

Abstract
The proper identification of differentially methylated CpGs is central in most epigenetic studies. The Illumina HumanMethylation450 BeadChip is widely used to quantify DNA methylation; nevertheless, the design of an appropriate analysis pipeline faces severe challenges due to the convolution of biological and technical variability and the presence of a signal bias between Infinium I and II probe design types. Despite recent attempts to investigate how to analyze DNA methylation data with such an array design, it has not been possible to perform a comprehensive comparison between different bioinformatics pipelines due to the lack of appropriate data sets having both large sample size and sufficient number of technical replicates. Here we perform such a comparative analysis, targeting the problems of reducing the technical variability, eliminating the probe design bias and reducing the batch effect by exploiting two unpublished data sets, which included technical replicates and were profiled for DNA methylation either on peripheral blood, monocytes or muscle biopsies. We evaluated the performance of different analysis pipelines and demonstrated that: (1) it is critical to correct for the probe design type, since the amplitude of the measured methylation change depends on the underlying chemistry; (2) the effect of different normalization schemes is mixed, and the most effective method in our hands were quantile normalization and Beta Mixture Quantile dilation (BMIQ); (3) it is beneficial to correct for batch effects. In conclusion, our comparative analysis using a comprehensive data set suggests an efficient pipeline for proper identification of differentially methylated CpGs using the Illumina 450K arrays.
Keywords: technical variability, DNA methylation, microarray, Illumina 450K, normalization
Introduction
Epigenome-wide association studies represent an opportunity to investigate disease-associated epigenetic variation in common human diseases.1 DNA methylation is the most studied form of epigenetic modification and it results from the addition of a methyl group to cytosine (5-mC) in the context of CpG dinucleotides. CpG methylation has been not only observed during development or differentiation and in association with diseases, but it has also been proposed as a prerequisite to unravel disease pathogenesis and understanding complex phenotypes.2,3 In the past few years, a number of technologies to measure 5-mC profiles on a genome-wide scale became rapidly available, conveniently subdivided into two groups. The first group identifies enriched DNA methylation by making use of methylation-sensitive restriction enzymes4,5 or immunoprecipitation with an antibody against 5-mC (MeDIP techniques6,7). The second group relies on bisulfite-based treatment to convert unmethylated cytosines to uraciles, (i.e., MethylC-Seq,8 RRBS-seq9). In both cases, microarrays and sequencing approaches have been adopted.10,11 Sequencing-based methods allow comprehensive DNA methylation evaluation and assignment of specific states to specific alleles and can interrogate DNA methylation in repetitive sequences.
Array based profiling approaches represent a common option for genome-wide DNA methylation studies, allowing an analysis of a larger number of samples at an affordable cost. The sample size is especially relevant when considering that, in many cases, changes in DNA methylation are mild and the biological variability may be high. Illumina Infinium HumanMethylation450 BeadChip (450K) is based on the Infinium Technology and contains more than 480,000 probes, targeting 99% of genes and 96% of CpG island regions.12,13 Initial studies showed a strong correlation with the previous Illumina Infinium HumanMethylation27 BeadChip12 and high concordance between biological replicates of a cell line.14 Furthermore, it has been demonstrated that Illumina 450K can detect differences of 20% in methylation with 99% confidence.12 However, the analysis of the 450K arrays is complicated by the inclusion of two different bead types associated to two different chemical assays, Infinium I and Infinium II. Infinium I considers two bead types (methylated and unmethylated) for the same CpG locus, both sharing the same color channel, whereas Infinium II utilizes a single bead type and two color channels.12 Dedeurwaerder et al.15 showed that Infinium II assays have larger variance and are less sensitive for the detection of extreme methylation values, which is probably associated to the dual-channel read-out, thus rendering the Infinium I assay a better estimator of the true methylation state. A “peak-based correction” method that rescales the Infinium II data on the basis of the Infinium I data was first developed, although a number of recent works highlighted potential problems with this method.15-17 Afterwards, SWAN (subset-quantile within array normalization) introduced sub-quantile normalization for the methylated and unmethylated channels separately, assuming that the distribution of the probes with similar number of CpGs should be similar, irrespective of probe type.16 A second sub-quantile normalization method (SQN) considered different probe categories to obtain quantiles and correct for the probe-type bias.17 Finally, a novel model-based normalization algorithm (Beta Mixture Quantile dilation, BMIQ) was recently developed and compared favorably with other methods.18
A proper normalization is key in the analysis first to avoid any enrichment toward any probe type in the differential methylation analysis, and second because identifying contiguous differentially methylated regions from single CpG sites requires the avoidance of technical variation especially for those sites positioned within the tested regions.19 There have been several efforts to develop new methodologies, and comparisons have been made using small data sets. To the best of our knowledge, there is yet no comprehensive comparison between methodologies in large number of samples that evaluates in detail the technical variability over the pipeline and the effect in the identification of differential methylated probes.
To address the above-mentioned problems, we therefore compared different analysis pipelines and performed a careful evaluation of the variability along each step of the pipelines. Specifically, here we focused on the resolution of the variability into its technical and biological constituents, when methylation arrays are “contaminated” either with a bias between Infinium I and Infinium II probe design types or a batch effect. We used two independent data sets with extensive level of biological and technical replication and rigorous design, in order to assess (1) the reduction of the technical variability during normalization, (2) the removal of probe design type bias, (3) the effective reduction of batch effect and (4) the identification of differentially methylated (DM) sites. We identified the pipelines that were suitable to reduce the technical variability and highlighted that only those incorporating a probe-type correction step provided consistent measurements for DM analysis. In particular, BMIQ, a newly develop algorithm,18 provided simultaneous correction for probe design and reduction of technical variability, in a consistent and reproducible way.
Results
In the current work, we focused on evaluating the changes in technical variability and the effect of different normalization pipelines, which should mainly consist in eliminating or reducing the variability introduced during the experimental process, while maintaining the biological variation between conditions. Six different pipelines were considered (Fig. 1), each including a quality control step after which probes and samples were selected, followed by a normalization step (see Materials and Methods). The final process consisted in the identification of DM CpG sites.

