCAATA |
| |
|
A2B_F_in |
biotin-TTATGTTAGGTAAGTTGTTTTTAAAATAGTAT |
| |
|
A2B_R_in |
AAACTAAAAACCAAATCCCTTCTATCA |
| A2B_Pyro1 | AACTCTTAATATAAAAATTTTCTTCCCA |
Public data processing
DNA methylation data generated by Iwamoto et al.,22 used to validate CETS model prediction was downloaded from GEO accession. Raw Cel files were processed using the AffyTiling package in R to obtain quantile normalized M values representative of methylation enriched and depleted samples per replicate. Neuronal proportion prediction was performed using the CETS package inputing the mean ratio of methylated to unmethylated signals per replicate at 3,841 probes overlapping the top 10,000 CETS markers. DNA methylation β value data generated by Gibbs et al.26 used for brain region specific analysis was downloaded from GEO accession GSE15745.
Statistical analysis
All statistical tests were performed in R (www.r-project.org). Using an Anderson-Darling test from the nortest package, all distributions derived from microarray data rejected the null hypothesis of normality and were subsequently evaluated with non-parametric tests. All statistical tests performed were two tailed and a p < 0.05 is considered significant. Unless otherwise specified, ± denotes the standard error of the mean. CETS model predictions of bulk DNA neuronal proportions excluded neuronal and non-neuronal DNA methylation profiles of the same sample for in silico matrix generation as per standard bootstrapping techniques. Data are located online under GEO accession GSE41826.
Supplementary Material
Author Contributions
Study design: Z.K.; laboratory experiments: J.G.; statistical analysis: Z.K. and M.A.
Acknowledgments
Human tissue was obtained from the NICHD Brain and Tissue Bank for Developmental Disorders and the University of Maryland, Baltimore, MD. This work was funded by NIMH 1R21MH094771-01. We would like to further thank The Solomon R. and Rebecca D. Baker Foundation for their generous support of this research.
Glossary
Abbreviations:
- CETS
cell epigenotype specific
- CGI
CpG island
- CHARM
comprehensive high-throughput arrays for relative methylation
- DMR
differentially methylated region
- FACS
fluorescence activated cell sorting
- GO
gene ontology
- HM27
Illumina Infinium Human Methylation 27
- HM450
Illumina Infinium Human Methylation450 Beadchip
- LCM
laser capture microdisection
- MDD
major depressive disorder
- tDMR
tissue-specific differentially methylated region
- TSS
transcription start site
- UTR
untranslated region
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/23924
References
- 1.Akbarian S. The molecular pathology of schizophrenia--focus on histone and DNA modifications. Brain Res Bull. 2010;83:103–7. doi: 10.1016/j.brainresbull.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mill J, Tang T, Kaminsky Z, Khare T, Yazdanpanah S, Bouchard L, et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet. 2008;82:696–711. doi: 10.1016/j.ajhg.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegmund KD, Connor CM, Campan M, Long TI, Weisenberger DJ, Biniszkiewicz D, et al. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One. 2007;2:e895. doi: 10.1371/journal.pone.0000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sabunciyan S, Aryee MJ, Irizarry RA, Rongione M, Webster MJ, Kaufman WE, et al. GenRED Consortium Genome-wide DNA methylation scan in major depressive disorder. PLoS One. 2012;7:e34451. doi: 10.1371/journal.pone.0034451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohgane J, Yagi S, Shiota K. Epigenetics: the DNA methylation profile of tissue-dependent and differentially methylated regions in cells. Placenta. 2008;29(Suppl A):S29–35. doi: 10.1016/j.placenta.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 6.Nagase H, Ghosh S. Epigenetics: differential DNA methylation in mammalian somatic tissues. FEBS J. 2008;275:1617–23. doi: 10.1111/j.1742-4658.2008.06330.x. [DOI] [PubMed] [Google Scholar]
- 7.Sakamoto H, Kogo Y, Ohgane J, Hattori N, Yagi S, Tanaka S, et al. Sequential changes in genome-wide DNA methylation status during adipocyte differentiation. Biochem Biophys Res Commun. 2008;366:360–6. doi: 10.1016/j.bbrc.2007.11.137. [DOI] [PubMed] [Google Scholar]
- 8.Suzuki M, Sato S, Arai Y, Shinohara T, Tanaka S, Greally JM, et al. A new class of tissue-specifically methylated regions involving entire CpG islands in the mouse. Genes Cells. 2007;12:1305–14. doi: 10.1111/j.1365-2443.2007.01136.x. [DOI] [PubMed] [Google Scholar]
- 9.Irizarry RA, Ladd-Acosta C, Carvalho B, Wu H, Brandenburg SA, Jeddeloh JA, et al. Comprehensive high-throughput arrays for relative methylation (CHARM) Genome Res. 2008;18:780–90. doi: 10.1101/gr.7301508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–3. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji H, Ehrlich LI, Seita J, Murakami P, Doi A, Lindau P, et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010;467:338–42. doi: 10.1038/nature09367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nifosì F, Toffanin T, Follador H, Zonta F, Padovan G, Pigato G, et al. Reduced right posterior hippocampal volume in women with recurrent familial pure depressive disorder. Psychiatry Res. 2010;184:23–8. doi: 10.1016/j.pscychresns.2010.05.012. [DOI] [PubMed] [Google Scholar]
- 14.Ekman CJ, Lind J, Ryden E, Ingvar M, Landen M. Manic episodes are associated with grey matter volume reduction - a voxel-based morphometry brain analysis. Acta Psychiatr Scand. 2010;122:507–15. doi: 10.1111/j.1600-0447.2010.01586.x. [DOI] [PubMed] [Google Scholar]
- 15.Cotter D, Hudson L, Landau S. Evidence for orbitofrontal pathology in bipolar disorder and major depression, but not in schizophrenia. Bipolar Disord. 2005;7:358–69. doi: 10.1111/j.1399-5618.2005.00230.x. [DOI] [PubMed] [Google Scholar]
- 16.Uranova NA, Vostrikov VM, Orlovskaya DD, Rachmanova VI. Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: a study from the Stanley Neuropathology Consortium. Schizophr Res. 2004;67:269–75. doi: 10.1016/S0920-9964(03)00181-6. [DOI] [PubMed] [Google Scholar]
- 17.Matigian N, Windus L, Smith H, Filippich C, Pantelis C, McGrath J, et al. Expression profiling in monozygotic twins discordant for bipolar disorder reveals dysregulation of the WNT signalling pathway. Mol Psychiatry. 2007;12:815–25. doi: 10.1038/sj.mp.4001998. [DOI] [PubMed] [Google Scholar]
- 18.Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, Ferretti RE, Leite RE, et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol. 2009;513:532–41. doi: 10.1002/cne.21974. [DOI] [PubMed] [Google Scholar]
- 19.Inta D, Monyer H, Sprengel R, Meyer-Lindenberg A, Gass P. Mice with genetically altered glutamate receptors as models of schizophrenia: a comprehensive review. Neurosci Biobehav Rev. 2010;34:285–94. doi: 10.1016/j.neubiorev.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 20.Moncrieff J. A critique of the dopamine hypothesis of schizophrenia and psychosis. Harv Rev Psychiatry. 2009;17:214–25. doi: 10.1080/10673220902979896. [DOI] [PubMed] [Google Scholar]
- 21.De Berardis D, Conti CM, Serroni N, Moschetta FS, Olivieri L, Carano A, et al. The effect of newer serotonin-noradrenalin antidepressants on cytokine production: a review of the current literature. Int J Immunopathol Pharmacol. 2010;23:417–22. doi: 10.1177/039463201002300204. [DOI] [PubMed] [Google Scholar]
- 22.Iwamoto K, Bundo M, Ueda J, Oldham MC, Ukai W, Hashimoto E, et al. Neurons show distinctive DNA methylation profile and higher interindividual variations compared with non-neurons. Genome Res. 2011;21:688–96. doi: 10.1101/gr.112755.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reimand J, Kull M, Peterson H, Hansen J, Vilo J. g:Profiler--a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35(Web Server issue):W193-200. doi: 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Numata S, Ye T, Hyde TM, Guitart-Navarro X, Tao R, Wininger M, et al. DNA methylation signatures in development and aging of the human prefrontal cortex. Am J Hum Genet. 2012;90:260–72. doi: 10.1016/j.ajhg.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 2010;6:e1000952. doi: 10.1371/journal.pgen.1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi JK. Contrasting chromatin organization of CpG islands and exons in the human genome. Genome Biol. 2010;11:R70. doi: 10.1186/gb-2010-11-7-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74–9. doi: 10.1038/nature10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glatt SJ, Cohen OS, Faraone SV, Tsuang MT. Dysfunctional gene splicing as a potential contributor to neuropsychiatric disorders. Am J Med Genet B Neuropsychiatr Genet. 2011;156B:382–92. doi: 10.1002/ajmg.b.31181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, et al. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003;114:75–86. doi: 10.1016/S0092-8674(03)00477-X. [DOI] [PubMed] [Google Scholar]