Figure 1. The workflow to compare pipelines. Six pipelines were considered in this study, as explained in the main text. Using data set A, we run all the pipelines, selecting the one to be tested with data set B, which also allows the evaluation of the correction for batch effect.
A pipeline was positively scored whether (1) technical replicates clustered together, (2) the correlation increased while (3) the average absolute difference between technical replicates decreased and (4) the bias between Infinium probe types decreased as well (for a detailed description see Materials and Methods section). Furthermore, we analyzed the number of DM sites that we were able to detect under different scenarios. In the sections below we will briefly describe the data sets, then the pipelines that we studied will be presented, evaluated and compared. Lastly, we will investigate the batch effect and its removal. Here, we define a batch as a subgroup of samples or experiments exhibiting a systematic non-biological difference that is not correlated with the biological variables under study.
Data sets
In order to analyze the pattern of technical variability reduction, we took advantage of data set A, which showed a large level of biological and technical replication and contrasted conditions with expected consistent differences in methylation. Specifically, we contrasted DNA methylation profiles obtained from monocytes (n = 43) or peripheral blood (PB, n = 53). Importantly, samples were randomly assigned to the slides and were processed together, to avoid additional bias in the variability. Clustering analysis showed that major differences exist between the two biological groups and that technical replicates cluster together even before any normalization, as evident from multidimensional and hierarchical clustering (Fig. S1A and B). The dispersion of the PB is higher as compared with that of monocytes, as expected, reflecting the heterogeneous vs. homogeneous composition of these sample types (Fig. S1A).
We studied the reduction of batch effect using data set B, which consisted of paired samples from healthy volunteers before and after supervised exercise training. Here major differences were manifest at the inter-individual level, as opposed to data set A, and minor differences between biological conditions (Fig. S1C and D). The definition of a batch was based on the qualitative assessment of differences between samples linked to processing day, slide or position, using a multidimensional scaling (MDS) plot. We noticed that major differences were related to the processing day. Hereafter, we will refer to these groups of arrays with the same processing day simply as batches.
We performed biological and technical validations using two publicly available data sets. We replicated DM sites between monocytes and PB using data set C,20 while matched 450K and bisulfite pyrosequencing (BPS) data from data set D15 were used to check the performance of selected normalization methods for the estimation of the percentage methylation.
Testing different pipelines using dataset A
Patterns of variability reduction in absence of batch effect
We exploited data set A as a valuable resource for analyzing the technical variability reduction along the normalization steps. Actually, nine samples had at least one technical replicate and biological groups were represented to a satisfactory degree. Importantly, because (1) the assignment to arrays was randomized, (2) we did not observe any potential known confounder driving a clustering plot (i.e., slide or array) and (3) we used data from samples processed together in the same array facility, we assumed that negligible source of variation came from factors unrelated to the biological variables, or in other words that the variation between conditions was fairly larger than the variability introduced by any potential batch effect. Actually, principal component analysis (PCA) showed that the first two components, accounting for ~20% of the variability, were highly correlated with the contrasted groups (monocytes vs. PB), while the components associated with slide and array accounted for a smaller part (Fig. S3).
We first considered a lumi-based pipeline, as specifically explained in Methods section. After quality control, 473,097 out of 485,577 probes were filtered in (97.4%) and 95 out of 96 samples remained. One sample was excluded because of overall low intensity and abnormal methylation M profile (not shown). Filtered-out probes included 65 SNPs (from Illumina manifest), 11,648 probes on Chr X or Y and 767 probes with detection p value > 0.01 in > 5% of the samples. The probe intensities appeared to be properly normalized (Fig. S4), except for a small region of low intensity values for one subject, which however was consistent between technical replicates, hence putatively originating from sample-specific characteristics. Next, we observed that the distance between technical replicates generally decreased and a global increase of correlation was observed after normalization, although the amplitude was small and the correlation already high before normalization (Fig. 2A and 5). The M-values after normalization showed an effective reduction of the noise, evaluated by the absolute difference between technical replicates, although to a different extent for different ranges of M-value (Fig. 2B−D), with the best performance for low methylated positions. Looking at the density plots multiple peaks may be identified originating from the different density profiles of type I vs. type II Infinium probes (Fig. 3A and B). We employed here a color adjustment followed by a quantile normalization (QN) on the pooled signal intensities of methylated and unmethylated probes, before calculating methylation estimates (β or M values). QN is a common procedure in high throughput data analysis to reduce between-sample variation and it optimally centered the signal between arrays, correcting for influence of the position on the slide (Fig. S4). However, it did not reduce the probe-type bias, as expected (Fig. 3B). Therefore, we included a further step by performing BMIQ (beta mixture quantile dilation) on quantile-normalized data. BMIQ is a recently developed model-based correction method, which has been proven to optimally eliminate the bias between probe types.18 Indeed, we verified that the bias was effectively reduced (Figs. 3C and 6B), and the technical variability not amplified, since the deviation between technical replicates was not increased after BMIQ or in some cases was further reduced as compared with QN (Fig. 6A). Additionally, even when BMIQ alone was applied to raw data, we were able to detect a reduction of technical variability for six out of nine pairs (Fig. 6A) and a removal of probe type bias (Fig. 3D).

Figure 2. Evaluation of the lumi pipeline. (A) The difference in sample correlation (upper panel) or Euclidean distance (lower panel) between quantile normalization (QN) and raw data. For convenience, only samples represented by two or three technical replicates are shown. A gray bar indicates the samples from monocytes, whereas a black bar denotes the samples from peripheral blood (PB). Replicates belonging to the same pair are consecutively located on each row or column. The color code indicates a decrease (blue) or an increase (yellow) in correlation or distance. (B) The absolute deviation between technical replicates in raw, color adjusted and QN data shows the consistent reduction of the technical variability after normalization. (C and D) The logarithmic ratio between the variability after QN and the variability on raw data are shown for PB (C) and monocytes (D). For each probe, we calculated the average M-values and the corresponding mean absolute difference between technical replicates. The log2 ratio between QN and raw data was used to check the performance of the normalization in reducing the variability and the presence of possible bias for sites with low or high levels of methylation. The red line indicates the loess fitting.

Figure 5. Minfi pipeline incorporate an adjustment for probe design type, which is obtained with SWAN. The raw (A), and SWAN (B) densities of M-values are shown either for each sample, or as the average density for Infinium I or Infinium II probe design.