- 31.Beffert U, Weeber EJ, Durudas A, Qiu S, Masiulis I, Sweatt JD, et al. Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron. 2005;47:567–79. doi: 10.1016/j.neuron.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 32.Bark IC, Hahn KM, Ryabinin AE, Wilson MC. Differential expression of SNAP-25 protein isoforms during divergent vesicle fusion events of neural development. Proc Natl Acad Sci U S A. 1995;92:1510–4. doi: 10.1073/pnas.92.5.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Collins CE, Young NA, Flaherty DK, Airey DC, Kaas JH. A rapid and reliable method of counting neurons and other cells in brain tissue: a comparison of flow cytometry and manual counting methods. Front Neuroanat. 2010;4:5. doi: 10.3389/neuro.05.005.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li JZ, Vawter MP, Walsh DM, Tomita H, Evans SJ, Choudary PV, et al. Systematic changes in gene expression in postmortem human brains associated with tissue pH and terminal medical conditions. Hum Mol Genet. 2004;13:609–16. doi: 10.1093/hmg/ddh065. [DOI] [PubMed] [Google Scholar]
- 35.Birdsill AC, Walker DG, Lue L, Sue LI, Beach TG. Postmortem interval effect on RNA and gene expression in human brain tissue. Cell Tissue Bank. 2011;12:311–8. doi: 10.1007/s10561-010-9210-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu K, Li S, Bodhinathan K, Meyers C, Chen W, Campbell-Thompson M, et al. Enhanced expression of Pctk1, Tcf12 and Ccnd1 in hippocampus of rats: Impact on cognitive function, synaptic plasticity and pathology. Neurobiol Learn Mem. 2012;97:69–80. doi: 10.1016/j.nlm.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaminsky Z, Tochigi M, Jia P, Pal M, Mill J, Kwan A, et al. A multi-tissue analysis identifies HLA complex group 9 gene methylation differences in bipolar disorder. Mol Psychiatry. 2012;17:728–40. doi: 10.1038/mp.2011.64. [DOI] [PubMed] [Google Scholar]
- 38.Horvath S, Zhang Y, Langfelder P, Kahn RS, Boks MP, van Eijk K, et al. Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biol. 2012;13:R97. doi: 10.1186/gb-2012-13-10-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar A, Gibbs JR, Beilina A, Dillman A, Kumaran R, Trabzuni D, et al. Age-associated changes in gene expression in human brain and isolated neurons. Neurobiol Aging. 2013;34:1199–209. doi: 10.1016/j.neurobiolaging.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–11. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- 41.Weyer A, Schilling K. Developmental and cell type-specific expression of the neuronal marker NeuN in the murine cerebellum. J Neurosci Res. 2003;73:400–9. doi: 10.1002/jnr.10655. [DOI] [PubMed] [Google Scholar]
- 42.Sturrock RR. A comparison of quantitative histological changes in different regions of the ageing mouse cerebellum. J Hirnforsch. 1990;31:481–6. [PubMed] [Google Scholar]
- 43.Sturrock RR. Changes in neuron number in the cerebellar cortex of the ageing mouse. J Hirnforsch. 1989;30:499–503. [PubMed] [Google Scholar]
- 44.Sturrock RR. Age related changes in Purkinje cell number in the cerebellar nodulus of the mouse. J Hirnforsch. 1989;30:757–60. [PubMed] [Google Scholar]
- 45.Khaitovich P, Muetzel B, She X, Lachmann M, Hellmann I, Dietzsch J, et al. Regional patterns of gene expression in human and chimpanzee brains. Genome Res. 2004;14:1462–73. doi: 10.1101/gr.2538704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ladd-Acosta C, Pevsner J, Sabunciyan S, Yolken RH, Webster MJ, Dinkins T, et al. DNA methylation signatures within the human brain. Am J Hum Genet. 2007;81:1304–15. doi: 10.1086/524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matevossian A, Akbarian S. Neuronal nuclei isolation from human postmortem brain tissue. J Vis Exp. 2008 doi: 10.3791/914. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.