Figure 3. Lumi pipeline does not incorporate an adjustment for probe design type, which is obtained with BMIQ. The raw (A), QN normalized (B) and QN + BMIQ (C) densities of M-values are shown either for each sample, or as the average density for Infinium I or Infinium II probe design. (D) BMIQ alone is also suitable for eliminating the probe design type bias.

Figure 6. Elimination of probe design bias using BMIQ. (A) The absolute deviation between technical replicates after QN, QN + BMIQ or BMIQ show the consistent reduction of the technical variability after adjusting for probe design type. To calculate the difference, β-values were considered in this case. (B) The mean absolute difference between probe pairs (as defined in method section) shows that the reduction of the technical noise due to different design type, is optimally obtained using BMIQ, which is superior to SWAN, SQN or GS.
Afterward, we exploited a minfi-based pipeline on the same data set, to verify the effect of different preprocessing and normalization processes. After quality, 473,115 out of 485,577 probes were filtered in (97.4%) and 95 samples remained. One sample was excluded because of overall bad control plots and anomalous methylation profile (not shown). Filtered-out probes included 65 SNPs (from Illumina manifest), 11,289 probes on Chr X or Y and 1,108 probes with minfi detection P value > 0.01 in > 5% samples. After SWAN (subset within-array normalization), the bias between probe types was reduced, the M-value density got effectively normalized (Fig. 5) and the samples correctly clustered according to sample type. SWAN is a within-sample normalization procedure, however it has been proposed for reduction of between-samples technical variability.16 We tested this hypothesis on our data set, showing that a very narrow decrease of the deviation between technical replicates occurred after SWAN, independently of M-value (Fig. 4B−D). At the same time, the correlation structure was maintained and we observed that on average the correlation increased within conditions but decreased between conditions (Fig. 4A; Fig. S6).

Figure 4. Evaluation of the minfi pipeline. (A) The difference in sample correlation (upper panel) or Euclidean distance (lower panel) between SWAN and raw data. For convenience, only samples represented by two or three technical replicates are shown. Color codes and positions of the samples are the same as Figure 2. (B) The absolute deviation between technical replicates in raw and SWAN-normalized data shows the reduction of the technical variability after normalization. (C and D) The logarithmic ratio between the variability after SWAN and the variability on raw data are shown for PB (C) and monocytes (D). For each probe, we calculated the average M-values and the corresponding mean absolute difference between technical replicates, as explained in Figure 2.
We also evaluated the pipeline described by Touleimat and Tost17 that we denoted here as SQN, referring to the subset quantile normalization performed within. After quality control, 473,864 autosomal probes out of 485,577 probes were filtered in (97.6%) and one sample was excluded. The algorithm performed SQN on the methylation estimates (β values) using the “relation to CpG” to calculate the reference quantiles, i.e., probes were categorized according to their position with respect to CpG islands as provided by Illumina (CpG Island, S shore, S shelf, N shore, N shelf or outside).17 The distribution of the methylation estimates (β and M values) resulted overlapping for all samples (Fig. S7A). However, although the bias between probe types was actually reduced (Fig. 6B; Fig. S7B), the technical variability was abnormally amplified, which we could evaluate by increased average absolute difference, decreased correlation and increased distance between technical replicates after normalization (Fig. S7C−F).
Finally, we evaluated the control normalization and background subtraction performed on GenomeStudio (Illumina) and denoted here as GS. 473,097 autosomal probes out of 485,577 probes were filtered in (97%) and one sample was excluded. The distribution of the methylation estimates (β and M values) resulted normalized (Fig. S8A), whereas the bias between probe types was not eliminated (Fig. S8B). Moreover, the technical variability was not reduced, which we could evaluate by increased average absolute difference, decreased correlation and increased distance between technical replicates after normalization (Fig. S8C−F).
Comparative analysis between the different pipelines
In order to compare the performance of the pipelines, we considered three benchmarks: (1) the reduction of the bias between Infinium I and II, (2) the reduction of the variability between technical replicates and (3) the ability of identifying DM probes.
Reduction of the probe bias in the 450K array
The capability of reducing the probe type bias was evaluated through the analysis of the deviation between pairs of adjacent probes, one from each design and within 200 bp of each other. As shown in Figures 3B and 6B, between-sample normalization techniques, like QN on signal intensity, cannot correct for this bias when reducing sample-to-sample dispersion. Nevertheless, the most effective correction was obtained with BMIQ alone or in combination with QN, which were both effective in reducing the bias with a bigger extent as compared with the other methods considered here, in agreement to what was originally reported (Fig. 6B).18 The advantage was also shown by the comparison of the densities of M values, after stratification for probe type, which revealed no difference in the amplitudes after correction (Fig. 3C and D).
Reduction of the technical variability
We evaluated the reduction of the technical variability as one of the key goals to achieve, after the pre-processing and normalization steps. We carefully assessed how close the technical replicates resulted after normalization, by evaluating their average absolute difference, as proposed for Illumina 27K arrays.21 Overall, we assessed that the best performance was obtained after performing lumi-based QN + BMIQ or BMIQ alone. Actually, the impact on the variability reduction was minor after SWAN and unfavorable after SQN or GS, when evaluating the discrepancy between technical replicate pairs (Fig. 2, 4 and 6; Figs. S7 and S8).
Differential methylation analysis
Initially, we compared the different pipelines for their ability of detecting DM between monocytes vs. PB. Thus, we obtained the difference in M values as effect size (ΔM), p values and FDR-corrected p values. We addressed the problem of identifying statistically significant change in methylation, in the presence of probe type bias. Therefore, we used volcano plots to compare p-values and ΔM (Fig. 7A; Fig. S9). We observed that Type II probes have overall smaller amplitude in methylation change, the ΔM appeared more compressed without any probe design adjustment and two distinct populations appeared in a volcano plot. However, BMIQ, SWAN, SQN or GS corrected this bias although to a different extent (Fig. 7A; Fig. S9).

Figure 7. The analysis of DM sites is influenced by the normalization process. (A) Volcano plots show the p value vs. the difference in methylation as calculated by limma. Comparable amplitude in methylation difference is obtained only after adjusting with QN + BMIQ, BMIQ but not with QN only. (B) Spearman correlation between P-values obtained after different normalization options. The correlation was calculated progressively including probes from the ranked list of CpG sites, as described in Methods section. (C) Venn diagram showing the number of DM sites obtained after different pipelines. The threshold for claiming DM was FDR Adjusted p < 0.05 AND absolute(ΔM) > 1. (D) PCA of all probes denotes the pattern of variability. The color indicates the smoothed density (black = low, yellow = high). The first PC accounts for the average methylation level, while the second PC indicates the direction of the methylation change in monocytes as compared with PB. DM sites are indicated as dots, with different colors for increased (green) or decreased (white) methylation. There is no sign of different behavior of type I or type II probes.
We initially compared the distribution of p values obtained with different approaches and looked at the correlation between them. Without a reference method to count true positives and true negatives on the basis of a threshold, we could initially verify that a strong correlation for the top-ranked items existed, which are more likely to be true positives. Hence, selecting top-down the significant sites as explained in the method section, the Spearman correlation reached a maximum (ρ > 0.95), before starting to decrease when counting also non-significant sites, representing noise. However, SQN performed differently, and the maximum pairwise correlation with the other pipelines was always lower (Fig. 7B; Fig. S10). This behavior possibly suggests that the number of false positives was different after different pipelines. To test this hypothesis, without a known list of true DM sites, we fitted a latent class analysis as described in Methods section,22,23 to predict the class membership of each CpG site as being DM or not. We exploited the output of the six different pipelines to obtain six binary variables specifying if the site was DM of not-DM, using two simple thresholds: (1) FDR adjusted p < 0.05 or (2) FDR adjusted p < 0.05 and |ΔM| > 1. We selected the second threshold because we showed that the specificity reached the maximum at the expense of dropping the sensitivity is some cases, which however was still the highest using BMIQ alone or in combination with QN (Fig. S11).
In principle, a feature selection based on the dual threshold of adjusted P-value < 0.05 and |ΔM| > 1 ensures a more realistic selection of probes, as it is currently done on gene expression analysis with microarrays. Table 1 and Figure 7C report the number of DM sites. Indeed, after correction for probe type with BMIQ, we obtained homogeneous fold changes, irrespective of probe design type, and more sites were reported as DM because of the improved estimation of the fold change for type II probes. This explained why we obtained more DM position after the application of the BMIQ algorithm. The threshold was not completely arbitrary, but chosen on the basis of previous observations, as described below (see Du et al.24). In contrast to M-values, β-values have non-constant variance, hence making difficult to choose a fixed threshold. However, Bibikova et al.12 recommend a Δβ of 0.2 when analyzing Infinium450k arrays to detect DM sites with 99% confidence. The relationship between M and β values is not linear, therefore a Δβ = 0.2 cannot be directly translated in ΔM. However, for intermediate methylated sites (0.2 < M < 0.8), an approximated linear relationship exists24 and Δβ = 0.2 roughly corresponds to ΔM = 1.33, which represents a threshold for maximum specificity. Hence, ΔM = 1 represented a compromise between increasing the true positive rate and not decreasing the detection rate. We provided replication of DM sites using data from an independent cohort (data set C)20 and running the lumi:QN + BMIQ pipeline. We found that that 88% of the DM CpGs in data set A were also DM in data set C (Figs. S12 and S13). Moreover, the use of the stringent threshold increased the percentage of replicated sites, thus confirming that it reduced the false positive rate.
Table 1. Number of differentially methylated CpG sites resulting from the indicated pipelines (columns) and thresholds (rows).
| Threshold | lumi:QN | lumi:QN + BMIQ | lumi:BMIQ | minfi:SWAN | SQN | GS |
|---|---|---|---|---|---|---|
| Adjusted p < 0.05 |
215,822 |
203,159 |
157,646 |
162,113 |
219,288 |
171,898 |
| Adjusted p < 0.05 AND | ΔM | > 1 |
14,746 | 24,171 | 24,465 | 17,387 | 19,247 | 17,752 |
We expected a considerable number of CpG sites to be specifically down-methylated in monocytes when compared with PB, because of the multiple blood cell types contained in this reference group. Actually, monocyte-specific CpG hypomethylation should be observed in promoters and likely on enhancers, as a prerequisite for lineage-specific gene expression.25 Therefore, in order to better visualize the pattern of variability in CpG sites, we performed PCA on M-values obtained after lumi:QN + BMIQ (Fig. 7D) observing that, without scaling the data, the first two components accounted for 98% of the variance and captured the average methylation level and the DM sites between monocytes and PB. Moreover, we gained a global vision of the relative DNA methylation of monocytes with respect to PB. Intermediate-high methylated sites increased or decreased their methylation levels, while intermediate-low methylated sites exclusively decreased their methylation in monocytes. Examples of specific methylation patterns of monocytes/lymphocytes associated genes are given in Figure S13. We then analyzed in detail the associated categories of each probe, using the provided Illumina Manifest File annotation and the DM sites (Fig. S14). Indeed, we observed a greater relative fraction of enhancer and DNase hypersensitivity sites (DHS) among DM sites, as compared with non-DM sites. Moreover, when comparing the link with annotated regulatory regions, we detected enrichment in groups classified as “cell type specific” but a deprivation of “promoter associated.” At the same time, the relative number of DM sites in CpG Islands (CGI) was also lower. As CGIs are enriched in promoters, it is possible that cell-specific CpG methylation is preferentially observed in enhancers, in agreement with the observation that enhancers show a better tissue specificity as compared with promoters.26 This observation deserves further clarification. Moreover, while CGIs have been shown to co-localize with the promoters of all constitutively expressed genes and approximately 40% of those displaying a tissue restricted expression profile, non-CGI promoters are generally associated with restricted expression.27,28 Indeed, when we selected only the DM probes in promoters (TSS200), a decreased fraction was located on CGI, as compared with non-DM, but an increased fraction was outside, including GCI shores, which have been described to have tissue-specific DNA methylation.29-31 This observation could explain why we are observing less DM sites in CGI when comparing monocytes vs. PB, i.e., a defined cell type vs. a heterogeneous cell population.
Validation of selected pipeline
Comparison with bisulfite pyrosequencing (BPS)
We compared matched BPS-450K data from data set D,15 in order to check the agreement of the methylation values with an independent technique. Hence, we applied the lumi:QN + BMIQ pipeline on data set D and we calculated the bias from BPS values for 8 type II and 6 type I probes for which BPS data were reported and QC criteria were met. We verified that the deviation from BPS was reduced for type II probes after the selected pipeline (Fig. S15), confirming that the correction obtained with BMIQ for type II probes was actually advantageous.
Efficacy on removing strong batch effect
To this point we analyzed patterns of variability reduction in a data set that is not dominated by known strong batch effects. Because this kind of artifacts has been shown to have a substantial effect on high throughput experiments and in particular with Illumina methylation arrays,21 we asked whether we could highlight and reduce this additional layer of confounding variability, in particular when biological factors are admixed with non-random assignment of samples to arrays or batches. To this end, we analyzed the variability on data set B, where two groups of samples processed on different days were present. The main effect was attributable to subgroups of samples processed on the same day. More importantly, one biological factor was almost completely confounded with the batch, because one group was totally represented by male samples and the other has a majority of females. The first three principal components on raw data were highly associated with the processing group or the slide, together accounting for more than 40% of the variability (Fig. S3). On this data set we applied the lumi:QN + BMIQ pipeline, since this previously resulted in the best performance. We observed that technical replicates on different batches did not cluster together, although the pairs of samples on the same batch (corresponding to the same subject before and after exercise training) were closely clustered together (Fig. S16). Clearly, a strong portion of the variability is dominated by non-biological source of variation, which however was reduced after batch adjustment. Indeed, we showed a reduction of the deviation from zero (Fig. 8A) and an increase in correlation between replicates (Fig. 8B) after correcting for the batch with the ComBat function. Moreover, a single group for all the replicates from the same subject was identified on a MDS plot (Fig. S16). Hence, we showed that it is possible to reduce the amount of variation due to non-biological factors, even in the presence of “contaminated” contrasts, where a sub-structure of the subjects could be potentially influential. However, whether the removal is complete or partial, it remained to be properly evaluated, because of the overlapping biological-technical factors.
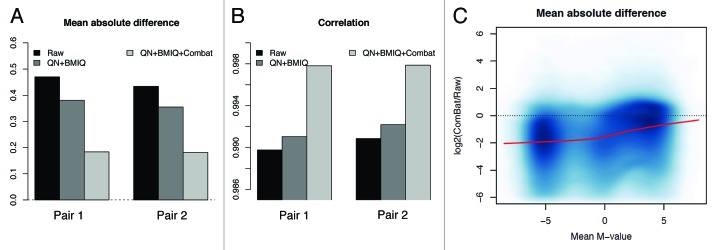
Figure 8. The elimination of unwanted batch effect further reduces the technical variability. After correcting for batch effect, we observed an increase of correlation (A) and a decrease of the absolute deviation (B) between technical replicates. (C) The logarithmic ratio between the variability after QN + BMIQ + ComBat and the variability on raw data are shown, calculated as explained in Figure 2.
Discussion
The principal conclusions may be summarized as follows. We observed that: (1) the effect of different normalization strategies is variable, the most effective being lumi:QN + BMIQ; (2) it is critical to correct for the probe design type, and BMIQ resulted the optimal method; (3) it is beneficial to correct for batch effects due to the intrinsic proprieties of highly parallel methylation profiling with Illumina arrays. In this section, first we will discuss the relevance of the data sets and the normalization methods used in this paper. Second, the correction for probe design type and batch effect will be examined and finally a few notes on differential methylation will be given.
The relevance of DNA methylation as a regulatory mechanism is well established and its possible clinical application is an important research topic. A paramount example is represented by deregulation of DNA methylation in association with cancer, where methylation in gene body or Transcription Start Sites of tumor suppressor genes and de-regulation of DNA methylation machinery may occur.32-35 Eventually, DNA methylation has been also considered as a marker for solid cancer diagnostics.36,37 Likewise, this epigenetic mark has been associated with other non-cancer diseases (reviewed in ref. 37), including autoimmune and neurological diseases. While the biological and clinical relevance of DNA methylation is undoubtedly recognized, the technical platforms used for its assessment and the analysis pipelines are rapidly evolving. In particular, Illumina Infinium 450K Human Methylation arrays, an extension of the previous 27K Human Methylation platform, are currently widely employed; although a consensus on analysis pipelines has not been reached, mainly because the introduction of two probe types on the same array has made the analysis process more complex. Therefore, in this work we described the comparison of analysis approaches for Illumina Infinium 450K arrays. The significance of this effort mainly relies on the data sets used, which we exploited as a resource to test the disentanglement of technical error, and on the detailed comparison of selected analysis methods. While the exact design, the biological conditions and the analysis of significance of the experimental data are beyond the scope of this publication and will be the topic of upcoming works, we took advantage of the level of technical replication as a reliable safeguard to test for the ability of removal of unwanted variation. To our knowledge, data set A is actually unique for the number of technical replicates of clinical DNA samples profiled on Illumina 450K arrays. As already pointed out, all DNA samples were processed together, and the position on the arrays was completely randomized to account for slide and array position effect. Hence, given the substantial differences between the contrasted groups and the absence of any strong batch effect, we assumed that this scenario was optimal to compare pipelines and normalization methods.
Importantly, here we performed QN on signal intensity and not on the methylation estimate. Although it has been noted that the assumptions of QN for methylation data might not hold,38 given the potential global differences in the distribution of the methylation estimates of contrasted groups, lumi-based QN normalization is nevertheless performed on the signal intensity, rather than β or M values. Hence, when comparing similar tissues and excluding sex chromosomes (as in our case), the fluorescence intensity distribution appeared to be comparable and QN resulted a valid option as proposed for Illumina Human Methylation27 platform.21 Additionally, the artificial relationship between the signal intensity and the position on the slide was properly eliminated after QN (Fig. S4), indicating that for this data set this normalization was beneficial. Careful examination of the signal and considerations on the study design should guide the application of this level of normalization, in order not to undermine the integrity of the biological signal.
The different performance of different probe designs in terms of methylation estimates was previously shown,12,15 and methods have been developed to deal with this bias.15-18 We focused on the parallel comparison of different approaches and pipelines, with the goal of providing an unbiased evaluation of the available tools. In our analysis the reduction of the technical variability was considered our main evaluation parameter. We considered the deviation between technical replicates and the bias between different probe types on the same sample as sources of unwanted variation and hence scored as positive any pipeline leading to a simultaneous reduction of both errors. On our data set, we validated BMIQ as the most proficient algorithm to reduce the probe design bias also in combination with QN. This result further validates the initial evaluation of BMIQ performance.18 In addition, we noticed that SQN reduced the probe type bias, but at the expense of an increased variability between technical replicates. This is most likely due to the effect of applying subset quantile normalization to the methylation estimates and not on signal intensities, and hence constraining the β values to have the same distribution. Indeed, this led to a reduction of the probe type bias as originally described,17 but without maintaining or decreasing the original distance between technical replicates.
It should be pointed out that in this manuscript we mainly define a batch a subgroup of samples that were processed together, on the same day or in a very short time. However, this definition should not be taken as absolute, because a clear definition of a batch results from careful examination of the data set, in order to identify what is an appropriate batch variable other than the processing group, as the slide or the array (i.e., position on the slide). We did not evaluate other popular approaches that do not use information on the source of unwanted variation, the exemplar being surrogate variable analysis,39 or simpler methods as regressing with the principal components, as the main focus here was highlighting the requirement for batch effect correction, rather than evaluating methods. Specifically, one biological factor in data set B (the gender) was confounded with the main batch variable, the processing subgroup, and the presence of technical replicates was helpful in elucidating the decrease in variation. Although we observed an improvement, we cannot completely exclude that part of the variability is still accounted for by batch effect. Hence, the finest safeguard against this source of unwanted variation is a careful study design, coupled with a random assignment of the samples to the arrays, the inclusion of a method to account for batch effect and the presence of technical replicates, one for each processing subgroup.
The ultimate goal of most experimental designs is to provide a list of DM sites, obtained after a proper normalization. Hence, we tested the performance in achieving a number of DM sites after the selected pipelines. It should be noted that a rigorous evaluation should include an assessment of the specificity and sensitivity. However, this situation requires the availability of a reference method or data sets, in order to count true and false positives, as well as true and false negatives. For Illumina methylation data, public data set often report the independent validation of few loci, which are insufficient for a proper assessment of the specificity and sensitivity. Here we contrasted different DNA sources from two different cell populations, potentially harboring consistent biological differences. The consequences arising from the presence of different white blood cells in the estimation of methylation profiles have been recently recognized and analyzed,20 pointing out the existence of reliable differences in the methylation levels for specific loci in different blood cells. We believed that the expected biological difference, the satisfactory presence of both technical and biological replicates, and the randomized assignment represented ideal settings to verify the performance of different pipelines in identifying DM sites. The use of latent class analysis22,23 to predict the unknown true state of CpG sites led to the proposal of sensitivity and specificity for each pipeline, although some limitation should be considered. One main assumption of latent class models is conditional independency of the methods being tested, which in our case might be violated because correlation exists between the outputs of different pipelines. However, when we further tested a model that included correlations among pipelines by adding a random effect, with the stringent threshold we obtained maximum sensitivity and specificity by using lumi:QN + BMQ or lumi:BMIQ.
In conclusion, to our knowledge this work represented the first attempt to systematically compare bioinformatics pipelines for the analysis of Illumina 450K methylation data, considering sufficient amount of technical and biological replication to score the results with a data-driven approach. We carefully examined the reduction of technical variability, validating BMIQ as the optimal normalization methods, also in combination with QN if required. We suggested guidelines to the evaluation of current and upcoming new pipelines, providing also a valuable reference data set for performance testing.
Materials and Methods
Profiling of DNA methylation
The DNA methylation profiles used in this study were generated from the Infinium HumanMethylation450K BeadChip, an array-based detection of methylation status of 485,577 sites on bisulfite-treated DNA.12,14 The same laboratory processed all the samples (BEA core facility, Karolinska Institute).
M-values to evaluate methylation levels
Throughout this study, we mostly evaluated methylation levels with M-values rather than β-values. β-values are defined as the ratio of the methylated probe intensity and the total signal intensity. They range from 0 to 1 and have the clear advantage of representing the percentage methylation for each site, although their variance has been shown to be non-constant,24 a result that we also observed in our data set. M-values represent the log2 ratio of the intensities of methylated and unmethlylated probes. When M-values were calculated instead, an approximate homoscedasticity was obtained (Fig. S2), providing an advantage for most commonly used analysis methods, which assume constant variance. Specifically, we considered that using M-value represented a better alternative to β-values for analyzing the reduction of the technical variability and the performance of the analysis pipelines. We noticed however that the variability was slightly influenced by the computation method of the M-value for each pipeline, as revealed by the modest increase in the variability in the low end of the scatter plot in one case (Fig. S2).
Metrics for evaluation of the performance of the bioinformatics pipelines
An assessment of the performance was obtained evaluating the patterns of technical variability reduction, using several metrics: (1) hierarchical clustering (HCL) with Euclidean distance and average linkage, (2) clustering using Multidimensional Scaling (MDS), (3) the Pearson correlation, (4) the average absolute difference between technical replicates, (5) the density profile of M-values, specifically examining for differences between Infinium probe types, (6) Principal Component Analysis (PCA) and (7) the absolute difference between adjacent probe pairs, one from each design and within 200bp of each other (within probe clusters as described in Teschendorff et al.).18
Data set A (technical replicates data set)
This data set consisted of 85 samples. DNA from two cohorts was isolated either from Peripheral Blood (PB) or from CD14+ monocytes sorted from PB. Specifically, 50 samples from PB and 36 samples from monocytes were randomly assigned to 8 BeadChips with technical replicates and processed in one run (a total of 96 DNA samples). Eight samples were technically replicated in pairs, while one sample was represented in a trio of replicates (GEO accession GSE43976)
Data set B (paired sample/batch effect data set)
This data set consisted of paired samples from 17 healthy volunteers participating in the EpiTrain study. DNA was extracted from muscle biopsies from vastus lateralis before and after a 3 mo period of supervised exercise training. Two batches were present: one BeadChip was processed in one day, while two BeadChips were processed together in a separate day. One subject had one technical replicate pair per condition, with one of the two members of the pair for each batch. DNA samples from the same subject were hybridized on the same BeadChip.
Data set C (validation data set)
This is a subset of the data set studied by Reinius et al. (GEO accession GSE35069).20 Only samples from whole blood (n = 6) or CD14+ monocytes (n = 6) were considered, in order to validate the DM probes in an independent cohort.
Data set D (validation data set)
Data from Dedeurwaerder et al. (GEO accession GSE29290)15 were used to validate the methylation estimates with bisulfite pyrosequencing (BPS). Six samples were considered, including HCT116 wild-type (WT) and double-knockout (DKO) cell lines. Matched BPS data were available for 15 CpG sites.
Lumi-based pipeline
We used GenomeStudio (Illumina) to generate final reports containing signal intensities and detection P-values, which then were used as input files for this pipeline. No background subtraction or control normalization was applied within GenomeStudio. Bioconductor “lumi” package24,40 was used for quality control and normalization. Within this package, methylation levels β and M are defined as follows:
| (1) |
| (2) |
where IM and IU represent the fluorescence intensity originating form methylated or unmethylated CpG locus, respectively, and α is a constant. Notably, for Infinium I design, IM and IU are derived from two different bead type, while for Infinium II design IM and IU signals originate from the same bead type, but using different colors to discriminate methylated vs. unmethylated locus. M values were always considered, unless otherwise specified.
The assays measuring 65 SNPs (from Illumina manifest) were first removed, then sample outliers were removed by inspecting (1) the overall signal intensity, (2) the distribution of M-values, (3) the number of detected sites and (4) the relationship between sample after hierarchical clustering or multidimensional scaling. Probes on chromosomes X and Y were removed to eliminate potential artifacts originating from the presence of a different proportion of males and females. Probes with a detection p value > 0.01 exceeding 5% of the samples were also filtered out. Color-bias adjustment (Col.Adj) and quantile normalization (QN) were performed on signal intensities as implemented in lumi. Briefly, the QN works on total signal intensity, assuming that the distributions of the pooled methylated and unmethylated probes are similar for different samples. We then performed probe type bias adjustment beginning with QN or raw data and using beta-mixture quantile normalization (BMIQ) on β-values.18
The minfi pipeline
We used raw *.idat files as a starting point for this pipeline, based on the Bioconductor library “minfi.”38 Within this package, β and M values are defined as follows:
| (3) |
| (4) |
where IM and IU represent the fluorescence intensity originating form methylated or unmethylated CpG locus, respectively, and γ is a constant. Hence, the following relationship holds:
| (5) |
We would like to remark that it is slightly different to the M-value defined in the lumi pipeline, where the constant is added to both numerator and denominator. Here the outliers were removed by inspecting the overall signal intensity, the distribution of M-values and the control probe profile. Probes with a minfi detection p value > 0.01 exceeding 5% of the samples were removed, as well as those on chromosomes X and Y and the assays measuring 65 SNPs (from Illumina manifest). Next, subset-quantile within array normalization (SWAN) was performed.16 The assumption is that the distribution of intensities of probes with the same number of CpGs in the probe body should be similar regardless of design type.
The subset quantile normalization (SQN) pipeline
The pipeline described by Touleimat and Tost17 was applied here on GenomeStudio (Illumina) final reports containing signal intensities and detection p values. The algorithm performed (1) probe quality filtering based on detection p value, (2) removal of probes on sex chromosomes, (3) color bias adjustment and (4) SQN on the methylation estimates (β values) using the “relation to CpG” to calculate the reference quantiles.
Correction of the batch effect
We used either MDS or PCA to detect patterns of variability originating from a subset of samples. Next, we evaluated the association of the first five principal components with batch effect (processing day, slide or array), using a Wilcoxon or Kruskal-Wallis test. Finally, we applied the ComBat function in the R library sva,39 to account for and to eliminate biases in the M-values. The ComBat function performs an empirical Bayes adjustment to reduce batch effect. Batch effect correction was only applied to data set B.
Determination of differentially methylated (DM) sites
DM sites were defined using limma package on M values.40 β values were transformed to M values using Eq. 4, when required. To account for the high correlation between technical replicates, we estimated the average correlation using linear mixed models for each gene and accounted for this prior weight in the linear model when for testing DM. Using limma, we could estimate the effect size (difference in M-values or ΔM), p values and FDR corrected p-values for the contrast of interest. Here we compared monocytes vs. PB using autosomal data. CpG sites were selected as DM if adjusted p value < 0.05 and |ΔM| > 1. To compare the distribution of p values obtained after different normalizations, we partitioned the lists in blocks of 250 probes and calculated the Spearman correlation progressively including them from the top to the bottom of the list. This procedure was repeated for each pipeline taking that pipeline as a reference (n), then ordering the probes in ascending order according to the corresponding p value and finally calculating the Spearman correlation with the p values obtained with another pipeline (k).
In order to compare DM sites obtained using pipelines, we exploited a latent class analysis approach, using the R library randomLCA.41 Two threshold were selected: (1) FDR adjusted p < 0.05 or (2) FDR adjusted p < 0.05 and |ΔM| > 1. Using the above thresholds, we created manifest binary variables indicating if a site was considered DM (1) or not-DM (0), then we counted the frequencies of the observed patterns comparing pipelines and then we fitted first a two latent class model (2LC) and then a two latent class model with random effect (2LCR) which includes correlation among pipelines.22,23 We finally estimated the sensitivity and specificity values for each pipeline using the conditional outcome probabilities for the positive and negative classes.
Supplementary Material
Acknowledgments
This work was supported by grants from the Swedish Research Council (J.T., M.J. and S.R.); the Swedish Association of Persons with Neurological Disabilities (M.J. and S.R.; The Swedish Brain Foundation (M.J. and S.R.); AFA Insurance (T.E. and M.A.); Heller Research Fellowship (A.E.T.); FP7 SYNERGY-COPD (F.M., D.G. and J.T.); BILS (D.G.), Karolinska Institutet (J.T.), and Stockholm County (J.T.). We thank Dr Nizar Touleimat and Dr Jörg Tost for kindly providing the code for running the SQN pipeline.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/24008
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/24008
References
- 1.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12:529–41. doi: 10.1038/nrg3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feinberg AP. Epigenomics reveals a functional genome anatomy and a new approach to common disease. Nat Biotechnol. 2010;28:1049–52. doi: 10.1038/nbt1010-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petronis A. Epigenetics as a unifying principle in the aetiology of complex traits and diseases. Nature. 2010;465:721–7. doi: 10.1038/nature09230. [DOI] [PubMed] [Google Scholar]
- 4.Lan X, Adams C, Landers M, Dudas M, Krissinger D, Marnellos G, et al. High resolution detection and analysis of CpG dinucleotides methylation using MBD-Seq technology. PLoS One. 2011;6:e22226. doi: 10.1371/journal.pone.0022226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serre D, Lee BH, Ting AH. MBD-isolated Genome Sequencing provides a high-throughput and comprehensive survey of DNA methylation in the human genome. Nucleic Acids Res. 2010;38:391–9. doi: 10.1093/nar/gkp992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borgel J, Guibert S, Weber M. Methylated DNA immunoprecipitation (MeDIP) from low amounts of cells. Methods Mol Biol. 2012;925:149–58. doi: 10.1007/978-1-62703-011-3_9. [DOI] [PubMed] [Google Scholar]
- 7.Mohn F, Weber M, Schübeler D, Roloff TC. Methylated DNA immunoprecipitation (MeDIP) Methods Mol Biol. 2009;507:55–64. doi: 10.1007/978-1-59745-522-0_5. [DOI] [PubMed] [Google Scholar]
- 8.Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766–70. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Irizarry RA, Ladd-Acosta C, Carvalho B, Wu H, Brandenburg SA, Jeddeloh JA, et al. Comprehensive high-throughput arrays for relative methylation (CHARM) Genome Res. 2008;18:780–90. doi: 10.1101/gr.7301508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. 2010;28:1097–105. doi: 10.1038/nbt.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288–95. doi: 10.1016/j.ygeno.2011.07.007. [DOI] [PubMed] [Google Scholar]
- 13.Bibikova M, Le J, Barnes B, Saedinia-Melnyk S, Zhou L, Shen R, et al. Genome-wide DNA methylation profiling using Infinium® assay. Epigenomics. 2009;1:177–200. doi: 10.2217/epi.09.14. [DOI] [PubMed] [Google Scholar]
- 14.Sandoval J, Heyn HA, Moran S, Serra-Musach J, Pujana MA, Bibikova M, et al. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics. 2011;6:692–702. doi: 10.4161/epi.6.6.16196. [DOI] [PubMed] [Google Scholar]
- 15.Dedeurwaerder S, Defrance M, Calonne E, Denis H, Sotiriou C, Fuks F. Evaluation of the Infinium Methylation 450K technology. Epigenomics. 2011;3:771–84. doi: 10.2217/epi.11.105. [DOI] [PubMed] [Google Scholar]
- 16.Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012;13:R44. doi: 10.1186/gb-2012-13-6-r44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Touleimat N, Tost J. Complete pipeline for Infinium(®) Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics. 2012;4:325–41. doi: 10.2217/epi.12.21. [DOI] [PubMed] [Google Scholar]
- 18.Teschendorff AE, Marabita F, Lechner M, Bartlett T, TegnEr J, Gomez-Cabrero D, et al. A Beta-Mixture Quantile Normalisation method for correcting probe design bias in Illumina Infinium 450k DNA methylation data. Bioinformatics (Oxford, England) 2012 doi: 10.1093/bioinformatics/bts680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaffe AE, Murakami P, Lee H, Leek JT, Fallin MD, Feinberg AP, et al. Bump hunting to identify differentially methylated regions in epigenetic epidemiology studies. Int J Epidemiol. 2012;41:200–9. doi: 10.1093/ije/dyr238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén S-E, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7:e41361. doi: 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun Z, Chai HS, Wu Y, White WM, Donkena KV, Klein CJ, et al. Batch effect correction for genome-wide methylation data with Illumina Infinium platform. BMC Med Genomics. 2011;4:84. doi: 10.1186/1755-8794-4-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qu Y, Tan M, Kutner MH. Random effects models in latent class analysis for evaluating accuracy of diagnostic tests. Biometrics. 1996;52:797–810. doi: 10.2307/2533043. [DOI] [PubMed] [Google Scholar]
- 23.Hui SL, Zhou XH. Evaluation of diagnostic tests without gold standards. Stat Methods Med Res. 1998;7:354–70. doi: 10.1191/096228098671192352. [DOI] [PubMed] [Google Scholar]
- 24.Du P, Zhang X, Huang C-C, Jafari N, Kibbe WA, Hou L, et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidl C, Klug M, Boeld TJ, Andreesen R, Hoffmann P, Edinger M, et al. Lineage-specific DNA methylation in T cells correlates with histone methylation and enhancer activity. Genome Res. 2009;19:1165–74. doi: 10.1101/gr.091470.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–9. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Illingworth RS, Bird AP. CpG islands--‘a rough guide’. FEBS Lett. 2009;583:1713–20. doi: 10.1016/j.febslet.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 28.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 29.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doi A, Park I-H, Wen B, Murakami P, Aryee MJ, Irizarry R, et al. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–3. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768–75. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rideout WM, 3rd, Coetzee GA, Olumi AF, Jones PA. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science. 1990;249:1288–90. doi: 10.1126/science.1697983. [DOI] [PubMed] [Google Scholar]
- 33.Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424–33. doi: 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–83. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 36.Heichman KA, Warren JD. DNA methylation biomarkers and their utility for solid cancer diagnostics. Clin Chem Lab Med. 2012;50:1707–21. doi: 10.1515/cclm-2011-0935. [DOI] [PubMed] [Google Scholar]
- 37.Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet. 2012;13:679–92. doi: 10.1038/nrg3270. [DOI] [PubMed] [Google Scholar]
- 38.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nat Rev Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 39.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007;3:1724–35. doi: 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–8. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 41.Beath K. randomLCA: Random Effects Latent Class Analysis. 2011. http://CRAN.R-project.org/package=randomLCA
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.